

Abstract
Gangliosides are the main glycolipids of neuronal plasma membranes. Their surface patterns are generated by coordinated processes, involving biosynthetic pathways of the secretory compartments, catabolic steps of the endolysosomal system, and intracellular trafficking. Inherited defects in ganglioside biosynthesis causing fatal neurodegenerative diseases have been described so far almost exclusively in mouse models, whereas inherited defects in ganglioside catabolism causing various clinical forms of GM1- and GM2-gangliosidoses have long been known. For digestion, gangliosides are endocytosed and reach intra-endosomal vesicles. At the level of late endosomes, they are depleted of membrane-stabilizing lipids like cholesterol and enriched with bis(monoacylglycero)phosphate (BMP). Lysosomal catabolism is catalyzed at acidic pH values by cationic sphingolipid activator proteins (SAPs), presenting lipids to their respective hydrolases, electrostatically attracted to the negatively charged surface of the luminal BMP-rich vesicles. Various inherited defects of ganglioside hydrolases, e.g., of β-galactosidase and β-hexosaminidases, and of GM2-activator protein, cause infantile (with tetraparesis, dementia, blindness) and different protracted clinical forms of GM1- and GM2-gangliosidoses. Mutations yielding proteins with small residual catabolic activities in the lysosome give rise to juvenile and adult clinical forms with a wide range of clinical symptomatology. Apart from patients' differences in their genetic background, clinical heterogeneity may be caused by rather diverse substrate specificities and functions of lysosomal hydrolases, multifunctional properties of SAPs, and the strong regulation of ganglioside catabolism by membrane lipids. Currently, there is no treatment available for neuronal ganglioside storage diseases. Therapeutic approaches in mouse models and patients with juvenile forms of gangliosidoses are discussed.
Introduction
Gangliosides had been discovered by Ernst Klenk in the 1930s (Klenk, 1937, 1939) when he analyzed postmortem brain tissues of patients with Tay–Sachs disease, a fatal infantile form of amaurotic idiocy. At that time the clinical diagnosis “amaurotic idiocy” comprised an inherited disease characterized by high numbers of lipid-laden cells in the nervous system and the visceral organs, mental retardation, and impaired vision or blindness. With the help of biochemical, enzymatic, molecular genetic, morphological, and animal studies, the clinical picture of amaurotic idiocy has been resolved in the last five decades into two major groups of diseases at the molecular level: lysosomal ganglioside storage diseases, comprising GM1-gangliosidoses and 4 forms of GM2-gangliosidoses, and 10 forms of neuronal ceroid lipofuscinoses poorly characterized so far (Jalanko and Braulke, 2009).
Gangliosides are typical components of neuronal plasma membranes. The first ganglioside structure, that of ganglioside GM1 (Fig. 1), was elucidated by Kuhn and Wiegand (1963). Six major and many minor ganglioside species have been identified in mammalian brains (Yu et al., 2011). Inherited defects in ganglioside metabolism can cause fatal neurodegenerative diseases. Best known so far are inherited defects in lysosomal ganglioside catabolism, the GM1- and GM2-gangliosidoses.
Figure 1.
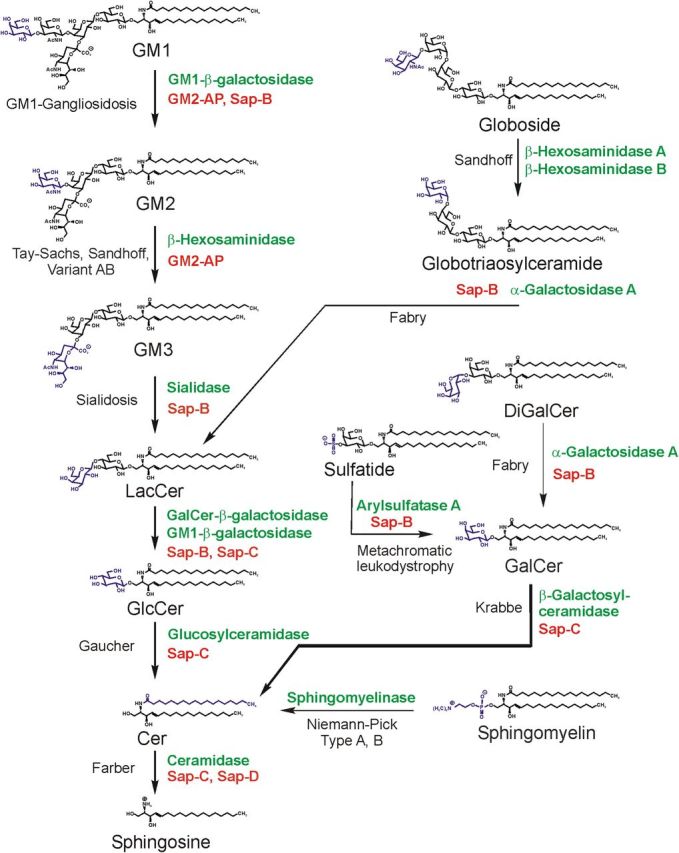
Pathway of lysosomal sphingolipid degradation. The eponyms of known metabolic diseases and those of SAPs (red) necessary for in vivo degradation are indicated. Hydrolases are given in green. Heterogeneity in the lipid part of the sphingolipids is not indicated. Variant AB, Variant AB of GM2-gangliosidosis [deficiency of GM2-activator protein (GM2-AP)]; Sap, sphingolipid activator protein (modified from Sandhoff and Kolter, 1995).
Up to now, no defects in ganglioside biosynthesis have been described in humans with the exception of two reports on three patients with an infantile onset refractory epilepsy (Simpson et al., 2004; Fragaki et al., 2013), caused by a deficiency of ganglioside GM3-synthase. It is associated with psychomotor delay, blindness, deafness, and respiratory chain dysfunction. In addition, one patient with a genetically undisclosed basis of infantile GM2-synthase deficiency, a ganglioside GM3 storage and loss of higher gangliosides has been described, passed away at age 3.5 months (Fishman et al., 1975).
However, blocks in ganglioside biosynthesis generated in gene-manipulated mice cause a broad spectrum of pathological phenotypes (Proia, 2003).
Ganglioside biosynthesis and its defects in genetically manipulated mice
Glycolipids are expressed in a cell-type- and stage-specific manner, gangliosides being the main glycolipids of neuronal surfaces. Their surface patterns are generated by coordinated processes, involving biosynthetic and catabolic steps and intracellular trafficking.
Their hydrophobic membrane anchors, the ceramide residues, are formed in the endoplasmic reticulum (ER), glycosylation of ceramide takes place at the Golgi and trans-Golgi network (TGN), and their constitutive degradation mainly in the endolysosomal compartment (Kolter and Sandhoff, 1999). After formation of glucosylceramide (GlcCer) from ceramide (Cer) at the cytosolic surface of Golgi membranes and its flip to the luminal surface of Golgi membranes, GlcCer gets converted to lactosylceramide (LacCer). At the luminal surfaces of Golgi and TGN membranes, LacCer is sialylated to form small precursor gangliosides (GM3, GD3, and GT3). They are then converted by membrane-bound promiscuous glycosyltransferases to complex ganglioside structures, which reach cellular surfaces by vesicular transport of the secretory pathway. This concept of “combinatorial ganglioside biosynthesis” (Kolter et al., 2002) allows the generation of cell-specific patterns on neuronal surfaces depending on the availability of precursor molecules, expression of biosynthetic enzymes and their regulation. Generation and analysis of mutant mice with defects in the pathways of ganglioside biosynthesis supported this model (Kolter et al., 2002; Sandhoff and Kolter, 2003). Mutant mice defective in b-series gangliosides were viable and without apparent neurological abnormalities (Kawai et al., 2001; Okada et al., 2002) and reach an almost normal life span. Mice defective in GalNAc-transferase, missing the major brain gangliosides, were viable but suffered from instability of brain structures with detachment of myelin sheets from axonal membranes (Schnaar, 2010) and male infertility. A combined defect of GM3-synthase and GM2-synthase causes an early fatal disease. Here, the absence of all a-series and b-series gangliosides triggers the formation of 0-series gangliosides, which are not observed in the wild-type brain. Their additional synthesis, however, cannot compensate the loss of all the major neuronal ganglioside structures. Surprising was that the total ganglioside content in the brains of the various mutant mice stayed almost unchanged at the wild-type level (Sandhoff, 2012).
The ubiquitous block of GlcCer synthesis, the precursor of the main glycosphingolipids (GSLs), is lethal in mice at embryonic day 6 (Yamashita et al., 1999). However, its cell-type-specific knock out in neurons (Jennemann et al., 2005), hepatocytes (Jennemann et al., 2010), or keratinocytes (Jennemann et al., 2007) still allowed complete embryogenesis but with the birth of fatally sick animals. Apparently, some complex glycolipids derived from GlcCer such as the stage-specific embryonic antigens (Kannagi et al., 1983; Eggens et al., 1989) involved in cellular adhesion processes are needed at early embryonic stages for development of a multicellular organism (Kolter et al., 2000).
Gangliosides have been discovered as GSLs of neuronal membranes by lipid analysis of Tay–Sachs disease (Klenk, 1937, 1939), but their complex chemical structure was elucidated only later in the 1960s (Kuhn and Wiegandt, 1963). Their storage in neuronal lysosomes paved the way for the analysis of their cellular metabolism and lysosomal catabolism (Sandhoff et al., 1969; Sandhoff, 1977)
Clinical phenotypes, correlations to pathological and biochemical phenotypes
All types of gangliosidoses are caused by inherited defects in ganglioside catabolism (Fig. 1) and can manifest with severe, extremely variable clinical symptoms (Table 1) in almost any age group, although most patients have an early infantile onset and a late infantile or juvenile, fatal end of their disease, which is dominated by the central nervous involvement (Fig. 2A) (Lyon et al., 1996a). The cerebral degeneration is marked by the lipid storage present in neurons with the ultrastructurally characteristic “membranous cytoplasmic bodies,” which are secondary, “storage” lysosomes (Fig. 2B). These storage bodies distend the neurons (Fig. 2C), which eventually are lost, but the exact reason is unclear (for example, cell inactivity by ganglioside-induced impairment of synaptic transmission is discussed). The peripheral and autonomous nerve systems are also involved, but with minor clinical relevance, though lysosomal storage bodies are demonstrable at places.
Table 1.
The main clinical hallmarks and symptoms in gangliosidoses
| Disease | Manifestation | Regular symptoms/signs | Facultative symptoms/signs | Remarks |
|---|---|---|---|---|
| GM1-gangliosidosis | Infantilea,b | Dysmorphic face, severe dysostosis, hepatomegaly, unable to sit, muscle hypotonia, later on spasticity, visual failure, foam cells in bone marrow, oligosacchariduria, mucopolysacchariduria | Cherry-red macular spotc, nystagmus, seizures, splenomegaly, joint contractures, hematologic signs, vacuolated blood lymphocytesd | Rapid or 1–2 year downhill course with psychomotor deterioration, general dystrophy, feeding problems |
| Juvenilea | By 4 years or later: dysarthria, extrapyramidal signs including dystonia | Slight intellectual impairment, mild bone changes, foam cells in bone marrow | Some patients have also parts or attenuated forms of the symptoms listed for the infantile disease, other patients belong to the chronic group | |
| Adulta | As in juvenile disease, but with more insidious onset | Psychosis | Disease is chronic rather than with only adult manifestation | |
| Chronic | Combines late infantile, juvenile, and sometimes adult manifestations in which the above listed symptoms are partly or gradually developed | |||
| GM2-gangliosidosis, variants B, B1 and 0 | Infantilee | Loss of head/trunk control, smiling, by about 6 months, cherry-red macular spotc, high startle response to noise, muscle hypotonia by 1 year, epilepsy, tetraparesis with some spasticity, dementia, blindness | Megalencephaly, visceral involvement and oligosacchariduria only in variant 0 | Downhill course mostly less rapid than in early GM1-gangliosidosis |
| Juvenile/subacute | Onset by 3 to 6 years, dysarthria, loss of speech, spastic paraparesis, later on paraplegyf, pyramidal signsf, cerebellar ataxia, mental deterioration | Seizures, irritability, psychiatric signs, denervation muscle atrophy (EMG), areflexia, cherry red macular spotc | Clinical variability even in siblingsg, above remark to juvenile GM1-gangliosidosis here also apt | |
| Adult | Clumsiness, lower motor neuron disease, denervation muscle atrophy, cerebellar ataxia | Psychosis | High clinical variability, oldest patient was 76 years | |
| Chronic | See remark to chronic GM1-gangliosidosis | |||
| GM2-gangliosidosis, variant AB | Most as in variants B, B1, 0 | Cherry red macular spotc | No adult patients known |
aGrouping of GM1-gangliosidosis patients as type I, II, and III of the disease according to the age at clinical onset is questionable by the often ambiguous onset or insidious or chronic manifestation through all, except the early infantile, age groups (Lyon et al, 1996b).
bEarlier named “generalized gangliosidosis.”
cSee Fig. 2D.
dSee Fig. 2E.
eEarlier named “infantile amaurotic idiocy”; relatively high prevalence of variant B (Tay–Sachs disease) in Ashkenazim.
fSee Fig. 2A.
gMany patients are Ashkenazim, but a genetic isolate of variant B1 is in Spain.
Figure 2.

Clinical and morphological observations in gangliosidoses. A, Near end stage in a Spanish 13-year-old patient with GM2-gangliosidosis variant B1 (photograph courtesy of H.H. Goebel, Mainz). B, GM2-gangliosidosis variant 0 in a 23-week-old fetus; and part of a cortical neuron; early stage of membranous cytoplasmic bodies (two clusters marked by crosses), i.e., lipid storing secondary lysosomes, ×36,000 (courtesy W. Schlote, Tübingen). C, The large cells are neurons distended by lysosomal storage in the medulla oblongata of a 1.5-year-old patient with GM1-gangliosidosis. The small dark bodies are nuclei from increased numbers of glial cells (“reactive gliosis”). Cresyl violet stain ×300 (courtesy of H.U. Benz, Tübingen). D, Cherry red macular spot in an infantile patient with GM2-gangliosidosis variant B (Tay–Sachs disease). The dark red spot in the middle of the light central area is secondary to lipid storage in neuronal cells in this area; the storing cells have lost their processes that normally cover the fovea centralis. The fovea normally appears as yellow, but is changed to the red macular spot showing the color of the choroidea behind the retina. E, Lymphocytes with clusters of vacuoles (storage lysosomes) in blood smear of an early infantile patient with GM1-gangliosidosis, indicating the “generalized storage disorder.” May-Grünwald stain; ×1500 (courtesy of H.U. Benz, Tübingen).
In addition to the neuronal involvement, an extraneural involvement (Table 1) is striking in infantile and partly present in juvenile patients with GM1-gangliosidosis; even an intrauterine manifestation with hydrops fetalis is possible, and a dysmorphic (gargoyle-like) face is regular in early infantile patients. Juvenile and adult GM1-gangliosidosis patients have a predominant or almost pure neurologic picture (Suzuki et al., 2001). Extraneural manifestations are absent in GM2-gangliosidosis with the exception of variant 0, also called Sandhoff disease, in which slight visceromegaly is frequent (Gravel et al., 2001) and some involvement of bones and even the heart can occur (Venugopalan and Joshi, 2002).
The clinical phenotypes in the gangliosidoses can, though with some caveats, be correlated to the different types and numbers of substrates, which are accumulated in these diseases. Examples of the substrates of the acid hydrolases whose deficiencies define the diseases are gangliosides, asialogangliosides, other glycolipids, specific oligosaccharides and, in case of acid β-galactosidase, mucopolysaccharides. In the deficiencies, the patterns of accumulated substrates can be explained in a simplified format as follows. The main substrates of the acid β-galactosidase, which are deficient in GM1-gangliosidosis, are GM1-ganglioside (Fig. 1), specific oligosaccharides, and keratan sulfate, so that this disease is marked not only by ganglioside storage (whose main pathologic correlate is neuronal degeneration) but also by oligosaccharidosis and mucopolysaccharidosis, with the extraneuronal clinical involvement due to the latter. The main substrate of the β-hexosaminidase A deficient in GM2-gangliosidosis variant B (also called Tay–Sachs disease) or suppressed in the GM2-activator-deficient variant AB, is GM2-ganglioside, so the disease is “only” a ganglioside storing disorder. The main substrates of the β-hexosaminidases A and B deficient in GM2-gangliosidosis variant 0 are GM2-ganglioside and oligosaccharides, so the disease includes gangliosidosis and oligosaccharidosis, with some visceral involvement due to the latter.
Another correlation can, though with some reasonable doubts, be assumed between the time (as patients' age) of onset or manifestation, or the duration (chronicity) of the disease, and the rudimentary catabolic functions of the defective (if not completely absent) acid hydrolases in the patients. This “residual activity hypothesis” postulates higher residual functions for late starting/manifesting or chronic forms of gangliosidoses than for early (infantile) disease forms (see Protracted clinical forms of gangliosidoses and the threshold theory, below). Although the low residual, probably pathophysiologically relevant activities of the defective hydrolases cannot always reliably be assessed under laboratory conditions in vitro, that hypothesis seemed to be acceptable for many patients who were studied enzymatically. As to those residual functions, it is interesting that in late manifesting GM1-gangliosidosis the almost only substrate relevant for the biochemical phenotype is the ganglioside accumulation, particularly in neurons, while for the breakdown of other substrates the residual functions seem to be nearly sufficient.
However, in a certain type of genetic acid β-galactosidase deficiency, also a sufficient catabolism of GM1-ganglioside is possible, whereas the breakdown of other substrates is severely impaired, and the resulting disease is not gangliosidosis but mucopolysaccharidosis IV (Morquio) type B (Suzuki et al., 2001). This indicates different substrate (e.g., the ganglioside/activator protein complex) binding sites, protein-folding-relevant sites, or subdomains in the enzyme, which are required for the catabolism of different substrates, and suggests that these sites are differentially or alternatively defective in different patients who have then very variable diseases caused by their β-galactosidase defects. On the other hand, a special GM1-ganglioside-storing disease (associated with oligosaccharide storage) results from an indirect deficiency of acid β-galactosidase (and sialidase, and a specific sulfatase) in galactosialidosis, which is caused by the genetic deficiency of the so-called protective protein (identical to carboxypeptidase A) (d'Azzo et al., 2001). When this protein is defective, the functional complex of the normal protein with acid β-galactosidase and sialidase (and the sulfatase) cannot be formed, and these enzymes, though having a normal structure, are subject to rapid decay. The resulting clinical phenotype is very similar to that in classic GM1-gangliosidosis (and to that in the genetic sialidase defect, sialidosis). So an enzymatic diagnosis of GM1-gangliosidosis has always to be confirmed by demonstrating the presence of normal sialidase activity, or by molecular gene analysis.
Summary outline of clinical symptomatology
The main symptomatology of the gangliosidoses is given as a partially simplified summary in Table 1. Clinical details should be found in the special literature (Lyon et al., 1996a, b; Gravel et al., 2001; Suzuki et al., 2001). Pathologists have found that in gangliosidosis the brain is sometimes slightly infiltrated with inflammatory cells considered to be a response to the dysfunction of the lipid-laden neurons and to the abundance of antigenically acting gangliosides. Here follow some supplementary clinical remarks. Neuroimaging has no essential role in the characterization of gangliosidoses, but in late disease stages cerebral and cerebellar atrophy can often be documented and slight T2-weighted hyperintensities may appear earlier in the brainstem and white matter (Wilken et al., 2008). Magnetic resonance spectroscopy (N-acetyl aspartate, myoinositol, and other signals) may be helpful in documenting neuronal loss and inflammation (Assadi et al., 2008). The prenatal and postnatal laboratory diagnosis of gangliosidoses is usually made enzyme biochemically (e.g., in blood samples), and/or molecularly. Many aspects of genotype–phenotype correlation in these diseases were described in the special literature. Therapeutic strategies such as bone marrow transplantation (BMT), use of drugs with substrate reducing, chaperon-like and anti-inflammatory effects, and intracerebral or intrathecal enzyme replacement are under investigation (see Therapeutic approaches, below).
Pathway of ganglioside degradation
Topology of endocytosis and lysosomal digestion of gangliosides
Lysosomes are intracellular stomachs. They degrade macromolecules and release their components as nutrients into the cytosol for salvage pathways and energy metabolism. Macromolecules and membrane components reach the lysosomal compartment for digestion by autophagy (Florey and Overholtzer, 2012), phagocytosis (Delves et al., 2006; Florey and Overholtzer, 2012), and by endocytotic pathways (Kolter and Sandhoff, 2005). Whereas water-soluble lysosomal hydrolases can attack water-soluble macromolecules directly, the digestion of gangliosides, GSLs, and membranes needs a more complex cooperation between soluble hydrolases, lipid binding and transfer proteins, and luminal intra-endolysosomal vesicles (Kolter and Sandhoff, 2005; Kolter and Sandhoff, 2010). Due to the lipid phase problem, water-soluble glycosidases hardly attack amphiphilic GSLs as components of vesicular membranes, but need the help of membrane perturbing and lipid binding proteins, the sphingolipid activator proteins (SAPs).
Our view of the topology of lysosomal ganglioside digestion
For their digestion, gangliosides of cellular surfaces reach luminal intra-endolysosomal vesicles or intra-endosomal membranes (IMs) (Fig. 3; Burkhardt et al., 1997; Möbius et al., 1999), which are generated during endocytosis. Luminal vesicles are formed by successive steps of vesicle budding and fission controlled by the endosomal-sorting complex proteins (Wollert and Hurley, 2010). The vesicles are prepared for lysosomal digestion by a lipid-sorting process beginning at the level of endosomes (Kolter and Sandhoff, 2005; Gallala et al., 2011). Cholesterol is sorted out by two sterol binding proteins, NPC-2 and NPC-1 (Abdul-Hammed et al., 2010), and sphingomyelin is degraded by acid sphingomyelinase (ASM; V. Oninla, B. Breiden, K. Sandhoff, unpublished observations). Anionic bis(monoacylglycero)phosphate (BMP) that stimulates ganglioside catabolic steps substantially is formed from phosphatidyl glycerol in the IMs (Gallala and Sandhoff, 2011; Gallala et al., 2011). Whereas the lysosomal perimeter membrane seems to be quite resistant against lysosomal degradation, IMs are digested with the help of lipid binding proteins (SAPs) and hydrolytic enzymes. Perimeter membranes are protected by a thick glycocalyx enriched in polylactosamine structures, and by a high cholesterol level (Hay, 1989; Appelqvist et al., 2012), the chaperon HSP70 (Kirkegaard et al., 2010), and presumably also by a high lateral pressure attenuating the interaction with GM2AP, a lipid binding protein essential for ganglioside catabolism (Giehl et al., 1999).
Figure 3.

Proposed topology of endocytosis and lysosomal lipid and membrane digestion (Gallala et al., 2011). “A section of the plasma membrane is internalized by way of coated pits or caveolae. These membrane patches include GSLs (red) and receptors such as epidermal growth factor receptor (EGFR; blue). These vesicles fuse with the early endosomes, which mature to late endosomes. Endosomal perimeter membranes form invaginations, controlled by ESCRT proteins (Wollert and Hurley, 2010), which bud off, forming intra-endosomal vesicles. Lipid sorting occurs at this stage. The pH of the lumen is ∼5. At this pH, ASM is active and degrades sphingomyelin of the intra-endosomal vesicles to ceramide, whereas the perimeter membrane is protected against the action of ASM by the glycocalyx facing the lumen. This decrease in sphingomyelin, coupled with the increase in ceramide, facilitates the binding of cholesterol to NPC2 and its transport to the perimeter membrane of the late endosome where it is transferred to NPC1 (Kwon et al., 2009). This protein mediates the export of cholesterol through the glycocalyx, eventually reaching cholesterol binding proteins in the cytosol. Ultimately, late endosomes fuse with lysosomes. The GSLs are harbored in intralysosomal vesicles that face the lumen of the lysosome, and are degraded by hydrolases with the assistance of SAPs. The products of this degradation are exported to the cytosol or loaded on CD1b immunoreceptors and exported to the plasma membrane for antigen presentation. Gradients of pH in the lysosol, and intra-endolysosomal vesicle content of cholesterol (Chol), BMP, sphingomyelin (SM, hypothetical), and ceramide (Cer, hypothetical) are shown” (modified from Kolter and Sandhoff, 2010).
Biochemistry of ganglioside catabolism and its diseases
Ganglioside catabolism proceeds in a stepwise procedure (Fig. 1) at the surface of luminal intralysosomal vesicle and membrane structures. These are enriched with anionic BMP, which attracts polycationic proteins, glycosidases, and SAPs, to the ganglioside-containing membrane surfaces in the acidic intralysosomal environment. Whereas GM1 hydrolyzing β-galactosidase and GM2 splitting HexA bind to these anionic surfaces, they do not attack the membrane bound ganglioside substrates in the absence of membrane perturbing lipid binding and transfer proteins. The first case of GM1 storage was described in 1963 as a special form of infantile amaurotic idiocy (Jatzkewitz and Sandhoff, 1963). Hydrolysis of vesicle-bound GM1 by lysosomal ganglioside β-galactosidase needs the presence of GM2AP or saposin B (Sap-B; Wilkening et al., 2000). Anionic lipids like BMP in the GM1 carrying vesicles stimulate GM1 hydrolysis efficiently. Inherited defects of the GM1 degrading β-galactosidase cause GM1-gangliosidoses (Okada and O'Brien, 1968).
The nonspecific and versatile GM1 degrading β-galactosidase occurs as part of a lysosomal multi-enzyme complex, containing sialidase, cathepsin A (the so-called protective protein) (d'Azzo et al., 2001), and N-acetylaminogalacto-6-sulfate sulfatase (Pshezhetsky and Ashmarina, 2001). Due to changes of the substrate specificity of variant patient enzymes, an inherited defect of GM1-β-galactosidase may also lead to a major accumulation of galactose containing keratan sulfate and oligosaccharides, a phenotype called Morquio syndrome, type B (mucopolysaccharidosis IV B) (Neufeld and Muenzer, 2001; Okumiya et al., 2003).
Hydrolysis of GM2 is facilitated at anionic vesicular surfaces by the cooperation of two proteins, HexA (α,β) and the membrane perturbing and lipid binding protein GM2AP (Fig. 4) (Kolter and Sandhoff, 2006). At acidic pH values, the latter one is a protonated cationic amphiphilic protein and can bind to the anionic, BMP-rich membrane surfaces, and lift a ganglioside molecule (Fig. 4). A HexA binding helix of the GM2AP facilitates formation of the Michaelis–Menten complex for the degradation of GM2, the Tay–Sachs ganglioside, and glycolipid GA2. Inherited defects of either of the proteins result in fatal neurodegenerative diseases (Kolter and Sandhoff, 2006). Tay–Sachs disease is caused by mutations in the HexA gene with loss of HexA (α,β) and S (α,α) activity. HexA mutations that still allow the formation of the heterodimeric HexA (α,β) and affect only its activity against anionic substrates result in B1 variant of GM2-gangliosidosis (Kytzia et al., 1983; Kytzia and Sandhoff, 1985). On the other hand, inherited defects in HexB gene cause the loss of HexA (α,β) and HexB (β,β) activities in Sandhoff disease. Here, the combined deficiency of both major hexosaminidase isoenzymes causes the additional storage of neutral glycolipids, especially of globoside in the visceral organs, and of oligosaccharides. The rare defect of GM2AP results in AB-variant of GM2-gangliosidoses (Sandhoff et al., 1989; Sandhoff and Kolter, 2003). Genetically manipulated knock-out mice are available as models for these diseases to study their course and pathogenesis, and to investigate therapeutic approaches.
Figure 4.

Mechanism of GM2-AP-Liftase. A, Model for the interaction of GM2-activator protein with luminal lysosomal membranes (IM) in the degradation of ganglioside GM2 (modified from Wendeler et al., 2004a). “GM2-AP interacts with the membrane by dipping the two exposed hydrophobic loops, V90–W94 and V153–L163, into the apolar part of the membrane. Ganglioside GM2 is recognized by specific sites at the rim of the cavity. In the open protein conformation, the large hydrophobic area reaching from the apolar phase of the membrane to the activator's cavity lowers the energy barrier for lipids leaving the membrane in an upward direction. After the ceramide tail has arrived inside the activator's cavity, the inward-bending of the hydrophobic loop V153–L163 is favored, and the conformation changes to the closed form (Wright et al., 2003). This folding in of the hydrophobic loop leaves a more polar patch close to the membrane. The activator is then anchored only by the loop V90–W94. It may now rotate slightly upward to expose all polar patches more fully to the solvent, and it may also leave the membrane and interact with the degrading enzyme. The photoaffinity label analogs C(n = 5,1)-TPD-GM2 were photo-incorporated specifically into the region V153–L163” (Sandhoff, 2012). B, BMP and GM2-AP stimulate the hydrolysis of LUV-bound ganglioside GM2. The degradation of ganglioside GM2 inserted into LUVs doped with 0 mol % BMP (○), 5 mol % BMP(□), 10 mol % BMP (·), 20 mol % BMP (▴), or 25 mol % BMP (■) was measured in the presence of increasing concentrations of GM2-AP (0–2.2 μm) (Werth et al., 2001).
Protracted clinical forms of gangliosidoses and the threshold theory
Complete or nearly complete loss of GM1 or GM2 catabolic activities results in fatal infantile forms of storage diseases. However, allelic mutations, which produce proteins with some residual catabolic activities, give rise to protracted clinical forms often described as late infantile, juvenile, or chronic diseases (Sandhoff et al., 1989) with a wide range of clinical symptomatology. At the biochemical level, the clinical heterogeneity is paralleled by a variation of the extent and the pattern of glycolipid accumulation and by different levels of residual catabolic activities detected in cultures of patients' fibroblasts (Fig. 5). Catabolic activities were assayed in cell cultures using the physiological, radiolabeled lipid substrates instead of the water-soluble synthetic substrates, which serve widely for diagnostic purposes. The water-soluble substrates may well lead to erroneous data because of the lipid phase problem and the absent control by GM2AP (and other activators) of the catabolism of physiological lipid substrates such as GM2 ganglioside. According to Figure 5, GM2 cleavage by HexA was almost absent in fibroblasts of infantile patients, and significantly higher in those of juvenile and adult patients (Kytzia et al., 1984; Leinekugel et al., 1992). This observation can well be modeled on the basis of a greatly simplified kinetic model (Conzelmann and Sandhoff, 1983, 1991; Leinekugel et al., 1992) presented in Figure 5. A nice correlation was observed between the predicted and the measured lipid substrate turnover, and also between the decrease of the ganglioside GM2 turnover and the clinical course of the disease. Ten to 20% of normal GM2 cleaving activity appear already compatible with normal life. Similar observations were reported for metachromatic leukodystrophy (MLD), Gaucher, Sandhoff, and ASM-deficient Niemann–Pick disease (Kudoh et al., 1983; Kytzia et al., 1984; Leinekugel et al., 1992; Graber et al., 1994).
Figure 5.

Residual catabolic activity correlates with clinical forms of GM2 gangliosidoses (modified from Sandhoff, 2012). A, Steady-state substrate concentration as a function of enzyme concentration and activity (Conzelmann and Sandhoff, 1983). The model underlying this theoretical calculation assumes influx of the substrate into the lysosomal compartment at a constant rate (vi) and its subsequent utilization by the catabolic enzyme. Blue line = [S]eq steady-state substrate concentration; ………… = theoretical threshold of enzyme activity; ——- = critical threshold value, taking limited solubility of substrate into account; red line = turnover rate of substrate (flux rate). B, To experimentally verify the basic assumptions for this model, studies were performed in cell culture (Leinekugel et al., 1992). The radiolabeled substrate ganglioside GM2 was added to cultures of skin fibroblasts with different activities of β-hexosaminidase A and its uptake and turnover measured. The correlation between residual enzyme activity and the turnover rate of the substrate was essentially as predicted: degradation rate of ganglioside GM2 increased steeply with residual activity, to reach the control level at a residual activity of ∼10–15% of normal. All cells with an activity above this critical threshold had a normal turnover. Comparison of the results of these feeding studies with the clinical status of the donor of each cell line basically confirmed our notions but also revealed the limitations of the cell culture approach.
Of course, additional factors induced by the mutation and the storage process in the disease will also contribute to the clinical course and disease pathogenesis, e.g., misregulation of proteins and intracellular trafficking, formation of lytic and toxic compounds (Nilsson et al., 1982; Igisu and Suzuki, 1984), depletion of precursor pools, alterations of composition and function of membranes, mislocalized storage compounds like GM1, which activate ER-chaperons and trigger neuronal apoptosis (Tessitore et al., 2004), and altered regulation of endolysosomal proteins and enzymes by an increased load of lipids (see below). Also the formation of meganeurites and an increase in synaptic spines may well disturb the connectivity in the nervous system (Purpura and Suzuki, 1976).
A genetic mutation and loss of a catabolic activity may not only result in accumulation of nondegradable substrates but also affect the transcription rate of many other genes. Mutations in the HexB gene and the subsequent loss of HexA and HexB activity in genetically manipulated mice trigger a significant downregulation of ∼300 transcripts and a significant upregulation of another ∼250 transcripts including genes of the immune system (Myerowitz et al., 2002).
Genetically manipulated mice are not always faithful copies of the human disease. In contrast to Tay–Sachs patients, HexA knock-out mice reach an almost normal life span and hardly get sick. The reason seems to be a small difference in the substrate specificity of murine and human sialidase. Whereas human sialidase is almost inactive, murine sialidase has a minor but significant cleaving activity on the storage compound ganglioside GM2 and thereby opens a catabolic bypass (Sandhoff and Jatzkewitz, 1967; Sango et al., 1995). Mutant mice develop only a minor storage process in some neurons, e.g., in the pyramidal cells of the cortex (Taniike et al., 1995). Apparently, the catabolic activity of the bypass (sequential action of murine sialidase to convert GM2 to GA2, and hex B to convert GA2 to LacCer) is sufficient in almost all neurons of the cerebral cortex except in the murine pyramidal cells and in a few others. These cells probably have a rather high biosynthetic rate for gangliosides, thereby slightly exceeding their maximal catabolic capacity (Vmax of the catabolic system; Fig. 5) in case of hex A knocked out. Further pathogenetic cascades activated in these diseases, altered lipid trafficking, altered autophagy, and induction of inflammation, have been reviewed (Vitner et al., 2010; Xu et al., 2010).
Many proteins involved in ganglioside metabolism are nonspecific
The inherited defect of an endolysosomal protein usually affects only one step in ganglioside and sphingolipid catabolism. However, a more careful investigation of the proteins revealed that many of them have rather promiscuous functions and, therefore, have a broader effect on metabolism than originally expected. For glycosidases this has been known for a long time, and it was no surprise to find out that GM1-β-galactosidase hydrolyzes many other β-galactosides in addition to ganglioside GM1 and GA1 (Kolter and Sandhoff, 2006). Mutations in the β-galactosidase gene can even change the enzyme's substrate specificity and affect the catabolism of GSLs and glycosaminoglycans differently.
It was also no surprise to realize that human β-hexosaminidases have broad and partially overlapping substrate specificities. Again some mutations in the HexA gene may also change substrate specificity of the patients' HexA. Those that disrupt enzyme activity only against anionic substrates (like GM2) and not against neutral substrates (like globoside) cause B1 variant of GM2-gangliosidosis (Kytzia et al., 1983), whereas those that affect HexA activity against all substrates including neutral substrates cause Tay–Sachs disease (variant B of GM2-gangliosidosis). Active β-hexosaminidases are the homodimers HexB (β,β) and HexS (α,α), and the heterodimer HexA (α,β). Each of them carries two active sites at the domain interfaces (Maier et al., 2003; Lemieux et al., 2006). Therefore, mutations affecting the dimer formation produce inactive monomeric subunits causing infantile diseases. The broad and overlapping specificity of murine hexosaminidases A, B, and S is highlighted by mice lacking both subunits of β-hexosaminidase. They store gangliosides and glycosaminoglycans and display phenotypic, pathologic, and biochemical features of mucopolysaccharidoses, not seen in Tay–Sachs and Sandhoff disease (Sango et al., 1996).
The first activator protein described, the sulfatide activator protein, is now called Sap-B. It was discovered as an essential cofactor for the lysosomal catabolism of sulfatides (Mehl and Jatzkewitz, 1964). Its deficiency causes a late infantile form of MLD with a pronounced sulfatide storage (Mehl and Jatzkewitz, 1964; Stevens et al., 1981). Sap-B combines with sulfatides, and the formed stoichiometric soluble complex then forms a Michaelis–Menten complex with arylsulfatase A (Mehl and Jatzkewitz, 1964; Mraz et al., 1976). Though this suggested a high lipid binding specificity of Sap-B, we now know that the human Sap-B is not specific for sulfatide binding, but rather a promiscuous lipid binding protein (Gärtner et al., 1983; Sun et al., 2008). It even stimulates the hydrolysis of bacterial lipids by bacterial hydrolases (Li et al., 1988). Also the other saposins have broad functions (Doering et al., 1999) and are rather unspecific lipid binding proteins, like the GM2AP (Wendeler et al., 2004b). Even the glycosyltransferases involved in the biosynthesis of complex gangliosides at Golgi membranes are rather versatile. There are only one GalNAc-, one Gal-, and two sialyl-transferases available in Golgi membranes to convert the precursors LacCer, GM3, GD3, and GT3 into the different complex gangliosides of the 0-, a-, b-, and c-series (Kolter et al., 2002).
Membrane lipids are regulators of ganglioside catabolism
Promiscuous functions of endolysosomal proteins and enzymes make it difficult to understand the precise molecular pathology of the diseases caused by a defect of any of these proteins. The complexity is further increased by the strong lipid dependency of many endolysosomal proteins, given that the lipid composition of many organelle membranes is mostly unknown, especially in patients' tissues. In vitro experiments have shown that catabolic steps at surfaces of substrate carrying liposomes—simulating luminal endolysosomal vesicles—not only need low pH values, enzymes, and SAPs for significant degradation rates, but also a sufficient concentration of anionic lipids such as BMP in the liposomal membranes.
Both, GM2AP and Sap-B, allow that β-galactosidase hydrolyzes GM1. Therefore, neither a defect of GM2AP nor of Sap-B causes a GM1 accumulation, since the other one left efficiently facilitates the reaction. However, the reaction is rather slow in neutral liposomes and is stimulated 2- to 9-fold in the presence of an anionic phospholipid such as BMP, phosphatidylinositol, dolicholphosphate, and phosphatidylserine (Wilkening et al., 2000). Even at acidic, lysosomal pH values, these phospholipids convey negative charges to the vesicular surfaces, which attract and bind the protonated and positively charged hydrolases and SAPs to catalyze the hydrolytic reaction. This view also applies to the degradation of liposomal ganglioside GM2 by HexA in the presence of GM2AP. GM2AP is essential for the enzymatic hydrolysis of ganglioside GM2, even in the presence of high BMP concentrations. Its absence causes the fatal neurodegenerative AB variant of GM2-gangliosidosis in vivo. Still, the reaction is stimulated by anionic BMP >100-fold (Werth et al., 2001) (Fig. 4B). Also downstream reactions (Fig. 1) are stimulated by anionic membrane lipids. At concentrations of anionic phospholipids >10 mol% in the liposomal membranes, the cleavage of GlcCer by β-glucosidase (Wilkening et al., 1998) and that of ceramide by acid ceramidase (Linke et al., 2001) proceeds even in the absence of a SAP.
Therapeutic approaches
Currently, there is no treatment available for patients with ganglioside storage diseases (van Gelder et al., 2012). A main obstacle is the blood–brain barrier, which prevents the passage of therapeutic enzymes and proteins into the brain. For the prevention of substrate storage, substrate influx into the lysosomal compartment must not exceed this compartment's catabolic capacity (see Protracted clinical forms of gangliosidoses and the threshold theory, above; Conzelmann and Sandhoff, 1983, 1991; Leinekugel et al., 1992). This means that therapeutic methods should either increase the catabolic rate above the threshold or decrease the influx rate of substrates below the maximum catabolic capacity of the affected cells, e.g., by reducing the cellular substrate biosynthetic rate (Fig. 6). Therapeutic strategies in use such as enzyme replacement therapy (ERT) for adult Gaucher disease and those under study in animal models such as BMT yielded so far no substantial and long-lasting improvements in mouse models with GM1- and GM2-gangliosidosis. However, intracerebroventricular administration of a modified recombinant HexB (Matsuoka et al., 2011) and a highly phosphomannosylated recombinant human HexA reduced the level of storage compounds (Tsuji et al., 2011) in the HexA- and HexB-deficient mouse brain efficiently.
Figure 6.

Intracellular metabolic flux of sphingolipids. vi = the influx rate of the substrate into the lysosome; for further information see the text. Modified from Kolter and Sandhoff, 1999).
Additional approaches using low molecular compounds penetrating the blood–brain barrier have been evaluated in murine models of gangliosidoses, but so far without substantial success. They include the use of chemical chaperons, substrate analogs, or active site inhibitors (Matsuda et al., 2003; Tropak et al., 2004) to stabilize variant patient hydrolases during their biosynthesis and folding, and thereby enhance their hydrolytic activity as introduced by Fan et al. (1999).
Another approach is the substrate reduction therapy. It uses inhibitors of the biosynthetic pathway, such as iminosugars of the nojirimycin type (Butters et al., 2005; Fantur et al., 2012; Suzuki et al., 2012) and reduces the influx of substrates into the lysosomes (Kolter and Sandhoff, 1999). However, treatment of two Tay–Sachs patients with N-butyldeoxynojirimycin (Miglustat) did not arrest their clinical deterioration (Bembi et al., 2006), whereas a stable neurological picture was reported for two patients with Sandhoff disease after Miglustat treatment (Tallaksen and Berg, 2009; Wortmann et al., 2009).
However, reducing the ganglioside biosynthesis with inhibitors of the ceramide glucosyltransferase, N-butyldeoxynojirimycin or N-butyldeoxygalactonojirimycin, led to an increased survival of Sandhoff mice up to 2 months (Andersson et al., 2004; Jeyakumar et al., 2004).
In a clinical Phase I/II trial, treatment of late onset GM2-gangliosidosis patients with a presumed chaperon of β-hexosaminidases, pyrimethamine, for a 4 month period enhanced their leukocyte HexA levels, but clinical efficacy could not be assessed (Clarke et al., 2011).
A combination of different methods for patients with significant residual enzymatic activity, e.g., substrate reduction together with enzyme enhancement therapy, nonsteroidal anti-inflammatory drugs, and antioxidants is expected to yield better results.
Gene therapy
To circumvent the blood–brain barrier, functional copies of the mutant gene were directly inserted into murine brain cells with the help of retroviral and lentiviral vectors (Biffi and Naldini, 2005). The functional enzyme should be stably overexpressed in the transfected mutant cells and secreted, thereby cross-correcting adjacent cells (Arfi et al., 2005; Martino et al., 2005).
Intracerebral injection of an adenoviral vector encoding the β-subunit of HexA increased HexA activity to nearly normal levels in HexB-deficient mice (Bourgoin et al., 2003). Also, the intracranial inoculation of recombinant adeno-associated viral (AAV) vectors encoding both, human α- and β Hex-subunit genes, resulted in an almost generalized correction of the catabolic lysosomal defect in the same mice (Cachón-González et al., 2006). Animals survived more than a year with delayed onset of the disease, reduced ganglioside storage and inflammation, preserved motor function, and rescue of acute neurodegenerative illness (Sargeant et al., 2012).
AAV-mediated neonatal gene delivery by intracerebroventricular injection seems to be an effective approach to treat GM1-gangliosidoses. It resulted in a widespread appearance of the missing β-galactosidase activity in the murine brain and a normalization of all GSL levels (Broekman et al., 2007; Baek et al., 2010).
Though these results are promising, it will take many more investigations and progress to develop a real gene therapy for GSL and sphingolipid storage diseases.
Conclusions and outlook
To identify the molecular basis of amaurotic idiocy of the lipid accumulating type, it was important to isolate the storage compounds and clarify their chemical structures. This allowed a search for the defective catabolic steps to identify the enzymes and lipid binding proteins involved, their genomic structures, and the mutations causing the different gangliosidoses. The fact that the main storage compounds (gangliosides GM1 and GM2) are preferentially synthesized in neuronal cells is crucial for the start of neurodegenerative diseases and complicates all therapeutic approaches. So far, efficient ERT is not feasible, since the blood–brain barrier prevents cerebral uptake of therapeutic proteins. Also genetic approaches that appeared to be promising in mouse models are still not reliable (Cachón-González et al., 2006).
There is no simple model to understand the complex pathogenesis of monogenetic gangliosidoses. The primary lipid storage affects the endolysosomal system of neurons and obviously does not trigger an effective feedback inhibition of the storage compounds' biosynthesis. However, the storage may well impair the turnover of membranes and macromolecules, affecting neuronal metabolism and function.
Degradation of gangliosides requires the interaction of rather unspecific hydrolases and lipid binding proteins at surfaces of intraluminal vesicles and membrane structures. These complex interactions, including the substantial dependency of many catabolic steps on lipid binding cofactors (SAPs) and the lipid composition of the intraluminal, lysosomal membranes, seem to prevent an exact and quantitative understanding of the degradative sphingolipid pathways.
We also do not know to what extent the lipid load of the affected lysosomes disturbs their primary functions as cellular stomachs (Tamboli et al., 2011) to degrade macromolecules and release the products into the cytosol as nutrients for biosynthetic pathways and energy metabolism. One should also keep in mind that the cellular uptake of essential factors, for example, the uptake of vitamin B12 involving several proteins, and that of Fe2+ via transferrin and the transferrin receptor, may well be impaired by the lipid load causing a traffic jam in the endolysosomal system (Jeyakumar et al., 2009). The storage of GSLs in gangliosidoses also triggers side effects such as the accumulation of hydrophobic material including phospholipids and hydrophobic protein fragments in the storage granules (“membranous cytoplasmic bodies”) (Samuels et al., 1963), but also transmembrane spans of mitochondrial ATP synthase and amyloid-β-peptide (Keilani et al., 2012). As in multiple sclerosis, functionally impaired neurons give rise to an immune response, and are prone to digestion by microglia and invaded macrophages (Wada et al., 2000; Wekerle, 2005).
Although we have learned a lot about the function of gangliosides, their biochemistry, enzymology, and cell biology, we are still far away from a real understanding of the molecular and cellular pathogenesis of ganglioside storage diseases and the development of therapeutic approaches. However, the successful dissection of the clinical picture of amaurotic idiocy into 5 different ganglioside storage diseases involving 4 different genes, and a group of 10 so far not extensively analyzed neuronal ceroidlipofuscinoses, may encourage the search for similar clinical entities of monogenetic diseases but with causative diversity, among the high number of endemic inherited diseases, the molecular basis of which still has to be elucidated.
Footnotes
Editor's Note: Disease Focus articles provide brief overviews of a neural disease or syndrome, emphasizing potential links to basic neural mechanisms. They are presented in the hope of helping researchers identify clinical implications of their research. For more information, see http://www.jneurosci.org/misc/ifa_minireviews.dtl.
This work was supported by the Deutsche Forschungsgemeinschaft. Dedicated on the occasion of her 60th birthday to Ingeborg Krägeloh-Mann, professor of Neuropediatrics, Tübingen.
References
- Abdul-Hammed M, Breiden B, Adebayo MA, Babalola JO, Schwarzmann G, Sandhoff K. Role of endosomal membrane lipids and NPC2 in cholesterol transfer and membrane fusion. J Lipid Res. 2010;51:1747–1760. doi: 10.1194/jlr.M003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Smith D, Jeyakumar M, Butters TD, Borja MC, Dwek RA, Platt FM. Improved outcome of N-butyldeoxygalactonojirimycin-mediated substrate reduction therapy in a mouse model of Sandhoff disease. Neurobiol Dis. 2004;16:506–515. doi: 10.1016/j.nbd.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Appelqvist H, Sandin L, Björnström K, Saftig P, Garner B, Ollinger K, Kågedal K. Sensitivity to lysosome-dependent cell death is directly regulated by lysosomal cholesterol content. PLoS One. 2012;7:e50262. doi: 10.1371/journal.pone.0050262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arfi A, Bourgoin C, Basso L, Emiliani C, Tancini B, Chigorno V, Li YT, Orlacchio A, Poenaru L, Sonnino S, Caillaud C. Bicistronic lentiviral vector corrects beta-hexosaminidase deficiency in transduced and cross-corrected human Sandhoff fibroblasts. Neurobiol Dis. 2005;20:583–593. doi: 10.1016/j.nbd.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Assadi M, Baseman S, Janson C, Wang DJ, Bilaniuk L, Leone P. Serial 1H-MRS in GM2 gangliosidoses. Eur J Pediatr. 2008;167:347–352. doi: 10.1007/s00431-007-0469-0. [DOI] [PubMed] [Google Scholar]
- Baek RC, Broekman ML, Leroy SG, Tierney LA, Sandberg MA, d'Azzo A, Seyfried TN, Sena-Esteves M. AAV-mediated gene delivery in adult GM1-gangliosidosis mice corrects lysosomal storage in CNS and improves survival. PLoS One. 2010;5:e13468. doi: 10.1371/journal.pone.0013468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bembi B, Marchetti F, Guerci VI, Ciana G, Addobbati R, Grasso D, Barone R, Cariati R, Fernandez-Guillen L, Butters T, Pittis MG. Substrate reduction therapy in the infantile form of Tay-Sachs disease. Neurology. 2006;66:278–280. doi: 10.1212/01.wnl.0000194225.78917.de. [DOI] [PubMed] [Google Scholar]
- Biffi A, Naldini L. Gene therapy of storage disorders by retroviral and lentiviral vectors. Hum Gene Ther. 2005;16:1133–1142. doi: 10.1089/hum.2005.16.1133. [DOI] [PubMed] [Google Scholar]
- Bourgoin C, Emiliani C, Kremer EJ, Gelot A, Tancini B, Gravel RA, Drugan C, Orlacchio A, Poenaru L, Caillaud C. Widespread distribution of beta-hexosaminidase activity in the brain of a Sandhoff mouse model after coinjection of adenoviral vector and mannitol. Gene Ther. 2003;10:1841–1849. doi: 10.1038/sj.gt.3302081. [DOI] [PubMed] [Google Scholar]
- Broekman ML, Baek RC, Comer LA, Fernandez JL, Seyfried TN, Sena-Esteves M. Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus-mediated gene delivery. Mol Ther. 2007;15:30–37. doi: 10.1038/sj.mt.6300004. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Hüttler S, Klein A, Möbius W, Habermann A, Griffiths G, Sandhoff K. Accumulation of sphingolipids in SAP-precursor (prosaposin)-deficient fibroblasts occurs as intralysosomal membrane structures and can be completely reversed by treatment with human SAP-precursor. Eur J Cell Biol. 1997;73:10–18. [PubMed] [Google Scholar]
- Butters TD, Dwek RA, Platt FM. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology. 2005;15:43R–52R. doi: 10.1093/glycob/cwi076. [DOI] [PubMed] [Google Scholar]
- Cachón-González MB, Wang SZ, Lynch A, Ziegler R, Cheng SH, Cox TM. Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proc Natl Acad Sci U S A. 2006;103:10373–10378. doi: 10.1073/pnas.0603765103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JT, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA, Tropak MB. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants) Mol Genet Metab. 2011;102:6–12. doi: 10.1016/j.ymgme.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann E, Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci. 1983;6:58–71. doi: 10.1159/000112332. [DOI] [PubMed] [Google Scholar]
- Conzelmann E, Sandhoff K. Biochemical basis of late-onset neurolipidoses. Dev Neurosci. 1991;13:197–204. doi: 10.1159/000112160. [DOI] [PubMed] [Google Scholar]
- d'Azzo A, Andria G, Strisciulio P, Galjaard H. Galactosialidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited diseases. Ed 8. New York: McGraw-Hill; 2001. pp. 3811–3826. [Google Scholar]
- Delves P, Martin S, Burton D, Roit I. Roit's essential immunology. Ed 11. Malden, MA: Blackwell Publishing; 2006. [Google Scholar]
- Doering T, Holleran WM, Potratz A, Vielhaber G, Elias PM, Suzuki K, Sandhoff K. Sphingolipid activator proteins are required for epidermal permeability barrier formation. J Biol Chem. 1999;274:11038–11045. doi: 10.1074/jbc.274.16.11038. [DOI] [PubMed] [Google Scholar]
- Eggens I, Fenderson B, Toyokuni T, Dean B, Stroud M, Hakomori S. Specific interaction between Lex and Lex determinants. A possible basis for cell recognition in preimplantation embryos and in embryonal carcinoma cells. J Biol Chem. 1989;264:9476–9484. [PubMed] [Google Scholar]
- Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med. 1999;5:112–115. doi: 10.1038/4801. [DOI] [PubMed] [Google Scholar]
- Fantur KM, Wrodnigg TM, Stütz AE, Pabst BM, Paschke E. Fluorous iminoalditols act as effective pharmacological chaperones against gene products from GLB(1) alleles causing GM1-gangliosidosis and Morquio B disease. J Inherit Metab Dis. 2012;35:495–503. doi: 10.1007/s10545-011-9409-2. [DOI] [PubMed] [Google Scholar]
- Fishman PH, Max SR, Tallman JF, Brady RO, Maclaren NK, Cornblath M. Deficient Ganglioside Biosynthesis: a novel human sphingolipidosis. Science. 1975;187:68–70. doi: 10.1126/science.803227. [DOI] [PubMed] [Google Scholar]
- Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol. 2012;22:374–380. doi: 10.1016/j.tcb.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragaki K, Ait-El-Mkadem S, Chaussenot A, Gire C, Mengual R, Bonesso L, Bénéteau M, Ricci JE, Desquiret-Dumas V, Procaccio V, Rötig A, Paquis-Flucklinger V. Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency. Eur J Hum Genet. 2013;21:528–534. doi: 10.1038/ejhg.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallala HD, Sandhoff K. Biological function of the cellular lipid BMP—BMP as a key activator for cholesterol sorting and membrane digestion. Neurochem Res. 2011;36:1594–1600. doi: 10.1007/s11064-010-0337-6. [DOI] [PubMed] [Google Scholar]
- Gallala HD, Breiden B, Sandhoff K. Regulation of the NPC2 protein-mediated cholesterol trafficking by membrane lipids. J Neurochem. 2011;116:702–707. doi: 10.1111/j.1471-4159.2010.07014.x. [DOI] [PubMed] [Google Scholar]
- Gärtner S, Conzelmann E, Sandhoff K. Activator protein for the degradation of globotriaosylceramide by human alpha-galactosidase. J Biol Chem. 1983;258:12378–12385. [PubMed] [Google Scholar]
- Giehl A, Lemm T, Bartelsen O, Sandhoff K, Blume A. Interaction of the GM2-activator protein with phospholipid-ganglioside bilayer membranes and with monolayers at the air-water interface. Eur J Biochem. 1999;261:650–658. doi: 10.1046/j.1432-1327.1999.00302.x. [DOI] [PubMed] [Google Scholar]
- Graber D, Salvayre R, Levade T. Accurate differentiation of neuronopathic and nonneuronopathic forms of Niemann-Pick disease by evaluation of the effective residual lysosomal sphingomyelinase activity in intact cells. J Neurochem. 1994;63:1060–1068. doi: 10.1046/j.1471-4159.1994.63031060.x. [DOI] [PubMed] [Google Scholar]
- Gravel R, Kaback MM, Proia RL, Sandhoff K, Suzuki K, Suzuki K. The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited diseases. Ed 8. New York: McGraw-Hill; 2001. pp. 3827–3876. [Google Scholar]
- Hay ED. Extracellular matrix, cell skeletons, and embryonic development. Am J Med Genet. 1989;34:14–29. doi: 10.1002/ajmg.1320340107. [DOI] [PubMed] [Google Scholar]
- Igisu H, Suzuki K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 1984;224:753–755. doi: 10.1126/science.6719111. [DOI] [PubMed] [Google Scholar]
- Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009;1793:697–709. doi: 10.1016/j.bbamcr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Jatzkewitz H, Sandhoff K. On a biochemically special form of infantile amaurotic idiocy. Biochim Biophys Acta. 1963;70:354–356. doi: 10.1016/0006-3002(63)90764-9. [DOI] [PubMed] [Google Scholar]
- Jennemann R, Sandhoff R, Wang S, Kiss E, Gretz N, Zuliani C, Martin-Villalba A, Jäger R, Schorle H, Kenzelmann M, Bonrouhi M, Wiegandt H, Gröne HJ. Cell-specific deletion of glucosylceramide synthase in brain leads to severe neural defects after birth. Proc Natl Acad Sci U S A. 2005;102:12459–12464. doi: 10.1073/pnas.0500893102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennemann R, Sandhoff R, Langbein L, Kaden S, Rothermel U, Gallala H, Sandhoff K, Wiegandt H, Gröne HJ. Integrity and barrier function of the epidermis critically depend on glucosylceramide synthesis. J Biol Chem. 2007;282:3083–3094. doi: 10.1074/jbc.M610304200. [DOI] [PubMed] [Google Scholar]
- Jennemann R, Rothermel U, Wang S, Sandhoff R, Kaden S, Out R, van Berkel TJ, Aerts JM, Ghauharali K, Sticht C, Gröne HJ. Hepatic glycosphingolipid deficiency and liver function in mice. Hepatology. 2010;51:1799–1809. doi: 10.1002/hep.23545. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Smith DA, Williams IM, Borja MC, Neville DC, Butters TD, Dwek RA, Platt FM. NSAIDs increase survival in the Sandhoff disease mouse: synergy with N-butyldeoxynojirimycin. Ann Neurol. 2004;56:642–649. doi: 10.1002/ana.20242. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Williams I, Smith D, Cox TM, Platt FM. Critical role of iron in the pathogenesis of the murine gangliosidoses. Neurobiol Dis. 2009;34:406–416. doi: 10.1016/j.nbd.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Kannagi R, Cochran NA, Ishigami F, Hakomori S, Andrews PW, Knowles BB, Solter D. Stage-specific embryonic antigens (SSEA-3 and -4) are epitopes of a unique globo-series ganglioside isolated from human teratocarcinoma cells. EMBO J. 1983;2:2355–2361. doi: 10.1002/j.1460-2075.1983.tb01746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H, Allende ML, Wada R, Kono M, Sango K, Deng C, Miyakawa T, Crawley JN, Werth N, Bierfreund U, Sandhoff K, Proia RL. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J Biol Chem. 2001;276:6885–6888. doi: 10.1074/jbc.C000847200. [DOI] [PubMed] [Google Scholar]
- Keilani S, Lun Y, Stevens AC, Williams HN, Sjoberg ER, Khanna R, Valenzano KJ, Checler F, Buxbaum JD, Yanagisawa K, Lockhart DJ, Wustman BA, Gandy S. Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-beta peptide. J Neurosci. 2012;32:5223–5236. doi: 10.1523/JNEUROSCI.4860-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C, Kinnunen PK, Nylandsted J, Jäättelä M. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–553. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- Klenk E. Die Fettstoffe des Gehirns bei Amaurotischer Idiotie und Niemann-Pick'scher Krankheit. Ber Ges Physiol. 1937;96:659–660. [Google Scholar]
- Klenk E. Niemann-Pick'sche Krankheit und Amaurotische Idiotie. Hoppe-Seyler's Z Physiol Chem. 1939;262:128–143. doi: 10.1515/bchm2.1939.262.3-5.128. [DOI] [Google Scholar]
- Kolter T, Sandhoff K. Sphingolipids—their metabolic pathways and the pathobiochemistry of neurodegenerative diseases. Angew Chem Int Ed Engl. 1999;38:1532–1568. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1532::AID-ANIE1532>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Sphingolipid metabolism diseases. Biochim Biophys Acta. 2006;1758:2057–2079. doi: 10.1016/j.bbamem.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010;584:1700–1712. doi: 10.1016/j.febslet.2009.10.021. [DOI] [PubMed] [Google Scholar]
- Kolter T, Magin TM, Sandhoff K. Biomolecule function: no reliable prediction from cell culture. Traffic. 2000;1:803–804. doi: 10.1034/j.1600-0854.2000.011007.x. [DOI] [PubMed] [Google Scholar]
- Kolter T, Proia RL, Sandhoff K. Combinatorial ganglioside biosynthesis. J Biol Chem. 2002;277:25859–25862. doi: 10.1074/jbc.R200001200. [DOI] [PubMed] [Google Scholar]
- Kudoh T, Velkoff MA, Wenger DA. Uptake and metabolism of radioactively labeled sphingomyelin in cultured skin fibroblasts from controls and patients with Niemann-Pick disease and other lysosomal storage diseases. Biochim Biophys Acta. 1983;754:82–92. doi: 10.1016/0005-2760(83)90084-X. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Wiegandt H. Die Konstitution der Ganglio-N-tetraose und des Gangliosids GI. Chem Ber. 1963;96:866–880. doi: 10.1002/cber.19630960329. [DOI] [Google Scholar]
- Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kytzia HJ, Sandhoff K. Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. J Biol Chem. 1985;260:7568–7572. [PubMed] [Google Scholar]
- Kytzia HJ, Hinrichs U, Maire I, Suzuki K, Sandhoff K. Variant of GM2-gangliosidosis with hexosaminidase A having a severely changed substrate specificity. EMBO J. 1983;2:1201–1205. doi: 10.1002/j.1460-2075.1983.tb01567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kytzia HJ, Hinrichs U, Sandhoff K. Diagnosis of infantile and juvenile forms of GM2 gangliosidosis variant 0. Residual activities toward natural and different synthetic substrates. Hum Genet. 1984;67:414–418. doi: 10.1007/BF00291402. [DOI] [PubMed] [Google Scholar]
- Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88:513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MN. Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis. J Mol Biol. 2006;359:913–929. doi: 10.1016/j.jmb.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SC, Sonnino S, Tettamanti G, Li YT. Characterization of a nonspecific activator protein for the enzymatic hydrolysis of glycolipids. J Biol Chem. 1988;263:6588–6591. [PubMed] [Google Scholar]
- Linke T, Wilkening G, Sadeghlar F, Mozcall H, Bernardo K, Schuchman E, Sandhoff K. Interfacial regulation of acid ceramidase activity. Stimulation of ceramide degradation by lysosomal lipids and sphingolipid activator proteins. J Biol Chem. 2001;276:5760–5768. doi: 10.1074/jbc.M006846200. [DOI] [PubMed] [Google Scholar]
- Lyon G, Adams RD, Kolodny EH. Early infantile progressive metabolic encephalopathies: clinical problems and diagnostic considerations. In: Lyon G, Adams RD, Kolodny EH, editors. Neurology of hereditary metabolic diseases of children. Ed 2. New York: McGraw-Hill; 1996a. pp. 45–123. [Google Scholar]
- Lyon G, Adams RD, Kolodny EH. Childhood and adolescent hereditary metabolic disorders. In: Lyon G, Adams RD, Kolodny EH, editors. Neurology of hereditary metabolic diseases of children. Ed 2. New York: McGraw-Hill; 1996b. pp. 177–281. [Google Scholar]
- Maier T, Strater N, Schuette CG, Klingenstein R, Sandhoff K, Saenger W. The X-ray crystal structure of human beta-hexosaminidase B provides new insights into Sandhoff disease. J Mol Biol. 2003;328:669–681. doi: 10.1016/S0022-2836(03)00311-5. [DOI] [PubMed] [Google Scholar]
- Martino S, Marconi P, Tancini B, Dolcetta D, De Angelis MG, Montanucci P, Bregola G, Sandhoff K, Bordignon C, Emiliani C, Manservigi R, Orlacchio A. A direct gene transfer strategy via brain internal capsule reverses the biochemical defect in Tay-Sachs disease. Hum Mol Genet. 2005;14:2113–2123. doi: 10.1093/hmg/ddi216. [DOI] [PubMed] [Google Scholar]
- Matsuda J, Suzuki O, Oshima A, Yamamoto Y, Noguchi A, Takimoto K, Itoh M, Matsuzaki Y, Yasuda Y, Ogawa S, Sakata Y, Nanba E, Higaki K, Ogawa Y, Tominaga L, Ohno K, Iwasaki H, Watanabe H, Brady RO, Suzuki Y. Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis. Proc Natl Acad Sci U S A. 2003;100:15912–15917. doi: 10.1073/pnas.2536657100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka K, Tamura T, Tsuji D, Dohzono Y, Kitakaze K, Ohno K, Saito S, Sakuraba H, Itoh K. Therapeutic potential of intracerebroventricular replacement of modified human beta-hexosaminidase B for GM2 gangliosidosis. Mol Ther. 2011;19:1017–1024. doi: 10.1038/mt.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehl E, Jatzkewitz H. Eine Cerebrosidsulfatase aus Schweineniere. Hoppe-Seylers Z Physiol Chem. 1964;339:260–276. doi: 10.1515/bchm2.1964.339.1.260. [DOI] [PubMed] [Google Scholar]
- Möbius W, Herzog V, Sandhoff K, Schwarzmann G. Intracellular distribution of a biotin-labeled ganglioside, GM1, by immunoelectron microscopy after endocytosis in fibroblasts. J Histochem Cytochem. 1999;47:1005–1014. doi: 10.1177/002215549904700804. [DOI] [PubMed] [Google Scholar]
- Mraz W, Fischer G, Jatzkewitz H. Low molecular weight proteins in secondary lysosomes as activators of different sphingolipid hydrolases. FEBS Lett. 1976;67:104–109. doi: 10.1016/0014-5793(76)80879-4. [DOI] [PubMed] [Google Scholar]
- Myerowitz R, Lawson D, Mizukami H, Mi Y, Tifft CJ, Proia RL. Molecular pathophysiology in Tay-Sachs and Sandhoff diseases as revealed by gene expression profiling. Hum Mol Genet. 2002;11:1343–1350. doi: 10.1093/hmg/11.11.1343. [DOI] [PubMed] [Google Scholar]
- Neufeld EF, Muenzer J. The Mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. Ed 8. New York: McGraw-Hill; 2001. pp. 3421–3452. [Google Scholar]
- Nilsson O, Månsson JE, Håkansson G, Svennerholm L. The occurrence of psychosine and other glycolipids in spleen and liver from the three major types of Gaucher's disease. Biochim Biophys Acta. 1982;712:453–463. doi: 10.1016/0005-2760(82)90272-7. [DOI] [PubMed] [Google Scholar]
- Okada M, Itoh Mi M, Haraguchi M, Okajima T, Inoue M, Oishi H, Matsuda Y, Iwamoto T, Kawano T, Fukumoto S, Miyazaki H, Furukawa K, Aizawa S, Furukawa K. b-series Ganglioside deficiency exhibits no definite changes in the neurogenesis and the sensitivity to Fas-mediated apoptosis but impairs regeneration of the lesioned hypoglossal nerve. J Biol Chem. 2002;277:1633–1636. doi: 10.1074/jbc.C100395200. [DOI] [PubMed] [Google Scholar]
- Okada S, O'Brien JS. Generalized gangliosidosis: beta-galactosidase deficiency. Science. 1968;160:1002–1004. doi: 10.1126/science.160.3831.1002. [DOI] [PubMed] [Google Scholar]
- Okumiya T, Sakuraba H, Kase R, Sugiura T. Imbalanced substrate specificity of mutant beta-galactosidase in patients with Morquio B disease. Mol Genet Metab. 2003;78:51–58. doi: 10.1016/S1096-7192(02)00199-3. [DOI] [PubMed] [Google Scholar]
- Proia RL. Glycosphingolipid functions: insights from engineered mouse models. Philos Trans R Soc Lond B Biol Sci. 2003;358:879–883. doi: 10.1098/rstb.2003.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pshezhetsky AV, Ashmarina M. Lysosomal multienzyme complex: biochemistry, genetics, and molecular pathophysiology. Prog Nucleic Acid Res Mol Biol. 2001;69:81–114. doi: 10.1016/S0079-6603(01)69045-7. [DOI] [PubMed] [Google Scholar]
- Purpura DP, Suzuki K. Distortion of neuronal geometry and formation of aberrant synapses in neuronal storage disease. Brain Res. 1976;116:1–21. doi: 10.1016/0006-8993(76)90245-6. [DOI] [PubMed] [Google Scholar]
- Samuels S, Korey SR, Gonatas J, Terry RD, Weiss M. Studies in Tay-Sachs disease. IV. Membranous cytoplasmic bodies. J Neuropath Exp Neurol. 1963;22:81–97. doi: 10.1097/00005072-196301000-00005. [DOI] [PubMed] [Google Scholar]
- Sandhoff K. The biochemistry of sphingolipid storage diseases. Angew Chem Int Ed Engl. 1977;16:273–285. doi: 10.1002/anie.197702733. [DOI] [PubMed] [Google Scholar]
- Sandhoff K. My journey into the world of sphingolipids and sphingolipidoses. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88:554–582. doi: 10.2183/pjab.88.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhoff K, Jatkewitz H. A particle-bound sialyl lactosidoceramide splitting mammalian sialidase. Biochim Biophys Acta. 1967;141:442–444. doi: 10.1016/0304-4165(67)90124-9. [DOI] [PubMed] [Google Scholar]
- Sandhoff K, Kolter T. Glykolipide der Zelloberfläche-Biochemie ihres Abbaus. Naturwissenschaften. 1995;82:403–413. doi: 10.1007/BF01133673. [DOI] [PubMed] [Google Scholar]
- Sandhoff K, Kolter T. Biosynthesis and degradation of mammalian glycosphingolipids. Philos Trans R Soc Lond B Biol Sci. 2003;358:847–861. doi: 10.1098/rstb.2003.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhoff K, Jatzkewitz H, Peters G. Die infantile amaurotische Idiotie und verwandte Formen als Gangliosid-Speicherkrankheiten. Naturwissenschaften. 1969;56:356–362. doi: 10.1007/BF00596925. [DOI] [PubMed] [Google Scholar]
- Sandhoff K, Conzelmann E, Neufeld EF, Kaback MM, Suzuki K. The GM2 gangliosidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic basis of inherited disease. New York: McGraw-Hill; 1989. pp. 1807–1839. [Google Scholar]
- Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald MP, Crawley JN, Sandhoff K, Suzuki K, Proia RL. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet. 1995;11:170–176. doi: 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- Sango K, McDonald MP, Crawley JN, Mack ML, Tifft CJ, Skop E, Starr CM, Hoffmann A, Sandhoff K, Suzuki K, Proia RL. Mice lacking both subunits of lysosomal β-hexosaminidase display gangliosidosis and mucopolysaccharidosis. Nat Genet. 1996;14:348–352. doi: 10.1038/ng1196-348. [DOI] [PubMed] [Google Scholar]
- Sargeant TJ, Drage DJ, Wang S, Apostolakis AA, Cox TM, Cachón-González MB. Characterization of inducible models of Tay-Sachs and related disease. PLoS Genet. 2012;8:e1002943. doi: 10.1371/journal.pgen.1002943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL. Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett. 2010;584:1741–1747. doi: 10.1016/j.febslet.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, Wang H, Wiznitzer M, Gurtz K, Verganelaki A, Pryde A, Patton MA, Dwek RA, Butters TD, Platt FM, Crosby AH. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- Stevens RL, Fluharty AL, Kihara H, Kaback MM, Shapiro LJ, Marsh B, Sandhoff K, Fischer G. Cerebroside sulfatase activator deficiency induced metachromatic leukodystrophy. Am J Hum Genet. 1981;33:900–906. [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Witte DP, Ran H, Zamzow M, Barnes S, Cheng H, Han X, Williams MT, Skelton MR, Vorhees CV, Grabowski GA. Neurological deficits and glycosphingolipid accumulation in saposin B deficient mice. Hum Mol Genet. 2008;17:2345–2356. doi: 10.1093/hmg/ddn135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Oshima A, Nanba E. β-Galactosidase deficiency (β-galactosidosis): GM1 gangliosidosis and Morquio B disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited diseases. Ed 8. New York: McGraw-Hill; 2001. pp. 3755–3809. [Google Scholar]
- Suzuki Y, Ichinomiya S, Kurosawa M, Matsuda J, Ogawa S, Iida M, Kubo T, Tabe M, Itoh M, Higaki K, Nanba E, Ohno K. Therapeutic chaperone effect of N-octyl 4-epi-beta-valienamine on murine G(M1)-gangliosidosis. Mol Genet Metab. 2012;106:92–98. doi: 10.1016/j.ymgme.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Tallaksen CM, Berg JE. Miglustat therapy in juvenile Sandhoff disease. J Inherit Metab Dis. 2009;32(Suppl 1):S289–S293. doi: 10.1007/s10545-009-1224-7. [DOI] [PubMed] [Google Scholar]
- Tamboli IY, Hampel H, Tien NT, Tolksdorf K, Breiden B, Mathews PM, Saftig P, Sandhoff K, Walter J. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Aβ generation. J Neurosci. 2011;31:1837–1849. doi: 10.1523/JNEUROSCI.2954-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniike M, Yamanaka S, Proia RL, Langaman C, Bone-Turrentine T, Suzuki K. Neuropathology of mice with targeted disruption of Hexa gene, a model of Tay-Sachs disease. Acta Neuropathol. 1995;89:296–304. doi: 10.1007/BF00309622. [DOI] [PubMed] [Google Scholar]
- Tessitore A, del P Martin M, Sano R, Ma Y, Mann L, Ingrassia A, Laywell ED, Steindler DA, Hendershot LM, d'Azzo A. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell. 2004;15:753–766. doi: 10.1016/j.molcel.2004.08.029. [DOI] [PubMed] [Google Scholar]
- Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D. Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff Patients. J Biol Chem. 2004;279:13478–13487. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji D, Akeboshi H, Matsuoka K, Yasuoka H, Miyasaki E, Kasahara Y, Kawashima I, Chiba Y, Jigami Y, Taki T, Sakuraba H, Itoh K. Highly phosphomannosylated enzyme replacement therapy for GM2 gangliosidosis. Ann Neurol. 2011;69:691–701. doi: 10.1002/ana.22262. [DOI] [PubMed] [Google Scholar]
- van Gelder CM, Vollebregt AA, Plug I, van der Ploeg AT, Reuser AJ. Treatment options for lysosomal storage disorders: developing insights. Expert Opin Pharmacother. 2012;13:2281–2299. doi: 10.1517/14656566.2012.729039. [DOI] [PubMed] [Google Scholar]
- Venugopalan P, Joshi SN. Cardiac involvement in infantile Sandhoff disease. J Paediatr Child Health. 2002;38:98–100. doi: 10.1046/j.1440-1754.2002.00765.x. [DOI] [PubMed] [Google Scholar]
- Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem. 2010;285:20423–20427. doi: 10.1074/jbc.R110.134452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci U S A. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wekerle H. Immune pathogenesis of multiple sclerosis. Neurol Sci. 2005;26(Suppl 1):S1–S2. doi: 10.1007/s10072-005-0386-9. [DOI] [PubMed] [Google Scholar]
- Wendeler M, Hoernschemeyer J, Hoffmann D, Kolter T, Schwarzmann G, Sandhoff K. Photoaffinity labelling of the human GM2-activator protein. Eur J Biochem. 2004a;271:614–627. doi: 10.1111/j.1432-1033.2003.03964.x. [DOI] [PubMed] [Google Scholar]
- Wendeler M, Hoernschemeyer J, John M, Werth N, Schoeniger M, Lemm T, Hartmann R, Kessler H, Sandhoff K. Expression of the GM2-activator protein in the methylotrophic yeast Pichia pastoris, purification, isotopic labeling, and biophysical characterization. Protein Expr Purif. 2004b;34:147–157. doi: 10.1016/j.pep.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Werth N, Schuette CG, Wilkening G, Lemm T, Sandhoff K. Degradation of membrane-bound ganglioside GM2 by beta -hexosaminidase A. Stimulation by GM2 activator protein and lysosomal lipids. J Biol Chem. 2001;276:12685–12690. doi: 10.1074/jbc.M007970200. [DOI] [PubMed] [Google Scholar]
- Wilken B, Dechent P, Hanefeld F, Frahm J. Proton MRS of a child with Sandhoff disease reveals elevated brain hexosamine. Eur J Paediatr Neurol. 2008;12:56–60. doi: 10.1016/j.ejpn.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Wilkening G, Linke T, Sandhoff K. Lysosomal degradation on vesicular membrane surfaces. Enhanced glucosylceramide degradation by lysosomal anionic lipids and activators. J Biol Chem. 1998;273:30271–30278. doi: 10.1074/jbc.273.46.30271. [DOI] [PubMed] [Google Scholar]
- Wilkening G, Linke T, Uhlhorn-Dierks G, Sandhoff K. Degradation of membrane-bound ganglioside GM1. Stimulation by bis(monoacylglycero)phosphate and the activator proteins SAP-B and GM2-AP. J Biol Chem. 2000;275:35814–35819. doi: 10.1074/jbc.M006568200. [DOI] [PubMed] [Google Scholar]
- Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464:864–869. doi: 10.1038/nature08849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortmann SB, Lefeber DJ, Dekomien G, Willemsen MA, Wevers RA, Morava E. Substrate deprivation therapy in juvenile Sandhoff disease. J Inherit Metab Dis. 2009;32(Suppl 1):S307–S311. doi: 10.1007/s10545-009-1261-2. [DOI] [PubMed] [Google Scholar]
- Wright CS, Zhao Q, Rastinejad F. Structural Analysis of Lipid Complexes of GM2-Activator Protein. J Mol Biol. 2003;331:951–964. doi: 10.1016/S0022-2836(03)00794-0. [DOI] [PubMed] [Google Scholar]
- Xu YH, Barnes S, Sun Y, Grabowski GA. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J Lipid Res. 2010;51:1643–1675. doi: 10.1194/jlr.R003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Wada R, Sasaki T, Deng C, Bierfreund U, Sandhoff K, Proia RL. A vital role for glycosphingolipid synthesis during development and differentiation. Proc Natl Acad Sci U S A. 1999;96:9142–9147. doi: 10.1073/pnas.96.16.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RK, Tsai YT, Ariga T, Yanagisawa M. Structures, biosynthesis, and functions of gangliosides–an overview. J Oleo Sci. 2011;60:537–544. doi: 10.5650/jos.60.537. [DOI] [PMC free article] [PubMed] [Google Scholar]