

Abstract
In layer 5 neocortical pyramidal neurons, backpropagating action potentials (bAPs) firing at rates above a critical frequency (CF) induce supralinear Ca2+ influx and regenerative potentials in apical dendrites. Paired temporally with an EPSP, this Ca2+ influx can result in synaptic plasticity. We studied the actions of neuropeptide Y (NPY), an abundant neocortical neuropeptide, on Ca2+ influx in layer 5 pyramidal neurons of somatosensory neocortex in Sprague Dawley and Wistar rats, using a combination of somatic and dendritic intracellular recordings and simultaneous Ca2+ imaging. Ca2+ influx induced by trains of bAPs above a neuron's CF was inhibited by NPY, acting only at the distal dendrite, via Y1 receptors. NPY does not affect evoked synaptic glutamate release, paired synaptic facilitation, or synaptic rundown in longer trains. Extracellular Cs+ did not prevent NPY's postsynaptic effects, suggesting it does not act via either G-protein-activated inwardly rectifying K+ conductance (GIRK) or hyperpolarization-activated, cyclic nucleotide-gated channels. NPY application suppresses the induction of the long-term depression (LTD) normally caused by pairing 100 EPSPs with bursts of 2 bAPs evoked at a supracritical frequency. These findings suggest that distal dendritic Ca2+ influx is necessary for LTD induction, and selective inhibition of this distal dendritic Ca2+ influx by NPY can thus regulate synaptic plasticity in layer 5 pyramidal neurons.
Introduction
Neocortical pyramidal neurons integrate and store information via changes in the strength of synaptic connections throughout their dendritic arbor (Gilbert et al., 2001; Heynen and Bear, 2001). The elaborate dendritic structure of neocortical layer 5 (L5) pyramidal neurons (PNs) spans all cortical layers and is compartmentalized for distinct information processing (Larkum et al., 2001; London and Häusser, 2005). The most distal aspect of L5 PN dendrites, the apical tuft, receives mainly feedback information from distal cortical areas (Douglas and Martin, 2004) and is important in associative learning and attention (Sjöström and Häusser, 2006; Gilbert and Sigman, 2007). A region of the apical dendrite usually just proximal to the apical tuft is capable of initiating Ca2+-dependent APs, which can propagate toward the soma and alter somatic activity. Distal Ca2+-APs are required for some long-term synaptic changes (Golding et al., 2002), but can also propagate to the soma and result in bursts of APs (Schiller et al., 1997; Larkum et al., 1999a; Schwindt and Crill, 1999). Dendritic Ca2+ APs have been shown previously to be essential for the induction of long-term potentiation (LTP) in L5 PNs (Kampa et al., 2006); however, while Ca2+ influx is often required for the induction of long-term depression (LTD; Nevian and Sakmann, 2006; Froemke et al., 2010), the necessity for a distal Ca2+ AP for induction of LTD in these neurons remains unclear (Lisman and Spruston, 2010).
Neuropeptide Y (NPY) is expressed in all lamina of the cerebral cortex (Hendry et al., 1984), predominantly in nonpyramidal, GABAergic interneurons (Hendry et al., 1984; Köhler et al., 1987). In neocortical layers 1–2, NPY-expressing neurons with horizontally oriented projections comprise different populations (Hendry et al., 1984) that release NPY into L1 (Karagiannis et al., 2009), including the distal apical dendrites of L5 pyramidal neurons. Y1 receptors are concentrated mainly in neocortical layers 1–3 (Dumont et al., 1996) and are localized largely on dendritic processes (Kopp et al., 2002). The only reported presynaptic action of NPY on L5 pyramidal cell inputs results in prolonged increases in GABAergic synaptic transmission, decreases in AMPA receptor-mediated excitatory transmission, or both (Bacci et al., 2002). Despite the abundant evidence for NPY receptors on dendrites of L5 PNs (Kopp et al., 2002), their physiological significance has yet to be determined.
We studied responses of somatosensory L5 PNs from adult rats to trains of backpropagating action potentials (bAPs) at different frequencies, using either simultaneous whole-cell recordings from dendrites and somata, or whole-cell somatic recordings with or without simultaneous dendritic Ca2+ imaging. We found that NPY inhibits Ca2+-dependent APs in distal apical dendrites, and acts only postsynaptically on adult L5 PNs, primarily on the distal dendrite near the Ca2+ spike initiation zone. To study effects of NPY on synaptic plasticity, we elicited LTD by temporally pairing a layer 1 EPSP and a pair of somatically induced bAPs [above the critical frequency (CF)] in L5 PNs. LTD was never observed with NPY present, but was elicited in the same cell after washout of NPY, indicating a role for NPY in regulating neocortical synaptic plasticity.
Materials and Methods
Slice preparation.
Young adult [postnatal day 25 (P25)–P40] male Wistar or Sprague Dawley rats were decapitated and whole brains were removed and immersed in cold (0–4°C) artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 MgCl, 10 or 25 glucose, 2 CaCl2, pH 7.4, saturated with 95% O2, 5% CO2 (carbogen). All extracellular solutions had an osmolarity of 300 ± 2 mOsm. Parasagittal slices (300 μm) of the somatosensory cortex were cut with a vibrating slicer and maintained in carbogenated ACSF at 37°C for 15 min, brought gradually to room temperature (24°C), and kept there until the slice was needed. L5 pyramidal neurons were chosen after visually identifying the presence of an intact distal apical dendrite, using either infrared differential interference contrast or oblique illumination (Pérez-Garci et al., 2006). All animal procedures were performed in accordance with Canadian Council of Animal Care guidelines (protocol approved by the University of Alberta Health Sciences Laboratory Animal Committee) and relevant international laws and policies (EEC Council Directive 86/609, OJ L 358, 1, December 12, 1987; Guide for the Care and Use of Laboratory Animals, 7th Edition, U.S. National Research Council, 1996).
Electrophysiology.
Dual and single whole-cell current-clamp experiments were performed with ACSF (33–34°C) constantly perfused over the slice in a recording chamber at the focus of a fixed-stage microscope (Zeiss FS2+; Hamilton et al., 2010; Pérez-Garci et al., 2006). In some experiments, recording pipettes (soma, 5–10 MΩ; dendrite, 15–20 MΩ) were pulled from thick-walled borosilicate glass (1511-M; Freidrick and Dimmock), while in other experiments they were pulled from thin-walled borosilicate tubing (5–6 MΩ; TW150F; WPI). Pipettes were filled with a solution containing the following (in mm): 105 K-gluconate, 10 HEPES, 30 KCl, 2 MgCl2, 2 MgATP, 2 Na2ATP, 0.3 GTP, and 0.2% biocytin or neurobiotin, pH 7.25 ± 0.05. In imaging experiments, pipettes also contained Oregon Green BAPTA-1 (OGB-1; 100 μm; Invitrogen) for Ca2+ imaging, and often also Alexa 594 (10 μm; Invitrogen) to enable the visualization of the dendritic tree. Dendritic and somatic recordings were made with Axoclamp-2B amplifiers (Molecular Devices) or Dagan BVC-700 amplifiers. Trains of four APs used to determine CFs were evoked with square-wave, depolarizing current pulses (1 ms, 1–5 nA) from the somatic pipette at frequencies from 20–200 Hz. Extracellular stimulation of layer 2/3 was at low (25%), medium (50%), and high intensities (75%) relative to maximal EPSP responses. All L1 stimulation was at 50% of maximal intensity. Dendritic current injection was in the shape of a double exponential, as described previously (Larkum et al., 1999b), and applied either alone or 5 ms after the start of somatic current injection in pairing experiments.
Ca2+ imaging.
Intracellular Ca2+ imaging experiments began with the loading of L5 PNs with OGB-1 (100 μm) and Alexa 594 (10 μm) for ∼45 min via a somatic pipette containing otherwise the same intracellular solution as above. Fluorescence was measured with a 512 × 512 back-illuminated frame transfer CCD camera (Pentamax, Micromax, or EMCCD; Roper). Images were binned at 2 × 2 or 4 × 4 and acquired at frame rates of ∼40 Hz.
Immunohistochemistry.
After recording, the pipette was slowly removed from the neuron, and then slices were carefully removed from the recording chamber, fixed in buffered 4% paraformaldehyde for at least 48 h, and washed in a potassium phosphate buffer solution (KPBS) containing KH2PO4 (3.57 mm), K2HPO4 (anhydrous; 16.43 mm), and NaCl (15.4 mm), and then dehydrated in 25% sucrose for 48–72 h. Slices were then incubated in streptavidin-conjugated Alexa 555 (Invitrogen), 2% normal goat serum (Rockland), and 0.3% Triton X-100 (Sigma) for 2 h. After washout with KPBS, slices were mounted on slides with Prolong Gold antifade reagent (Invitrogen) and imaged with an upright confocal microscope (Zeiss LSM 510).
LTD protocol.
The LTD protocol consisted of 100 pairings of an EPSP, evoked by a bipolar stimulating electrode placed in L1, followed 25 ms later with a pair of APs evoked at a supracritical frequency; pairings were repeated at 1 Hz. The critical frequency for each L5 PN was calculated, and the pairing frequency used in the protocol was an average of 56 ± 11 Hz (n = 13) higher than the CF. There was no difference in pairing frequencies used in the NPY versus non-NPY LTD experiments (p = 0.5; unpaired t test). Stimulus–response curves for EPSP amplitude were established before the experiment began; EPSP amplitudes were set at ∼50% of maximum. EPSPs were recorded once every minute. L5 PNs were maintained with DC current at their initial resting membrane potential if necessary. Cells in which the membrane potential shifted >5 mV were discarded.
Drugs.
Unless indicated otherwise, all compounds were applied via the bath. In some experiments, kynurenic acid (1 mm; Sigma), picrotoxin (100 μm), and (3-aminopropyl)(cyclohexylmethyl)phosphinic acid (CGP 46381) (20 μm; Tocris Bioscience) were applied via the bath. Synthetic human NPY was purchased from Peptidec Technologies. The Y1-receptor agonist F7P34[NPY] (a gift from Dr. A. Beck-Sickinger, University of Leipzig, Leipzig, Germany) was made by solid-state synthesis as described previously (El Bahh et al., 2005). The Y1 receptor antagonist (R)-N-[[4-(aminocarbonylaminomethyl)-phenyl]methyl]-N2-(diphen ylacetyl)-argininamide trifluoroacetate (BIBO 3304) was a gift from Dr. H. Doods (Boehringer Ingelheim, Biberach, Germany). For localized application, NPY (1 μm) and Alexa 594 (1 μm) were dissolved in ACSF and loaded into a borosilicate glass puffer pipette. The pipette was moved into position (distally, or at the soma) under visual guidance using a motorized manipulator (Siskiyou), and control recordings were taken before drug application commenced. Pressure ejection of the NPY-containing fluorescent solution was observed periodically for the duration of the 5 min application.
Statistical analysis.
Analysis of the CF was performed as described previously (Larkum et al., 1999a). Briefly, to calculate the CF at the soma, we measured the amplitude at a single time point after the last AP evoked at the cell body for all frequencies tested (20–200 Hz; Fig. 1). This is referred to as the afterdepolarization (ADP) amplitude, which provides similar values in calculating CF as does the use of the ADP voltage–time integral (∫V * t; Potez and Larkum, 2008). To calculate the dendritic CF, we measured the ∫V * t of all four depolarizations above the resting potential (Fig. 1), a more sensitive measure than the amplitude because dendritic depolarizations also broaden as they reach the CF (Fig. 1B; Larkum et al., 1999a). The somatic ADP amplitude, ∫V * t, or peak Ca2+ transient (in imaging experiments) was plotted against frequency, and a sigmoidal curve was fitted to the data. The CF was defined as the frequency at half-maximal amplitude of this curve (Fig. 1E; Larkum et al., 1999a).
Figure 1.
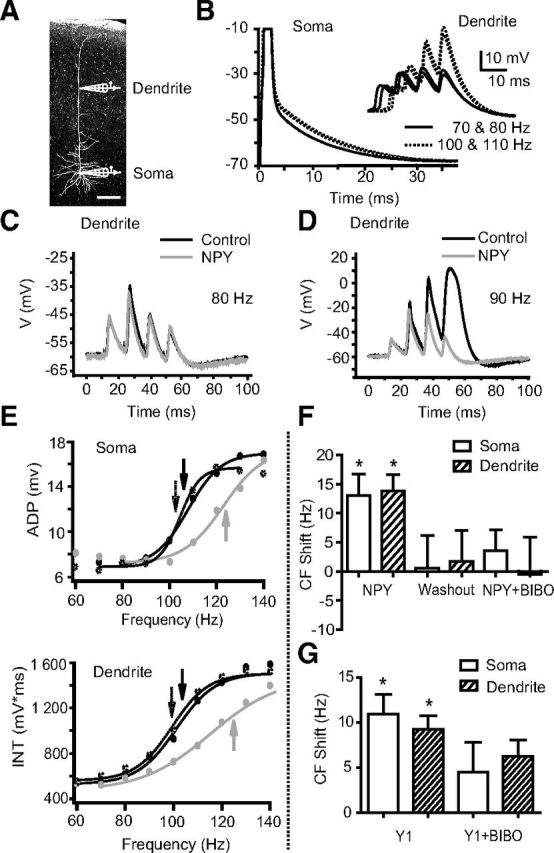
NPY increases the CF in L5 PNs. A, A neurobiotin-filled L5 PN with representative somatic and dendritic (>500 μm from the soma) recording electrodes. Scale bar, 200 μm. B, CFs were studied using trains of four somatic APs (1 ms square pulses of 1–5 nA current injections at the soma). ADPs of the fourth AP recorded at the soma from trains of different frequencies are shown superimposed. Inset, Events recorded simultaneously at the distal dendrite of this neuron. Above the CF, supralinear responses were seen at the soma and dendrite (dotted lines). C, D, Compared to control responses (black traces), bath application of NPY (1 μm, gray traces) had little effect on dendritic responses to AP trains below the CF (80 Hz). Above the CF (∼90 Hz in this cell), NPY decreases the third and fourth dendritic depolarizations. E, Somatic ADP amplitudes (top) or the voltage-time integral (INT) for simultaneous dendritic recordings (bottom) from a representative L5 PN are plotted versus frequency (60–140 Hz shown here). Sigmoidal fits were used to determine the CF (arrows). NPY (1 μm; gray trace) increases the CF compared to control (black trace); this effect reverses upon washout (∼20 min; dotted trace). F, The average shifts in CF relative to control were calculated at the soma and dendrite after NPY application (n = 6), after washout of NPY and application of the Y1 antagonist BIBO 3304 (n = 6), and after a subsequent application of NPY in the presence of BIBO 3304 (n = 3). G, The average shifts in CF relative to control were calculated for the soma and dendrite after Y1 agonist application (F7P34[NPY], 1 μm; n = 3), and then with the agonist applied again after washout in the presence of BIBO 3304. *p < 0.05 (one-sample t test). Error bars indicate ±SEM.
All data are presented as mean ± SEM. unless indicated otherwise, with n being the number of neurons analyzed. Because usually only one to two slices per animal contained fully intact dendrites, we rarely studied more than two neurons per animal. For comparisons of group values, Student's t tests were used with α values of 0.05. One-sample t tests were used to compare means with the hypothetical value of 0 or 100. We used multiple t tests and one-way ANOVAs with Dunnett's multiple comparison test.
Results
NPY1 receptor activation increases the CF
We examined the effect of bath-applied NPY on Ca2+ electrogenesis using simultaneous whole-cell recordings at the soma and distal (>500 μm from the soma) apical dendrite of L5 PNs. Trains of 4 APs at 20–200 Hz were evoked by somatic depolarizations at 20 s intervals (Fig. 1A,B; Larkum et al., 1999a, 2007; Williams and Stuart, 2000; Potez and Larkum, 2008). Under control conditions, above a CF of APs we observed a supralinear increase in dendritic membrane potential responses (quantified by calculating the voltage–time integral, ∫V * t, of the dendritic depolarization; see Materials and Methods). Supralinear activity was observed reliably in each neuron examined (Fig. 1B,D–F), as described previously (Larkum et al., 1999a). Somatic AP trains at frequencies that elicited a regenerative response seen at the dendritic electrode also caused marked increases in the amplitudes of the ADPs recorded at the soma. This is consistent with earlier reports that distal regenerative activity can propagate, causing nonlinear behavior at the soma at the same CF as in the dendrite (Larkum et al., 1999a,b, 2001; Pérez-Garci et al., 2006). In paired dendritic and somatic recordings, the somatic CF was not significantly different from the CF recorded at the distal dendrite (soma, 117 ± 8 Hz; dendrite, 113 ± 7 Hz; p > 0.2; n = 6).
Although bath application of NPY (1 μm) had no significant effects on dendritic or somatic membrane potential, ADP amplitude, or dendritic ∫V * t at sub-CF frequencies, it increased the bAP frequency needed to elicit a distal regenerative response. The CF recorded at the dendritic electrode (Fig. 1D,F,G) was increased in the presence of NPY by 14 ± 3 Hz (p < 0.005; n = 6). This effect reversed upon washout of NPY (Fig. 1G). NPY increased the CF recorded simultaneously at the somatic electrode (Fig. 1E) by 13 ± 4 Hz (p < 0.02; n = 6); this also reversed upon washout (CF shift vs control, −1 ± 5 Hz; n = 6; Fig. 1G). The NPY-mediated shifts in CF were not significantly different between dendrite and soma (p > 0.5; n = 6). At the dendrite, NPY decreased the ∫V * t elicited by bAPs at the original CF, and at the soma, NPY decreased the ADP amplitude (Table 1). Interestingly, bath application of NPY has no effect on ADP amplitude in L5 PNs from younger animals (P14–P21; data not shown), consistent with the reported absence of significant dendritic potentials in L5 PNs of animals this age (Zhu, 2000). Together, these results indicate that NPY can reversibly inhibit Ca2+ electrogenesis and increase the CF in mature L5 PNs.
Table 1.
NPY increases the CF and decreases the ADP measured both at the soma (amplitude) of L5 PNs and at their dendrites (V* t)
| Parameter | Soma | Dendrite | |
|---|---|---|---|
| +NPY | CF shift (Hz) | 13 ± 4; p < 0.02; n = 6 | 14 ± 3; p < 0.005; n = 6 |
| ADP (amplitude or integral, % control) | −36.4 ± 11.7%; p < 0.04; n = 6 | −30.3 ± 4.0%; p < 0.0006; n = 6 | |
| Washout | CF shift (Hz) | −1 ± 5; p = 0.8; n = 6 | −1 ± 5 Hz; p = 0.8; n = 6 |
| ADP (amplitude or dendritic integral, % control) | −7.6 ± 21.3%; p = 0.7; n = 6 | 5.3 ± 8.2%; p = 0.6; n = 6 |
Four subtypes of NPY receptor (Y1, Y2, Y4, and Y5) are found in the brain (Michel et al., 1998; El Bahh et al., 2005). Given the evidence for Y1 receptors in neocortex (Kopp et al., 2002), we tested whether the Y1-specific antagonist BIBO 3304 (1 μm) could prevent the actions of NPY (1 μm) on the CF of NPY-sensitive L5 PNs. While NPY caused the previously described shift in somatic and dendritic CF, reapplication of the agonist in the presence of BIBO 3304 resulted in no significant change in CF, at either the dendrite (CF shift, −1 ± 6 Hz; p > 0.9; n = 3) or the soma (CF shift, 4 ± 4 Hz; p > 0.4; n = 3; Fig. 1G). In an additional set of simultaneous dendritic and somatic recordings in L5 PNs, we applied the highly selective Y1-receptor agonist F7P34 [NPY]. Consistent with the antagonist experiment, bath application of the Y1 agonist reversibly shifted the CF at both the soma (11 ± 2 Hz; p < 0.05, n = 3) and at the distal dendrite (9 ± 2 Hz; p < 0.03, n = 3; Fig. 1H). These results indicate that the Y1 receptor mediates the action of NPY in L5 PNs.
NPY does not act through GABA or glutamate neurotransmission
Synaptic inputs to L5 PN dendrites, especially GABA inputs, have been shown to affect dendritic Ca2+ electrogenesis (Larkum et al., 1999a; Pérez-Garci et al., 2006; Murayama et al., 2009). Furthermore, earlier work in juvenile rats indicated significant effects of NPY directly on synaptic inputs to L5 PNs (Bacci et al., 2002). We therefore tested the possibility that NPY may affect either glutamate or GABA synaptic inputs to L5 PNs by testing NPY's action after blocking glutamate and GABA receptors on L5 PNs. In L5 PNs, distal GABAB receptors inhibit Ca2+ spikes (Pérez-Garci et al., 2006) by directly inhibiting dendritic long-lasting voltage-dependent calcium channels (VDCCs; Cav1; Breton and Stuart, 2012; Pérez-Garci et al., 2013). Therefore, to also determine whether NPY acted via the GABAB system, we applied NPY (1 μm) in the presence of the selective GABAB receptor antagonist CGP 46381 (20 μm), in addition to kynurenic acid (1 mm) to block ionotropic glutamate receptors and picrotoxin (100 μm) to block GABAA receptors. In these experiments, we measured only the somatic ADPs, as it has been shown previously to be a reliable indicator of supracritical distal dendritic activity in L5 PNs (Potez and Larkum, 2008). Across all frequencies tested (20–200 Hz), we saw no effect of the synaptic blockers on either the ADP or the CF compared with control responses (data not shown). Under these conditions, NPY still shifted the CF from 110 ± 8 Hz to 133 ± 6 Hz (n = 5; p < 0.01; Fig. 2A,B), an effect not significantly different from that seen in the absence of the synaptic blockers (antagonists, CF shift, 13 ± 4 Hz; p > 0.05; Fig. 2). Thus, NPY's effect on dendritic electrogenesis does not appear to be mediated by actions on GABAergic or glutamatergic inputs.
Figure 2.
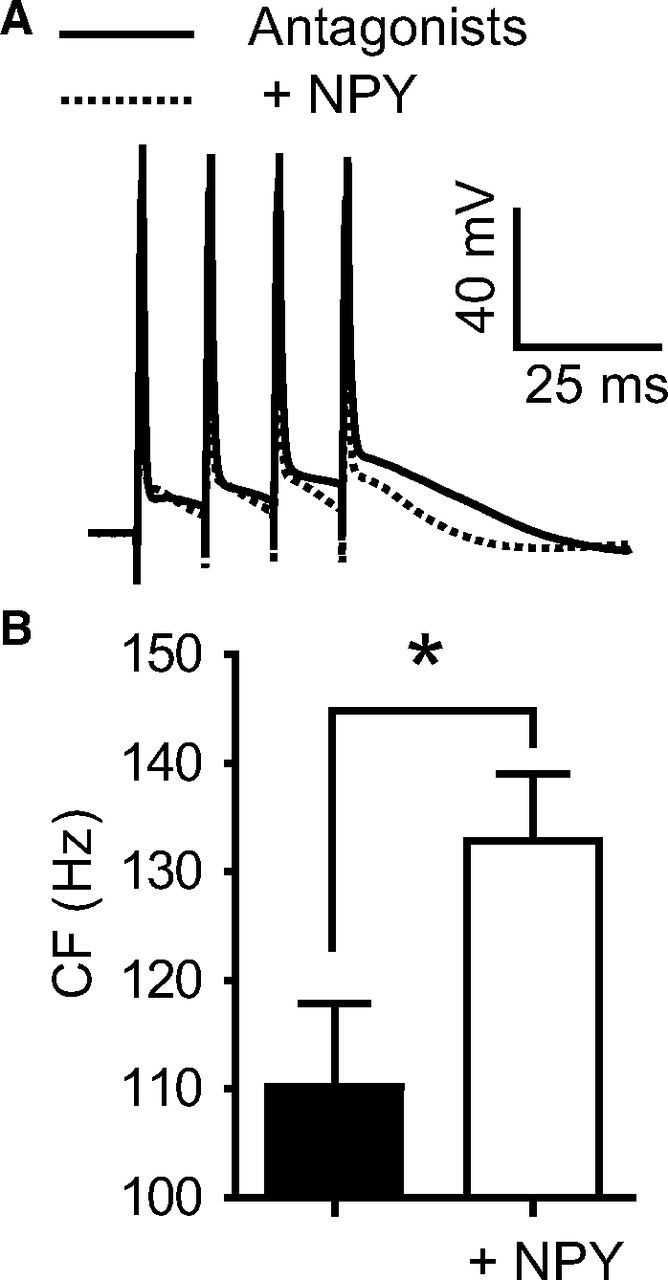
NPY increases the CF with glutamate and GABA synaptic transmission blocked. A, Bath application of kynurenic acid (1 mm) and picrotoxin (100 μm) to block glutamate- and GABA-gated ionotropic receptors, respectively, and the selective GABAB receptor antagonist CGP 46381 (20 μm; Antagonists, black trace) did not alter the CF compared to control (data not shown). With these antagonists present, 1 μm NPY (dotted trace) still inhibits the ADP recorded at the soma at the CF. B, NPY significantly increases the average CF in the presence of these antagonists (n = 7). These actions reverse upon washout of NPY (data not shown) *p < 0.05 (paired t test). Error bars indicate ±SEM.
NPY does not act through Ih or GIRK channels
Modulation of dendritic Ih or G-protein-activated inwardly rectifying K+ conductance (GIRK) can have profound effects on the integration of input to the soma (Stuart and Spruston, 1998; Korngreen and Sakmann, 2000; Berger et al., 2001; Kole et al., 2006). We did not observe a change in membrane potential at the somatic or dendritic electrode after bath addition of NPY (Fig. 1). However, to conclusively determine whether NPY may be acting on Ih and/or GIRK channels, we added 10 mm CsCl and 1 mm kynurenic acid to the bath. The kynurenic acid was applied to prevent the considerable elevation in glutamatergic transmission that otherwise would occur with a high concentration of bath applied CsCl. After observing the anticipated robust ADP amplitudes and lower CFs under these conditions compared with control experiments (50 ± 13 Hz; n = 5), we added NPY (1 μm) and still observed a significant shift in CF of 26 ± 6 Hz (n = 4; p < 0.05; Fig. 3).
Figure 3.
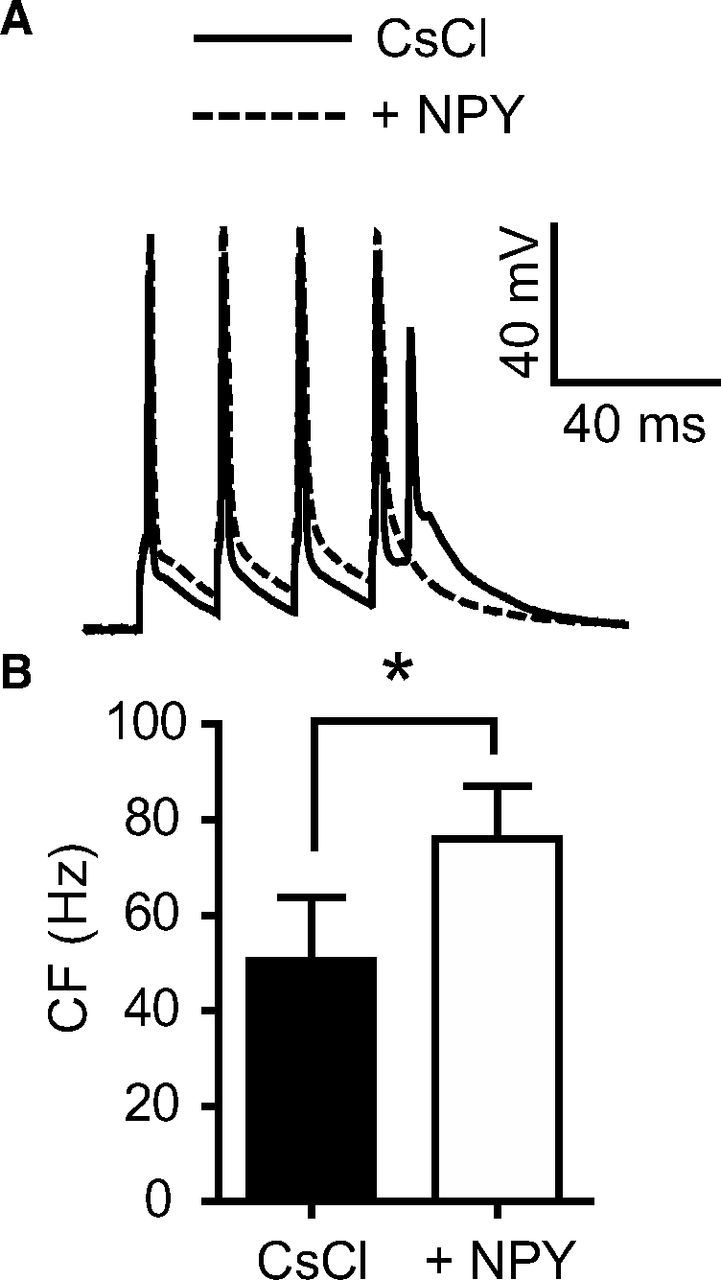
NPY increases the CF with Ih and GIRK channels blocked. A, CsCl (10 mm) was bath applied for 15 min, to block Ih and GIRK channels, along with kynurenic acid (1 mm) to reduce excessive excitability. The addition of NPY (1 μm) to the bath shifted the CF and decreased the size of the ADP. Shown here are 60 Hz traces. B, NPY shifted the average CF from 51 ± 13 Hz (CsCl) to 76 ± 11 (NPY; n = 4) *p < 0.05 (paired t test). Error bars indicate ±SEM.
NPY acts at distal dendritic sites
In L5 PNs, a markedly higher density of postsynaptic Y1 receptors is seen in distal, relative to proximal, dendrites (Kopp et al., 2002). Y1 receptors are also found postsynaptically in granule cells of the dentate gyrus, where they inhibit somatic and dendritic VDCCs (McQuiston et al., 1996; Hamilton et al., 2010). We therefore tested whether Y1 receptors located on distal dendrites in L5 PNs mediate this effect. We recorded from L5 PN somata with intracellular pipette solutions containing Alexa 594 (10 μm) and the Ca2+-sensitive dye Oregon Green BAPTA-1 (100 μm) to permit simultaneous recording of Ca2+ transients and somatic Vm using the same AP frequency sweep protocol as above. A patch pipette for focal NPY applications was filled with bath solution containing NPY (1 μm) and Alexa 594 (1 μm) to visualize the ejection of solution toward the cell (Fig. 4A). With the applicator pipette in position, but without ejecting any peptide, we obtained stable recordings of distal dendritic Ca2+ transients, along with Vm ADP responses (distal dendrite CF, 93 ± 10 Hz; somatic CF, 91 ± 9 Hz; p > 0.2; n = 6). When NPY was then ejected onto the distal dendrite (between the first distal branch point and the end of the tuft), we observed a significant and reversible CF shift at the dendrite (9.3 ± 2 Hz; p < 0.0005; n = 6) and at the soma (11 ± 1 Hz; p < 0.005; n = 6; Fig. 4B,C). Distal NPY application also significantly and reversibly decreased the peak Ca2+ transient at the control CF by 18 ± 4% (p < 0.02; n = 6) and the somatic ADP amplitude at the soma (by 36.8 ± 8.6%; p < 0.008; n = 6).
Figure 4.

NPY acts at the distal dendrite and not at the soma. A, L5 PNs were filled with the fluorescent Ca2+ indicator dye OGB-1 (100 μm) and Alexa 594 (10 μm) via a somatic patch pipette. NPY (1 μm in ACSF) is shown being ejected onto the distal dendrite from a pipette also containing Alexa 594 (1 μm). The box is representative of the region of distal Ca2+ imaging. The arrow (bottom) indicates the direction of perfusate flow. Scale bar, 50 μm. B, The ADP amplitude at the soma and peak Ca2+ currents in the distal dendrite were recorded simultaneously, before and after a distal application of NPY (left). Inset, Bottom left, Representative Ca2+ transients recorded near the control CF. After NPY washed out, the applicator pipette was repositioned to apply NPY at the soma of the same neuron (right). C, The CFs, calculated from the somatic ADP (black) and distal dendritic peak Ca2+ currents (gray), were affected only when NPY was applied to the distal dendrite (NPY Distal; n = 6). The effect of distal NPY application reversed with washout (Wash Distal; n = 6). Subsequent application of NPY at the soma (NPY soma) had no effect on the CF measured at either the soma or distal dendrite (n = 4). *p < 0.05 (difference from 0, one-sample t test). Error bars indicate ±SEM.
Once the NPY effect had washed out (p > 0.9 vs pre-NPY controls), we moved the applicator pipette to eject NPY onto the soma of four of the six neurons tested. Notably, NPY application to the soma of these cells had no significant effects on the distal dendritic CF, the somatic CF (Fig. 4B,C; n = 4), the peak Ca2+ transient, nor the ADP (p > 0.3 for all measures; n = 4). Since the somatic application also included coverage of a large part of the proximal basal dendrites of each neuron, the results suggest that NPY also does not act there to alter the CF. These data indicate that NPY acts at distal dendritic, but not somatic, sites to affect dendritic Ca2+ electrogenesis.
Dendrosomatic coupling is inhibited by NPY
An EPSP timed to occur appropriately with a bAP can result in a supralinear dendritic Ca2+ spike (Larkum et al., 1999b; Stuart and Häusser, 2001) that involves a combination of Na+, K+ (Stuart and Häusser, 2001), and distal dendritic Ca2+ currents (Koester and Sakmann, 1998). Based on our observations of NPY actions on L5 PN dendrites, we hypothesized that the threshold for a synaptically evoked, supralinear response would also increase with the application of NPY.
To test the hypothesis that NPY inhibits regenerative Ca2+ influx postsynaptically, we performed simultaneous recordings from the soma and distal apical dendrite, and then simulated an EPSP with injection of a current waveform at the dendritic electrode. Increasing dendritic depolarizations resulted in a nonlinear increase in dendritic potential and a regenerative potential (Fig. 5A; n = 2). This response was reduced by application of NPY (Fig. 5A). In separate experiments, we paired such a dendritic depolarization with a single AP evoked simultaneously at the soma. This pairing has been shown to elicit a maximal response in L5 PNs when the two stimuli are nearly simultaneous (Larkum et al., 1999b). Progressive increases in the amplitude of the dendritic current waveform revealed a robust, nonlinear increase in dendritic potential, which precedes but is amplified by the arrival of the bAP. This response results in an additional (or in some cases multiple) AP recorded at the soma (Fig. 5B).
Figure 5.

NPY inhibits the threshold for dendritic spike generation and EPSP-bAP pairing. A, Recordings were made at the distal dendrite (>500 μm from soma; left) and soma (right) of an L5 PN as in Figure 1. Current injection of increasing amplitudes (see Materials and Methods) applied at the dendrite elicited nonlinear dendritic potentials, the largest of which in turn induced action potentials at the soma (black). In the presence of NPY (2 μm), responses to the same dendritic stimulus amplitudes were substantially attenuated. Bi–Biv, Somatic APs were elicited at the soma with a square-wave current injection (Sinj; 4 nA, 1 ms) and paired (Δt = 5ms) with a subthreshold distal EPSP waveform injected at the dendritic electrode (Dinj). Subthreshold pairings (dendrite, 1.2 nA; soma, 4 nA) caused no regenerative activity in the dendrite (i). By increasing Dinj amplitude in 100 pA steps, Ca2+ spikes were observed at, and above, a pairing threshold (ii; Dinj, 1.3 nA) and resulted in an additional AP at the soma (ii–iv; black traces). Bath application of NPY (1 μm; gray) had no effect on subthreshold pairing (i), but inhibited the supralinear activity above the pairing threshold, and the additional spike at the soma (ii, iii). NPY's effect was overcome with stronger dendritic depolarization (iv). C, The dendritic ∫V*, normalized to the pairing threshold before (control, black) and after NPY application (gray; n = 4). D, The ∫V * t of the somatic responses recorded simultaneously with those in C (n = 4), also normalized to the pairing threshold before (control, black) and after NPY application (gray; n = 4). *p < 0.05 (control versus NPY, paired t test). Error bars indicate ±SEM.
To quantify this response, we defined “pairing threshold” as the amount of dendritic current injected that first caused a repeatable, supralinear dendritic response. In the recording in Figure 5B, the pairing threshold was 1.3 nA of dendritic current and resulted in a spike doublet at the soma. We repeated the experiment in the presence of 1 μm NPY. As expected, NPY had no effect on dendritic or somatic responses below the pairing threshold. However, NPY increased the amount of dendritic current required to achieve pairing threshold. In the four neurons we tested, NPY significantly reduced the dendritically recorded ∫V * t at currents at and above the control pairing threshold (Figs. 5B,C). In the presence of NPY, the dendritic current injection necessary to achieve pairing threshold increased by 225 ± 63 pA (p < 0.05; n = 4). NPY also inhibited the appearance of a second somatic AP at and above the pairing threshold (Figs. 5B,D). The modulation of Ca2+-dependent regenerative activity is consistent with NPY affecting the integrative properties of L5 PN dendrites.
NPY does not alter synaptic glutamate release onto L5 PNs
To determine whether NPY affects presynaptic inputs on L5 PNs, we stimulated L2/3 with a bipolar extracellular electrode using paired stimuli (interspike interval, 10 ms) at low, medium, and high intensities (see Materials and Methods). When NPY (1 μm) was applied via the bath, we observed no effect on EPSP amplitude recorded at L5 PN somata in all experiments (Fig. 6A,B). Specifically, neither the amplitudes of the first or second EPSP nor the paired-pulse ratio was affected by NPY application at low, medium, or high stimulus intensities (low, n = 5; medium, n = 4; high, n = 5; p > 0.05, one-way ANOVA). Finally, we tested whether NPY affected synaptic amplitude during prolonged stimulus trains (see NPY inhibits LTD in L5 PNs, below) using 100 medium-intensity EPSPs at 1 Hz to L1, repeating this synaptic protocol in the absence and then the presence of 1 μm NPY. We found no significant difference in amplitude between EPSPs at the beginning, during, and end of the train, either in the control or in the presence of NPY. (Fig. 6C,D). Together, these data indicate that NPY has no impact on presynaptic neurotransmitter release onto distal dendrites of L5 PNs.
Figure 6.
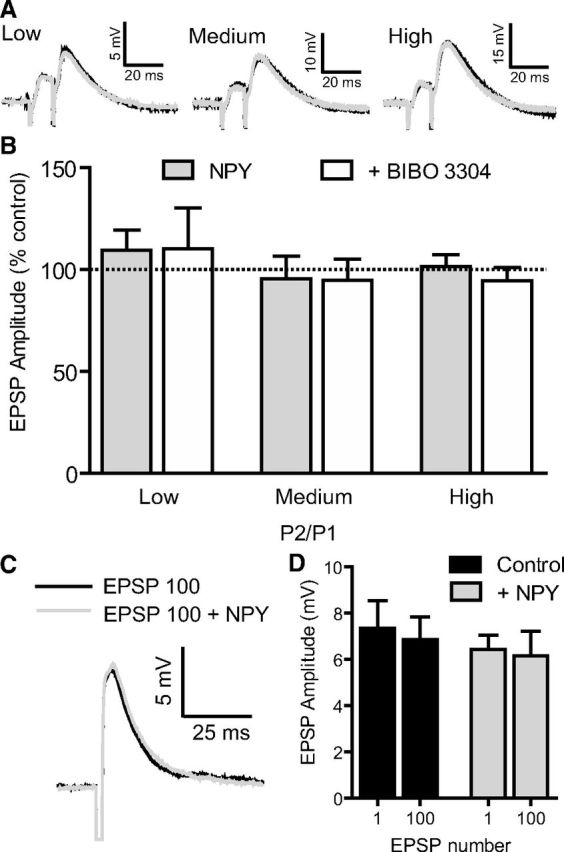
NPY does not alter presynaptic glutamate release. A, A bipolar stimulating electrode was placed in L2/3 and used to elicit EPSPs with a paired-pulse stimulus (10 ms interstimulus interval) with the stimulus strength set to elicit responses of low (25%), medium (50%), and high (75%) amplitudes, relative to maximal EPSP responses. B, EPSP amplitude of the first pulse (P1), second pulse (P2), and paired-pulse ratio (P2/P1) was not affected by NPY (1 μm) nor Y1 antagonist BIBO 3304 (1 μm) application for low (p = 0.7523; n = 5), medium (p = 0.6592; n = 4), or high (p = 0.1997; n = 5) levels of stimulation (one-way ANOVA with Dunnett's multiple comparison test). C, Single EPSP trace before and after train of 100 EPSPs at 1 Hz (black line). After NPY (1 μm) application, the rundown protocol was repeated. The 100th EPSP after NPY application is shown in gray. D, NPY (1 μm) did not change the average EPSP amplitude for the 1st or 100th EPSP (n = 4; p = 0.8397, one-way ANOVA). Error bars indicate ±SEM.
NPY inhibits LTD in L5 PNs
The induction of LTD relies on a variety of mechanisms in L5 PNs (Markram et al., 2011) including a location-dependent relationship between the site of synaptic stimulation and sign of plasticity (Sjöström and Häusser, 2006). In these neurons, proximal synapses are prone to LTP, whereas distal synapses (where NPY is seen to inhibit Ca2+ electrogenesis) seem to preferentially undergo LTD (Birtoli and Ulrich, 2004; Sjöström and Häusser, 2006; Czarnecki et al., 2007). To test whether the modulation of dendritically initiated Ca2+ spikes can influence LTD, we used a protocol consisting of a low-frequency (1 Hz) pairing of a presynaptic stimulus followed 25 ms later with the minimum two APs needed to invoke a supracritical bAP frequency (Fig. 7A), consistent with predictions of LTD induction in models (Clopath et al., 2010). We positioned a stimulating electrode in L1 and used inputs that had an EPSP rise time (measured at the soma) of at least 3 ms (average rise time, 4.3 ± 0.5 ms), which is an indicator of synapses located on the distal portion of the apical dendrite (Sjöström and Häusser, 2006). Our spike-timing-dependent plasticity (STDP) protocol was well within the temporal window for LTD, after accounting for the few milliseconds needed for the burst of APs to propagate to the distal tuft (Stuart and Sakmann, 1994). Using this STDP protocol we observed a reliable induction of LTD (Fig. 7B); the average reduction of EPSP amplitude to 65 ± 1% of control was observed over the last 15 min of recording (n = 7). This is consistent with observations of LTD induction in pairs of synaptically connected L2/3 and L5 PNs using a similar protocol (Letzkus et al., 2006).
Figure 7.
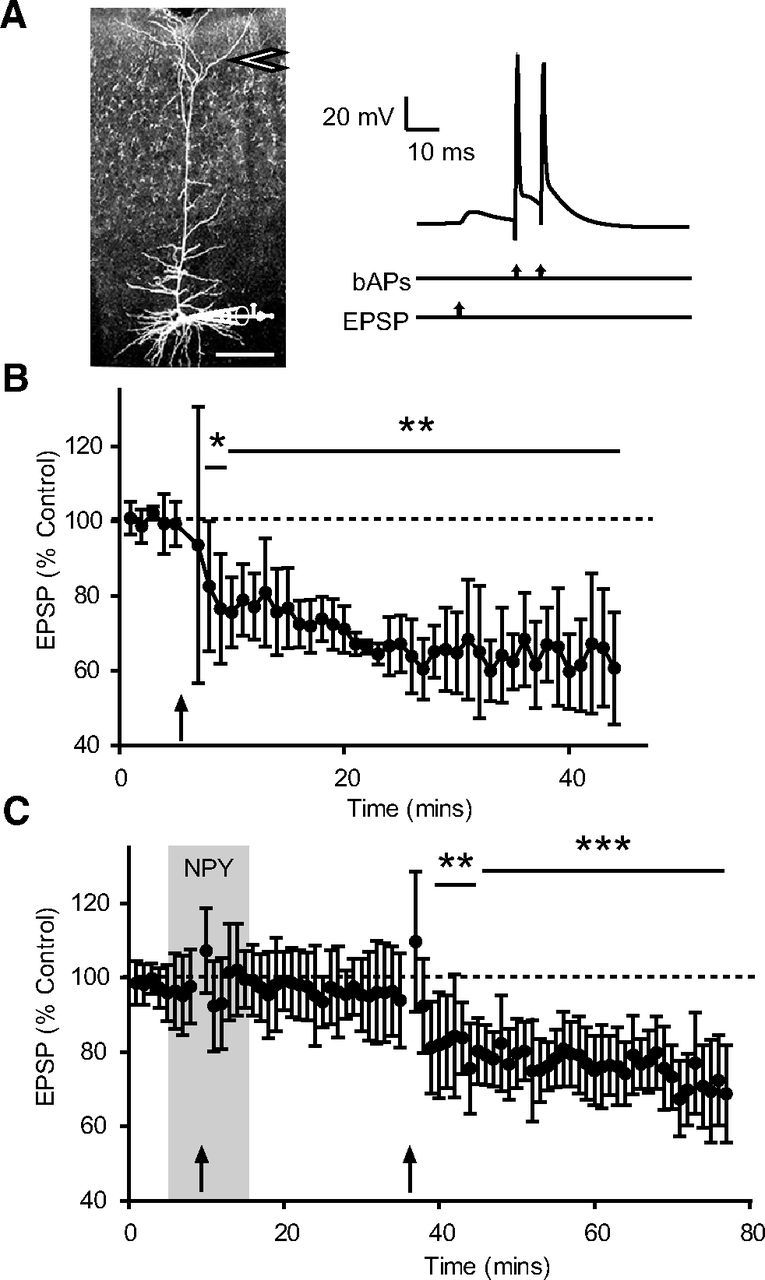
NPY inhibits induction of LTD. A, Representative somatic recording configuration with bipolar stimulation electrode positioned in L1 (arrow). Scale bar, 200 μm. The LTD protocol consisted of 100 trials of an EPSP paired 25 ms later with a burst of two somatically evoked APs; pairings were repeated at 1 Hz. The intraburst AP frequency was above the CF for each L5 PN studied. B, EPSP amplitude during the experiment, normalized (% Control) to baseline values. The pairing protocol (arrow) reliably induced LTD, with the average EPSP amplitude reduced to 65 ± 1% of control over the last 15 min of recording (n = 7). C, In another set of experiments, NPY was bath applied beginning 5 min before the onset of the pairing protocol, and for a further 5 min afterward. There was no change in EPSP amplitude 25 min after the pairing protocol. Following 20 min washout of NPY, the pairing protocol was administered again; this resulted in LTD and an average reduction in EPSP amplitude to 74 ± 1% control over the last 15 min of recording (n = 6). *p < 0.05; **p < 0.01; ***p < 0.001 (difference from 100, one-sample t test). Error bars indicate ±SEM.
We hypothesized that if LTD at these synapses requires the distal Ca2+ electrogenesis caused by supracritical bAPs, then NPY's actions should disrupt it. Therefore, in another group of L5 PNs, we performed the LTD protocol in the presence of NPY. NPY (1 μm) was applied via the bath for 5 min before initiating the LTD protocol and remained present for an additional 5 min after the protocol ended. NPY did not have any effect on baseline EPSP amplitude (control, 7.7 ± 0.5 mV; NPY, 7.5 ± 0.4 mV; n = 17; p > 0.7) after 10 min of application. After 20 min of washout, no LTD was observed in any neuron tested (EPSP amplitude was 95 ± 1% of control; n = 6; Fig. 7C). After NPY washout, we repeated the LTD protocol and reliably observed LTD induction in all neurons tested (Fig. 7C). In the absence of NPY, the LTD protocol resulted in a stable reduction in EPSP amplitude (to 74 ± 1% of control; p < 0.01; n = 6) over the last 15 min of recording.
Discussion
When synchronized with EPSPs, AP backpropagation can result in LTD depending on numerous factors, including the timing and frequency of presynaptic and postsynaptic APs, dendritic depolarization, the presence of a dendritic spike, synapse location, activation of NMDA receptors or VDCCs, and actions of neuromodulators (Heynen and Bear, 2001; Kampa et al., 2006; Letzkus et al., 2006; Clopath et al., 2010; Hamilton et al., 2010; Lisman and Spruston, 2010; Markram et al., 2011). Ultimately, these factors determine the [Ca2+]i dynamics that are needed for plasticity (Graupner and Brunel, 2012). LTD induction in neocortical PNs requires a VDCC-dependent rise in [Ca2+]i in postsynaptic spines sufficient to bind to proteins on metabotropic glutamate receptors, thereby contributing to a downstream retrograde endocannabinoid signal mediated by a phospholipase C-dependent pathway (Bender et al., 2006; Nevian and Sakmann, 2006). Blockade of VDCC-mediated Ca2+ influx with Ni2+ or chelation of intracellular Ca2+ with BAPTA prevents the activation of this pathway, and thus of LTD (Birtoli and Ulrich, 2004); however, it is unknown whether intrinsic neuromodulators can affect the induction of LTD in L5 PNs. Here, Ca2+ spikes initiated in the distal apical dendrite by bAP trains at rates above a CF were inhibited by activation of NPY receptors located on distal dendrites, which in turn inhibited the induction of LTD. Thus, Ca2+ influx at the distal apical dendritic initiation zone (Larkum et al., 2009) is crucial to this form of plasticity, and the localized postsynaptic action of NPY can prevent its occurrence.
Our findings here in L5 PNs are consistent with earlier reports that either multiple APs above a CF or nearly simultaneous presynaptic and postsynaptic activity will induce regenerative events in the distal apical dendrite (Koester and Sakmann, 1998; Larkum et al., 1999a,b), and that these distal dendritic Ca2+ spikes propagate to the soma (Larkum et al., 2001, 2009). Since Y1 receptors are concentrated on the dendrites of L5 PNs (Kopp et al., 2002), and the most readily excitable, NPY-expressing neurons reside in L1 (Karagiannis et al., 2009), we hypothesized that these postsynaptic receptors would decrease Ca2+-influx, thereby suppressing dendritic regenerative activity and resulting in an increase in the CF, similar to the actions of Y1 receptors in the dentate gyrus (Hamilton et al., 2010). We indeed observed that NPY, via the Y1 receptor, suppressed regenerative activity in the apical dendrite and increased the CF. Because local application of NPY to the apical dendrite, but not the soma, resulted in significant changes in CF, similar to those seen with bath application, we can reasonably conclude that NPY acts predominantly at the distal apical dendrite.
Earlier work indicated that NPY application to L5 PNs from young (P13–P21) rats causes a gradual and prolonged facilitation of evoked GABAergic IPSC inputs, and a gradual, prolonged inhibition of evoked excitatory EPSC amplitudes (Bacci et al., 2002). The effect on GABA inputs was attributed to an inhibition of GABAergic input to L5 interneurons, with consequent disinhibition of GABA cells innervating L5 PNs (Bacci et al., 2002). While activation of either GABAA, or GABAB, receptors also inhibits dendritic Ca2+ spikes in L5 PNs (Larkum et al., 1999b; Pérez-Garci et al., 2006), the effect of NPY seen here was not changed by blockade of GABAA and GABAB receptors with picrotoxin and CGP 46381, respectively. Furthermore, NPY did not alter evoked excitatory synaptic transmission, studied at a variety of intensities, nor did it either alter short-term synaptic plasticity or affect synaptic rundown at the frequencies used in LTD induction. Finally, NPY's action was not unusually persistent; the inhibition of regenerative activity seen during NPY application washed out within 10–30 min. We therefore consider it unlikely that the effect of NPY on the CF, pairing threshold, and LTD was mediated by prolonged effects on synaptic transmission onto L5 PNs. Our experiments were conducted in animals considerably older than those in which the synaptic actions of NPY were described previously (Bacci et al., 2002), largely because dendritic electrogenesis only is fully expressed in adult animals (Zhu, 2000; Larkum and Zhu, 2002). It is possible that differences between our findings and those of Bacci et al. (2002) could result from developmental changes, in which presynaptic actions of NPY seen in younger animals are superseded by the actions we describe here. In separate experiments (data not shown), we were able to replicate the NPY-mediated potentiation of Ca2+-dependent miniature GABA responses in L5 PNs from young (P 14–P20) rats reported by Bacci et al. (2002), but did not observe NPY-mediated changes in spontaneous GABA synaptic events. As the actions of NPY on dendritic electrogenesis remained robust when GABAergic transmission was blocked, we consider the actions of NPY studied here to be independent of the GABA system.
NPY receptors can increase GIRK conductances (Rhim et al., 1997, Chee et al., 2010) or block a hyperpolarization-activated cation conductance (Ih; Giesbrecht et al., 2010); either effect might indirectly inhibit Ca2+ electrogenesis (Berger et al., 2001). NPY's effect on the CF persisted in the presence of a concentration of CsCl (10 mm) sufficient to completely block these channels, making these mechanisms unlikely. However, NPY receptors do inhibit N-type and other VDCCs in the hippocampus, at both presynaptic and postsynaptic sites, and the predominant postsynaptic actions of NPY in hippocampus are via Y1 receptors (McQuiston and Colmers, 1996;McQuiston et al., 1996; Qian et al., 1997). The present findings are entirely consistent with an inhibition of VDCCs at the calcium spike initiation zone on the distal apical dendrite (Pérez-Garci et al., 2006; Murayama et al., 2009; Palmer et al., 2012), similar to its actions on VDCCs seen in DGCs of the hippocampus (Hamilton et al., 2010). Because the regenerative event in L5 PN dendrites requires the cooperation of several conductances including for Na+ (Larkum et al., 1999b), we cannot readily exclude the possibility that there is an additional effect of NPY on Na+ influx; however, we consider this much less likely given the widespread actions of NPY receptors on neuronal VDCCs and the lack of effect of NPY on sub-CF bAPs.
In the neocortex, top-down information is conveyed to L1, while bottom-up thalamic input arrives mainly onto L4 stellate cells that then innervate proximal dendrites of L5 PNs (Douglas and Martin, 2004). Here, the application of NPY onto the distal apical dendrite of an L5 PN inhibited Ca2+ spikes and decreased its sensitivity to nearly simultaneous presynaptic and postsynaptic input. The STDP model proposed previously by Clopath et al. (2010) predicts that LTD induction requires a low level of postsynaptic depolarization to trigger an increase in postsynaptic [Ca2+]i and the subsequent release of retrograde messengers. This suggests that the induction of a regenerative dendritic Ca2+ potential, paired with modest levels of synaptic excitation, is the key determinant of plasticity (Kampa et al., 2006; Graupner and Brunel, 2012). Because NPY has conclusively no effects on evoked glutamate release under our conditions, its actions are ideal to examine not only the role of dendritic Y1 receptors themselves, but also to unambiguously determine the role of dendritic Ca2+ influx in LTD induction. By decreasing Ca2+ influx at firing rates above the CF, NPY will decrease the amount of dendritic depolarization and reduce VDCC-mediated Ca2+ influx, consistent with the models of Clopath et al. (2010) and Graupner and Brunel (2012), thus preventing the VDCC-induced endocannabinoid signaling (Nevian and Sakmann, 2006); both actions would suppress LTD induction.
GABA input to distal dendrites of L5 PNs also inhibits activity-dependent Ca2+ electrogenesis (Pérez-Garci et al., 2006; Breton and Stuart, 2012); however, synaptic GABA release only suppresses BAC activity briefly, transiently preventing plasticity. The longer-lasting actions of NPY, whose release requires more intense stimulation, should suppress plasticity over a more prolonged period. Neuromodulators can alter synaptic learning rules by altering the threshold for plasticity (Graupner and Brunel, 2012), as seen here with NPY. Neocortical NPY-expressing neurons are almost exclusively GABAergic (Karagiannis et al., 2009); therefore, cells such as Martinotti-like neurons, which can release GABA at low levels of activity and NPY with higher levels of activity, are poised to alter the [Ca2+]i threshold for plasticity in a dynamic manner, depending on their activation level. Selective stimulation of Martinotti-like neurons using targeted expression of optogenetic or Designer Receptors Exclusively Activated by Designer Drugs (DREADD) proteins could be used to determine the impact of endogenous NPY release on active dendritic properties of L5 PNs.
Our results suggest that NPY can dramatically influence the integration of top-down and bottom-up inputs in L5 PNs, particularly when in the plasticity-prone, depolarizing up state. Consistent with the prediction of a factor critical for the induction of plasticity (Lisman and Spruston, 2010), regenerative Ca2+ activity at the distal apical dendritic initiation zone not only is important for the integration of converging synaptic input (Larkum et al., 1999b, 2009), but is also poised to prominently regulate the induction of LTD at distal synapses. Because the Martinotti-like cells of L1 and L2 are likely to be driven by excitatory inputs from L1, it is possible that the top-down input governs the suppression of LTD by NPY.
Footnotes
This work was supported by Canadian Institutes for Health Research (CIHR) Operating Grant MT10520 and the CIHR Team on the Neurobiology of Obesity (Grant OTG 88592; W.F.C.). The University of Alberta Hospitals Foundation provided generous equipment support for this project. W.F.C. is especially grateful to Dr. Bert Sakmann for his support and encouragement in the initiation of this work as a sabbatical guest in his laboratory. We are grateful to Dr. Annette Beck-Sickinger (Leipzig) for the generous gift of the NPY receptor-selective agonists, and Dr. Henri Doods (Boehringer-Ingelheim) for the gift of BIBO 3304. T.J.H. was supported in part by a bursary from the Centre for Neuroscience, University of Alberta. W.F.C. is a Medical Scientist of the Alberta Heritage Foundation for Medical Research.
The authors declare no competing financial interests.
References
- Bacci A, Huguenard JR, Prince DA. Differential modulation of synaptic transmission by neuropeptide Y in rat neocortical neurons. Proc Natl Acad Sci U S A. 2002;99:17125–17130. doi: 10.1073/pnas.012481899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger T, Larkum ME, Lüscher HR. High I(h) channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. J Neurophysiol. 2001;85:855–868. doi: 10.1152/jn.2001.85.2.855. [DOI] [PubMed] [Google Scholar]
- Birtoli B, Ulrich D. Firing mode-dependent synaptic plasticity in rat neocortical pyramidal neurons. J Neurosci. 2004;24:4935–4940. doi: 10.1523/JNEUROSCI.0795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton J, Stuart G. Somatic and dendritic GABAB receptors regulate neuronal excitability via different mechanisms. J Neurophysiol. 2012;8:2810–2818. doi: 10.1152/jn.00524.2012. [DOI] [PubMed] [Google Scholar]
- Chee MJ, Myers MG, Jr, Price CJ, Colmers WF. Neuropeptide Y suppresses anorexigenic output from the ventromedial nucleus of the hypothalamus. J Neurosci. 2010;30:3380–3390. doi: 10.1523/JNEUROSCI.4031-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clopath C, Büsing L, Vasilaki E, Gerstner W. Connectivity reflects coding: a model of voltage-based STDP with homeostasis. Nat Neurosci. 2010;13:344–352. doi: 10.1038/nn.2479. [DOI] [PubMed] [Google Scholar]
- Czarnecki A, Birtoli B, Ulrich D. Cellular mechanisms of burst firing-mediated long-term depression in rat neocortical pyramidal cells. J Physiol. 2007;578:471–479. doi: 10.1113/jphysiol.2006.123588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas RJ, Martin KA. Neuronal circuits of the neocortex. Annu Rev Neurosci. 2004;27:419–451. doi: 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- Dumont Y, Fournier A, St-Pierre S, Quirion R. Autoradiographic distribution of [125I]Leu31,Pro34]PYY and [125I]PYY3–36 binding sites in the rat brain evaluated with two newly developed Y1 and Y2 receptor radioligands. Synapse. 1996;22:139–158. doi: 10.1002/(SICI)1098-2396(199602)22:2<139::AID-SYN7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- El Bahh B, Balosso S, Hamilton T, Herzog H, Beck-Sickinger AG, Sperk G, Gehlert DR, Vezzani A, Colmers WF. The anti-epileptic actions of neuropeptide Y in the hippocampus are mediated by Y and not Y receptors. Eur J Neurosci. 2005;22:1417–1430. doi: 10.1111/j.1460-9568.2005.04338.x. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Letzkus JJ, Kampa BM, Hang GB, Stuart GJ. Dendritic synapse location and neocortical spike-timing-dependent plasticity. Front Synaptic Neurosci. 2010;2:29. doi: 10.3389/fnsyn.2010.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesbrecht CJ, Mackay JP, Silveira HB, Urban JH, Colmers WF. Countervailing modulation of Ih by neuropeptide Y and corticotrophin-releasing factor in basolateral amygdala as a possible mechanism for their effects on stress-related behaviors. J Neurosci. 2010;30:16970–16982. doi: 10.1523/JNEUROSCI.2306-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert CD, Sigman M. Brain states: top-down influences in sensory processing. Neuron. 2007;54:677–696. doi: 10.1016/j.neuron.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Gilbert CD, Sigman M, Crist RE. The neural basis of perceptual learning. Neuron. 2001;31:681–697. doi: 10.1016/S0896-6273(01)00424-X. [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Graupner M, Brunel N. Calcium-based plasticity model explains sensitivity of synaptic changes to spike pattern, rate, and dendritic location. Proc Natl Acad Sci U S A. 2012;6:3991–3996. doi: 10.1073/pnas.1109359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton TJ, Wheatley BM, Sinclair DB, Bachmann M, Larkum ME, Colmers WF. Dopamine modulates synaptic plasticity in dendrites of rat and human dentate granule cells. Proc Natl Acad Sci U S A. 2010;107:18185–18190. doi: 10.1073/pnas.1011558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry SH, Jones EG, Emson PC. Morphology, distribution, and synaptic relations of somatostatin- and neuropeptide Y-immunoreactive neurons in rat and monkey neocortex. J Neurosci. 1984;4:2497–2517. doi: 10.1523/JNEUROSCI.04-10-02497.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heynen AJ, Bear MF. Long-term potentiation of thalamocortical transmission in the adult visual cortex in vivo. J Neurosci. 2001;21:9801–9813. doi: 10.1523/JNEUROSCI.21-24-09801.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol. 2006;574:283–290. doi: 10.1113/jphysiol.2006.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis A, Gallopin T, Dávid C, Battaglia D, Geoffroy H, Rossier J, Hillman EM, Staiger JF, Cauli B. Classification of NPY-expressing neocortical interneurons. J Neurosci. 2009;29:3642–3659. doi: 10.1523/JNEUROSCI.0058-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci U S A. 1998;95:9596–9601. doi: 10.1073/pnas.95.16.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler C, Schultzberg M, Radesäter AC. Distribution of neuropeptide Y receptors in the rat hippocampal region. Neurosci Lett. 1987;75:141–146. doi: 10.1016/0304-3940(87)90287-4. [DOI] [PubMed] [Google Scholar]
- Kole MH, Hallermann S, Stuart GJ. Single Ih channels in pyramidal neuron dendrites: properties, distribution, and impact on action potential output. J Neurosci. 2006;26:1677–1687. doi: 10.1523/JNEUROSCI.3664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp J, Xu ZQ, Zhang X, Pedrazzini T, Herzog H, Kresse A, Wong H, Walsh JH, Hökfelt T. Expression of the neuropeptide Y Y1 receptor in the CNS of rat and of wild-type and Y1 receptor knock-out mice. Focus on immunohistochemical localization. Neuroscience. 2002;111:443–532. doi: 10.1016/S0306-4522(01)00463-8. [DOI] [PubMed] [Google Scholar]
- Korngreen A, Sakmann B. Voltage-gated K+ channels in layer 5 neocortical pyramidal neurones from young rats: subtypes and gradients. J Physiol. 2000;525:621–639. doi: 10.1111/j.1469-7793.2000.00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ. Signaling of layer 1 and whisker-evoked Ca2+ and Na+ action potentials in distal and terminal dendrites of rat neocortical pyramidal neurons in vitro and in vivo. J Neurosci. 2002;22:6991–7005. doi: 10.1523/JNEUROSCI.22-16-06991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Kaiser KM, Sakmann B. Calcium electrogenesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc Natl Acad Sci U S A. 1999a;96:14600–14604. doi: 10.1073/pnas.96.25.14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999b;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J Physiol. 2001;533:447–466. doi: 10.1111/j.1469-7793.2001.0447a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Waters J, Sakmann B, Helmchen F. Dendritic spikes in apical dendrites of neocortical layer 2/3 pyramidal neurons. J Neurosci. 2007;27:8999–9008. doi: 10.1523/JNEUROSCI.1717-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Nevian T, Sandler M, Polsky A, Schiller J. Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science. 2009;325:756–760. doi: 10.1126/science.1171958. [DOI] [PubMed] [Google Scholar]
- Letzkus JJ, Kampa BM, Stuart GJ. Learning rules for spike timing-dependent plasticity depend on dendritic synapse location. J Neurosci. 2006;26:10420–10429. doi: 10.1523/JNEUROSCI.2650-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Spruston N. Questions about STDP as a general model of synaptic plasticity. Front Synaptic Neurosci. 2010;2:140. doi: 10.3389/fnsyn.2010.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London M, Häusser M. Dendritic computation. Annu Rev Neurosci. 2005;28:503–532. doi: 10.1146/annurev.neuro.28.061604.135703. [DOI] [PubMed] [Google Scholar]
- Markram H, Gerstner W, Sjöström PJ. A history of spike-timing-dependent plasticity. Front Synaptic Neurosci. 2011;3:4. doi: 10.3389/fnsyn.2011.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Colmers WF. Neuropeptide Y2 receptors inhibit the frequency of spontaneous but not miniature EPSCs in CA3 pyramidal cells of rat hippocampus. J Neurophysiol. 1996;76:3159–3168. doi: 10.1152/jn.1996.76.5.3159. [DOI] [PubMed] [Google Scholar]
- McQuiston AR, Petrozzino JJ, Connor JA, Colmers WF. Neuropeptide Y1 receptors inhibit N-type calcium currents and reduce transient calcium increases in rat dentate granule cells. J Neurosci. 1996;16:1422–1429. doi: 10.1523/JNEUROSCI.16-04-01422.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, Quirion R, Schwartz T, Westfall T. XVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacol Rev. 1998;50:143–150. [PubMed] [Google Scholar]
- Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, Larkum ME. Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature. 2009;457:1137–1141. doi: 10.1038/nature07663. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent-plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Schulz JM, Murphy SC, Ledergerber D, Murayama M, Larkum ME. The cellular basis of GABAB-mediated interhemispheric inhibition. Science. 2012;335:989–993. doi: 10.1126/science.1217276. [DOI] [PubMed] [Google Scholar]
- Pérez-Garci E, Gassmann M, Bettler B, Larkum ME. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron. 2006;50:603–616. doi: 10.1016/j.neuron.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Pérez-Garci E, Larkum ME, Nevian T. Inhibition of dendritic Ca2+ spikes by GABAB receptors in cortical pyramidal neurons is mediated by a direct Gi/o-betagamma subunit interaction with Cav1 channels. J Physiol. 2013;591:1599–1612. doi: 10.1113/jphysiol.2012.245464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potez S, Larkum ME. Effect of common anesthetics on dendritic properties in layer 5 neocortical pyramidal neurons. J Neurophysiol. 2008;99:1394–1407. doi: 10.1152/jn.01126.2007. [DOI] [PubMed] [Google Scholar]
- Qian J, Colmers WF, Saggau P. Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: modulation of presynaptic Ca2+ entry. J Neurosci. 1997;17:8169–8177. doi: 10.1523/JNEUROSCI.17-21-08169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim H, Kinney GA, Emmerson PJ, Miller RJ. Regulation of neurotransmission in the arcuate nucleus of the rat by different neuropeptide Y receptors. J Neurosci. 1997;17:2980–2989. doi: 10.1523/JNEUROSCI.17-09-02980.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol. 1997;505:605–616. doi: 10.1111/j.1469-7793.1997.605ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt P, Crill W. Mechanisms underlying burst and regular spiking evoked by dendritic depolarization in layer 5 cortical pyramidal neurons. J Neurophysiol. 1999;81:1341–1354. doi: 10.1152/jn.1999.81.3.1341. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Häusser M. A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron. 2006;51:227–238. doi: 10.1016/j.neuron.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Häusser M. Dendritic coincidence detection of EPSPs and action potentials. Nat Neurosci. 2001;4:63–71. doi: 10.1038/82910. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature. 1994;367:69–72. doi: 10.1038/367069a0. [DOI] [PubMed] [Google Scholar]
- Stuart G, Spruston N. Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J Neurosci. 1998;18:3501–3510. doi: 10.1523/JNEUROSCI.18-10-03501.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Backpropagation of physiological spike trains in neocortical pyramidal neurons: Implications for temporal coding in dendrites. J Neurosci. 2000;20:8238–8246. doi: 10.1523/JNEUROSCI.20-22-08238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ. Maturation of layer 5 neocortical pyramidal neurons: amplifying salient layer 1 and layer 4 inputs by Ca 2+ action potentials in adult rat tuft dendrites. J Physiol. 2000;526:571–587. doi: 10.1111/j.1469-7793.2000.00571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]