

Abstract
Neural stem cells (NSCs) reside in vivo in hypoxic environments, and NSC proliferation is enhanced in vitro under hypoxic conditions. Various adaptive responses to hypoxia are mediated by hypoxia-inducible factors (HIFs), a family of basic helix–loop–helix Per-Arnt-Sim (PAS) transcription factors. Necdin, a MAGE (melanoma antigen) family protein, is expressed abundantly in postmitotic neurons and possesses potent antimitotic and antiapoptotic activities. We here report that hypoxia induces degradation of the necdin protein in primary NSCs by HIF-mediated ubiquitin-proteasome system. Necdin was expressed in primary NSCs prepared from the ganglionic eminences of mouse embryos. Hypoxia enhanced neurosphere formation of NSCs, in which the necdin protein level was significantly reduced. Primary NSCs prepared from necdin-deficient mice exhibited higher rates of proliferation and apoptosis than those from wild-type mice in normoxia, whereas there were no significant differences in the proliferation and apoptosis rates between necdin-deficient and wild-type NSCs in hypoxia. HIF-2α was predominantly expressed in hypoxic NSCs, where expression of HIF-responsive genes was upregulated. HIF-2α interacted with necdin via its PAS domain, which enhanced necdin ubiquitination. Lentivirus-mediated expression of the PAS domain in primary NSCs promoted necdin degradation and enhanced NSC proliferation in normoxia, whereas a small-molecule inhibitor of HIF-2α translation stabilized the necdin protein and reduced NSC proliferation in hypoxia. These results suggest that oxygen tension regulates the necdin protein level in NSCs through HIF-2α-mediated proteasomal degradation to modulate their proliferation and apoptosis.
Introduction
The mammalian brain is the most metabolically active organ and requires large amounts of oxygen for its physiological function. Oxygen availability is crucial for the energy metabolism in neurons, where a large amount of ATP is used for their function and survival. However, the oxygen levels in the brain are considerably lower than the atmospheric levels (Ereciska and Silver, 2001). In embryonic brain in vivo, oxygen tensions are particularly low in the ventricular zone, where neural stem cells (NSCs) reside (Mutoh et al., 2012). An accumulating body of evidence suggests that hypoxia enhances the expansion of embryonic NSCs or neuronal progenitors in vitro (Morrison et al., 2000; Studer et al., 2000; Chen et al., 2007, 2010; Zhao et al., 2008; Clarke and van der Kooy, 2009; Rodrigues et al., 2010). These observations raise the possibility that hypoxia is indispensable for the high proliferation activity of NSCs.
Cellular responses to hypoxia are often mediated by hypoxia-inducible factors (HIFs), which regulate oxygen-dependent transcription of various genes involved in oxygen and energy homeostasis (Greer et al., 2012). HIF-1α and HIF-2α are basic helix–loop–helix Per-Arnt-Sim (PAS) transcription factors that share a high degree of sequence identity. HIF-1α influences the expansion and cell fate switching of NSCs during development (Zhao et al., 2008; Panchision, 2009; Mazumdar et al., 2010; Mutoh et al., 2012). However, the molecular mechanisms whereby HIF proteins are involved in the regulation of NSC proliferation remain elusive at the present time.
Necdin is a pleiotropic protein expressed abundantly in postmitotic neurons (Maruyama et al., 1991; Uetsuki et al., 1996). Necdin interacts with many regulatory proteins and serves as a hub of protein–protein interaction networks (Lavi-Itzkovitz et al., 2012). Ectopic expression of necdin strongly suppresses the proliferation of cell lines (Hayashi et al., 1995; Taniura et al., 1998, 2005; Kuwako et al., 2004). Necdin, like retinoblastoma protein, targets cellular E2F transcription factors (Taniura et al., 1998; Kobayashi et al., 2002). Furthermore, necdin suppresses oxidative damage-induced neuronal apoptosis by interacting with p53 (Taniura et al., 1999; Hasegawa and Yoshikawa, 2008). Necdin also interacts with HIF-1α and promotes its degradation (Moon et al., 2005; Friedman and Fan, 2007), suggesting that the interaction between necdin and HIF-1α affects cell proliferation and apoptosis under hypoxic conditions.
Several lines of evidence indicate that necdin is expressed in multipotent stem cells or committed progenitors of mesodermal/mesenchymal origin, such as mesoangioblast stem cells (Brunelli et al., 2004), brown adipocyte precursors (Tseng et al., 2005), skeletal muscle satellite cells (Deponti et al., 2007), hematopoietic stem cells (Kubota et al., 2009; Liu et al., 2009), and mesenchymal stem cells or preadipocytes in white adipose tissues (Fujiwara et al., 2012). However, little is known about the expression and function of necdin in mammalian NSCs.
Using primary NSCs prepared from mouse embryos, we demonstrate that hypoxia promotes degradation of the necdin protein in primary NSCs via HIF-2α-mediated ubiquitination to enhance their proliferation. The present findings provide insights into the molecular mechanism that controls the proliferation rates of NSCs under different oxygen tensions.
Materials and Methods
Animals.
Necdin gene (Ndn) mutant mice (Ndntm1Ky) were generated and maintained as described previously (Kuwako et al., 2005). Heterozygous male mice (Ndn+/−) [>20 generations in ICR (Institute for Cancer Research) background] were crossed with wild-type female mice (Ndn+/+) to obtain wild-type (Ndn+m/+p) and paternal Ndn-deficient (Ndn+m/−p) littermates. Genotypes of all mice were analyzed by PCR for mutated Ndn locus. Experiments using gene-targeted mice were approved by the Recombinant DNA and Animal Experiment Committees of the Institute for Protein Research, Osaka University, and performed in accordance with institutional guidelines and regulations.
NSCs.
Primary NSCs were prepared from striatal primordia [also known as ganglionic eminences (GEs)] of mice at embryonic day (E) 14.5 (Reynolds et al., 1992). GEs from wild-type and necdin-deficient (Ndn+m/−p) mice were dissected, incubated for 5 min at 37°C in Ca2+/Mg2+-free glucose-supplemented HBSS with 0.05% trypsin, dissociated in DMEM supplemented with 10% fetal bovine serum, and centrifuged at 200 × g for 3 min. The cell pellet was resuspended and incubated for 6 d at 37°C under humidified 5% CO2 conditions in DMEM/F12 medium (Invitrogen) supplemented with 14 mm sodium bicarbonate, 1 mm [scap] N-acetyl-l-cysteine, 33 mm d(+)-glucose, 1 mg/ml bovine serum albumin (Sigma-Aldrich), 2 mm l-glutamine, 20 ng/ml EGF (PeproTech), 20 ng/ml FGF (PeproTech), and B27 (Invitrogen). Floating spheres were collected and used as primary NSCs for the following experiments. For cultures in hypoxic conditions, primary NSCs were incubated at 37°C under 2% O2/5% CO2 conditions in a multigas incubator (MCO-5M, Panasonic). For differentiation, primary neurospheres were dissociated with StemPro Accutase (Invitrogen) and plated onto poly-l-ornithine-coated coverslips in growth factor-deprived DMEM/F12 medium for 5 d.
Immunostaining.
Frozen 12 μm-thick tissue sections were prepared from ICR mouse embryos at E14.5 and immunostained as described previously (Kuwajima et al., 2004). For immunocytochemistry, primary NSCs, dispersed NSCs, and differentiated cells were fixed with 10% formalin solution at room temperature for 20 min and then permeabilized with methanol at room temperature for 20 min. Samples were incubated at 4°C overnight with primary antibodies and fluorescence dye-conjugated secondary antibodies at room temperature for 90 min. Primary antibodies used are as follows: rabbit polyclonal antibodies against necdin (NC243; 1:1000), nestin (ST-1; 1:000) (Aizawa et al., 2011), guinea pig polyclonal antibody against necdin (GN1; 1:1000) (Kuwako et al., 2005), and Dlx2 (GDlx2, 1:3000; Kuwajima et al., 2006), mouse monoclonal antibodies against Sox2 (1:500; R&D Systems), βIII-tubulin (1:1000; Promega), and GFAP (1:1000; gift from Dr. Seiichi Haga, Tokyo Metropolitan Institute of Medical Science). Secondary antibodies are as follows: anti-rabbit, anti-guinea pig, and anti-mouse IgGs conjugated with cyanine 3 and cyanine 2 (1:500; Jackson ImmunoResearch). Chromosomal DNA was detected with 3.3 μm Hoechst 33342 (Sigma-Aldrich). The images were observed with a fluorescence microscope (MZ16 F, Leica Microsystems) or a fluorescence microscope (BX-50-34-FLAD1, Olympus Optical), taken by charge-coupled device camera system (DP70, Olympus Optical), and processed using Adobe PhotoShop CS5.
Western blot analysis.
Cells were homogenized with a lysis buffer containing 10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 10% glycerol, and a protease inhibitor mixture (Complete, Roche). The protein concentration was determined by the Bradford method (Bio-Rad). Proteins (10 μg/lane) were separated by 10% SDS-PAGE, and electroblotted to polyvinylidene difluoride membranes (Immobilon, Millipore). Membranes were blocked with skim milk in PBS/0.05% Tween 20, and incubated with primary antibodies against necdin (NC243; 1:3000), actin (JLA20; 1:200; Developmental Studies Hybridoma Bank), β-tubulin (1:1000; MP Biomedicals), Sox2 (1:500; R&D System), CD133 (1:500; eBioscience), HIF-1α (1:500; R&D Systems), HIF-2α (sc-28706; 1:400; Santa Cruz Biotechnology). Flag (M2; 1:500; Sigma-Aldrich), Myc (9E10; 1:10), proliferating cell nuclear antigen (PCNA; PC10; 1:500; Santa Cruz Biotechnology), ubiquitin (FK2; 1:1000; Enzo Life Sciences), and Cdc2 (sc-54; 1:300; Santa Cruz Biotechnology). The membrane was incubated with horseradish peroxidase-conjugated IgG (Cappel). Proteins were visualized by Western Lightning Plus-ECL (PerkinElmer). Signal intensities were quantified with an image analyzer (LAS-1000 Plus, Fuji Film) and ImageJ 1.44 software. Lysates of hypoxic HEK293A cells treated with 100 μm CoCl2 and 10 μm MG132 (Peptide Institute) for 12 h were used as controls of HIF proteins.
Neurosphere assay.
The efficiency of neurosphere formation was determined on the basis of the study described previously (Reynolds and Weiss, 1996). Briefly, primary NSCs were treated with StemPro Accutase, dispersed into single cells, and resuspended in DMEM/F12 medium with the supplements described above. Dispersed cells (150 cells per well) were plated in 96-well plates (Thermo Fisher Scientific) and cultured for 5 d in normoxia or hypoxia. Neurospheres were observed with an inverted microscope (Olympus IX70), and images were captured. The diameters of individual neurospheres were measured using ImageJ 1.44 software. Neurospheres of >20 μm in diameter were counted and divided into three categories (20–40, 40–60, and >60 μm in diameter). To inhibit endogenous HIF-2α expression, NSCs cultured for 6 d were treated with HIF-2α Translation Inhibitor (Calbiochem; Zimmer et al., 2008) under hypoxic conditions for 3 d (for Western blot analysis) or for 5 d (for neurosphere assay).
Purification of NSCs.
NSCs were purified using anti-CD133 antibody-coupled microbeads because CD133 (also known as prominin) has often been used to isolate neural stem cells (Uchida et al., 2000; Pfenninger et al., 2007). After dissection, cells were centrifuged at 300 × g for 10 min and washed with PBS containing 0.5% BSA (AlbuMAX, Invitrogen) and 2 mm EDTA. Pellets were suspended with 80 μl of wash buffer and mixed with anti-CD133-coupled microbeads (2 μl for 1 × 106 cells; Miltenyi Biotec). The mixture was incubated for 15 min at 4°C and separated on a magnetic column (Miltenyi Biotec). Cells bound to the microbeads (CD133+) and unbound cells (CD133−) were resuspended in DMEM/F12 containing 2 mm l-glutamine, B27, 20 ng/ml EGF, and 20 ng/ml FGF, incubated for 5 d, and subjected to the neurosphere assay.
Cell proliferation assay.
For BrdU incorporation assay, primary NSCs (6 d culture) were incubated in hypoxic conditions for 24 h, labeled with 10 μm BrdU for 4 h, dissociated into single cells, and plated onto the poly-l-ornithine-coated coverslips in 24-well plates (5 × 104 cells per well). BrdU incorporation assay was performed as described previously (Taniura et al., 1999; Fujiwara et al., 2012). Cells were incubated with mouse monoclonal anti-BrdU antibody (1:30, Roche) and anti-nestin (ST-1; 1:1000) antibody for 1 h at room temperature, and incubated with second antibody against IgGs conjugated with cyanine 2 and cyanine 3, respectively, and counterstained with Hoechst 33324. BrdU-labeled cells among nestin-immunopositive cells were counted by fluorescence microscopy. For cell proliferation assay in vivo, pregnant mice were injected intraperitoneally with ethynyl deoxyuridine (EdU; Invitrogen; 50 mg/kg body weight) at gestational day 14.5, and embryonic forebrain tissues were fixed 2 h later. Incorporated EdU was detected using Click-iT EdU Alexa Fluor Imaging Kit (Invitrogen). The fluorescence intensity was quantified by fluorescence microphotometry as described previously (Hasegawa et al., 2012). Briefly, fluorescence images (12-bit digital monochrome images) were captured with a CCD camera (CoolSNAP monochrome, Nippon Roper). EdU fluorescence intensity in the GE ventricular/subventricular zone was analyzed using fluorescence image analysis software (Fluoroimage Cool V, Mitani). The background intensity of the equal-sized adjacent area without EdU signals was subtracted from the intensity of EdU signal-containing area.
Apoptosis assay.
Nuclear DNA fragmentation was analyzed by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) visualized with Texas Red (Gavrieli et al., 1992; Uetsuki et al., 1999). Primary NSCs (6 d culture) were dispersed and incubated in normoxic (20% O2) or hypoxic conditions (2% O2) for 24 h. NSCs were dispersed, plated onto poly-l-ornithine-coated coverslips in 24-well plates (5 × 104 cells per well), fixed 6 h after plating, and subjected to TUNEL analysis. Chromosomal DNA was counterstained with 3.3 μm Hoechst 33342. TUNEL analysis in brain tissues was performed as described previously (Kuwako et al., 2005). TUNEL-positive cells in the proliferative zone of dorsal GE were counted and quantified as the number in the equal-sized area.
Quantitative RT-PCR.
Total RNA was extracted from NSCs with phenol and guanidine thiocyanate mixture (TRI Reagent, Molecular Research Center), and genomic DNA was digested with RQ1 RNase-free DNase (Promega). cDNA was synthesized from total RNA (2 μg) using Transcriptor First Strand cDNA Synthesis Kit (Roche). cDNA (10 ng) was used as a template for PCR. Primers used for real-time PCR are as follows: HIF-1α (forward, 5′-ggacaagtcaccacagga-3′; reverse, 5′-ggagaaaatcaagtcgtg-3′); HIF-2α (forward, 5′-ccaaggggcaggtggtat-3′; reverse, 5′-cacgtcgttcttctcgatctca-3′); Vegf (forward, 5′-ccccgacgagatagagta-3′; reverse, 5′-gctttggtgaggtttgat-3′); Glut1 (forward, 5′-tcaacacggccttcactg-3′; reverse, 5′-cacgatgctcagataggacatc-3′); Cyclin D1 (forward, 5′-ccaacaacttcctctcctgc-3′; reverse, 5′- agaccagcctcttcctccac-3′); erythropoietin (forward, 5′-catctgcgacagtcgagttctg-3′; reverse, 5′-cacaacccatcgtgacattttc-3′); Cdc2 (forward, 5′-caaaatagagaaaattggagaaggtac-3′; reverse, 5′-agattttgaggttttaagtctctgtga-3′). RT-PCR products were quantified using a real-time PCR instrument (LightCycler, Roche) and FastStart DNA MasterPLUS SYBR Green I kit (Roche). Melting curves were analyzed to confirm a single species of each PCR product. α-Actin cDNA was used as an internal standard to quantify the relative expression of each cDNA. Values of HIF-1α and HIF-2α were corrected with PCR efficiencies using equal amounts of full-length HIF-1α and HIF-2α cDNAs.
Coimmunoprecipitation.
HEK293A cells were transfected with expression vectors and harvested 24 h later. Cell lysates were subjected to coimmunoprecipitation assay as described previously (Hasegawa et al., 2012). cDNA encoding mouse full-length HIF-2α was cloned from NSCs cultured under hypoxic conditions. For Myc-tagged protein expression, full-length HIF-2α, the PAS domain (HIF-2α 97-341), TAD (transactivation domain; HIF-2α 490-874), and ODD (oxygen-dependent degradation) domain (HIF-2α 516-686) were subcloned into 6xMyc-pcDNA3.1. cDNA encoding the N-terminal domain (necdin 1–100) and the MAGE (melanoma antigen) homology domain (necdin 101–300) of necdin were subcloned into p3xFLAG-CMV14 (Sigma-Aldrich). Expression vectors for necdin and Myc-tagged p53 were described previously (Taniura et al., 1999). The cells were harvested 24 h after transfection and lysed in the lysis buffer. Proteins in the lysates (150 μg) were incubated at 4°C for 2 h with antibodies against Flag (M2; 1:50; Sigma-Aldrich), Myc (9E10; 1:4) and necdin (NC243; 1:100), pelleted with protein A-Sepharose (GE Healthcare), and detected by Western blotting. For detection of endogenous complex of necdin and HIF-2α, nuclear lysates (1 mg) of E14.5 mouse forebrain were incubated with guinea pig anti-necdin antibody (GN1; 1:100; Kuwako et al., 2005), pelleted with Dynabeads Protein A (Invitrogen), separated by 7.5% SDS-PAGE, and detected by Western blotting with antibodies against HIF-2α, PCNA (PC10; Santa Cruz Biotechnology; 1:500) and necdin (NC243; 1:3000).
In vitro binding assay.
pMALC2 plasmids carrying cDNAs for HIF-2α and its deletion mutants were constructed for the MBP fusion proteins. In vitro binding assay was performed as described previously (Hasegawa and Yoshikawa, 2008). Briefly, MBP-HIF-2α fusion proteins were affinity purified with amylose resin (New England Biolabs). Purified MBP-HIF-2α fusion protein (2 μg) bound to amylose resin was incubated with purified His-tagged necdin (200 ng) at 4°C for 30 min in 0.5 ml of binding buffer (20 mm Tris-HCl, pH 7.5, 200 mm NaCl, and 1 mm EDTA). The resin was washed with the binding buffer, and bound proteins were eluted with 20 mm maltose. His-tagged necdin was detected by Western blotting.
Ubiquitination assay.
HEK293A cells were transfected with expression vectors for Myc-tagged proteins, necdin, and Flag-ubiquitin by the calcium phosphate method and harvested 24 h later following treatment with 10 μm MG132 for 3 h before harvest. Flag-ubiquitin-conjugated necdin was immunoprecipitated with anti-necdin antibody (NC243; 1:100) and detected by immunoblotting with anti-Flag monoclonal antibody (M2; 1:500; Sigma-Aldrich). For detecting endogenous necdin ubiquitination, NSCs were infected with lentivirus vectors, incubated for 6 d, and harvested. Ubiquitin-conjugated necdin was immunoprecipitated with anti-necdin antibody (NC243; 1:100), separated by 10% SDS-PAGE, and detected by Western blotting with anti-ubiquitin antibody (FK2).
Recombinant lentiviruses.
Recombinant lentiviruses were produced by HEK293FT cells transfected with viral DNA and two helper plasmids as described previously (Miyoshi et al., 1998; Fujiwara et al., 2012). cDNAs for Flag-tagged HIF-2α PAS and ODD domains were subcloned into pENTR1A entry vector (Invitrogen) to construct the destination vector CSII-CMV-RfA-PAS-IRES2-DsRed-Express2, in which DsRed-Express2 (Clontech) was inserted as an expression indicator. For viral infection, lentiviral particles (multiplicity of infection, 30) were added to the dissociated GE cell suspension or isolated CD133+ NSCs, which were subsequently incubated for 6 d.
Statistical analysis.
Statistical significance was tested using an unpaired Student's t test or one-way ANOVA followed by Tukey's post hoc test. A significance of p < 0.05 was required for rejection of the null hypothesis.
Results
Necdin is expressed in primary NSCs
We first examined the expression patterns of necdin and Dlx2, a homeodomain protein required for GABAergic interneuron differentiation, in the GEs of E14.5 mouse by immunohistochemistry (Fig. 1A). Necdin was strongly expressed in the septum and cortical marginal zone, whereas the necdin immunoreactivity was very weak in the GE and neocortical ventricular zone, where most of NSCs are located. Double-immunostaining analysis revealed that the GEs contained strongly Dlx2-immunoreactive cells. These expression patterns of necdin and Dlx2 in mouse forebrain are consistent with the previous observations (Kuwajima et al., 2006).
Figure 1.
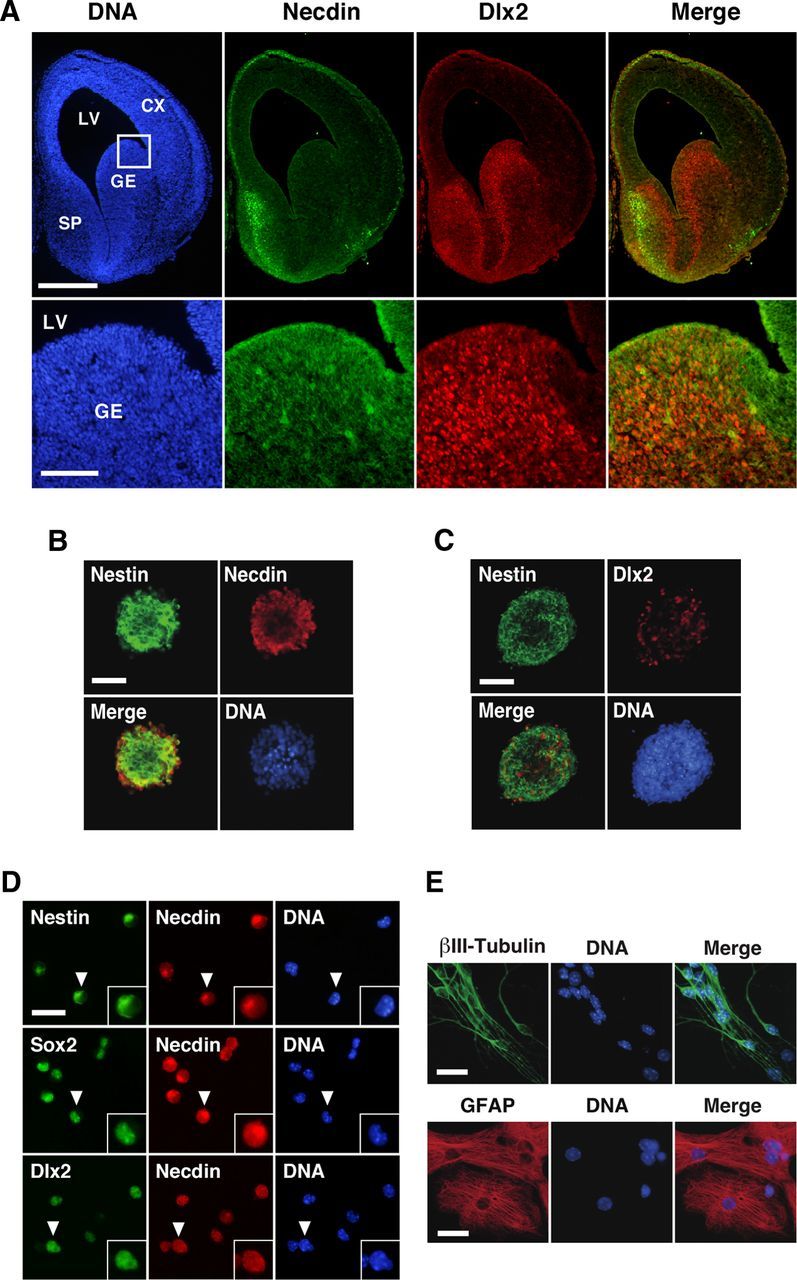
Expression patterns of necdin in embryonic brain and primary NSCs. A, Double immunostaining for necdin and Dlx2. Frozen forebrain sections of E14.5 mouse embryo were double-stained for necdin and Dlx2. Necdin (green) and Dlx2 (red) images were merged (yellow). CX, cortex; LV, lateral ventricle; SP, septum. B, C, Immunostaining for nestin, necdin, and Dlx2 in primary NSCs. NSCs (6 d culture) in primary neurospheres were immunostained for nestin (green) and necdin (red; B) or Dlx2 (red; C), and two images were merged (yellow). D, Expression of necdin in NSC markers. Primary neurospheres were dispersed, fixed 2 h later, and double-stained for necdin and nestin, Sox2, or Dlx2. Insets, magnified images of representative cells (arrowheads). Quantitative data are in Results. E, Immunostaining for βIII-tubulin and GFAP in differentiated cells. Primary cultured NSCs were induced to differentiate in the growth factor-deprived medium for 5 d. The cells were immunostained for βIII-tubulin (green) and GFAP (red). Chromosomal DNA was stained with Hoechst 33342 (DNA; A–E). Scale bars: A, 500 μm (top), 100 μm (bottom); B, C, 50 μm; D, E, 25 μm.
We then examined the expression pattern of necdin in NSCs prepared from the GEs in E14.5 mouse forebrain. Primary NSCs, which were prepared by culturing the GE cells in the presence of growth factors for 6 d, were immunopositive for both necdin and nestin, a neural stem cell marker, and most of the necdin-immunopositive cells overlapped with nestin-immunopositive cells (Fig. 1B). NSCs expressing both Dlx2 and nestin were also observed (Fig. 1C). To characterize the necdin-expressing NSCs, primary neurospheres were dispersed and double-stained for necdin and nestin, Sox2 (another NSC marker), or Dlx2 (Fig. 1D). Necdin was expressed in almost all primary NSCs, whereas fewer cells were immunopositive for nestin, Sox2, and Dlx2 than were immunopositive for necdin (necdin, 97 ± 1%; nestin, 86 ± 2%; Sox2, 91 ± 2%; Dlx2, 67 ± 1%; 150 cells analyzed; mean ± SEM; n = 3). In most of the NSCs, the necdin immunoreactivity in the cytoplasm was stronger than that in the nucleus. The population of necdin+/Sox2+ cells was the largest among those of double-positive populations (necdin+/Sox2+, 94 ± 3%; necdin+/nestin+, 88 ± 2%; necdin+/Dlx2+, 68 ± 1%; 150 cells analyzed; n = 3). Upon withdrawal of the growth factors from the culture medium, most of the GE-derived NSCs differentiated into neurons and astrocytes as judged by the expression of βIII-tubulin and GFAP, respectively (Fig. 1E), indicating that these NSCs are multipotent. These results suggest that necdin is expressed in embryonic NSCs capable of differentiating into neuronal and glial lineages.
Necdin deficiency and hypoxia enhance neurosphere formation
We then examined whether the necdin protein level was changed in NSCs cultured in hypoxic conditions. Western blot analysis revealed that a ∼42 kDa necdin protein was detected in the NSCs (Fig. 2A). Primary NSCs, which were cultured for 6 d at atmospheric 20% O2, were incubated for another 3 d at 2% (hypoxia) and 20% O2 (normoxia). The necdin protein level in hypoxic NSCs was significantly reduced to 42% of those in normoxic NSCs (Fig. 2B,C). On the other hand, the necdin protein was undetected in NSCs prepared from paternal Ndn-mutant mice. We then compared the expansion rates of wild-type NSCs cultured under different O2 conditions by neurosphere assay. The total neurosphere number of necdin-deficient NSCs was significantly larger than that of wild-type NSCs in normoxia, whereas no significant difference in the total cell number was noted between wild-type and necdin-null NSCs in hypoxia (Fig. 2D,E), suggesting that endogenous necdin suppresses NSC proliferation only in normoxia. Analysis of neurosphere size distribution revealed that necdin-deficient NSCs had four times the frequency of large neurospheres (>60 μm in diameter) compared with wild-type NSCs. In contrast, no significant difference in each size group was noted between necdin-null and wild-type NSCs in hypoxia (Fig. 2F).
Figure 2.
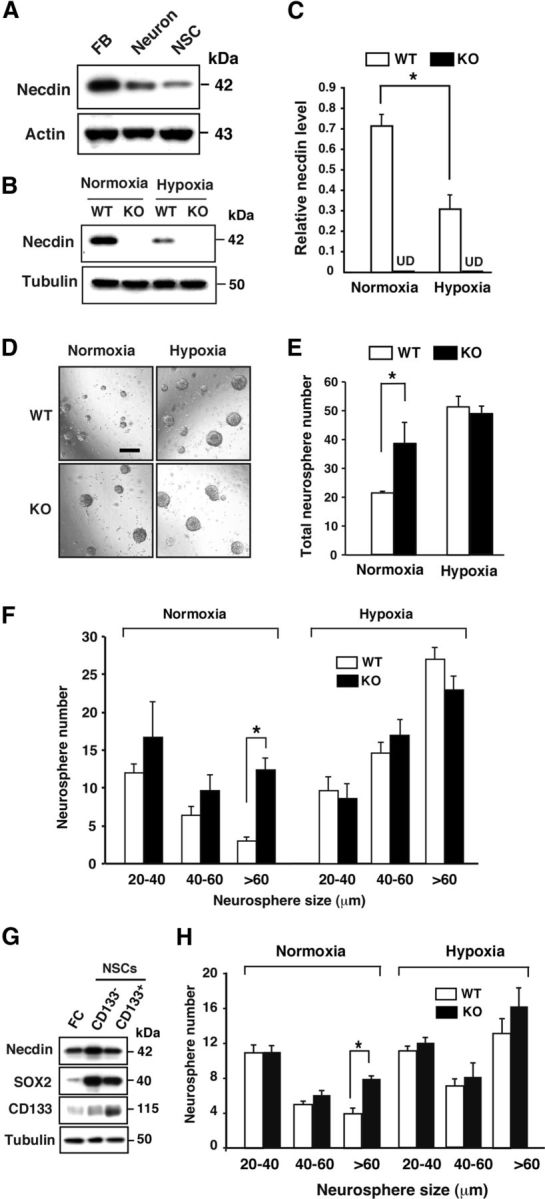
Necdin deficiency and hypoxia enhance neurosphere formation. A, Necdin expression in primary NSCs. Expression of necdin and actin in NSCs was analyzed by Western blotting using E14.5 mouse forebrain (FB) and primary neurons (Neuron) as positive controls. B, C, Necdin protein levels in normoxic and hypoxic NSCs. Primary NSCs (6 d culture) prepared from wild-type (WT) and Ndn-mutant mice (KO) were incubated for 3 d in 20% O2 (Normoxia) and 2% O2 (Hypoxia). The cell lysates were analyzed by Western blotting using antibodies to necdin and β-tubulin (B). The necdin level was quantified by densitometry and normalized to the β-tubulin level (C). UD, undetectable. D–F, Neurosphere formation assay. NSCs (6 d culture) were subjected to neurosphere formation assay in normoxia or hypoxia for 5 d. Representative images of NSCs from WT and KO mice are shown (D). Scale bar, 200 μm. The total number of neurospheres (>20 μm in diameter; E) and the numbers of three groups (20–40, 40–60, >60 μm in diameter; F) were presented. G, Expression of necdin and Sox2 in CD133+ NSCs. Dispersed forebrain cells (FC) were purified using anti-CD133 antibody-coupled microbeads, and unbound (CD133−) and bound (CD133+) cells were cultured for 6 d to obtain NSCs. Protein levels were analyzed by Western blotting. H, Neurosphere assay. CD133+ NSCs were subjected to neurosphere assay in normoxia or hypoxia for 5 d. The numbers of three groups (20–40, 40–60, >60 μm in diameter) were presented. C, E, F, H, mean ± SEM (n = 3). *p < 0.05.
To ascertain whether endogenous necdin modulates proliferation of NSCs, we purified NSCs using anti-CD133 antibody-coupled microbeads, cultured for 6 d, and subjected to the neurosphere assay. CD133+ NSCs showed a much higher neurosphere formation efficiency than CD133− NSCs (CD133− NSCs, 3 ± 0; CD133+ NSCs, 17 ± 2; p < 0.01), which is in agreement with the previous findings that CNS-derived CD133+ cells show characteristics of neural stem cells (Uchida et al., 2000; Pfenninger et al., 2007). Necdin and Sox2 were strongly expressed in both CD133+ NSCs and CD133− NSCs (Fig. 2G). Analysis of neurosphere size distribution revealed a significant increase in neurosphere formation was seen only in large neurospheres (>60 μm in diameter) of necdin-deficient CD133+ NSCs under normoxic conditions (Fig. 2H). These results suggest that hypoxia-induced reduction of endogenous necdin protein level facilitates the proliferation of NSCs.
Necdin-deficient NSCs show high proliferation and apoptosis rates
Necdin-deficient NSCs had more BrdU-positive cells when cultured under normoxic conditions, whereas there was no significant difference between wild-type and necdin-deficient NSCs cultured under hypoxic conditions (Fig. 3A). This indicates that both hypoxia and necdin deficiency enhance the proliferation rates of NSCs. On the other hand, the number of TUNEL-positive apoptotic cells was also increased in necdin-deficient NSCs in normoxia (Fig. 3B). Hypoxia markedly decreased the numbers of apoptotic cells in both wild-type and necdin-deficient NSCs. There was no significant difference in apoptotic cell population between wild-type and necdin-deficient NSCs in hypoxia. These results suggest that hypoxia-induced reduction of endogenous necdin protein level facilitates the proliferation of NSCs.
Figure 3.
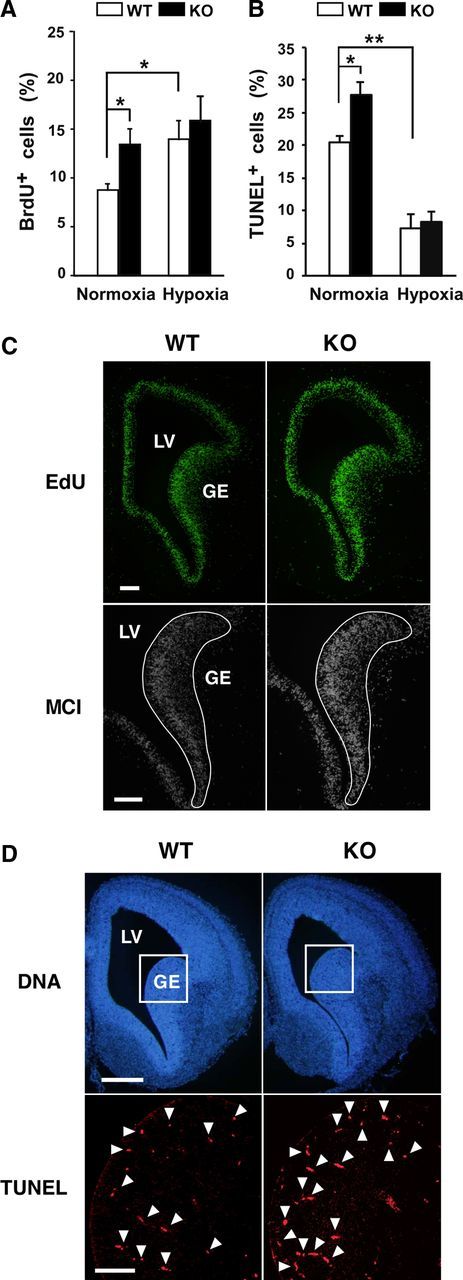
Necdin-deficient NSCs show high rates of proliferation and apoptosis. A, BrdU incorporation assay. Primary NSCs prepared from WT and KO mice were incubated in normoxia or hypoxia for 24 h, labeled with BrdU for 4 h, and stained for BrdU and chromosomal DNA. BrdU-positive cells (>150 cells analyzed; n = 3) were counted. B, Apoptosis assay. Primary NSCs were incubated in normoxia or hypoxia for 5 d, and subjected to TUNEL assay. TUNEL-positive cells (>300 cells analyzed; n = 3) were counted. A, B, mean ± SEM (n = 3). *p < 0.05; **p < 0.01. C, EdU incorporation assay. Pregnant mice were injected intraperitoneally with EdU at gestational day 14.5, and embryonic forebrain tissues were prepared 2 h later. Incorporated EdU in the forebrain section was observed by fluorescence microscopy. Fluorescence intensity of 12-bit digital monochrome images (MCI) inside the solid line (1.1 × 105 μm2) was quantified by fluorescence microphotometry. D, Apoptosis assay. Apoptotic cells in forebrain sections of E14.5 mouse were detected by TUNEL. Arrowheads point to representative TUNEL+ cells. TUNEL+ cells inside the square area (4.8 × 104 μm2) shown in chromosomal DNA images (DNA) were counted. Quantitative data (C, D) are in Results. LV, lateral ventricle. Scale bars: C, 100 μm; D, 200 μm (top), 50 μm (bottom).
To examine whether proliferation and apoptosis rates are also increased in the GE in vivo, we performed the proliferation assay using EdU, a thymidine analog incorporated into nuclear DNA of proliferative cells. The EdU incorporation was increased in the proliferative zones of the GE and cerebral cortex at E14.5 (Fig. 3C). Quantification using fluorescence microphotometry revealed a significant increase of EdU incorporation in the GE proliferative zone of necdin-deficient mice (relative fluorescence density: wild-type, 1.0 ± 0.0; necdin-deficient, 1.3 ± 0.1; p < 0.025, n = 3). The number of TUNEL-positive cells in the GE was also significantly increased in necdin-deficient mice (wild type, 27 ± 1, necdin-deficient, 41 ± 2; p < 0.005; n = 3; Fig. 3D). These results suggest that necdin has both antimitotic and antiapoptotic effects on GE cells in vivo.
Primary NSCs predominantly express HIF-2α
To examine whether HIFs were expressed in these primary NSCs, expression levels of HIF-1α and HIF-2α in normoxic and hypoxic NSCs were analyzed by quantitative RT-PCR (Fig. 4A). In normoxic NSCs, the level of HIF-2α mRNA was 9.1 times that of HIF-1α mRNA after correction of PCR efficiencies. The HIF-1α mRNA level in hypoxic NSCs was significantly increased to 2.5 times that in normoxic NSCs. Western blot analysis of HIF-1α and HIF-2α revealed that only the HIF-2α protein was detected in primary NSCs (6 d culture) incubated for 3 d in hypoxia, in which the necdin level was decreased in wild-type NSCs (Fig. 4B).
Figure 4.
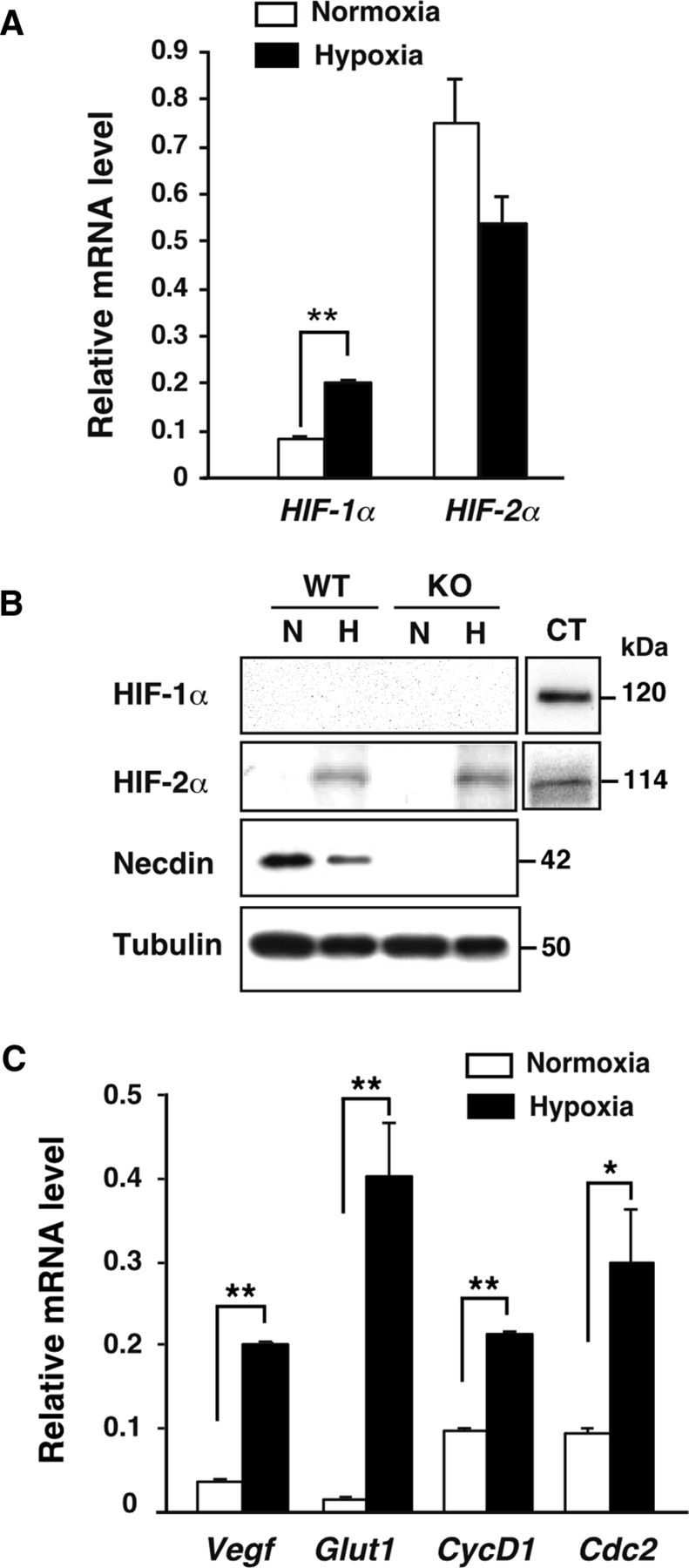
Expression of HIF-1α and HIF-2α in NSCs. A, HIF-1α and HIF-2α mRNA levels in NSCs. Primary NSCs (6 d culture) were incubated in 20% O2 (Normoxia) or 2% O2 (Hypoxia) for 3 d. HIF-1α and HIF-2α mRNA levels were analyzed by quantitative RT-PCR. Values were normalized with β-actin mRNA level and corrected with PCR efficiencies. B, Expression of HIF-1α, HIF-2α, and necdin proteins. Primary NSCs (6 d culture) prepared from wild-type (WT) and necdin-null mice (KO) were incubated in normoxia (N) or hypoxia (H) for 3 d. Protein levels were analyzed by Western blotting. CT (control), hypoxic HEK293A cell lysate. C, Expression of Vegf, Glut1, Cyclin D1 (CycD1), and Cdc2 mRNAs. Primary NSCs (6 d culture) were incubated in normoxia or hypoxia for 3 d. Expression of each mRNA was analyzed by quantitative RT-PCR. A, C, mean ± SEM (n = 3). *p < 0.05; **p < 0.01.
We then analyzed the expression levels of HIF-regulated genes in primary NSCs (6 d culture) that were incubated for another 3 d in normoxia or hypoxia. Expression of vascular endothelial growth factor (Vegf), glucose transporter 1 (Glut1), and cyclin D1 was significantly increased in hypoxic NSCs (5.3, 24.3, and 2.2 times those in normoxic NSCs, respectively) (Fig. 4C). Expression of erythropoietin, another HIF-regulated gene, was very weak compared with the expression levels of above genes, but was significantly increased in hypoxia (values relative to actin mRNA level: 0.0057 ± 0.0006 in normoxia; 0.0101 ± 0.0005 in hypoxia, p < 0.05). Expression of Cdc2, a gene downregulated by necdin (Kurita et al., 2006), in hypoxic NSCs was 3.2 times the normoxic level. These data suggest that the HIF-2α protein is stabilized and activates its downstream genes in hypoxic NSCs.
Necdin interacts with HIF-2α via the PAS domain
To examine the association between necdin and HIF-2α, we performed the coimmunoprecipitation assay using transfected HEK293A cells. We first examined the interactions between HIF-2α and necdin using p53 as a positive control. Necdin was coprecipitated with HIF-2α, which was conversely coprecipitated with necdin (Fig. 5A). HIF-2α bound to necdin via the MAGE homology domain (amino acids 101–300), whereas the N-terminal domain (amino acids 1–100), whose sequence is unique to necdin, failed to bind to HIF-2α (Fig. 5B).
Figure 5.

Necdin interacts with HIF-2α via the PAS domain. A, Interaction between necdin and HIF-2α. HEK293A cells were transfected with expression vectors for necdin, Myc-tagged HIF-2α (Myc-HIF-2α), and Myc-tagged p53 (Myc-p53, positive control), and lysates were immunoprecipitated (IP) and immunoblotted (IB) with anti-Myc (Myc) and anti-necdin (Necdin) antibodies. B, HIF-2α-binding domains of necdin. Expression vectors for Flag-tagged necdin full-length 1–325 (Flag-FL), N-terminal domain 1–100 (Flag-NT), and MAGE homology domain 101–300 (Flag-MHD) were cotransfected with HIF-2α cDNA into HEK293A cells, and cell lysates were immunoprecipitated and immunoblotted with anti-Myc (Myc) and anti-Flag (Flag) antibodies. C, Necdin-binding domains of HIF-2α. Combinations of expression vectors for necdin, Myc-tagged HIF-2α PAS domain (Myc-PAS), transactivation domain (Myc-TAD), oxygen-dependent degradation domain (Myc-ODD), and full-length (Myc-FL) were transfected into HEK293A cells, and cell lysates were immunoprecipitated and immunoblotted with anti-necdin and anti-Myc antibodies. D, In vitro binding assay. Bacterially synthesized MBP-HIF-2α domains immobilized on amylose resin were incubated with His-tagged necdin (His-necdin). Bound His-necdin was detected with anti-necdin antibody (top). Recombinant proteins were stained with Coomassie Brilliant Blue (bottom). Input, 10% His-necdin. E, Coimmunoprecipitation assay for endogenous complex containing necdin and HIF-2α. Forebrain nuclear lysates (Lysate; 1 mg) from E14.5 wild-type (WT) and necdin-deficient (KO) mice were immunoprecipitated with GN1 antibody (αNecdin IgG) and detected with antibodies against HIF-2α, PCNA, and necdin.
To locate the necdin-interacting domain of HIF-2α, deletion mutants, including the Myc-tagged PAS domain, ODD, TAD, and full-length HIF-2α, were coexpressed with necdin in HEK293A cells for coimmunoprecipitation assay (Fig. 5C). The HIF-2α PAS domain and full-length HIF-2α were coimmunoprecipitated with necdin, whereas neither ODD nor TAD bound to necdin. We then examined the direct interaction between necdin and HIF-2α PAS using MBP-fused HIF-2α domains (expressed in bacteria) and His-tagged necdin (expressed in insect cells; Fig. 5D). Necdin bound substantially to HIF-2α PAS domain and weakly to the HIF-2α ODD domain. We failed to prepare an intact TAD fragment in the bacterial expression system. These data indicate that necdin interacts directly with HIF-2α PAS domain.
To examine whether an endogenous complex containing necdin and HIF-2α is present under physiological conditions, coimmunoprecipitation assay was performed using nuclear extracts of E14.5 mouse forebrain. HIF-2α was coimmunoprecipitated with necdin, whereas PCNA, a negative control, failed to be coprecipitated with necdin. These data suggest that endogenous necdin forms a stable complex with HIF-2α in the mouse forebrain (Fig. 5E).
HIF-2α promotes necdin degradation via the proteasome pathway
To examine whether HIF-2α promotes necdin degradation, we transfected necdin and HIF-2α cDNAs into HEK293A cells and analyzed the necdin protein levels (Fig. 6A). HIF-2α reduced the necdin protein level in a dose-dependent manner (Fig. 6A, lanes 2–4). Using the proteasome inhibitor MG132, we also examined whether HIF-2α-dependent decrease in the necdin protein occurs by the proteasome degradation pathway (Fig. 6B). HIF-2α markedly decreased the necdin level (Fig. 6B, lane 4), whereas MG132 increased the levels of both necdin and HIF-2α (Fig. 6B, lane 2). These data suggest that HIF-2α targets necdin for proteasomal degradation.
Figure 6.

Necdin is degraded via the proteasome pathway. A, HIF-2α-dependent necdin degradation. Expression vectors for necdin (0.5 μg) and Myc-tagged HIF-2α cDNA (Myc-HIF-2α; amounts in micrograms) were transfected into HEK293A cells. Necdin, HIF-2α, and β-tubulin levels were analyzed by Western blotting. B, Proteasome-mediated degradation of necdin. HEK293A cells transfected with expression vectors for necdin and Myc-HIF-2α were harvested 24 h later following MG132 treatment for 3 h before harvest. Necdin, Myc, and β-tubulin were analyzed by Western blotting. DMSO, vehicle control. C, Time-dependent necdin degradation. NSCs were prepared and incubated for 24, 48, and 72 h in 2% O2 (Hypoxia) or for 72 h in 20% O2 (CT). Necdin, HIF-2α, and β-tubulin levels were analyzed by Western blotting. D, Effects of proteasome inhibition. Primary NSCs (6 d culture) were incubated in normoxia or hypoxia for 72 h, treated with (+) or without (−) MG132 for 24 h before harvest. Necdin, HIF-2α, and β-tubulin levels were analyzed by Western blotting.
We then analyzed endogenous levels of necdin and HIF-2α in hypoxic NSCs (Fig. 6C). When primary NSCs were incubated in hypoxia for 24, 48, and 72 h, the necdin protein level was markedly reduced at 72 h, when HIF-2α was clearly detectable (Fig. 6C, lane 4). The necdin protein level in hypoxic NSCs was increased when cultured in the presence of MG132 (Fig. 6D, lanes 3 and 4). The necdin level in normoxic NSCs was also increased by MG132 (Fig. 6D, lanes 1 and 2). These data suggest that necdin is targeted for proteasomal degradation in primary NSCs, and that necdin undergoes proteasomal degradation even in normoxia.
The HIF-2α PAS domain promotes necdin ubiquitination
We next examined whether HIF-2α promotes necdin ubiquitination, a critical step for proteasomal degradation. Expression vectors for Flag-ubiquitin, Myc-HIF-2α, and necdin were cotransfected into HEK293A cells, and ubiquitinated necdin was immunoprecipitated with anti-necdin antibody. Although necdin was slightly ubiquitinated in the absence of HIF-2α, its ubiquitination was markedly enhanced by HIF-2α (Fig. 7A). To examine whether the necdin-interacting PAS domain per se is capable of inducing necdin ubiquitination, the PAS domain and other HIF-2α domains were cotransfected with necdin into the HEK293A cells (Fig. 7B). Only the PAS domain markedly enhanced necdin ubiquitination, and the effect of the PAS domain was stronger than that of full-length HIF-2α.
Figure 7.

The HIF-2α PAS domain promotes necdin ubiquitination. A, HIF-2α-enhanced necdin ubiquitination. HEK293A cells transfected with expression vectors for Flag-tagged ubiquitin (Flag-ubiquitin), Myc-tagged HIF-2α (Myc-HIF-2α), and necdin were harvested 24 h later following MG132 treatment for 3 h before harvest. Cell lysates were immunoprecipitated with anti-necdin antibody and immunoblotted with anti-Flag antibody. B, PAS domain-mediated necdin ubiquitination. Cells transfected with expression vectors for Flag-ubiquitin, necdin, and Myc-HIF-2α domains (Fig. 5C for abbreviations) were treated and analyzed as above. C, PAS domain-mediated necdin ubiquitination in NSCs. NSCs were infected with Flag-tagged PAS-expressing lentivirus (PAS) or empty vector (DsRed), and cultured for 6 d in normoxia. Cell lysates were immunoprecipitated with anti-necdin antibody and immunoblotted with anti-ubiquitin antibody.
To examine whether the PAS domain promotes necdin ubiquitination in primary NSCs, we ectopically expressed the PAS domain in NSCs using lentivirus vectors (Fig. 7C). Polyubiquitinated necdin bands were detected even in PAS (−) control, suggesting that endogenous necdin is ubiquitinated in normoxic NSCs. When the PAS-expressing lentivirus was infected into primary NSCs, polyubiquitinated necdin bands at larger molecular sizes (>100 kDa) were enhanced.
The PAS domain promotes both necdin degradation and NSC proliferation
Because the PAS domain is resistant to oxygen-dependent degradation in normoxic conditions, we examined whether lentivirus-mediated expression of the PAS domain in normoxic NSCs induces changes similar to those seen in hypoxic NSCs (Fig. 8A). More than 90% of NSC spheres were infected with lentivirus vectors. The necdin protein level in PAS-expressing NSCs was significantly decreased to 58 and 54% of those in empty (DsRed) and ODD-expressing NSCs, respectively, in normoxic conditions, whereas the Cdc2 level in PAS-expressing NSCs was 1.6 and 1.4 times those in NSCs expressing DsRed and ODD, respectively (Fig. 8B,C). We then examined whether the PAS domain per se modulates neurosphere formation of CD133+ NSCs in normoxic conditions (Fig. 8D). The total neurosphere number of PAS-expressing NSCs was increased to 1.5 and 1.4 times those of NSCs expressing DsRed only and ODD, respectively (Fig. 8D). The numbers of small (20–40 μm in diameter) and large (>60 μm in diameter) neurospheres in the PAS-expressing NSCs were significantly larger than those in ODD-expressing NSCs (Fig. 8E). These data suggest that ectopic expression of the PAS domain mimics the HIF-2α-induced changes seen in hypoxic NSCs, such as ubiquitin-mediated degradation of necdin and enhanced NSC proliferation, even in normoxic conditions.
Figure 8.
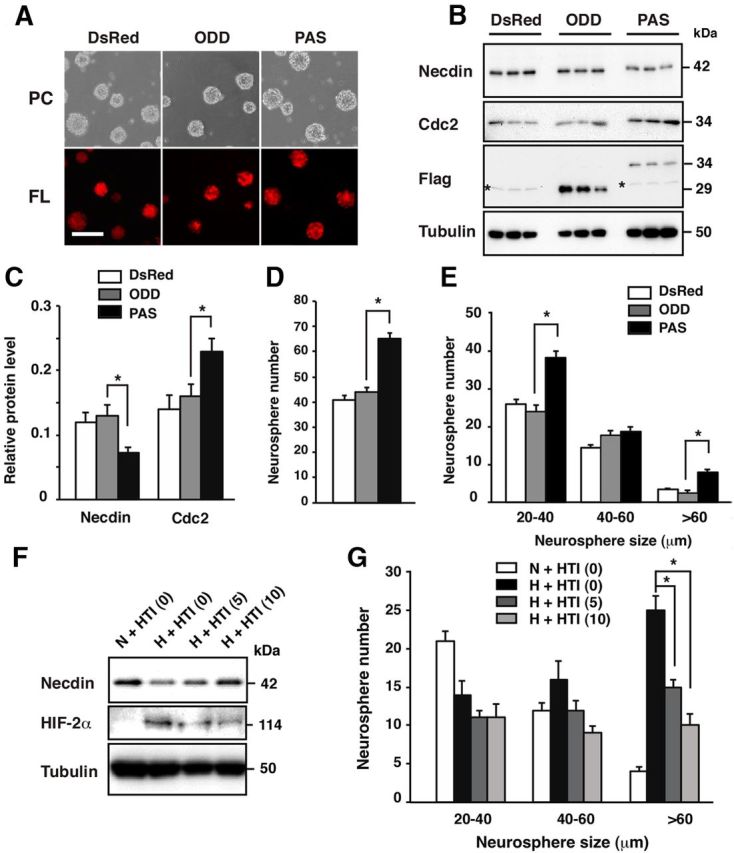
The HIF-2α PAS domain downregulates endogenous necdin level and facilitates NSC proliferation. A, Lentivirus-infected NSCs. NSCs were infected with lentiviruses expressing DsRed (DsRed, empty vector), Flag-tagged HIF-2α PAS domain (PAS), and Flag-tagged ODD (ODD), and cultured for 6 d. Images of phase-contrast (PC) and fluorescence (FL) of lentivirus-infected NSCs are shown. Scale bar, 100 μm. B, C, Expression of necdin and Cdc2 proteins. Lysates of infected NSCs were analyzed by Western blotting using antibodies to necdin, Cdc2, Flag, and β-tubulin (B). Asterisks indicate nonspecific bands. The protein levels were quantified and normalized to β-tubulin (C). D, E, Neurosphere assay. Lentivirus-infected CD133+ NSCs were subjected to neurosphere assay in normoxia. The total number of neurospheres (>20 μm in diameter; D) and the numbers of three groups (20–40, 40–60, >60 μm in diameter) were counted (E). F, G, Effects of HIF-2α translation inhibitor. Primary NSCs were treated with HIF-2α translation inhibitor (HTI) at 0, 5, and 10 μm for another 3 d in hypoxia (H). Control NSCs were cultured for 9 d in normoxia (N). HIF-2α and necdin were analyzed by Western blotting. G, Neurosphere assay. Primary NSCs were subjected to neurosphere assay in the presence of the translation inhibitor. The numbers of three groups were counted. C–E, G, mean ± SEM (n = 3). *p < 0.05.
To examine the effects of HIF-2α downregulation on the necdin protein level and proliferation rate of NSCs, NSCs were treated with a small-molecule HIF-2α translation inhibitor, which selectively reduces HIF-2α translation by enhancing the binding of Iron-Regulatory Protein 1 to the 5′-untranslated region of HIF-2α mRNA (Zimmer et al., 2008). This inhibitor, which reduced the HIF-2α level and stabilized the necdin protein in hypoxic NSCs (Fig. 8F), significantly reduced the number of large neurospheres (>60 μm in diameter) under hypoxic conditions (Fig. 8G). This suggests that downregulation of endogenous HIF-2α protein levels stabilizes the necdin protein and downregulates the proliferation rate of NSCs under hypoxic conditions.
Discussion
A considerable amount of the necdin protein was detected in NSCs when cultured in normoxia. Under these conditions, wild-type NSCs exhibited a lower expansion rate than necdin-deficient NSCs, suggesting that endogenous necdin suppresses NSC proliferation. Under hypoxic conditions, the expansion rate of necdin-deficient NSCs did not differ significantly from that of wild-type NSCs, in which the necdin protein level was markedly reduced. These findings implicate that hypoxia accelerates NSC proliferation, at least in part, through degradation of endogenous necdin. Because NSCs in developing brain reside in hypoxic environments (Mutoh et al., 2012), the necdin level in NSCs may be kept low in vivo. This suggests little or no difference in the proliferation rate between necdin-deficient NSCs and wild-type NSCs in vivo under physiological conditions. However, we found that EdU incorporation was significantly increased in the GE proliferative zone in vivo of necdin-deficient mice. These results suggest that degradation of the necdin protein in NSCs in vivo through the oxygen tension-dependent system is incomplete compared with necdin-null conditions. We assume that other complementary mechanisms that modulate the function of necdin are operative in NSCs.
Expansion of NSCs is attributed to increased proliferation, reduced apoptosis, or both. The present study has shown that the hypoxia-promoted expansion of wild-type NSCs is attributed to both increased proliferation and reduced apoptosis as judged by BrdU incorporation assay and TUNEL method, respectively (Fig. 3). Because hypoxia induces a marked reduction of apoptosis, it is suggested that reduced apoptosis of hypoxic NSCs makes a major contribution to their expansion. On the other hand, increased proliferation of necdin-deficient NSCs in normoxia is attributed to increased proliferation, but not to reduced apoptosis. On the contrary, necdin-deficient NSCs in normoxia showed a significant increase in apoptosis, indicating that necdin has also an antiapoptotic effect on GE NSCs. Thus, necdin has a dual function that suppresses proliferation and apoptosis of NSCs. In necdin-deficient NSCs, the increased rate of proliferation in normoxia can exceed the increased rate of concurrent apoptosis, resulting in the expansion of NSCs as represented by the increased number of large (>60 μm in diameter) neurospheres. If necdin had no antiapoptotic effect, expansion of necdin-deficient NSCs would be further augmented in normoxia. These findings suggest that necdin has a strong antimitotic activity in embryonic NSCs.
We found necdin-deficient mice, compared with wild-type mice, show higher rates of proliferation and apoptosis in the GE in vivo (Fig. 3C,D). We have previously reported that GE-derived GABAergic neurons exhibit abnormal differentiation in vivo in necdin-deficient mice at E14.5 (Kuwajima et al., 2006). Tangential migration of Dlx2-positive interneurons is also impaired in the forebrain of necdin-deficient mice at E14.5, and the number of cortical GABAergic interneurons is reduced at postnatal stages (Kuwajima et al., 2010). These mutant mice are highly susceptible to pentylenetetrazole-induced seizures. These findings suggest that hyperproliferation and increased apoptosis of NSCs at early stages of neurogenesis in the GE may lead to the abnormalities of GABAergic interneurons at later stages. Thus, endogenous necdin in GE NSCs may play an important role in normal development of GABAergic interneurons by suppressing both premature mitosis and apoptosis at early stages of neurogenesis.
Although >25 MAGE family genes have been identified in placental mammals, such as mouse and human (Barker and Salehi, 2002), nonmammalian species have only one MAGE gene (López-Sánchez et al., 2007). We have previously reported that Drosophila MAGE, a necdin-homologous MAGE protein, is expressed in neural stem cells (neuroblasts) and their progeny (ganglion mother cells and postmitotic neurons) during postembryonic neurogenesis (Nishimura et al., 2007). When Drosophila MAGE expression in developing mushroom bodies is downregulated by RNAi-mediated silencing, the population of ganglion mother cells in larval mushroom bodies is significantly increased (Nishimura et al., 2008). In these MAGE-knockdown flies, apoptosis in the neuronal progeny is enhanced during neuronal development. The present findings, together with the previous observations in Drosophila, indicate that necdin and its homologous MAGE are indispensable for the suppression of both proliferation and apoptosis of neuronal precursors. It has also been reported that the single MAGE genes in zebrafish (Danio rerio) and chicken (Gallus gallus) are expressed predominantly in the CNS proliferative zone and proliferating progenitors of projecting retinal neurons, respectively (Bischof et al., 2003; López-Sánchez et al., 2007). These observations raise the possibility that nonmammalian MAGE proteins also control both proliferation and apoptosis of NSCs and neuronal progenitors.
HIF-1α is expressed in a ubiquitous manner, whereas HIF-2α expression is restricted to specific cell types, such as vascular endothelial cells, neural crest cell derivatives, cardiomyocytes, and astrocytes (Wiesener et al., 2003). We found that HIF-2α mRNA is predominantly expressed in primary NSCs. The HIF-2α protein level was reduced in normoxia through oxygen-dependent proteolysis, but was stabilized in hypoxia. Although HIF-2α shares a significant sequence homology with well characterized HIF-1α, there is limited information on the expression and function of HIF-2α in NSCs. In zebrafish, HIF-2α is predominantly expressed in neural stem cell population of the neural tube during development, and morpholino-mediated knockdown of HIF-2α causes abnormal CNS development (Ko et al., 2011). Because necdin-homologous MAGE is expressed in developing zebrafish CNS (Bischof et al., 2003), the present findings may help explain the molecular mechanisms underlying the control of proliferation and differentiation of vertebrate NSCs through the interplay between HIF-2α and necdin or nonmammalian MAGE.
In hypoxic NSCs, expression levels of the HIF-regulated genes Vegf, Glut1, Cyclin D1, and erythropoietin were significantly increased, indicating that HIF-induced transcription is activated in hypoxia. It has previously been reported that expression of Vegf, erythropoietin, and Cyclin D1 is upregulated in NSCs under hypoxic conditions (Studer et al., 2000; Chen et al., 2010). The enhanced proliferation of NSCs is also supported by the increased expression of Cdc2 and Cyclin D1, both of which promote the cell cycle. It is noteworthy that Cdc2 expression is strictly downregulated by necdin in cerebellar granule neurons (Kurita et al., 2006). It has previously been shown that hypoxia-induced reduction of apoptosis contributes primarily to the enhanced expansion of hypoxic NSCs (Clarke and van der Kooy, 2009). Because Vegf significantly promotes the survival of embryonic neural stem cells (Wada et al., 2006), it is possible that the increased Vegf expression in hypoxic NSCs leads to the reduction of apoptosis.
Although there is a large amount of information about the molecular mechanisms underlying ubiquitin-dependent degradation of HIFs (Greer et al., 2012), little is known about the involvement of HIFs in the ubiquitin-dependent degradation of their target proteins. It has recently been reported that HIF-1α negatively controls regulatory T cell development by binding Foxp3 and targeting it for proteasomal degradation (Dang et al., 2011). Similarly, as shown presently, HIF-2α enhances the ubiquitination of necdin and induces its proteasomal degradation. Notably, necdin interacts preferentially with the PAS domain of HIF-2α. The PAS domains are present in many proteins and serve as important signaling modules that mediate the protein–protein interactions in response to various external and internal stimuli (Taylor and Zhulin, 1999). We are currently investigating whether the HIF-2α PAS domain serves as a platform that promotes necdin ubiquitination by recruiting a protein or a multiprotein complex possessing E3 ubiquitin ligase activity.
Necdin is expressed in stem cells or progenitors residing in non-neuronal tissues (Brunelli et al., 2004; Tseng et al., 2005; Deponti et al., 2007; Kubota et al., 2009; Liu et al., 2009; Fujiwara et al., 2012). Necdin is located in the nucleus of adipose tissue-derived mesenchymal stem cells in an undifferentiated state, and adipogenic stimuli downregulate the nuclear necdin level before the proliferation of committed adipocyte progenitors (Fujiwara et al., 2012), suggesting that nuclear necdin is degraded by proteolysis in response to adipogenic stimuli. Cell cycle-related proteins are often degraded in a stage-dependent manner by the ubiquitin-proteasome system. Similarly, the nuclear necdin level in NSCs is controlled in an oxygen tension-dependent manner by the ubiquitin-proteasome system. We have also found that necdin is expressed in NSCs residing in the ventricular/subventricular zone of the neocortex during embryonic development, and that both proliferation and apoptosis of these NSCs are increased in vivo in necdin-null mice (R. Minamide and K. Yoshikawa, unpublished observations). We assume that nuclear necdin level is strictly regulated in NSCs that occupy microenvironments containing different oxygen levels in the embryonic brain. Further studies on the post-translational modifications of the necdin protein will provide valuable insight into the physiological controls of the proliferation of NSCs during neurogenesis.
Footnotes
This work was supported by a Grant-in-Aid for Scientific Research B2 (24300134; to K.Y.) from the Japan Society for the Promotion of Science. We thank Drs. T. Ohkumo and H. Miyoshi for research materials and Ms. K. Imada for technical support.
The authors declare no competing financial interests.
References
- Aizawa T, Hasegawa K, Ohkumo T, Haga S, Ikeda K, Yoshikawa K. Neural stem cell-like gene expression in a mouse ependymoma cell line transformed by human BK polyomavirus. Cancer Sci. 2011;102:122–129. doi: 10.1111/j.1349-7006.2010.01775.x. [DOI] [PubMed] [Google Scholar]
- Barker PA, Salehi A. The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J Neurosci Res. 2002;67:705–712. doi: 10.1002/jnr.10160. [DOI] [PubMed] [Google Scholar]
- Bischof JM, Ekker M, Wevrick R. A MAGE/NDN-like gene in zebrafish. Dev Dyn. 2003;228:475–479. doi: 10.1002/dvdy.10398. [DOI] [PubMed] [Google Scholar]
- Brunelli S, Tagliafico E, De Angelis FG, Tonlorenzi R, Baesso S, Ferrari S, Niinobe M, Yoshikawa K, Schwartz RJ, Bozzoni I, Ferrari S, Cossu G. Msx2 and necdin combined activities are required for smooth muscle differentiation in mesoangioblast stem cells. Circ Res. 2004;94:1571–1578. doi: 10.1161/01.RES.0000132747.12860.10. [DOI] [PubMed] [Google Scholar]
- Chen HL, Pistollato F, Hoeppner DJ, Ni HT, McKay RD, Panchision DM. Oxygen tension regulates survival and fate of mouse central nervous system precursors at multiple levels. Stem Cells. 2007;25:2291–2301. doi: 10.1634/stemcells.2006-0609. [DOI] [PubMed] [Google Scholar]
- Chen X, Tian Y, Yao L, Zhang J, Liu Y. Hypoxia stimulates proliferation of rat neural stem cells with influence on the expression of cyclin D1 and c-Jun N-terminal protein kinase signaling pathway in vitro. Neuroscience. 2010;165:705–714. doi: 10.1016/j.neuroscience.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Clarke L, van der Kooy D. Low oxygen enhances primitive and definitive neural stem cell colony formation by inhibiting distinct cell death pathways. Stem Cells. 2009;27:1879–1886. doi: 10.1002/stem.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deponti D, François S, Baesso S, Sciorati C, Innocenzi A, Broccoli V, Muscatelli F, Meneveri R, Clementi E, Cossu G, Brunelli S. Necdin mediates skeletal muscle regeneration by promoting myoblast survival and differentiation. J Cell Biol. 2007;179:305–319. doi: 10.1083/jcb.200701027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ereciska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. 2001;128:263–276. doi: 10.1016/S0034-5687(01)00306-1. [DOI] [PubMed] [Google Scholar]
- Friedman ER, Fan CM. Separate necdin domains bind ARNT2 and HIF1alpha and repress transcription. Biochem Biophys Res Commun. 2007;363:113–118. doi: 10.1016/j.bbrc.2007.08.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara K, Hasegawa K, Ohkumo T, Miyoshi H, Tseng YH, Yoshikawa K. Necdin controls proliferation of white adipocyte progenitor cells. PLoS One. 2012;7:e30948. doi: 10.1371/journal.pone.0030948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J. 2012;31:2448–2460. doi: 10.1038/emboj.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J Neurosci. 2008;28:8772–8784. doi: 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Kawahara T, Fujiwara K, Shimpuku M, Sasaki T, Kitamura T, Yoshikawa K. Necdin controls Foxo1 acetylation in hypothalamic arcuate neurons to modulate the thyroid axis. J Neurosci. 2012;32:5562–5572. doi: 10.1523/JNEUROSCI.0142-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Matsuyama K, Takagi K, Sugiura H, Yoshikawa K. Arrest of cell growth by necdin, a nuclear protein expressed in postmitotic neurons. Biochem Biophys Res Commun. 1995;213:317–324. doi: 10.1006/bbrc.1995.2132. [DOI] [PubMed] [Google Scholar]
- Ko CY, Tsai MY, Tseng WF, Cheng CH, Huang CR, Wu JS, Chung HY, Hsieh CS, Sun CK, Hwang SP, Yuh CH, Huang CJ, Pai TW, Tzou WS, Hu CH. Integration of CNS survival and differentiation by HIF2alpha. Cell Death Differ. 2011;18:1757–1770. doi: 10.1038/cdd.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Taniura H, Yoshikawa K. Ectopic expression of necdin induces differentiation of mouse neuroblastoma cells. J Biol Chem. 2002;277:42128–42135. doi: 10.1074/jbc.M205024200. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Osawa M, Jakt LM, Yoshikawa K, Nishikawa S. Necdin restricts proliferation of hematopoietic stem cells during hematopoietic regeneration. Blood. 2009;114:4383–4392. doi: 10.1182/blood-2009-07-230292. [DOI] [PubMed] [Google Scholar]
- Kurita M, Kuwajima T, Nishimura I, Yoshikawa K. Necdin downregulates cdc2 expression to attenuate neuronal apoptosis. J Neurosci. 2006;26:12003–12013. doi: 10.1523/JNEUROSCI.3002-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima T, Taniura H, Nishimura I, Yoshikawa K. Necdin interacts with the Msx2 homeodomain protein via MAGE-D1 to promote myogenic differentiation of C2C12 cells. J Biol Chem. 2004;279:40484–40493. doi: 10.1074/jbc.M404143200. [DOI] [PubMed] [Google Scholar]
- Kuwajima T, Nishimura I, Yoshikawa K. Necdin promotes GABAergic neuron differentiation in cooperation with Dlx homeodomain proteins. J Neurosci. 2006;26:5383–5392. doi: 10.1523/JNEUROSCI.1262-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima T, Hasegawa K, Yoshikawa K. Necdin promotes tangential migration of neocortical interneurons from basal forebrain. J Neurosci. 2010;30:3709–3714. doi: 10.1523/JNEUROSCI.5797-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwako K, Taniura H, Yoshikawa K. Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J Biol Chem. 2004;279:1703–1712. doi: 10.1074/jbc.M308454200. [DOI] [PubMed] [Google Scholar]
- Kuwako K, Hosokawa A, Nishimura I, Uetsuki T, Yamada M, Nada S, Okada M, Yoshikawa K. Disruption of the paternal necdin gene diminishes TrkA signaling for sensory neuron survival. J Neurosci. 2005;25:7090–7099. doi: 10.1523/JNEUROSCI.2083-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi-Itzkovitz A, Tcherpakov M, Levy Z, Itzkovitz S, Muscatelli F, Fainzilber M. Functional consequences of necdin nucleocytoplasmic localization. PLoS One. 2012;7:e33786. doi: 10.1371/journal.pone.0033786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, Nimer SD. p53 Regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Sánchez N, González-Fernández Z, Niinobe M, Yoshikawa K, Frade JM. Single mage gene in the chicken genome encodes CMage, a protein with functional similarities to mammalian type II Mage proteins. Physiol Genomics. 2007;30:156–171. doi: 10.1152/physiolgenomics.00249.2006. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Usami M, Aizawa T, Yoshikawa K. A novel brain-specific mRNA encoding nuclear protein (necdin) expressed in neurally differentiated embryonal carcinoma cells. Biochem Biophys Res Commun. 1991;178:291–296. doi: 10.1016/0006-291X(91)91812-Q. [DOI] [PubMed] [Google Scholar]
- Mazumdar J, O'Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, Simon MC. O2 regulates stem cells through Wnt/beta-catenin signalling. Nat Cell Biol. 2010;12:1007–1013. doi: 10.1038/ncb2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Blömer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon HE, Ahn MY, Park JA, Min KJ, Kwon YW, Kim KW. Negative regulation of hypoxia inducible factor-1alpha by necdin. FEBS Lett. 2005;579:3797–3801. doi: 10.1016/j.febslet.2005.05.072. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci. 2000;20:7370–7376. doi: 10.1523/JNEUROSCI.20-19-07370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh T, Sanosaka T, Ito K, Nakashima K. Oxygen levels epigenetically regulate fate switching of neural precursor cells via hypoxia-inducible factor 1alpha-notch signal interaction in the developing brain. Stem Cells. 2012;30:561–569. doi: 10.1002/stem.1019. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Shimizu S, Sakoda JY, Yoshikawa K. Expression of Drosophila MAGE gene encoding a necdin homologous protein in postembryonic neurogenesis. Gene Expr Patterns. 2007;7:244–251. doi: 10.1016/j.modgep.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Sakoda JY, Yoshikawa K. Drosophila MAGE controls neural precursor proliferation in postembryonic neurogenesis. Neuroscience. 2008;154:572–581. doi: 10.1016/j.neuroscience.2008.03.075. [DOI] [PubMed] [Google Scholar]
- Panchision DM. The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol. 2009;220:562–568. doi: 10.1002/jcp.21812. [DOI] [PubMed] [Google Scholar]
- Pfenninger CV, Roschupkina T, Hertwig F, Kottwitz D, Englund E, Bengzon J, Jacobsen SE, Nuber UA. CD133 is not present on neurogenic astrocytes in the adult subventricular zone, but on embryonic neural stem cells, ependymal cells, and glioblastoma cells. Cancer Res. 2007;67:5727–5736. doi: 10.1158/0008-5472.CAN-07-0183. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci. 1992;12:4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues CA, Diogo MM, da Silva CL, Cabral JM. Hypoxia enhances proliferation of mouse embryonic stem cell-derived neural stem cells. Biotechnol Bioeng. 2010;106:260–270. doi: 10.1002/bit.22648. [DOI] [PubMed] [Google Scholar]
- Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 2000;20:7377–7383. doi: 10.1523/JNEUROSCI.20-19-07377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniura H, Taniguchi N, Hara M, Yoshikawa K. Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J Biol Chem. 1998;273:720–728. doi: 10.1074/jbc.273.2.720. [DOI] [PubMed] [Google Scholar]
- Taniura H, Matsumoto K, Yoshikawa K. Physical and functional interactions of neuronal growth suppressor necdin with p53. J Biol Chem. 1999;274:16242–16248. doi: 10.1074/jbc.274.23.16242. [DOI] [PubMed] [Google Scholar]
- Taniura H, Kobayashi M, Yoshikawa K. Functional domains of necdin for protein–protein interaction, nuclear matrix targeting, and cell growth suppression. J Cell Biochem. 2005;94:804–815. doi: 10.1002/jcb.20345. [DOI] [PubMed] [Google Scholar]
- Taylor BL, Zhulin IB. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev. 1999;63:479–506. doi: 10.1128/mmbr.63.2.479-506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YH, Butte AJ, Kokkotou E, Yechoor VK, Taniguchi CM, Kriauciunas KM, Cypess AM, Niinobe M, Yoshikawa K, Patti ME, Kahn CR. Prediction of preadipocyte differentiation by gene expression reveals role of insulin receptor substrates and necdin. Nat Cell Biol. 2005;7:601–611. doi: 10.1038/ncb1259. [DOI] [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetsuki T, Takagi K, Sugiura H, Yoshikawa K. Structure and expression of the mouse necdin gene. Identification of a postmitotic neuron-restrictive core promoter. J Biol Chem. 1996;271:918–924. doi: 10.1074/jbc.271.2.918. [DOI] [PubMed] [Google Scholar]
- Uetsuki T, Takemoto K, Nishimura I, Okamoto M, Niinobe M, Momoi T, Miura M, Yoshikawa K. Activation of neuronal caspase-3 by intracellular accumulation of wild-type Alzheimer amyloid precursor protein. J Neurosci. 1999;19:6955–6964. doi: 10.1523/JNEUROSCI.19-16-06955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Haigh JJ, Ema M, Hitoshi S, Chaddah R, Rossant J, Nagy A, van der Kooy D. Vascular endothelial growth factor directly inhibits primitive neural stem cell survival but promotes definitive neural stem cell survival. J Neurosci. 2006;26:6803–6812. doi: 10.1523/JNEUROSCI.0526-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesener MS, Jürgensen JS, Rosenberger C, Scholze CK, Hörstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt KU. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003;17:271–273. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- Zhao T, Zhang CP, Liu ZH, Wu LY, Huang X, Wu HT, Xiong L, Wang X, Wang XM, Zhu LL, Fan M. Hypoxia-driven proliferation of embryonic neural stem/progenitor cells-role of hypoxia-inducible transcription factor-1alpha. FEBS J. 2008;275:1824–1834. doi: 10.1111/j.1742-4658.2008.06340.x. [DOI] [PubMed] [Google Scholar]
- Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, Tolliday N, Lamb J, Pantopoulos K, Golub T, Iliopoulos O. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol Cell. 2008;32:838–848. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]