

Abstract
Bitter taste is a basic taste modality, required to safeguard animals against consuming toxic substances. Bitter compounds are recognized by G-protein-coupled bitter taste receptors (TAS2Rs). The human TAS2R10 responds to the toxic strychnine and numerous other compounds. The mechanism underlying the development of the broad tuning of some TAS2Rs is not understood. Using comparative modeling, site-directed mutagenesis, and functional assays, we identified residues involved in agonist-induced activation of TAS2R10, and investigated the effects of different substitutions on the receptor's response profile. Most interestingly, mutations in S853.29 and Q1755.40 have differential impact on stimulation with different agonists. The fact that single point mutations lead to improved responses for some agonists and to decreased activation by others indicates that the binding site has evolved to optimally accommodate multiple agonists at the expense of reduced potency. TAS2R10 shares the agonist strychnine with TAS2R46, another broadly tuned receptor. Engineering the key determinants for TAS2R46 activation by strychnine in TAS2R10 caused a loss of response to strychnine, indicating that these paralog receptors display different strychnine-binding modes, which suggests independent acquisition of agonist specificities. This implies that the gene duplication event preceding primate speciation was accompanied by independent evolution of the strychnine-binding sites.
Introduction
The mammalian sense of taste is critical for the evaluation of the chemical composition of ingested food. Specialized sensory cells devoted to detecting one of the five basic taste qualities (sweet, sour, salty, umami, and bitter) are localized in the oral cavity (Yarmolinsky et al., 2009). Bitter taste is mediated by numerous receptors, the TAS2Rs, belonging to the G-protein-coupled receptor superfamily (Adler et al., 2000; Matsunami et al., 2000). The human TAS2R gene family consists of ∼25 functional members, for most of which cognate bitter substances have been found (Meyerhof et al., 2010). The characterization of TAS2Rs revealed pronounced differences in their tuning breadths toward bitter agonists. Whereas some TAS2Rs are activated by few bitter compounds, most receptors reacted to several or even numerous substances (Meyerhof et al., 2010). Among those, TAS2R10 (Bufe et al., 2002), TAS2R14 (Behrens et al., 2004), and TAS2R46 (Brockhoff et al., 2007) stand out because each of them responds in vitro to a large fraction of hitherto tested bitter compounds. Each TAS2R is characterized by a unique pattern of cognate bitter substances, although numerous compounds activate several TAS2Rs (Meyerhof et al., 2010), summarized in the BitterDB database (Wiener et al., 2012). Interestingly, overlapping agonist spectra do not necessarily correspond to amino acid sequence similarities among the compared receptors (Behrens et al., 2009). Instead, sequence-dissimilar TAS2Rs may exhibit remarkable similarities in their response profiles. For example, TAS2R46 belongs to a subfamily of eight related lineage-specific TAS2Rs (Bufe et al., 2002; Shi et al., 2003) and responds to numerous bitter compounds, such as sesquiterpene lactones (e.g., parthenolide), the alkaloid strychnine, and the synthetic denatonium benzoate (Brockhoff et al., 2007; Meyerhof et al., 2010). Although TAS2R10 exhibits only 34% amino acid sequence identity with TAS2R46, it also responds to these compounds (Meyerhof et al., 2010). The overlap in agonist activation patterns could be a consequence of the extraordinarily broad tuning of both receptors and, hence, may occur on a random basis. However, it is also conceivable that the similar activation properties may originate from evolutionary conservation (e.g., both receptors originate from the same ancestral TAS2R gene and preserved some of its agonist profile). We have recently determined the structural requirements of TAS2R46 activation demonstrating that the receptor accommodates all agonists in a single binding pocket and that residues in transmembrane domain VII are particularly important for agonist selectivity (Brockhoff et al., 2010). In the present study, we investigated TAS2R10-agonist interaction to identify the basis for its exceptional breadth of tuning and to compare the residues involved in interactions with strychnine in TAS2R10 and in TAS2R46. By in silico modeling, point mutagenesis, and functional assays, we identified the putative agonist binding pocket of TAS2R10. Next, in silico agonist docking was performed with the chemically different compounds, strychnine, parthenolide, and denatonium benzoate, all of which activate both TAS2R10 and TAS2R46 (Meyerhof et al., 2010). The resulting binding modes were verified by mutagenesis and analyzed to provide details of molecular recognition and insights into the evolution of the binding site.
Materials and Methods
Bitter compounds.
Strychnine, chloramphenicol, papaverine, santonin, and denatonium benzoate were purchased from Sigma. Parthenolide and costunolide were available from a previous study (Brockhoff et al., 2007). Cucurbitacin B was purchased from CPS Chemie + Service.
Generation of expression constructs.
TAS2R10 constructs used in this study were generated on the basis of variant NP_07641, SNP T156; S231. The TAS2R10 coding sequence fused with an N-terminal sst3-tag and a C-terminal hsv-tag was cloned in the vector pcDNA5/FRT (Invitrogen) as described previously (Bufe et al., 2002). Site-directed mutagenesis of receptor cDNA was performed as described previously (Behrens et al., 2006). Oligonucleotides used for mutagenesis were synthesized by Eurofins Operon MWG and are listed in Table 1.
Table 1.
Oligonucleotides used for site-directed mutagenesis
| Construct | Oligonucleotide | Sequence (5′ to 3′) |
|---|---|---|
| TAS2R10-S85A | Forward | CTAATTGAATATATTGCTTACTTTTGGGTAATTG |
| Reverse | CAATTACCCAAAAGTAAGCAATATATTCAATTAG | |
| TAS2R10-S85T | Forward | GGTAACCTAATTGAATATATTACTTACTTTTGGGTAATTGG |
| Reverse | CCAATTACCCAAAAGTAAGTAATATATTCAATTAGGTTACC | |
| TAS2R10-S85V | Forward | GTAACCTAATTGAATATATTGTTTACTTTTGGGTAATTGG |
| Reverse | CCAATTACCCAAAAGTAAACAATATATTCAATTAGGTTAC | |
| TAS2R10-W88A | Forward | CTAATTGAATATATTAGTTACTTTGCGGTAATTGGTAATCAATCAAG |
| Reverse | CTTGATTGATTACCAATTACCGCAAAGTAACTAATATATTCAATTAG | |
| TAS2R10-W88F | Forward | GAATATATTAGTTACTTTTTCGTAATTGGTAATCAATCAAG |
| Reverse | CTTGATTGATTACCAATTACGAAAAAGTAACTAATATATTC | |
| TAS2R10-W88H | Forward | CCTAATTGAATATATTAGTTACTTTCATGTAATTGGTAATCAATCAAG |
| Reverse | CTTGATTGATTACCAATTACATGAAAGTAACTAAT ATATTCAATTAGG | |
| TAS2R10-V89A | Forward | GAATATATTAGTTACTTTTGGGCAATTGGTAATCAATCAAGTATGTGG |
| Reverse | CCACATACTTGATTGATTACCAATTGCCCAAAAGTAACTAATATATTC | |
| TAS2R10-N92A | Forward | GTTACTTTTGGGTAATTGGTGCTCAATCAAGTATGTGGTTTG |
| Reverse | CAAACCACATACTTGATTGAGCACCAATTACCCAAAAGTAAC | |
| TAS2R10-N92D | Forward | CTTTTGGGTAATTGGTGATCAATCAAGTATGTGGTTTG |
| Reverse | CAAACCACATACTTGATTGATCACCAATTACCCAAAAG | |
| TAS2R10-Q93A | Forward | GTTACTTTTGGGTAATTGGTAATGCATCAAGTATGTGGTTTGCCAC |
| Reverse | GTGGCAAACCACATACTTGATGCATTACCAATTACCCAAAAGTAAC | |
| TAS2R10-Q175A | Forward | GTATAAAAGTGAATACTTTATTAAAGCGATTTTGCTAAATCTGGGAGTC |
| Reverse | GACTCCCAGATTTAGCAAAATCGCTTTAATAAAGTATTCACTTTTATAC | |
| TAS2R10-Q175L | Forward | GTATAAAAGTGAATACTTTATTAAATTGATTTTGCTAAATCTGGGAGTC |
| Reverse | GACTCCCAGATTTAGCAAAATCAATTTAATAAAGTATTCACTTTTATAC | |
| TAS2R10-Q175N | Forward | GTATAAAAGTGAATACTTTATTAAAAATATTTTGCTAAATCTGGGAGTC |
| Reverse | GACTCCCAGATTTAGCAAAATATTTTTAATAAAGTATTCACTTTTATAC | |
| TAS2R10-Q175T | Forward | GTATAAAAGTGAATACTTTATTAAAACGATTTTGCTAAATCTGGGAGTC |
| Reverse | GACTCCCAGATTTAGCAAAATCGTTTTAATAAAGTATTCACTTTTATAC | |
| TAS2R10-Q175Y | Forward | GTGAATACTTTATTAAATACATTTTGCTAAATCTGGGAGTC |
| Reverse | GACTCCCAGATTTAGCAAAATGTATTTAATAAAGTATTCAC | |
| TAS2R10-L178A | Forward | CTTTATTAAACAGATTTTGGCAAATCTGGGAGTCATTTTC |
| Reverse | GAAAATGACTCCCAGATTTGCCAAAATCTGTTTAATAAAG | |
| TAS2R10-L178F | Forward | CTTTATTAAACAGATTTTGTTTAATCTGGGAGTCATTTTCTTC |
| Reverse | GAAGAAAATGACTCCCAGATTAAACAAAATCTGTTTAATAAAG | |
| TAS2R10-L178T | Forward | CTTTATTAAACAGATTTTGACAAATCTGGGAGTCATTTTCTTC |
| Reverse | GAAGAAAATGACTCCCAGATTTGTCAAAATCTGTTTAATAAAG | |
| TAS2R10-Y239A | Forward | CATCATCCTCTTTATCTTGGCTTTTATAGGCATGGCCATAG |
| Reverse | CTATGGCCATGCCTATAAAAGCCAAGATAAAGAGGATGATG | |
| TAS2R10-M263A | Forward | CTTATGTTTGGAGCGACAACCACAG |
| Reverse | CTGTGGTTGTCGCTCCAAACATAAG | |
| TAS2R10-M263E | Forward | CTTATGTTTGGAGAGACAACCACAGCC |
| Reverse | GGCTGTGGTTGTCTCTCCAAACATAAG | |
| TAS2R10-T266A | Forward | GTTTGGAATGACAACCGCAGCCATCTATCCCTG |
| Reverse | CAGGGATAGATGGCTGCGGTTGTCATTCCAAAC | |
| TAS2R10-M263E-T266A | Forward | GTTTGGAGAGACAACCGCAGCCATCTATC |
| Reverse | GATAGATGGCTGCGGTTGTCTCTCCAAAC | |
| Vector primer | CMV forward | CGCAAATGGGCGGTAGGCGTG |
| pCR3.1-BGH reverse | TAGAAGGCACAGTCGAGG |
The constructs generated by point mutagenesis are specified in the left column. Next, the nucleotide sequences for the forward and reverse primers are given in 5′ to 3′ direction.
Functional expression experiments.
HEK 293T-Gα16gust44 (Ueda et al., 2003) cells were transiently transfected with receptor constructs as described previously (Brockhoff et al., 2007). After incubation for ∼24 h, cells were loaded with Fluo4-AM (Invitrogen). Calcium responses of cells were measured after application of test substances using a Fluorometric Imaging Plate Reader (Molecular Devices). Test compounds were either first dissolved in DMSO and then diluted in C1 solution (130 mm NaCl, 5 mm KCl, 10 mm HEPES, pH 7.4, 2 mm CaCl2, and 10 mm glucose) not exceeding a final DMSO concentration of 1% (v/v) or directly dissolved in C1 solution. Data were collected from at least three independent experiments performed in duplicates. Fluorescence signals were corrected for responses of mock-transfected cells and normalized to background fluorescence. Dose–response relations and EC50 values were calculated in Sigma Plot 11 (SPSS) by nonlinear regression using the function: f(y) = ((a − d)/(1 + (x/EC50)nH) + d).
In silico modeling of TAS2R10.
All-atom homology model of TAS2R10 was based on the crystal structure of β2 adrenergic receptor–Gs-protein complex with bound agonist (PDB code 3SN6) (Rasmussen et al., 2011). Sequence alignment of TAS2R10 and the structural template was generated by the Expresso (Armougom et al., 2006) server (http://www.tcoffee.org/) and manually adjusted (Fig. 1A). The 3D model was generated using Modeler and refined by energy minimization using the Smart Minimizer algorithm and the CHARMM force field, as implemented in Discovery Studio 3.1 (Accelrys), followed by side-chain refinement using the SCWRL4 (Krivov et al., 2009) program and an additional round of energy minimization.
Figure 1.

Locating the TAS2R10 agonist binding pocket. A, The structure-based multiple sequence alignment of the modeled TAS2R10 receptor and the template crystal structure of β2 adrenergic receptor–Gs-protein complex with bound agonist (PDB code 3SN6) was generated by the Expresso server (http://www.tcoffee.org/). The most conserved residue in each helix is shaded yellow and is indicated by its Ballesteros-Weinstein numbering. Identical residues are in red, and similar residues are in blue. B, Top view of a TAS2R10 homology model based on the crystal structure of the agonist-bound state of the β-adrenergic receptor–Gs-protein complex (Rasmussen et al., 2011). The TMs are shown as ribbons. The circled area with a radius of ∼6Å was centered at N923.36 located at the bottom center of the putative agonist binding pocket. Amino acid residues whose side-chains were located within the indicated radius and oriented toward the extracellular side with respect to position N92 (highlighted and labeled) were subjected to alanine-scanning mutagenesis. C, Snake plot of TAS2R10. Residues corresponding to the most conserved positions in each TM (positions X.50 according to the Ballesteros-Weinstein numbering system) are indicated in black. Positions subjected to alanine-scanning mutagenesis are shown in gray and circled with white lines. TMs are numbered by Roman numerals. Membrane, N terminus (NH2), C terminus (COOH), extracellular (ec1-ec3), and intracellular loops (ic1-ic3) are labeled.
Throughout this article, transmembrane (TM) residues are identified by a superscript numbering system according to Ballesteros-Weinstein numbering method (Ballesteros and Weinstein, 1995); the residue corresponding to the Family A GPCRs' most conserved residue in a given TM is assigned the index X.50, where X is the TM number, and the remaining residues are numbered relative to this position, based on alignment with the Family A template (Fig. 1A). The location of a potential binding cavity was identified using the Q-SiteFinder server (Laurie and Jackson, 2005).
Small molecule docking to TAS2R10 model.
Docking of the agonists, strychnine, parthenolide, and denatonium, to the TAS2R10 binding pocket was performed using the “Flexible Docking” algorithm (Discovery Studio 3.1; Accelrys). For docking purposes, the pocket was defined using a 13Å sphere, centered at W883.32, which is highly conserved among TAS2Rs and was shown to be vital for TAS2R43, TAS2R30, and TAS2R46 activation (Pronin et al., 2004; Brockhoff et al., 2010) as well as other Family A GPCRs (Katritch et al., 2012). The side-chains of the following residues were allowed to move: S853.29, W883.32, V893.33, N923.36, Q933.37, N1434.58, K1745.39, Q1755.40, L1785.43, Y2396.51, M2637.39, and T2667.42. All docking experiments were conducted on a model without extracellular loops to reduce the uncertainty arising from inaccurately predicted loops (Yarnitzky et al., 2010 and references therein). Agonists were prepared for docking using the “LigPrep” module in Maestro Version 9.1.107 (Schrodinger).
Multiple sequence alignments of TAS2R10 and TAS2R46 sequences.
Alignments were generated using the TCoffee server (http://www.tcoffee.org/) (Notredame et al., 2000) and included protein sequences from human, chimpanzee, bonobo, gorilla, Bornean orangutan, rhesus monkey, hamadryas baboon, northern white-cheeked gibbon, and crab-eating macaque. The conservation of specific binding site positions in the different species was quantified using the ScoreCons server, using the normalized Shannon entropy scoring method, with 21 symbol types (20 standard amino acid types + 1 gap type), as described previously (Valdar, 2002). The multiple sequence alignments, showing the occurrence of amino acids types in these positions, were visualized using WebLogo3.0 (Crooks et al., 2004).
Immunocytochemistry.
As published previously (Behrens et al., 2009), HEK 293T Gα16gust44 cells were seeded onto poly-d-lysine-coated (10 μg/ml) glass coverslips and transfected with 500 ng cDNA construct per well, using 1.5 μl FuGene HDTM (Roche) according to the manufacturer's recommendations. Twenty-four hours later, cells were washed with warm PBS (37°C) and then cooled on ice for 30 min to block endocytosis. For cell surface staining, cells were incubated with biotin-conjugated concanavalin A (Sigma) for 1 h on ice. Next, cells were washed with ice-cold PBS, fixed with cold methanol/acetone (v/v = 1:1) for 2 min, and rinsed with PBS. Then, cells were incubated with 4% normal horse serum in PBS for 1 h at room temperature. For staining of the receptor proteins, mouse anti-HSV antiserum (Novagen, 1:15,000) was applied and left overnight at 4°C. After washing with PBS, Alexa488-conjugated goat anti-mouse antibody (Invitrogen, 1:2000) and streptavidin-Alexa633 (Invitrogen, 1:1000) were applied for 1 h at room temperature in PBS and 4% normal horse serum. Finally, cells were rinsed with deionized H2O and mounted with Fluorescent Mounting Medium (DakoCytomation). Receptor expression was analyzed by confocal laser scanning microscopy using a Leica TCS SP2 (Leica).
Results
Identification of a putative TAS2R10 agonist binding pocket
To locate the presumed agonist binding site of TAS2R10, we generated a 3D homology model of the receptor based on the structure of the active state of β2 adrenergic receptor, bound to its agonist and Gs-protein (Rasmussen et al., 2011) (Fig. 1B). Using an energy-based method (Laurie and Jackson, 2005), we identified an appropriately sized and energetically favorable area inside the TM bundle that would allow accommodation of TAS2R10 agonists. The putative binding site is located between the upper parts of TMs III to VII.
To verify the location of the predicted agonist binding pocket, residues whose side-chains point into the central cavity, were subjected to alanine-scanning mutagenesis (Fig. 1B,C). Ten point mutations were generated and the resulting receptors examined by calcium-imaging experiments in transiently transfected cells after administration of the structurally dissimilar TAS2R10 agonists: strychnine, parthenolide, and denatonium benzoate. From the dose–response relationships, threshold concentrations, maximal signal amplitudes, and EC50 values were calculated for each receptor mutant (Fig. 2A,B). We observed a complete loss of responsiveness for all agonists for 5 constructs (TAS2R10W88A, TAS2R10N92A, TAS2R10Q93A, TAS2R10Y239A, and TAS2R10M263A; for BW numbering, see Fig. 2A). Two constructs, TAS2R10V89A and TAS2R10T266A, exhibited decreased responses upon stimulation with all agonists. Interestingly, the sensitivity of construct TAS2R10L178A was increased for all agonists. The most complex activation patterns were observed for TAS2R10S85A and TAS2R10Q175A. The construct TAS2R10S85A showed reduced sensitivity for strychnine, elevated sensitivity for parthenolide, and a wild-type-like behavior when stimulated with denatonium benzoate. The construct TAS2R10Q175A exhibited elevated sensitivity for strychnine, a wild-type-like reaction upon parthenolide stimulation, and decreased responses to denatonium benzoate.
Figure 2.

Functional characterization of receptor constructs obtained by alanine-scanning mutagenesis. A, In the left columns, the mutated residue is indicated and its position according to the Ballesteros-Weinstein numbering system is given. For the agonists, strychnine, parthenolide, and denatonium benzoate, threshold concentrations (TH), maximal signal amplitudes as ΔF/F values (Max ampl), and EC50 concentrations are given. X = no statistically significant value above mock-control. B, Dose–response relations obtained for TAS2R10 mutants. Plots obtained for TAS2R10 wild-type (circles, solid lines), TAS2R10 mutants (squares, broken lines), and empty vector (stars, broken lines) are compared within each graph. y-Axis, relative changes in fluorescence (ΔF/F); x-axis, decadic logarithm of the agonist concentration given in μm.
Detailed analyses of TAS2R10 residues exhibiting agonist-selective effects
To determine the exact nature of interactions between agonists and receptor positions S853.29 and Q1755.40, we performed additional mutagenesis experiments (Fig. 3A,B) and in silico ligand docking of the three agonists (Fig. 4). The experimental data suggested a direct interaction of S853.29 with strychnine and a suboptimal sterical interaction with parthenolide. S853.29 was therefore mutated to Thr or Val. As evident from the dose–response relationship obtained for TAS2R10S85T (Fig. 3B), upon change of Ser to Thr, strychnine- or denatonium-induced activation was rather unchanged, whereas the receptor's parthenolide response was almost abolished. The mutant TAS2R10S85V exhibited reduced responses to all agonists tested (Fig. 3B). This indicates that interaction between strychnine and S85 relies on the formation of polar contacts present both in Ser and Thr. Indeed, the docking results suggest the formation of a hydrogen bond between residue S853.29 and the oxo group of strychnine (Fig. 4B). Other predicted interactions are also in agreement with the alanine mutagenesis results. Specifically, these are as follows: π-cation interactions between W883.32 and the partially positively charged N19 atom of strychnine; and another hydrogen bond formed between strychnine and Y2396.51 as well as van der Waals (vDW) contacts with V893.33 (Fig. 4B). The loss of S85 mutants' responses to parthenolide appears to be the result of steric hindrance because the interaction is impaired upon mutation to both bulkier residues, Thr and Val, independent of the presence of the hydroxyl group. Additional hydrogen bonds observed in the predicted structure of the parthenolide–receptor complex are formed with W883.32, N923.36, and the backbone of V893.33 (Fig. 4C).
Figure 3.

Detailed analysis of the agonist selective receptor positions S853.29 and Q1755.40. A, In the left columns, the mutated residue is indicated and its position according to the Ballesteros-Weinstein numbering system is given. For the agonists, strychnine, parthenolide, and denatonium benzoate, threshold concentrations (TH), maximal signal amplitudes as ΔF/F values (Max ampl), and EC50 concentrations are given. X = no statistically significant value above mock-control. B, Dose–response curves of constructs challenged with different concentrations of strychnine, parthenolide, and denatonium benzoate. Changes in fluorescence after agonist stimulation (ΔF/F; y-axis) were monitored and plotted together with the corresponding agonist concentration (decadic logarithm of the agonist concentration given in μm). Plots obtained for TAS2R10 wild-type (circles, solid lines), TAS2R10 mutants (squares, broken lines), and empty vector (stars, broken lines) are compared within each graph.
Figure 4.

Mode of interaction between TAS2R10 and strychnine, parthenolide, and denatonium benzoate. A, Homology model of TAS2R10 viewed perpendicular to the plasma membrane, with the extracellular side of the receptor shown on top and the intracellular side shown on the bottom of the figure. The structure is represented as ribbons and colored from the N-terminal (blue) to the C-terminal (red) amino acid sequence. The predicted binding site is shown in gray and is located among TMs III to VII. The inset shows an overlay of the three docked agonists in the binding site. B–D, The proposed docked conformations of strychnine (B), parthenolide (C), and denatonium (D) are shown in a side view. Agonists (strychnine, green; parthenolide, cyan; and denatonium, yellow) and receptor residues (light gray) involved in interactions are shown in stick representation. Dashed purple lines indicate hydrogen bonds; and orange lines, π-cation interactions. Part of TM IV is not shown for clarity.
Because experimental results for TAS2R10Q175A indicated that position 175 interacts with denatonium and is suboptimal for strychnine responsiveness, we introduced Leu, Tyr, or Asn residues at this position. When stimulated with denatonium benzoate, only the TAS2R10Q175N mutant exhibits maximal amplitudes similar to TAS2R10 wild-type, whereas the other mutants were almost inactive (Fig. 3B). This suggests the importance of the amide group, whereas the right shift of the dose–response curve suggests that the exact positioning of this moiety is also crucial for interaction. The introduction of a large hydrophobic residue, such as Leu, or the bulky ring of Tyr renders the corresponding mutants almost nonresponsive for denatonium benzoate. Indeed, the docking results of denatonium indicate formation of a hydrogen bond between the oxo group of the denatonium molecule and the amide group of Q1755.40 (Fig. 4D). Additional specific interactions are predicted with W883.32 and Y2396.51, and vDW contacts are predicted with V893.33.
The response to strychnine stimulation is comparable to the wild-type receptor for TAS2R10Q175N, is decreased for TAS2R10Q175L and TAS2R10Q175Y, and is increased for TAS2R10Q175A (Fig. 2B). This suggests that position 175 forms vDW contacts with strychnine. If the interactions of Q175 with the agonist were polar or ionic, Q175A mutation would have resulted in reduced interaction. The wild-type-like response of TAS2R10Q175N, even though the polar part is probably shifted in the mutant, supports the notion that the hydrophobic part of position 175 forms vDW contacts with strychnine. The contact with strychnine is suboptimal for the wild-type receptor, and even less compatible with the large Tyr or Leu, but optimized when the smaller Ala is introduced.
Further exploration of receptor–ligand complexes
To further verify the docked conformations of the agonists in the binding site, we performed additional experiments. The docking results suggested that W883.32 interacts with all tested agonists (Fig. 4). Our initial Ala mutation at this position caused a strong change in the nature of the amino acid side-chain, which could have affected the overall structural integrity of the mutant. Therefore, we generated the mutants TAS2R10W88F and TAS2R10W88H to investigate the effects of more subtle changes on receptor responses. Indeed, both exchanges led to a loss of function, supporting a crucial role of this amino acid position (Fig. 5A).
Figure 5.

Verification of agonist docking results. A, In the left columns, the mutated residue is indicated and its position according to the Ballesteros-Weinstein numbering system is given. For the agonists, strychnine, parthenolide, and denatonium benzoate, threshold concentrations (TH), maximal signal amplitudes as ΔF/F values (Max ampl), and EC50 concentrations are given. X = no statistically significant value above mock-control. B, Dose–response curves of constructs challenged with different concentrations of strychnine, parthenolide, and denatonium benzoate. Changes in fluorescence after agonist stimulation (ΔF/F; y-axis) were monitored and plotted together with the corresponding agonist concentration (decadic logarithm of the agonist concentration given in μm). Plots obtained for TAS2R10 wild-type (●), TAS210L178F (▴), TAS2R10L178T (■), TAS2R46 (×), and empty vector (★) are compared within each graph.
As our alanine-scanning and docking results indicated an important role of N923.36 for agonist interaction, we mutated this residue to Asp. Asp is negatively charged and not capable of acting as a hydrogen bond donor. Again, the loss of responsiveness seen for TAS2R10N92D attests to the crucial role of this proposed interaction (Fig. 5A).
Furthermore, the conserved position Y2396.51, corresponding to Y241 in TAS2R46, where it was shown previously to form a hydrogen bond with strychnine (Brockhoff et al., 2010), was found to be crucial for agonist interaction (Figs. 1, 4). The subtle change from Tyr to Phe in the construct TAS2R10Y239F led to a loss of responsiveness, confirming the importance of the hydroxyl group at this position (Fig. 5A).
L1785.43 was the sole position in our experiments for which a mutation to Ala led to increased responsiveness for the three tested agonists. This indicates a suboptimal fit between the native Leu178 and the tested agonists, which might be explained by the close location of this residue to all docked agonists (Fig. 4). To confirm this hypothesis, we tested the constructs TAS2R10L178F and TAS2R10L178T (Fig. 5A,B). As anticipated, introduction of the bulkier Phe at this position led to a loss of response to all agonists. The exchange to Thr, a polar amino acid slightly smaller than Leu, resulted in reduced agonist activation. Hence, activation of position 178 mutants is improved by mutation into a smaller hydrophobic residue, but not by changing it into a polar one.
To test whether the impact of position 178 is similar for the majority of cognate TAS2R10 agonists and whether changes in positions 85 and 175 lead to equally pronounced agonist selectivity in a wider range of TAS2R10 activating substances, we tested the tetracyclic triterpene cucurbitacin B, the sesquiterpene lactones santonin and costunolide, the alkaloid papaverine, and the antibiotic chloramphenicol on the corresponding receptor mutants (Fig. 6). Interestingly, cucurbitacin B activation of TAS2R10 is decreased by all mutations in positions 175 and 178. This indicates that position 178 also acts in an agonist-selective fashion, similarly to 85 and 175. All mutations at position 85, except S85V, show only minor effects on cucurbitacin B responsiveness. Both sesquiterpene lactones, santonin and costunolide, exhibit a very similar activation pattern compared with parthenolide, This corroborates our observations for the sesquiterpene type of agonists, namely, that the hydroxyl group of serine 85 is suboptimal, the rather neutral influence of position 175, and an improved activation when leucine 178 is replaced by alanine. Papaverine is only a weak agonist for the TAS2R10. However, mutating leucine 178 to alanine results in improved receptor activation by this compound. Similarly, chloramphenicol exhibits only weak agonistic properties; thus, the dramatic increases in potency and efficacy upon stimulation of the L178A underscore the strong steric hindrance caused by leucine 178 in the wild-type TAS2R10.
Figure 6.

Further characterization of the agonist-selective positions 85, 175, and 178. A, In the left columns, the mutated residue is indicated and its position according to the Ballesteros-Weinstein numbering system is given. For the agonists, cucurbitacin B, santonin, costunolide, papaverine, and chloramphenicol, threshold concentrations (TH), maximal signal amplitudes as ΔF/F values (Max ampl), and EC50 concentrations are given. X = no statistically significant value above mock-control. B, Dose–response curves of constructs challenged with different concentrations of cucurbitacin B, santonin, costunolide, papaverine, and chloramphenicol. Changes in fluorescence after agonist stimulation (ΔF/F; y-axis) were monitored and plotted together with the corresponding agonist concentration (decadic logarithm of the agonist concentration given in μm). Plots obtained for TAS2R10 wild-type and the indicated TAS2R10 mutants generated for receptor positions 85, 175, and 178 are compared within the graphs.
Comparative analysis of the strychnine-binding mode
An interesting evolutionary question is whether agonists shared by two different TAS2Rs show the same mode of interaction or whether they contact different receptor positions and/or residue types. An identical mode of interaction might suggest an evolutionary conserved feature, while a different binding mode may indicate the independent acquisition of responsiveness for this agonist. Previously, we demonstrated that, in the more strychnine-sensitive receptor TAS2R46, two residues in TM VII, E2657.39 and A2687.42 (compare Fig. 7E), were important determinants of agonist selectivity. We suspected that, if the strychnine-binding mode in TAS2R10 and TAS2R46 is similar, we may transform the less-sensitive strychnine receptor TAS2R10 into a more-sensitive strychnine receptor by transferring these residues from TAS2R46 into the TAS2R10 sequence. As seen in Figure 7A, the individual exchanges in these two positions strongly reduced the response of the mutants to all agonists, and to strychnine in particular. We also created the construct TAS2R10M263E T266A and tested it for strychnine responsiveness because such double mutation successfully transferred activation by strychnine to receptors TAS2R31 and TAS2R43 (Brockhoff et al., 2010). The TAS2R10 double-mutant showed no strychnine responses (and no responses to other agonists), indicating different strychnine-binding modes in the two receptors. These results are in agreement with the dissimilar docking modes of strychnine proposed for the two receptors (Fig. 7C,D). The impact of these mutations on TAS2R10 activation indicates important, albeit different, roles for positions 7.39 and 7.42 in TAS2R10 function as well.
Figure 7.

Comparison of the strychnine-binding modes in TAS2R10 and TAS2R46. A, B, Dose–response curves of constructs challenged with different concentrations of strychnine (top), parthenolide (middle), and denatonium benzoate (bottom). Changes in fluorescence after agonist stimulation (ΔF/F; y-axis) were monitored and plotted together with the corresponding agonist concentration (decadic logarithm of the agonist concentration given in μm). A, Dose–response curves obtained for TAS2R10 wild-type, TAS2R10M263E, TAS210M263E T266A, TAS2R10T266A, TAS2R46 wild-type, and empty vector are compared within each graph. B, Comparison of dose–response curves for TAS2R10 wild-type, TAS2R10Q175T, and empty vector. C, D, Strychnine-binding modes in the 7TM-bundle binding site of TAS2R10 (C) and TAS2RR46 (D). The receptor is represented as ribbons with TMs labeled in Roman numerals. The agonist is represented as green sticks. Interacting residues are shown as sticks and labeled. Dashed purple lines indicate hydrogen bonds; and orange lines, π-cation interactions. E, Amino acid sequence alignment of TAS2R10 and TAS2R46. The most conserved residue in each helix is shaded yellow and is indicated by its Ballesteros-Weinstein numbering. Identical residues are in red and similar residues are in blue.
Investigating position Q1755.40 (Figs. 2, 3), we noticed a significantly improved strychnine sensitivity of TAS2R10Q175A, indicating suboptimal interaction of the native Q175 with strychnine. Because TAS2R46 carries a smaller Thr residue at position 5.40, we suspected that, if strychnine is bound in the same fashion in both receptors, an exchange of Gln into Thr could increase the strychnine sensitivity of TAS2R10. However, the strychnine responsiveness of TAS2R10Q175T did not change, again disproving a conserved binding mode in the two receptors (Fig. 7B). Comparison of the identified binding site residues of TAS2R10 and TAS2R46 from human and primate species demonstrated that these residues are highly conserved among the different primate TAS2R10 orthologs but different from the TAS2R46 primate sequences (Fig. 8). These results suggest that the ability of the duplicated genes to recognize strychnine in two different ways has been acquired independently.
Figure 8.
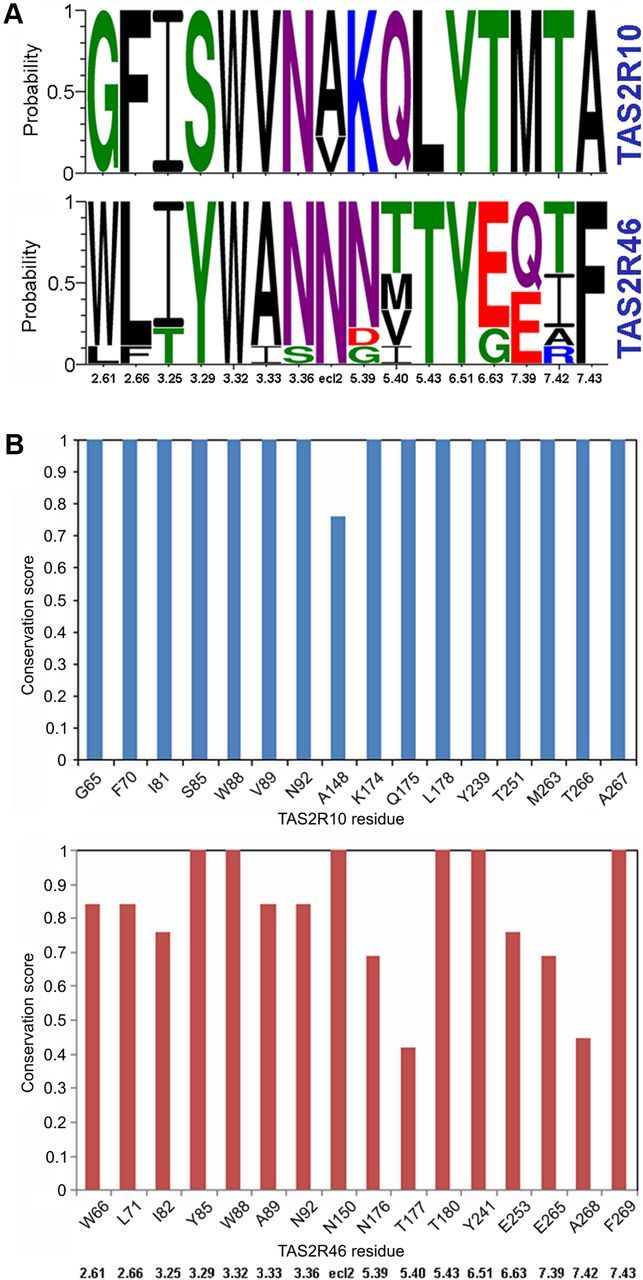
Sequence conservation analysis of TAS2R10 and TAS2R46. A, Conservation of TAS2R10 and TAS2R46 binding site residues in human and 8 primate protein sequences visualized using Weblogo. The y-axis shows the probability of each residue in a stack. The residue position is indicated on the x-axis by its Ballesteros-Weinstein number. Green represents polar residues; purple, neutral; blue, basic; red, acidic; and black, hydrophobic. Sequence logo generated by WebLogo3.0. B, Conservation of each of the binding site residues among the human and 8 primate sequences as calculated by the ScoreCons server. The residue position in human is indicated on the x-axis, and the corresponding BW number is shown below the plots. The y-axis shows the conservation score (maximal score of 1 corresponds to 100% sequence identity of the aligned positions).
Discussion
In the present study, we investigated the agonist binding site in human TAS2R10 by a combination of molecular methods and in silico analyses. We have identified several positions located in the binding pocket contributing to receptor-agonist interactions.
Because modification of receptor responses after point mutagenesis could also indicate misfolded or insufficiently expressed receptors, immunocytochemical analyses of all mutants were performed to identify changes in receptor expression (Fig. 9). None of the TAS2R10 mutants deviated substantially from the expression characteristics of the wild-type reference.
Figure 9.

Immunocytochemical staining of TAS2R10 constructs. HEK 293T Gα16gust44 cells were transiently transfected with constructs coding for TAS2R10 and TAS2R10 mutants. The receptors (green) were visualized using an anti-HSV tag antiserum. The cell surface (red) was stained with concanavalin A. For each construct (indicated in the top left corner), a series of three images is shown: left, receptor construct; middle, cell surface; right, overlay of receptor and cell surface staining. Scale bar, 50 μm. Images were taken with a confocal laser scanning microscope.
Two of the identified TAS2R10 positions are particularly interesting because they showed differential characteristics with respect to agonist interactions. Whereas the exchange of Ser at position 85 into Ala resulted in reduced strychnine responsiveness and improved activation by parthenolide, the third agonist, denatonium benzoate, was largely unaffected by this mutation. Similarly, exchanges of the residue at position 175 resulted in improved strychnine responsiveness and reduced denatonium responses while leaving activation by parthenolide unaffected. These observations demonstrate that the receptor binding pocket is not optimized for high-affinity interactions with a handful of ligands but is rather designed to accommodate many diverse agonists. Hence, the broad tuning of TAS2R10 comes with a trade-off resulting in rather low-affinity agonist interactions. Because relatively high agonist concentrations are necessary for the activation of most TAS2Rs, this feature could apply to additional TAS2Rs as well.
Another question addressed by our study is whether the existence of agonists common to two different TAS2Rs might point to an evolutionary conservation of pharmacological features, or, on the contrary, represent independent convergent evolutionary developments. Unlike other GPCR gene families, such as the somatostatin receptor gene family, in which all five sst receptors bind the common ligand sst-14 (Olias et al., 2004), indicating evolutionary conservation, TAS2R genes appear to have developed under positive selective pressure, driven by the benefits of diversification for enabling multiple bitter compound recognition (Soranzo et al., 2005; Shi and Zhang, 2006). Our mutagenesis experiments transferring residues important for strychnine interaction in TAS2R46 (Brockhoff et al., 2010) onto TAS2R10 demonstrated that these receptors differ in their strychnine-binding modes (Fig. 7). Hence, we conclude that the ability to be activated by strychnine was acquired twice independently during evolution.
In this context, it is tempting to speculate that the pharmacological modification of TAS2R46, including its strychnine responsiveness, has been enabled by an evolutionary more ancient activation profile of TAS2R10. Sequence analysis of TAS2R10 and TAS2R46 in human and primates shows high overall and binding site sequence similarity between orthologs in the different species. The similarity is much lower between the paralogs TAS2R10 and TAS2R46. The TAS2R10 binding site residues identified in the current study are conserved among humans and 8 analyzed primate species, suggesting that TAS2R10 receptors in these species are also likely to be activated by strychnine, whereas the TAS2R46 binding site among primates is more variable (Fig. 8; Tables 2, 3). These results suggest that gene duplication leading to the expansion of a primate-specific TAS2R subfamily (Shi et al., 2003) resulted in the development of a second, more strychnine-sensitive receptor in humans.
Table 2.
Comparison of the amino acid sequences of TAS2R10 and TAS2R46: sequence similarity between TAS2R10 and TAS2R46 orthologs
| Species | Full receptor sequence |
Binding site residues |
||
|---|---|---|---|---|
| TAS2R10a (%) | TAS2R46b (%) | TAS2R10a (%) | TAS2R46b (%) | |
| Chimpanzee | 98 | 97.1 | 100 | 93.8 |
| Bonobo | 98 | 97.1 | 100 | 87.5 |
| Gorilla | 97.7 | 97.4 | 100 | 81.2 |
| Bornean orangutan | 92.5 | 82.8 | 93.8 | 75 |
| Rhesus monkey | 91.2 | 84.5 | 100 | 62.5 |
| Hamadryas baboon | 90.2 | 84.5 | 93.8 | 68.8 |
| Northern white-cheeked gibbon | 93.5 | 83.4 | 100 | 81.2 |
| Crab-eating macaque | 90.9 | 86.1 | 100 | 75 |
| Average sequence similarity | 94 | 89.11 | 98.45 | 78.13 |
aSequence similarity to human TAS2R10.
bSequence similarity to human TAS2R46.
Table 3.
Comparison of the amino acid sequences of TAS2R10 and TAS2R46: sequence similarity between TAS2R10 and TAS2R46 paralogs in different species
| Species | Full receptor sequence (%) | Binding site residues (%) |
|---|---|---|
| Human | 33.9 | 25 |
| Chimpanzee | 34.2 | 31.2 |
| Bonobo | 34.2 | 31.2 |
| Gorilla | 33.5 | 25 |
| Bornean orangutan | 32.5 | 31.2 |
| Rhesus monkey | 33.5 | 31.2 |
| Hamadryas baboon | 36.5 | 18.8 |
| Northern white-cheeked gibbon | 33.8 | 25 |
| Crab-eating macaque | 34.2 | 18.8 |
| Average sequence similarity | 34.03 | 26.38 |
Recently, synthetic (Slack et al., 2010) and natural (Brockhoff et al., 2011) inhibitors of TAS2Rs were discovered, indicating that TAS2R antagonism is common rather than rare. In particular, the presence of natural inhibitors, which occur mixed with bitter agonists (Brockhoff et al., 2011), may require modification of receptor response profiles from time to time to avoid overlapping activities of agonists and antagonists.
Over the past few years, several experimental studies have addressed structural features of TAS2Rs important for agonist activation, showing the involvement of different combinations of residues located in TMs (Pronin et al., 2004; Biarnes et al., 2010; Brockhoff et al., 2010; Sakurai et al., 2010a,b; Upadhyaya et al., 2010; Singh et al., 2011). In the current work, positions S853.29 and Q1755.40, and L1785.43 stand out because of their agonist selectivity in line with the proposed idea of multiligand binding site optimization. Furthermore, the importance of 3.29 is in agreement with data obtained in other structure–function studies of TAS2Rs, all of which have experimentally identified important residues within TMIII (Biarnes et al., 2010; Brockhoff et al., 2010; Sakurai et al., 2010b; Upadhyaya et al., 2010). Moreover, in numerous family A GPCRs, critical contact points between ligands and receptors were identified (Katritch et al., 2012), with the exact points of contact ranging from close to the extracellular side (e.g., position 3.28 in the sphingosine 1-phosphate receptor 1) (Hanson et al., 2012) to deep within the lipid bilayer (e.g., 3.40 in histamine receptor) (Shimamura et al., 2011). Similarly, residues close to position 5.40 were found to be important for the activation of TAS2R46 (Brockhoff et al., 2010), TAS2R16 (Sakurai et al., 2010b), and other GPCRs (Katritch et al., 2012). This indicates a common spatial location of the binding site shared by TAS2Rs and class A GPCRs. Previously, we have identified two positions within TM VII of TAS2R46, E2657.39 and A2687.42, which exhibited pronounced agonist selectivity if swapped between related bitter receptors (Brockhoff et al., 2010). Although we have not observed the same effect when mutating the corresponding positions in the TAS2R10 (M2637.39 and T2667.42), the mutation of both positions in the TAS2R10 led to a loss of function, confirming the importance of TM VII residues for TAS2R function.
In summary, we have shown here that TAS2R10 interacts with its agonists by using a similar TM bundle cavity as seen for TAS2R46. Strikingly, the common agonist of both receptors, strychnine, activates TAS2R10 and TAS2R46 by a different binding mode, via somewhat different subsites, pointing to an evolutionary independent acquirement of strychnine responsiveness in the two receptors. Most importantly, the differential effects observed for some mutations upon agonist activation (e.g., improved activation by one agonist vs decreased activation by another) shed light on the evolutionary pressure of this receptor family to accommodate many agonists at the expense of the optimal affinities that might be achievable for individual agonists. Nevertheless, the impact of changes in the agonist-selective positions of TAS2R10 underscores the precision of bitter receptor compound interaction, suggesting that the broad tuning properties are achieved without sacrificing agonist selectivity.
Footnotes
This work was supported by the German Research Foundation Grant ME1024/2-3 to W.M. and intramural Hebrew University grant to M.Y.N.
W.M. and M.B. have filed patents on human bitter taste.
References
- Adler E, Hoon MA, Mueller KL, Chandrashekar J, Ryba NJ, Zuker CS. A novel family of mammalian taste receptors. Cell. 2000;100:693–702. doi: 10.1016/s0092-8674(00)80705-9. [DOI] [PubMed] [Google Scholar]
- Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, Keduas V, Notredame C. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006;34:W604–W608. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models of structure–function relations in G-protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Behrens M, Brockhoff A, Kuhn C, Bufe B, Winnig M, Meyerhof W. The human taste receptor hTAS2R14 responds to a variety of different bitter compounds. Biochem Biophys Res Commun. 2004;319:479–485. doi: 10.1016/j.bbrc.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Behrens M, Bartelt J, Reichling C, Winnig M, Kuhn C, Meyerhof W. Members of RTP and REEP gene families influence functional bitter taste receptor expression. J Biol Chem. 2006;281:20650–20659. doi: 10.1074/jbc.M513637200. [DOI] [PubMed] [Google Scholar]
- Behrens M, Brockhoff A, Batram C, Kuhn C, Appendino G, Meyerhof W. The human bitter taste receptor hTAS2R50 is activated by the two natural bitter terpenoids andrographolide and amarogentin. J Agric Food Chem. 2009;57:9860–9866. doi: 10.1021/jf9014334. [DOI] [PubMed] [Google Scholar]
- Biarnes X, Marchiori A, Giorgetti A, Lanzara C, Gasparini P, Carloni P, Born S, Brockhoff A, Behrens M, Meyerhof W. Insights into the binding of Phenyltiocarbamide (PTC) agonist to its target human TAS2R38 bitter receptor. PLoS One. 2010;5:12394. doi: 10.1371/journal.pone.0012394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhoff A, Behrens M, Massarotti A, Appendino G, Meyerhof W. Broad tuning of the human bitter taste receptor hTAS2R46 to various sesquiterpene lactones, clerodane and labdane diterpenoids, strychnine, and denatonium. J Agric Food Chem. 2007;55:6236–6243. doi: 10.1021/jf070503p. [DOI] [PubMed] [Google Scholar]
- Brockhoff A, Behrens M, Niv MY, Meyerhof W. Structural requirements of bitter taste receptor activation. Proc Natl Acad Sci U S A. 2010;107:11110–11115. doi: 10.1073/pnas.0913862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhoff A, Behrens M, Roudnitzky N, Appendino G, Avonto C, Meyerhof W. Receptor agonism and antagonism of dietary bitter compounds. J Neurosci. 2011;31:14775–14782. doi: 10.1523/JNEUROSCI.2923-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bufe B, Hofmann T, Krautwurst D, Raguse JD, Meyerhof W. The human TAS2R16 receptor mediates bitter taste in response to beta-glucopyranosides. Nat Genet. 2002;32:397–401. doi: 10.1038/ng1014. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG, Han GW, Kuhn P, Rosen H, Stevens RC. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie AT, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- Matsunami H, Montmayeur JP, Buck LB. A family of candidate taste receptors in human and mouse. Nature. 2000;404:601–604. doi: 10.1038/35007072. [DOI] [PubMed] [Google Scholar]
- Meyerhof W, Batram C, Kuhn C, Brockhoff A, Chudoba E, Bufe B, Appendino G, Behrens M. The molecular receptive ranges of human TAS2R bitter taste receptors. Chem Senses. 2010;35:157–170. doi: 10.1093/chemse/bjp092. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Olias G, Viollet C, Kusserow H, Epelbaum J, Meyerhof W. Regulation and function of somatostatin receptors. J Neurochem. 2004;89:1057–1091. doi: 10.1111/j.1471-4159.2004.02402.x. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Tang H, Connor J, Keung W. Identification of ligands for two human bitter T2R receptors. Chem Senses. 2004;29:583–593. doi: 10.1093/chemse/bjh064. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta(2) adrenergic receptor-Gs-protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Misaka T, Ueno Y, Ishiguro M, Matsuo S, Ishimaru Y, Asakura T, Abe K. The human bitter taste receptor, hTAS2R16, discriminates slight differences in the configuration of disaccharides. Biochem Biophys Res Commun. 2010a;402:595–601. doi: 10.1016/j.bbrc.2010.10.059. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Misaka T, Ishiguro M, Masuda K, Sugawara T, Ito K, Kobayashi T, Matsuo S, Ishimaru Y, Asakura T, Abe K. Characterization of the beta-d-glucopyranoside binding site of the human bitter taste receptor hTAS2R16. J Biol Chem. 2010b;285:28373–28378. doi: 10.1074/jbc.M110.144444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Zhang J. Contrasting modes of evolution between vertebrate sweet/umami receptor genes and bitter receptor genes. Mol Biol Evol. 2006;23:292–300. doi: 10.1093/molbev/msj028. [DOI] [PubMed] [Google Scholar]
- Shi P, Zhang J, Yang H, Zhang YP. Adaptive diversification of bitter taste receptor genes in Mammalian evolution. Mol Biol Evol. 2003;20:805–814. doi: 10.1093/molbev/msg083. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Pydi SP, Upadhyaya J, Chelikani P. Structural basis of activation of bitter taste receptor T2R1 and comparison with class A GPCRs. J Biol Chem. 2011;286:36032–36041. doi: 10.1074/jbc.M111.246983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack JP, Brockhoff A, Batram C, Menzel S, Sonnabend C, Born S, Galindo MM, Kohl S, Thalmann S, Ostopovici-Halip L, Simons CT, Ungureanu I, Duineveld K, Bologa CG, Behrens M, Furrer S, Oprea TI, Meyerhof W. Modulation of bitter taste perception by a small molecule hTAS2R antagonist. Curr Biol. 2010;20:1104–1109. doi: 10.1016/j.cub.2010.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soranzo N, Bufe B, Sabeti PC, Wilson JF, Weale ME, Marguerie R, Meyerhof W, Goldstein DB. Positive selection on a high-sensitivity allele of the human bitter-taste receptor TAS2R16. Curr Biol. 2005;15:1257–1265. doi: 10.1016/j.cub.2005.06.042. [DOI] [PubMed] [Google Scholar]
- Ueda T, Ugawa S, Yamamura H, Imaizumi Y, Shimada S. Functional interaction between T2R taste receptors and G-protein alpha subunits expressed in taste receptor cells. J Neurosci. 2003;23:7376–7380. doi: 10.1523/JNEUROSCI.23-19-07376.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya J, Pydi SP, Singh N, Aluko RE, Chelikani P. Bitter taste receptor T2R1 is activated by dipeptides and tripeptides. Biochem Biophys Res Commun. 2010;398:331–335. doi: 10.1016/j.bbrc.2010.06.097. [DOI] [PubMed] [Google Scholar]
- Valdar WS. Scoring residue conservation. Proteins. 2002;48:227–241. doi: 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- Wiener A, Shudler M, Levit A, Niv MY. BitterDB: a database of bitter compounds. Nucleic Acids Res. 2012;40:D413–D419. doi: 10.1093/nar/gkr755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarmolinsky DA, Zuker CS, Ryba NJ. Common sense about taste: from mammals to insects. Cell. 2009;139:234–244. doi: 10.1016/j.cell.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarnitzky T, Levit A, Niv MY. Homology modeling of G-protein-coupled receptors with X-ray structures on the rise. Curr Opin Drug Discov Devel. 2010;13:317–325. [PubMed] [Google Scholar]