

Abstract
The axonal and synaptic mechanisms underlying dysfunction and repair of the injured CNS are poorly understood. Unresolved issues include to what degree, when, and how the surviving neurons degenerate and the extent of synaptic remodeling both along the severed axon and in the nearby area. One of the main reasons is the lack of tools to study the complex asynchronous and dynamic features of individual lesioned axon responses in the intact brain. To address these issues, we combined two-photon microscopy and laser microsurgery to image the real-time reorganization of cortical circuitry at synaptic resolution for periods of up to 1 year in the brain of living mice. Injured cortical axons were eliminated proximally through a two-phase retraction process, which continued for at least 3 months postlesion and was independent of the presence of scar tissue. Remarkably, axons which later attempt to regenerate in both the mature and juvenile brain retracted less, raising the possibility that targeting retraction may improve the chances of axon regrowth after axotomy. Comparing prelesion and postlesion dynamics on the same axons over several days and weeks revealed that, although synapse formation rates were unaffected, boutons on injured axons were either rapidly and persistently lost, or extremely resistant, depending on cell-type and their prelesion structural dynamics. Our data suggest a lasting deficiency in synaptic output on surviving injured cortical axons and a surprising difference in the vulnerability of synaptic boutons after axotomy, which depend on cell-type and their recent history.
Introduction
Axonal damage is a primary feature of many neurological diseases (Raff et al., 2002; Medana and Esiri, 2003). When neurons lose synaptic connections as a result of axonal degeneration, synaptic remodeling on the surviving proximal side and nearby spared neurons could potentially compensate for lasting deficiencies in neuronal connectivity. On the other hand, additional synaptic loss on the proximal side could deepen the functional deficits associated with axonal injury. Understanding the mechanisms of synaptic responses to axonal damage could therefore be crucial in managing neurological injury.
After axonal damage to the distal side, the portion disconnected from the cell body, undergoes Wallerian degeneration, a regulated and intensively studied process of axon elimination (Gillingwater et al., 2006; Coleman and Freeman, 2010). In contrast, we know comparatively little about the response of the proximal side of the axon, i.e., the portion still connected to the cell body. Unresolved issues include to what degree, and how, the surviving axon eventually degenerates over extended periods of time, and the extent of synaptic remodeling both along the severed axon and in the nearby area, as most previous studies could not assess synaptic reorganization (Cafferty et al., 2008). Synapses on cortical axons come in two major forms, en passant boutons (EPBs)–swellings along the axon shaft, or as short 1–5 μm outgrowths extending from the axon shaft called terminaux boutons (TBs; De Paola et al., 2006). TBs are much more dynamic on axons of cortical layer (L) 6 neurons than on other cell types [∼50% turnover over a 24 d period compared with ∼10% for thalamocortical (TCA) neurons], suggesting the plasticity/stability of axonal boutons is intrinsically specified (De Paola et al., 2006). These results raise the question of whether the synaptic response to injury could also be cell type-dependent.
To study the mechanisms of axonal and synaptic reorganization after minimal injury, we have used a combination of two-photon microscopy and laser microsurgery to monitor the response of excitatory cortical circuitry for periods ranging from minutes up to 1 year in the brain of living mice.
We report three main findings: (1) The proximal side of injured cortical axons retracts within a few days and progressively continues to do so for at least 3 months postlesion. Unexpectedly, this retraction does not involve acute-axonal degeneration and infiltrating macrophages as previously seen after spinal cord injury. (2) Interestingly, axons that later attempt to regenerate in both the mature and juvenile brain retract less. (3) Axonal boutons on the surviving axons are either rapidly (hours) and persistently (up to 1 month) lost, or are extremely resistant, depending on cell-type and their prelesion structural dynamics.
Our data suggest the existence of cell-autonomous programs of axon and synaptic elimination, which operate in different neuronal populations of the adult brain. Targeting progressive axon retraction and synapse formation/elimination pathways on specific subsets of damaged neurons may provide new therapeutic strategies for counteracting the functional deficits associated with axonal injury.
Materials and Methods
Animals
Adult male (n = 48, 12–14 weeks) and juvenile mice (n = 13, 3–4 weeks) from Thy-1-GFP-M (Feng et al., 2000) and Thy1-GFP L15 (De Paola et al., 2003; with cytosolic and membrane targeted GFP, respectively) on the same C57BL/6 genetic background were used for all imaging experiments. Animals were housed with littermates, in standard individually ventilated caging and maintained in a 12 h light/dark cycle with access to food and water ad libitum. All experiments involving live animals were conducted by researchers holding a UK personal license and conducted in accordance with the Animals (Scientific Procedures) Act 1986 (UK) and associated guidelines.
Surgery
Cranial windows were surgically implanted overlying barrel cortex according to previously published methods (Holtmaat et al., 2009). Briefly, mice were anesthetized with a ketamine-xylazine intraperitoneal (i.p.) injection (0.083 mg/g ketamine, 0.0078 mg/g xylazine). The animals were then administered intramuscular dexamethasone (0.02 ml at 4 mg/ml) to limit inflammation response, and subcutaneous bupivacaine (1 mg/kg), a local anesthetic. Once the skull was exposed, a few drops of lidocaine (1% solution) were applied on its surface. The glass coverslip that seals the window was placed directly over the dura and the bone edges, with a thin layer of agarose in between, and sealed with dental cement. Mice were allowed to recover for 10–15 d before the start of the imaging protocol.
Imaging
A purpose built microscope equipped with a tunable Coherent Ti:Sapphire laser and PrarieView acquisition software was used for all in vivo imaging experiments. Mice were anesthetized with ketamine-xylazine (0.083 and 0.0078 mg/g, i.p.) and secured to a fixed support under the microscope. The eyes were coated with Lacri-lube (Allergan) to prevent dehydration, an underlying heat pad used to maintain body temperature (37°C) and rehydration administered with isotonic saline solution (i.p.) as required during long imaging sessions. Depth of anesthesia was closely monitored by regularly checking toe pinch and respiratory rate. An Olympus 4× with a 0.13 numerical aperture (NA) objective was used to identify characteristic blood vessel patterns and to reliably relocate previously imaged areas of the cortical neuropil. An Olympus 40× 0.80 NA water-immersion objective was used to acquire the images (typically 100 × 100 μm field of view, 512 × 512 pixels). A pulsed 910 nm laser beam was used never exceeding 70 mW on the back focal plane. Each imaging session typically lasted for 60–90 min, during which time up to 40 image stacks (1 μm step size) were collected. Where possible, 2–3 nonoverlapping axons were selected for lesioning in each preparation. For synaptic analysis in vivo we first identified long axonal stretches and then collected high-magnification images of the synapses along the axon shaft at 6 h, 1 d, 4 d, 8 d, and monthly intervals (see Fig. 1C). The amount of a terminal axon branch removed was ∼500 μm on average (range ∼150 μm up to >1000 μm; in case multiple endings were present, we considered the total length of the disconnected portion), leaving a minimum of 250 μm (range 250–2200 μm) of surviving axon for postlesion analysis (60 TB/axon and 30 EPB/axon on average).
Figure 1.

Laser mediated axotomy in the living mouse brain. A, Axons undergo stereotypical retraction from the lesion site, and swelling of the axon shaft in the surviving axon (proximal to cell body). Arrow indicates the lesion site. B, A binary mask (green) at a single focal plane showing only a small part of the selected axon (left) is used to target the high-energy laser to create an axonal lesion with characteristic fluorescent mark (arrow). C, Time course for repeated imaging prelesion and postlesion to enable analysis of synaptic dynamics. Dotted box highlights the 1 month time points considered. Scale bars: A, 10 μm; B, 5 μm.
Laser mediated microlesions in the intact mouse brain
A total of 107 axons were lesioned across all experimental conditions and observed for at least two consecutive time points postlesion (83 axons in the adult brain; and 24 axons in the juvenile brain). More than one-half of lesioned axons (48 axons in the adult brain; 12 axons in the juvenile brain) were observed chronically for >2 weeks and up to 1 year postlesion. Identified axons were severed by inducing a laser lesion with a single circular spot scan (a binary mask of 4 μm diameter, ∼600 mW at the back focal plane (i.e., 1 order of magnitude higher than the power used for imaging), 800 nm and 30 μs dwell time; Canty et al., 2013). We quantified the diameter of the first 10 lesions based on the fluorescent mark left and found very low variability (see Results for details). Lesions were induced in the upper layers of the cortex, to a depth of 50 μm beneath the pial surface. A dextran tracer (5 mg/ml solution, Texas Red, Invitrogen) was injected into the tail vein to label the vasculature and avoid targeting axonal lesions close to blood vessels. Lesions were made as far apart as possible (minimum 300 μm).
Cortical axons were imaged at variable intervals (see Fig. 1) before and after lesion for as long as window clarity was preserved. Animals were killed at defined time points postlesion (11 d, 1 m, 3 m, 6 m, and 12 m) or where the health of the mouse became compromised (maximum age postlesion 12 months). During one of the imaging sessions, overlapping low-magnification overviews of the labeled neuropil were collected to help identification of axons in the fixed brain (Canty et al., 2013).
At the conclusion of each experiment, mice were injected with a lethal dose of ketamine/xylazine and transcardially perfused with 40 ml of ice-cold 4% paraformaldehyde for 10 min. The brain was dissected, post-fixed overnight at 4°C in the same fixative, transferred to 0.01 m PBS containing 0.02% azide and stored at 4°C.
Data analysis
In vivo two-photon images were processed using Matlab (synaptic remodeling; MathWorks), ImageJ (length quantification, file conversions), Adobe Photoshop (section alignment, brightness and contrast, image rotation) or Neurolucida (axonal arbor tracing and Sholl analysis) as required. Figures were prepared using Microsoft Office Suite and Adobe Illustrator.
Axons were classified by morphological criteria, as previously described (De Paola et al., 2006). Axons were grouped as L6 if TB density was >0.07/μm. All other axons, those with fewer TBs and/or those with EPBs, were grouped together as L2/3/5 and TCA.
Axon retraction.
We measured the distance from the proximal tip to the lesion site at specific intervals postlesion (see Fig. 3B–G). Where growth was observed, the measurement was excluded from calculations of the average retraction. Of the 83 distinct axons lesioned in the adult brain, we obtained measurements for 57 axons at day 1, 42 axons at day 4, and 32 axons at day 8. For the comparison between neuron types (see Fig. 3F) we selected the time window with most data points, i.e., 28 ± 4 d.
Figure 3.
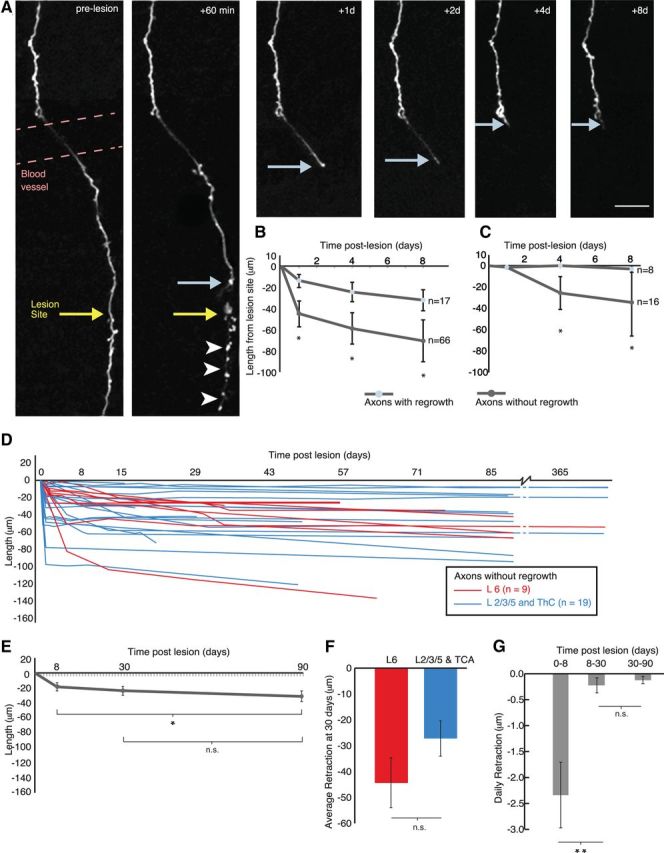
Real-time monitoring of injured cortical axons reveals a link between retraction and regrowth. A, Axon retraction following laser microlesion. The distal portion of the axon, disconnected from the cell body (below lesion site), degenerates (arrowheads). Light blue arrows indicates the proximal stump, yellow arrow indicates lesion site. B, Regrowing axons retract significantly less than nonregrowing ones in the adult brain at 1, 4, and 8 d postlesion. Severed nonregrowing axons retract away from the lesion site with significant retraction in the following 8 d. C, Severed axons in juvenile animals showed less retraction after axotomy than the adult animals and a significant difference between regrowing and nonregrowing ones (for regrowing axons, n = 8, average retraction was 1.2 ± 1.2 μm at 1 d postlesion; 0.0 ± 0.0 μm at 4 d postlesion; 3.2 ± 2.4 μm at 8 d postlesion; for nonregrowing axons, n = 16, average retraction was 1.4 ± 1.4 μm at 1 d postlesion; 25.5 ± 15.2 μm at 4 d postlesion; 34.4 ± 31.4 μm at 8 d postlesion). D, Chronic imaging for up to one year postlesion shows the relative stability of the position of the axonal stump after the initial retraction until 8 d (n = 28) in both L6 (red, n = 9) and L2/3/5 and TCA (blue, n = 19) nonregrowing axons. E, Progressive retraction over 3 months for 10 nonregrowing axons. F, No difference in retraction between L6 and L2/3/5 and TCA axons. G, Retraction is more rapid in the first week compared with the following 3 months. Error bar represents the SEM. *p < 0.05, **p < 0.01 (Wilcoxon signed-rank test); n.s., nonsignificant. Scale bar, 20 μm.
Axon regrowth.
We defined axons as attempting to regrow (regrowing axons) if the elongation from the cut stump reached the lesion site and exceeded twice the maximum noise measurement (i.e., >6 μm; the maximum difference in measurements between fiducial points over repeated imaging sessions is 3 μm; De Paola et al., 2006; Canty et al., 2013).
Axonal bouton quantification.
Two axonal populations were selected for this study, which were morphologically distinguishable (De Paola et al., 2006). TB rich axons whose cell bodies originate in cortical layer 6 (L6) form both axo-spinous and axo-dendritic synapses (Anderson and Martin, 2001) and are known to be highly dynamic in the adult brain (De Paola et al., 2006). TBs were tracked using both Thy1-GFP-L15 (membrane bound GFP expression; De Paola et al., 2003) and Thy1-GFP-M (cytosolic GFP expression). EPB rich axons whose cell bodies reside in either of L2, 3, 5, or the thalamus, mainly form axo-spinous synapses and are relatively stable in the adult brain (De Paola et al., 2006). EPBs were tracked using Thy1-GFP-M (cytosolic GFP expression; Feng et al., 2000).
Turnover ratio (TOR) between two imaging sessions a and b, is defined as (nG + nL)/(2 N), where nG and nL are the numbers of bouton gains and losses respectively, and N is the total number of boutons in session a.
Destabilization was measured by calculating the probability of destabilization (ProbDest = N boutons present between 0 and 8 d and then lost/total N present 0–8 d). The data in Figures 4C,G, 5, and 6 were normalized using the average of the prelesion values. The data in Figure 4E,F were normalized using the daily prelesion loss/gain fraction and densities (Table 1). The fraction of TBs which were newly formed in the week before the lesion and then lost before the end of the imaging paradigm (see Fig. 4I) was calculated by dividing by the total number of TBs which were newly formed in the week before the lesion.
Figure 4.
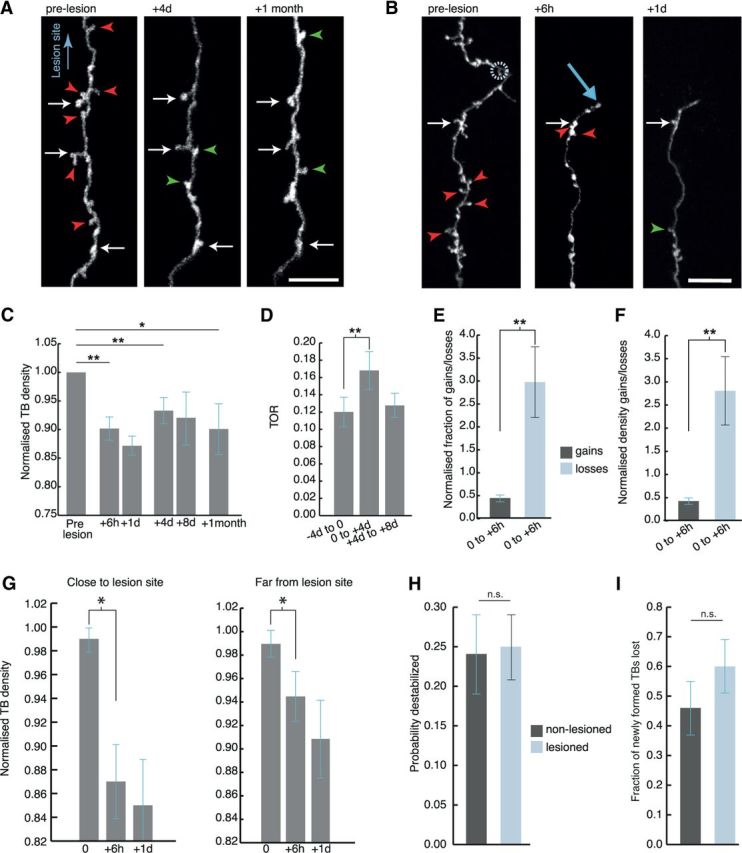
Rapid and persistent synaptic loss after axotomy on L6 axons. A, B, L6 axons demonstrate a rapid loss of TB synapses from as early as 6 h postlesion, which is maintained for at least 1 month. Synaptic loss is evident both further away from the lesion (A), and close to the lesion site (indicated by a circle in B); light blue arrow indicates axonal stump. C, When normalized to the average of pre lesion synaptic density, quantification of synaptic loss indicates a significant reduction in TB density along the surviving axon by 6 h (p = 0.004), which persists at 1 d (p = 0.002), 4 d (p = 0.007), and 1 month postlesion (p = 0.02). D, TB turnover (TOR) increased significantly (p = 0.007) in the first 4 d postlesion, and returned to baseline levels 4 d later. E, F, Further analysis indicated that the increased fraction (p = 0.004) and density (p = 0.003) of losses (pale blue) compared with synaptic gains (dark gray) was significant from 6 h postlesion. G, TB density significantly decreases both close (within 250 μm from the lesion site, day 0 = 0.99 ± 0.01, +6 h = 0.87 ± 0.04, +1 d = 0.85 ± 0.04; p = 0.02 between 0 and +6 h) and far from the lesion site (following 250 μm, day 0 = 0.99 ± 0.01, +6 h = 0.94 ± 0.02, +1 d = 0.91 ± 0.03; p = 0.03 between 0 and +6 h). Data normalized to prelesion density. H, TBs present for all the prelesion time points are not affected by the lesion. I, The fraction of TBs which were newly formed in the week before the lesion and then lost before the end of the imaging paradigm increases after the lesion. White arrows in A, B indicate stable synaptic boutons, red arrowheads indicate losses, and green arrowheads indicate gains. Error bar represents the SEM. *p < 0.05, **p < 0.01 (Wilcoxon signed-rank test); n.s., nonsignificant. Scale bars, 10 μm.
Figure 5.

Stability of L2/3/5 and TCA axonal boutons after axotomy. A, Representative example of in vivo time lapse imaging showing stable EPBs on L2/3/5 and TCA cells. B, D, Normalized synaptic density of EPBs does not significantly change at +1 d (p = 0.75), +4 d (p = 0.25), or +8 d (p = 0.57) postlesion. C, E, Similarly, there is no significant change in TOR of EPB synapses at either 1 or 4 d intervals postlesion. White arrows in A indicate stable synaptic boutons, red arrowheads indicate losses and green arrowheads indicate gains. n.s., nonsignificant. Scale bar, 10 μm (A).
Figure 6.

Absence of axon and synaptic reorganization in the surrounding neuropil. A, Individual axons were lesioned, often as close to few μm from nearby axons (inset) and the surrounding neuropil analyzed for up to 11 d postlesion. Pale gray arrows indicate intact axon before lesion, red arrowheads indicate the axonal stump, and blue arrowheads indicate previous trajectory of degenerated axonal segment. B, 10 and 30 μm analysis diameter bins (red circles) were created surrounding the lesion site. There was no significant difference in either total axonal length (C), data normalized to prelesion length, within 10, 30, and 100 μm or synaptic density (D), data normalized to prelesion density, detected in the surrounding circuitry after the lesion. n.s., Nonsignificant. Scale bars: A, 20 μm; B, 30 μm.
Table 1.
Rapid TB loss after laser axotomy
| Time from lesion | Normalized density | SEM | P value |
|---|---|---|---|
| Prelesion | 1.00 | ||
| +0.25 d | 0.90 | 0.02 | 0.004 (0–0.25 d) |
| +1 d | 0.87 | 0.02 | 0.002 (0–1 d) |
| +4 d | 0.93 | 0.02 | 0.007 (0–4 d) |
| +8 d | 0.92 | 0.04 | 0.63 (4–8 d) |
| +1 month | 0.90 | 0.04 | 0.02 (0–1 m) |
| Gain fractiona | |||
| 0 to +0.25 d | 0.44 | 0.07 | |
| Loss fractionb | |||
| 0 to +0.25 d | 2.97 | 0.77 | <0.01 (gain vs loss) |
| Gain densityc | |||
| 0 to +0.25 d | 0.43 | 0.07 | |
| Loss densityd | |||
| 0 to +0.25 d | 2.79 | 0.73 | <0.01 (gain vs loss) |
Normalized TB density and fraction or density of TB gains/losses for L6 axons at the time points indicated (n = 6 mice, 10 axons, 693 TBs).
aNormalized to daily gain fraction prelesion;
bnormalized to daily loss fraction prelesion;
cnormalized to daily gain density prelesion;
dnormalized to daily loss density prelesion.
TBs on L6 axons were manually traced and correlated between sessions using custom analysis software (Holtmaat et al., 2009; Grillo et al., 2013). TBs (1–5 μm) were annotated and scored as either stable, lost or gained according to stringent criteria modified from (Holtmaat et al., 2009). To be included for analysis, the surviving axon had to have a TB density of 0.07/μm or higher (60 TB/axon on average, 37 minimum) and be at least 250 μm in length (average, ∼500 μm). Twenty distinct axons were imaged and lesioned in total for this analysis, of which 12 could be used for the 4 d calculations (6 up to 8 d postlesion), 10 for the 6 h and 1 d and 8 were monitored also 1 month postlesion. A subset of 8 axons (6 mice) were imaged for five consecutive 4 d sessions but not lesioned, to assess destabilization in normal conditions. For the analysis of TB density along the axon we defined the area close to lesion as the first 250 μm and far from lesion the following 250 μm. All images were scored blind to the experimental condition and by at least two observers.
EPB rich axons were analyzed using EPBscore, a custom built software using a Matlab platform (MathWorks; Grillo et al., 2013, Song et al., unpublished observations). Briefly, 16-bit images were median filtered and saved and the resulting image segmented with an adjustable threshold. Processes were traced in 3D and an axon intensity profile generated. The median intensity value of all pixels along the axon profile was set as the axonal backbone estimation. Peaks in the axon profile were scored as boutons and measured as backbone units of intensity. Fiducial points were chosen by the operator to align ROIs over successive sessions to correlate boutons. EPBs had to be at least two times brighter than the backbone in at least one session to be included in the analysis, based on serial section electron microscopy reconstructions, which showed the smallest EPB that formed a structurally complete synapse was 1.92 times backbone intensity (Grillo et al., 2013). EPBs had to be present in at least two consecutive imaging sessions to be included in the analysis. The intensity values over time were exported into Microsoft Excel and postprocessed using Matlab (MathWorks) scripts. To be included for analysis, the surviving axon was at least 500 μm in length, and contained a minimum of 20 EPBs (30 on average).
All statistical analysis was performed either in the Matlab (MathWorks) suite or Microsoft Excel. Unless stated otherwise, all measurements are given as mean ± SEM. Wilcoxon signed-rank test was used for all the comparisons unless otherwise stated. Results were considered significant when p < 0.05. In the figures, *p < 0.05, **p < 0.01.
Results
Laser mediated axotomy in the living mouse brain
To assess synaptic reorganization after minimal injury in the brain, we used Thy1-GFP mice, which express GFP in a subset of L2/3/5/6 and TCA axons in either the cytosol (Feng et al., 2000) or cell membrane (De Paola et al., 2003). Axons were imaged individually through a cranial window and classified according to their morphology based on previous anatomical reconstructions and findings (De Paola et al., 2006; see Materials and Methods). To study synaptic reorganization over time scales including both immediate and more delayed responses to injury we used two-photon laser-mediated axotomy, which uniquely allows paired, statistically powerful, comparisons between prelesion and postlesion dynamics over extended periods of time within the same axons (O'Brien et al., 2009; Ylera et al., 2009; Allegra Mascaro et al., 2010; Farrar et al., 2012; Canty et al., 2013). Importantly, this localized lesion is not associated with a prominent glial scar and infiltrating macrophages (Ylera et al., 2009; Canty et al., 2013), allowing to tease apart intrinsic versus extrinsic determinants of axon and synaptic reorganization. We were able to transect axons within defined lengths away from the distal end in most neurons (for 58 of 83 axons, mean portion cut = 542 ± 38 μm, range ∼150–1289 μm). By manipulating the parameters of the excitation laser we were able to reproducibly induce localized axonal lesion (Fig. 1A), which resulted in the generation of a fluorescent mark (diameter = 4.13 ± 0.32 μm, n = 10 lesions) in the tissue (Figs. 1A, B, 2). Over the following 60 min, axons on the proximal side were observed to respond to the lesion in a stereotypical manner; rapidly showing signs of beading and blebbing close to the lesion site, formation of swollen end bulbs and recovery of axon shaft integrity within minutes postlesion (Figs. 1A, 2). These reactive changes, together with the eventual disappearance of the disconnected distal axon segment in all cases (107 in total; 83 in the adult brain and 24 in the juvenile brain), verified that the axons were severed in each case. The axonal segment proximal to the cell body survived the lesion in all cases and was observed for up to 12 months for signs of remodeling. Here we describe the short (minutes, hours), medium (days), and long-term (months) changes in cortical connectivity that ensued as a result of targeted axonal lesions in the upper layers of the cortical gray matter (Fig. 1C).
Figure 2.

Stereotypical short-term changes following axonal lesion. A, Reconstruction of an axon targeted for lesion. The boxed area in red is shown in B. B, In vivo imaging of the axon in A showing short-term changes after axotomy including blebbing close to the lesion site (arrow), thinning of axon shaft, retraction of the cut axonal stump from the lesion site and eventual swelling of the axonal stump. p, Proximal; d, distal. Scale bar, 5 μm.
Two-phases of cortical axon retraction
Severed axons in both the spinal cord (Kerschensteiner et al., 2005) and optic nerve (Knoferle et al., 2010) undergo a rapid (within minutes and hours) process of fragmentation called acute-axonal degeneration, at both the proximal and distal sides. In the cortical gray matter we show that this does not happen on the proximal side, which instead retracts from the lesion site over several days and weeks. We observed 30 severed axons over minutes and hours and found no evidence of proximal axon fragmentation (Fig. 2). We measured the length of the surviving axon, relative to the lesion site, at a variety of intervals postlesion for both regrowing (n = 17 distinct axons in total across all time points) and nonregrowing axons (n = 66 distinct axons in total across all time points). Regrowing and nonregrowing axons represent two fundamentally different axonal populations in the adult brain (Canty et al., 2013).
For nonregrowing axons, the proximal stump retracted 42.9 ± 11.3 μm away from the lesion site within the first 24 h of the lesion (Fig. 3B; n = 43 axons with 1 d time points). There was a significant further retraction at 4 d (59.3 ± 15.3 μm, n = 32; p < 0.001, paired comparison between 1 d and 4 d, n = 22 axons with both time points) and at 8 d postlesion (71.9 ± 20.2 μm, n = 24; p < 0.001, paired comparison between 1 d and 8 d, n = 19 axons; p = 0.05, paired comparison between 4 d and 8 d, n = 19 axons; Fig. 3A,B). Remarkably, axons which later attempt to regrow (see Materials and Methods; Canty et al., 2013) retracted significantly less than axons which did not: for regrowing axons, average retraction was 13.8 ± 5.5 μm at 1 d postlesion (n = 14 axons with 1 d time point); 22.2 ± 9.1 μm at 4 d postlesion (n = 10); 31.7 ± 10.2 μm at 8 d postlesion (n = 8); and 14.0 ± 9.8 μm at 90 d postlesion (n = 4; Fig. 3B; p = 0.009, Wilcoxon rank-sum test comparing 1 d time points between regrowing and nonregrowing axons, and p < 0.05 nonparametric ANOVA using the 1, 4, and 8 d time points in the two groups), suggesting that the retraction and the regrowth may be related processes. There was no apparent relationship between the amount cut and the subsequent retraction as we removed similar lengths in both regrowing (498 ± 68 μm, n = 12) and nonregrowing axons (565 ± 80 μm, n = 17, p = 0.55).
In addition, there was no discernible difference between different nonregrowing axon classes in the degree of retraction after 1 d (L6 axon retraction 57.8 ± 31.8, n = 14; L2/3/5 and thalamocortical axon retraction = 35.7 ± 7.3, n = 29; p = 0.83).
To further explore the mechanisms of retraction we investigated the process in juvenile mice (3–4 weeks), at a time when neurons are still shaping their axonal arbors (i.e., elongating and retracting) (Portera-Cailliau et al., 2005). After axotomy, axons in the developing brain retracted significantly less than in the adult brain (at 4 d postlesion retraction was 15.2 ± 9.8 μm for 21 developing axons compared with 50.4 ± 12.1 μm for the 42 adult axons which had a 4 d postlesion time point; p < 0.001, Wilcoxon rank-sum test). Moreover, in the juvenile animals, there was a significant difference in retraction between axons which showed regrowth (n = 8) and those that did not (n = 16; Fig. 3C; Mann–Whitney U test using the 4 and 8 d time points, which had most data: n = 8 regrowing and 13 nonregrowing axons at 4 d, and n = 6 regrowing and 6 nonregrowing axons at 8 d, Monte Carlo significance p = 0.04), providing further evidence for a link between the growth state of the neuron and the retraction after axotomy.
A subset of axonal stumps from nonregrowing axons were observed up to 12 months postlesion (Fig. 3D; n = 12 at 3 months, n = 5 at 6 months, n = 5 at 12 months) and showed a much slower, but progressive retraction over the first 3 months (Fig. 3E; average retraction 8 d = 18.7 ± 5.0 μm; 30 d = 23.6 ± 5.5; 90 d = 31.1 ± 6.7; p = 0.04, paired comparison between 8 d and 90 d for 11 axons which were monitored for both time points). We further compared the amount of retraction among different neuron classes after ∼30 d (see Materials and Methods) and found no difference (Fig. 3F; L6 axon retraction = 44.4 ± 9.6 μm, n = 9; L2/3/5 and thalamocortical axon retraction = 27.2 ± 6.8, n = 8; p = 0.23), confirming our findings from shorter time points.
The daily retraction during the first 8 d was significantly higher than for the following 8–30 and 30–90 d (Fig. 3G; average daily retraction 0–8 d = 2.3 ± 0.63 μm; 8–30 d = 0.22 ± 0.14; 30–90 d = 0.12 ± 0.07; n = 10 axons; p = 0.0098 between 0–8 and 8–30; p = 1 between 8–30 and 30–90). This suggests the existence of two phases of retraction: an acute phase in the first week where the majority of axon retraction occurs followed by a much slower phase over several months.
Rapid bouton loss or resistance after axotomy depend on cell type and their prelesion structural dynamics
We next looked for alterations in synaptic remodeling along the surviving axon shaft as this may inform us on potential compensatory mechanisms or further functional deficits associated with axotomy, which could not be studied with other models of axonal injury. In previous work, we demonstrated that the neuronal populations studied here, i.e., from L2/3/5/6 and thalamus, have stable synaptic densities along the axon shaft in the adult brain (De Paola et al., 2006). Interestingly, despite the stability of synaptic density over time, a fraction of the synapses are formed and eliminated, which is largely dependent on axon type (De Paola et al., 2006). L6 axons mainly form synapses as TBs, rather than EPBs (McGuire et al., 1984) and undergo sustained TB changes over a few days (De Paola et al., 2006). We therefore first examined the density of L6 TBs over periods of a week after the lesion. We found a decrease in the density of TBs 4 d after the lesion (Fig. 4A,C; Table 1; n = 10 mice, 12 axons, 664 starting TBs), with no further decrease at 8 d (n = 7 axons).
The decreased density of TBs could be due to decreased TB formation or increased TB elimination or both. To distinguish between these possibilities we imaged TB dynamics at 4 d intervals. The turnover (TOR) of TBs increased 4 d after the lesion (Fig. 4D; TOR −4 d to 0 = 0.12 ± 0.02; 0 to +4 d = 0.17 ± 0.02; p < 0.01) and returned to prelesion levels 4 d later (TOR +4 d to +8 d = 0.13 ± 0.01, p > 0.05).
To more precisely determine the onset of the loss we reduced the imaging interval to include 6 h (i.e., 0.25 d) and 1 d intervals. Interestingly, there was a rapid and significant decrease in synaptic density evident as early as 6 h postlesion (Fig. 4B,C; Table 1). Both the fraction (Fig. 4E; Table 1), and the density (Fig. 4F; Table 1) of TB losses within 6 h from the lesion increased more than twofold compared with the gains, which were not affected by the lesion. This rapid TB loss was most noticeable on the axon shaft closest to the lesion site (Fig. 4G; n = 10 axons close and 9 axons far from lesion site), but significant along the entire length analyzed (up to 500 μm, p < 0.03 between 0 and 6 h).
Are newly formed or relatively stable TBs more likely to be lost after axotomy? We defined destabilized boutons as TBs that were present during the first 3 imaging sessions prelesion (i.e., between 0 and 8 d) and then lost before the end of the imaging paradigm. Because relatively stable TBs (i.e., present for all the prelesion time points) are not affected [Fig. 4H; destabilization rates are 25 ± 0.04% vs 24 ± 0.05% for 6 lesioned (353 TBs) and 8 nonlesioned (380 TBs) axons, respectively, p = 0.75, Wilcoxon rank-sum]; this suggests that TBs, which were newly formed in the week before the lesion are preferentially lost. Indeed, the fraction of TBs which were newly formed in the week before the lesion and then lost before the end of the imaging paradigm (see Materials and Methods) increases compared with nonlesioned cases, although not significantly (Fig. 4I; fraction of newly formed TBs which were subsequently lost is 60 ± 0.09% vs 46 ± 0.09% for lesioned and nonlesioned axons, respectively, p = 0.35, Wilcoxon rank-sum).
To determine the potential time course for recovery of synaptic density we analyzed TB density 1 month postlesion (Fig. 4A,C;Table 1). The decrease in TB density was maintained for at least 1 month postlesion (n = 6 mice, 8 axons). Consistent with the sustained drop in density, the rate of TB gains and losses was comparable >1 month (Nloss/total = 22 ± 2%; Ngain/total = 20 ± 3%; p = 0.25). Our data therefore suggest a lasting deficiency in synaptic output on cortical injured axons that extends behind the retracted stump for at least 500 μm.
To find out whether bouton loss after axotomy is a general phenomenon affecting diverse neuron types, a second population of axons rich in EPB synapses with cell bodies located in L2/3/5 and the thalamus was included in this study (Fig. 5). A similar analysis was undertaken at daily and 4 d intervals pre and postlesion. Surprisingly, both the density (Fig. 5B,D) and TOR (Fig. 5C,E) are not affected 1 d (n = 5 mice, 6 axons; 161 starting EPBs; −1 d density, normalized to the average of prelesion values = 0.99 ± 0.04, day 0 = 1.01 ± 0.04, +1 d = 1.04 ± 0.04; p = 1; normalized 1 d TOR, 0 to +1 d = 0.99 ± 0.21; p = 0.75) and 4 d after the lesion (−4 d density, normalized to the average of prelesion values = 0.99 ± 0.02, day 0 = 1.01 ± 0.02, +4 d = 1.12 ± 0.08, +8 d = 1.10 ± 0.21; p = 0.25 between 0 and +4 d; normalized 4 d TOR, 0 to +4 d = 1.08 ± 0.10, +4 d to +8 d = 1.13 ± 0.32; p = 0.50 between prelesion and 0 to +4 d). Moreover, only 1 of 27 TBs were lost up to 4 d postlesion (n = 5 mice, 6 axons), a result (96% stability over 4 d) consistent with published data of nonlesioned axons; De Paola et al., 2006). This, together with the fact that the rates of both destabilized and newly formed EPBs are unaffected by the lesion (data not shown), indicates that both EPBs and TBs on a subset of cortical axons are remarkably resistant to the same insult which triggers synaptic bouton loss on other types of cortical axons. Overall these data reveal a surprising difference in the vulnerability of synaptic boutons after axotomy depending on cell type and, in the case of L6 axons, on their recent history.
Absence of axon and synaptic reorganization in the surrounding neuropil
We next wanted to determine whether any axon growth/retraction or synaptic remodeling occurs in the surrounding neuropil in response to the loss of connectivity caused by the laser-induced gray matter degeneration. Taking advantage of the variability in expression patterns of GFP between animals, we selected animals with densely labeled neuropil (Fig. 6), lesioned single axons and then monitored the labeled circuitry in the surrounding area 11 d postlesion (Fig. 6; n = 3 mice, n = 13 lesions). In most cases there were axons of passage within a few μm of the lesion center (Fig. 6A, inset; the minimum distance between the center of the lesion and the closest axon was 5.4 ± 0.6 μm). We undertook a detailed analysis of total axonal length and synaptic density in 10, 30, and 100 μm diameter bins surrounding the lesion site (Fig. 6B). In all cases we excluded the lesioned axon from the analysis, which typically retracted on the proximal side and disappeared distal to the lesion. We found that there was no significant change in total axon length-either within or outside a 10 or 30 μm diameter surrounding the lesion center (Fig. 6C; normalized axon length within 10 μm = 0.99 ±0.18, p > 0.05 compared with prelesion; within 30 μm = 0.99 ± 0.07; within 100 μm = 1.00 ± 0.01; p > 0.05). We also looked for changes in synaptic density in both TB and EPB rich axons surrounding the lesion center and found that there was no significant difference in either population (Fig. 6D; normalized TB density within 10 μm = 1.31 ± 0.23, p > 0.05; normalized EPB density within 10 μm = 1.08 ± 0.11, p > 0.05; normalized TB density within 30 μm = 1.10 ± 0.19, p > 0.05; normalized EPB density within 30 μm = 0.80 ± 0.15, p > 0.05) or within 100 μm of the lesion center (data not shown). This implies that the synaptic changes do not extend extensively to undamaged circuits around the lesion site. However, as they are not labeled, we cannot exclude a direct effect on the cells immediately postsynaptic to the damaged axon.
Discussion
Using a combination of two-photon live microscopy and laser mediated axotomy we discovered a rapid and lasting process of synaptic and axon elimination after minimal CNS lesions. Although in this paper we focused on the events which occurred on the proximal side of the axon, we consider axon regeneration and associated synaptic responses, and the elimination of the distal side in two separate reports (Canty et al., 2013; Canty et al., unpublished observations). The main features of proximal axon elimination are as follows: (1) A rapid retraction phase in the first week followed by a slower one over the next 3 months. (2) Axons which later attempt to regrow retract less. (3) Synaptic bouton loss depends on cell type and their prelesion structural dynamics. Our data identify new avenues and intervention windows to counteract the functional deficits associated with axon damage. They raise neutralizing synaptic loss and progressive axon retraction as potential therapeutic strategies in addition to the already intensively explored area of axon regeneration. In particular, although preventing axon and synaptic retraction in the first few days following axonal damage is likely to have the highest functional impact; our data suggest that interventions in the following weeks and months could still be beneficial.
Laser-mediated axotomy in the living mouse brain
We have used short pulses of high-energy femtosecond lasers (Yanik et al., 2004; O'Brien et al., 2009; Ylera et al., 2009; Allegra Mascaro et al., 2010; Farrar et al., 2012; Canty et al., 2013) directed through the viewing window to induce discrete lesions (Fig. 1). A potential disadvantage of laser-mediated lesions is that the localized damage caused may not necessarily model more complex types of axonal injury. For example, laser axotomy lacks a sustained immune response, which is common in other more clinically relevant settings including stroke, multiple sclerosis, and traumatic brain injury models. A decisive advantage of such high-precision laser-surgery compared with classic needle stick-mediated injury or other traumatic injury models is that axon and synaptic dynamics can uniquely be monitored before the lesion for several days. This allows both prelesion and postlesion dynamics to be directly compared within the same axons, increasing the statistical power of the analysis. We note that key features of axon retraction (Fig. 3), and rapid synaptic remodeling (Figs. 4, 5) discovered here, would have been missed by group averaging across animals (e.g., from fixed samples, data not shown). We expect this approach to be useful to test in vivo repair strategies in ways not previously possible in the mammalian brain.
Real-time monitoring of injured cortical axons reveals a link between retraction and regrowth
After an injury axonal loss of the surviving axon is a clinically important phenomenon as it is associated with removal of synaptic connections (Raff et al., 2002; Luo and O'Leary, 2005). We find that cortical axons, belonging to several neuronal populations, retract within a few days from the lesion and progressively continue to do so for at least 3 months postlesion (Fig. 3). These data are consistent with previous reports of spinal cord injury models (Seif et al., 2007; Kerschensteiner et al., 2005; Farrar et al., 2012). Importantly, our individual axon imaging approach allowed us to gain unique mechanistic insights into this process. First, most of the axon elimination occurs by retraction with no evidence of acute-axonal degeneration (Figs. 2, 3) as in the spinal cord (Kerschensteiner et al., 2005) and optic nerve (Knoferle et al., 2010). Second, we show that retraction occurs in two phases: an acute phase, where most of the axon retracts, and a slower but progressive one, which happens over several months (Fig. 3). Third, retraction remarkably depends on the growth state of the neuron, as regrowing axons retracted to a smaller extent (Fig. 3B,C), suggesting that targeting retraction may improve the chances of axon regrowth after axotomy. Finally, it can occur in the absence of scar tissue (Ylera et al., 2009; Canty et al., 2013) and infiltrating macrophages (Horn et al., 2008), suggesting it is a cell autonomous phenomenon.
Real-time monitoring of synaptic remodeling following minimal injury in the adult brain
Together with axon regeneration, synaptic reorganization on surviving axons or spared neurons could also play a compensatory role after axonal damage. Here we identify the extent and time frame of synaptic responses. Our results show that axonal transection causes a cell type-specific elimination of axonal boutons on surviving injured axons (Figs. 4, 5), whereas the surrounding excitatory circuitry does not seem to react notably (Fig. 6). EPB density and dynamics on injured L2/3/5 and thalamocortical axons are remarkably not affected up to 8 d postlesion (Fig. 5). It will be important in the future to investigate the mechanisms for the resistance of EPBs after axotomy. In contrast, both TB density and dynamics are altered by the loss of axonal integrity and connectivity (Fig. 4A–D,G). This is due to a rapid (hours) increase in both the fraction and the density of TB loss, whereas TB formation is unaffected (Fig. 4E,F). Why are boutons proximal to the lesion site lost? One possibility is that the postsynaptic dendrites are also damaged. However, the fact that laser mediated axotomy is restricted to few micrometers around the lesion site (Canty et al., 2013; Allegra Mascaro et al., 2013) makes it unlikely that the dendrites contacted by the lesioned axons, which are located tens to hundreds of micrometers away, are also affected. Rather, the increased loss of TBs, which were newly formed in the week before the lesion (Fig. 4H,I), indicates an active elimination process perhaps aimed at saving intracellular resources. These observations indicate that targeting pathways involved in synapse elimination or formation on the surviving damaged axons is a promising new strategy to counteract the debilitating effects of axonal injury.
Footnotes
This work was supported by the Medical Research Council. We Thank: Jana Kalz, Hamlata Dechwland, and Enrico Petretto for help with experiments and analysis; Pico Caroni, Carlos Portera-Cailliau, James Fawcett, and Martin Raff for comments on a previous version of the manuscript; and Anthony Lewis for help with the figures.
The authors declare no competing financial interests.
References
- Allegra Mascaro AL, Sacconi L, Pavone FS. Multi-photon nanosurgery in live brain. Front Neuroenergetics. 2010;2:21. doi: 10.3389/fnene.2010.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegra Mascaro AL, Cesare P, Sacconi L, Grasselli G, Mandolesi G, Maco B, Knott WG, Huang L, De Paola V, Strata P, Pavone FS. In vivo single branch axotomy induces GAP-43 dependent sprouting and synaptic remodeling in cerebellar cortex. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1219256110. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JC, Martin KA. Does bouton morphology optimize axon length? Nat Neurosci. 2001;4:1166–1167. doi: 10.1038/nn772. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, McGee AW, Strittmatter SM. Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci. 2008;31:215–220. doi: 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty AJ, Huang L, Jackson JS, Little GE, Knott GW, Maco B, De Paola V. In vivo single neuron axotomy triggers axon regeneration to restore synaptic density in specific cortical circuits. Nat Commun. 2013 doi: 10.1038/ncomms3038. in press. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paola V, Arber S, Caroni P. AMPA receptors regulate dynamic equilibrium of presynaptic terminals in mature hippocampal networks. Nat Neurosci. 2003;6:491–500. doi: 10.1038/nn1046. [DOI] [PubMed] [Google Scholar]
- De Paola V, Holtmaat A, Knott G, Song S, Wilbrecht L, Caroni P, Svoboda K. Cell type-specific structural plasticity of axonal branches and boutons in the adult neocortex. Neuron. 2006;49:861–875. doi: 10.1016/j.neuron.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Farrar MJ, Bernstein IM, Schlafer DH, Cleland TA, Fetcho JR, Schaffer CB. Chronic in vivo imaging in the mouse spinal cord using an implanted chamber. Nat Methods. 2012;9:297–302. doi: 10.1038/nmeth.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, Ingham CA, Parry KE, Wright AK, Haley JE, Wishart TM, Arbuthnott GW, Ribchester RR. Delayed synaptic degeneration in the CNS of Wlds mice after cortical lesion. Brain. 2006;129:1546–1556. doi: 10.1093/brain/awl101. [DOI] [PubMed] [Google Scholar]
- Grillo FW, Song S, Teles-Grilo Ruivo LM, Huang L, Gao G, Knott GW, Maco B, Ferretti V, Thompson D, Little GE, De Paola V. Increased axonal bouton dynamics in the aging mouse cortex. Proc Natl Acad Sci U S A. 2013;110:E1514–1523. doi: 10.1073/pnas.1218731110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Knöferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, Tonges L, Stadelmann C, Bruck W, Bahr M, Lingor P. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A. 2010;107:6064–6069. doi: 10.1073/pnas.0909794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- McGuire BA, Hornung JP, Gilbert CD, Wiesel TN. Patterns of synaptic input to layer 4 of cat striate cortex. J Neurosci. 1984;4:3021–3033. doi: 10.1523/JNEUROSCI.04-12-03021.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–530. doi: 10.1093/brain/awg061. [DOI] [PubMed] [Google Scholar]
- O'Brien GS, Martin SM, Söllner C, Wright GJ, Becker CG, Portera-Cailliau C, Sagasti A. Developmentally regulated impediments to skin reinnervation by injured peripheral sensory axon terminals. Current biology: CB. 2009;19:2086–2090. doi: 10.1016/j.cub.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C, Weimer RM, De Paola V, Caroni P, Svoboda K. Diverse modes of axon elaboration in the developing neocortex. PLoS Biol. 2005;3:e272. doi: 10.1371/journal.pbio.0030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Seif GI, Nomura H, Tator CH. Retrograde axonal degeneration “dieback” in the corticospinal tract after transection injury of the rat spinal cord: a confocal microscopy study. J Neurotrauma. 2007;24:1513–1528. doi: 10.1089/neu.2007.0323. [DOI] [PubMed] [Google Scholar]
- Yanik MF, Cinar H, Cinar HN, Chisholm AD, Jin Y, Ben-Yakar A. Neurosurgery: functional regeneration after laser axotomy. Nature. 2004;432:822. doi: 10.1038/432822a. [DOI] [PubMed] [Google Scholar]
- Ylera B, Ertürk A, Hellal F, Nadrigny F, Hurtado A, Tahirovic S, Oudega M, Kirchhoff F, Bradke F. Chronically CNS-injured adult sensory neurons gain regenerative competence upon a lesion of their peripheral axon. Curr Biol. 2009;19:930–936. doi: 10.1016/j.cub.2009.04.017. [DOI] [PubMed] [Google Scholar]