

Abstract
The transcription factor FoxP2 has been associated with the development of human speech but the underlying cellular function of FoxP2 is still unclear. Here we provide evidence that FoxP2 regulates genesis of some intermediate progenitors and neurons in the mammalian cortex, one of the key centers for human speech. Specifically, knockdown of FoxP2 in embryonic cortical precursors inhibits neurogenesis, at least in part by inhibiting the transition from radial glial precursors to neurogenic intermediate progenitors. Moreover, overexpression of human, but not mouse, FoxP2 enhances the genesis of intermediate progenitors and neurons. In contrast, expression of a human FoxP2 mutant that causes vocalization deficits decreases neurogenesis, suggesting that in the murine system human FoxP2 acts as a gain-of-function protein, while a human FoxP2 mutant acts as a dominant-inhibitory protein. These results support the idea that FoxP2 regulates the transition from neural precursors to transit-amplifying progenitors and ultimately neurons, and shed light upon the molecular changes that might contribute to evolution of the mammalian cortex.
Introduction
Speech and language disorders in humans have a substantial genetic component and likely involve many genes. The forkhead box transcription factor, FoxP2, has been widely studied in this regard since it is mutated and autosomal dominant in developmental verbal dyspraxia (Fisher et al., 1998), a syndrome involving significant speech and language deficits, as well as broader cognitive dysfunction such as autism spectrum disorder (Feuk et al., 2006). FoxP2 is highly conserved in vertebrates (Enard et al., 2002), and the mouse and human orthologs differ by three amino acid changes and an extra glutamine in the human poly-glutamine tract. One approach for understanding how FoxP2 might regulate speech and language involves modeling FoxP2 function in animals with defined verbal behaviors such as songbirds and mice. In this regard, mice carrying a null mutation in FoxP2 (Shu et al., 2005) and knock-in mice carrying either a humanized FoxP2 allele (Enard et al., 2009) or human FoxP2 mutations (Fujita et al., 2008) have been generated. Interestingly, mice where FoxP2 is perturbed displayed abnormalities in neonatal ultrasonic vocalization, a stereotypic behavior that is studied as a model for development of human speech and social cognition. However, these mice were also developmentally delayed and exhibited abnormalities in cerebellar development and motor function. It is not yet clear whether these neural abnormalities are the cause of the ultrasonic vocalization deficits and/or how these abnormalities relate to the impaired language development seen in humans.
These studies indicate that FoxP2 plays an important role in neural development, which is perhaps not surprising given the developmental roles played by other members of the same family. For example, FoxP1 defines columnar identity in the spinal cord (Rousso et al., 2008) and regulates outflow tract septation and myocardial proliferation during cardiac development (Wang et al., 2004). FoxP4 is required for cardiac development, regulating fusion of the bilateral cardiac primordia and subsequent formation of the primitive heart tube (Li et al., 2004b). In addition, FoxP1 and FoxP2 cooperatively regulate lung airway morphogenesis and differentiation and esophageal muscle development (Shu et al., 2007). Moreover, FoxP2 and FoxP4 were recently found to regulate neurogenesis in the chick and mouse spinal cord (Rousso et al., 2012). Thus, the FoxP family regulates development in part by regulating embryonic precursors. In this regard, several studies have shown that FoxP2 mRNA is expressed in the precursor zones of the developing brain (Shu et al., 2001; Ferland et al., 2003). Since we have previously shown that perturbations in embryonic neural precursor development likely occur in genetic syndromes that cause cognitive dysfunction (Gauthier et al., 2007; Wang et al., 2010), we hypothesized that FoxP2 mutations may, at least in part, cause cognitive dysfunction by dysregulating embryonic neurogenesis. Here, we provide evidence for this idea, showing that in the developing mouse cortex FoxP2 knockdown inhibits the transition from radial precursors to intermediate progenitors and neurons, and that a human FoxP2 mutant that causes verbal dyspraxia also inhibits neurogenesis, acting as a dominant-negative protein.
Materials and Methods
Animals.
All animal use was approved by the Animal Care Committee of the Hospital for Sick Children in accordance with the Canadian Council of Animal Care policies. CD1 mice were used for all experiments except the cell sorting, and were obtained from Charles River Laboratory. Sox2:EGFP mice (Ellis et al., 2004) were genotyped and maintained as described previously (Biernaskie et al., 2009). Embryos and neonatal pups of both sexes were used.
Primers and plasmids.
Two primer sets were used for FoxP2 mRNA. The first was FoxP2 forward (5′-CCAAACCATCTCCCAAACCT-3′), FoxP2 reverse (5′-TCTGAATGTCGCCTTCGTATG-3′), and the second was FoxP2 forward (5′-GCTAAGTAACCCTGGACTGATC-3′), FoxP2 reverse (5′-TCTTCATCCTCTGCAATCACG-3′). The nuclear enhanced green fluorescent protein (EGFP) expression plasmid was driven from the Ef1α (Eef1a; Mouse Genome Informatics) promoter (pEF-EGFP) and has been previously described (Barnabé-Heider et al., 2005). The piggybac (PB)-EGFP construct contains an EGFP reporter driven by the CAG promoter and flanked by inverted repeats, and is recognized by the PB transposase (Nagy et al., 2011). The FoxP2 shRNAs and the negative control were purchased from EZBiolab and made in the pGPU6-neo backbone. The targeted sequence was 5′-GCACTTTAAGCAGCCAATTAG-3′ for shFoxP2#1 and 5′-GCAAACCAGTGGATTGAAATC-3′ for shFoxP2#2. The sequence for the shRNA negative control was 5′-TTCTCCGAACGTGTCACGT-3′. All plasmids for the FoxP2 overexpression studies were cloned into pCMV-Tag4A and include pCMV-Tag4A-mouse FoxP2 cDNA (mFoxP2), pCMV-Tag4A-human FoxP2 cDNA (hFoxP2), or pCMV-Tag4A-human FoxP2-R553H (hFoxP2-R553H). The pCMV-Tag4A empty vector, human FoxP2, and human FoxP2-R553H were kindly provided by Dr. Genevieve Konopka (University of Texas Southwestern Medical Center; Konopka et al., 2009). The mouse FoxP2 construct was subcloned into the pCMV-Tag4A empty vector from an E13/14 mouse cortex cDNA library. FoxP1 plasmids in the pCMV-Tag2A backbone and FoxP4 plasmids in the pCMV-Tag3B backbone were kindly provided by Dr. Edward Morrissey (University of Pennsylvania School of Medicine; Li et al., 2004a). The overexpression plasmid for mouse FoxP2 used for experiments in Figure 1, B and G, was kindly provided by Dr. Zea Borok (University of Southern California; Zhou et al., 2008).
Figure 1.

Expression of FoxP2 in embryonic cortical precursors. A, RT-PCR analysis for FoxP2 mRNA in total RNA isolated from E12 murine cortex (E12; left), from E12 cortical precursor cultures 1 or 3 d after plating (1 or 3 days in vitro (DIV); left), or from Sox2:EGFP-positive precursors sorted from the E13 cortex (right). As controls, samples were generated in the absence of RT. Molecular weight markers are shown to the left of both sides. B, Western blot analysis of lysates of HEK293 cells transfected with expression constructs for flag-tagged FoxP1, flag-tagged FoxP2, or myc-tagged FoxP4, probed with antibodies for flag, myc, the Erk proteins as a control, or one of three different FoxP2 antibodies (Ab#1 is from Abcam, Ab#2 is from Sigma, and Ab#3 is from Santa Cruz Biotechnology). Note that only FoxP2 Ab#3 is specific for FoxP2. C, Western blot analysis of lysates of the embryonic cortex at various time points (top left), of E12 cortical precursors cultured for 1 d (1 DIV; top right), and of Sox2:EGFP-positive precursors sorted from the E13 cortex (bottom right), all probed with FoxP2 Ab#3. Top right, Includes flag-tagged FoxP2 that was overexpressed in HEK293 cells (FoxP2, right lane), which runs at a slightly higher molecular weight due to the triple-flag tag. The blot shown on the top left was reprobed for total Erk protein as a loading control (bottom left). Molecular weight markers are shown to the left of each blot. D, E, Confocal micrographs of coronal sections through the E12.5 cortex immunostained for FoxP2 (red) and Pax6 (D) or Tbr2 (E) (both in green; the right side shows the merged images). The bottom row of micrographs in each part shows higher magnification images. In D arrows denote cells that are positive for Pax6 and FoxP2, while in E they denote Tbr2-positive cells that express more robust levels of FoxP2. Scale bars: Top, 50 μm; bottom, 20 μm. F, Fluorescence micrograph of E12 cortical precursors cultured for 1 d, immunostained for FoxP2 (red) and Tbr2 (green) and counterstained with Hoechst 33258 (blue) to show cell nuclei. Arrows denote a Tbr2-positive intermediate progenitor that expresses higher levels of FoxP2, and arrowheads denote cells with low levels of nuclear FoxP2 that are negative for Tbr2. Scale bar, 20 μm. G, Left, Western blot analysis of lysates of HEK293 cells cotransfected with a flag-tagged murine FoxP2 overexpression construct either alone (Con) or with one of two different shRNAs for murine FoxP2 (sh1 and sh2), probed with FoxP2 Ab#3. Middle and right, Western blot analysis of lysates of HEK293 cells cotransfected with flag-tagged murine FoxP1, flag-tagged murine FoxP2, or myc-tagged murine FoxP4 ± FoxP2 shRNAs #1 or #2, and probed with antibodies for flag (middle) or myc (right). All blots were reprobed for total Erk as a loading control. Molecular weights are shown on the far left.
Cortical precursor cell cultures.
Cortical precursor cells were cultured as previously described (Barnabé-Heider et al., 2005). Briefly, cerebral cortices were dissected from embryonic day 12 (E12) CD1 mouse embryos in ice-cold HBSS (Invitrogen) and transferred to Neurobasal medium (Invitrogen) containing 500 μm l-glutamine (Cambrex Biosciences), 2% B27 supplement (Invitrogen), 1% penicillin-streptomycin (Invitrogen), and 40 ng/ml FGF2 (BD Biosciences). The tissue was mechanically triturated with a plastic pipette and plated onto 4-well chamber slides (Nunc) precoated with 2% laminin (BD Biosciences) and 1% poly-d-lysine (Sigma). Cells were plated at a density of 200,000 cells/well for 4-well chamber slides. Culture medium was not changed for the duration of the experiment. For transfections, 1–2 h after plating, 1 μg of total DNA (comprised of 0.25 μg of pEF-EGFP plasmid plus 0.75 μg of shRNA for knockdown experiments; 0.1 μg of pEF-EGFP plasmid plus 0.9 μg of pCMV-Tag4A empty vector, human FoxP2, mouse FoxP2, or human FoxP2-R553H plasmids for overexpression experiments) or 1.4 μg of total DNA (comprised of 0.1 μg of PB transposase plasmid plus 0.1 μg of PB-EGFP plasmid plus 0.3 μg of control or FoxP2 shRNA plasmid plus 0.9 μg of pCMV-Tag4A empty vector or human FoxP2 plasmid) and 2 μl of Fugene 6.0 (Roche) or 1 μl of Lipofectamine 2000 (Invitrogen) were mixed with 100 μl of Opti-MEM (Invitrogen), incubated at room temperature for 1 h, and added to the cultures. This resulted in a transfection of, at most, 1–3% of cells.
In utero electroporation.
In utero electroporation was performed as described previously (Barnabé-Heider et al., 2005; Gauthier et al., 2007) with E13/14 CD1 mice, injecting a 1:3 ratio of the nuclear EGFP plasmid with the shRNA or overexpression plasmids (total of 4 μg of DNA) and 0.5% trypan blue as a tracer. For the rescue experiments, DNA was mixed at a ratio of 1 μg of pEF-EGFP:1 μg of FoxP2 shRNA: 3 μg of human FoxP2 for a total of 5 μg of DNA per embryo. For knockdown experiments in which P3 animals were analyzed, DNA was mixed at a ratio of 1 μg of PBase:1 μg of PB-EGFP: 3 μg of FoxP2 shRNA for a total of 5 μg of DNA per embryo. The square electroporator CUY21 EDIT (TR Tech) was used to deliver five 50 ms pulses of 40–50 V with 950 ms intervals per embryo. Except for analysis of neonatal animals, all brains were dissected 3 d post-transfection in ice-cold HBSS. For analysis of neonatal animals, embryos were electroporated from separate litters but pups were born on the same day and brains were harvested at postnatal day 3 (P3). All electroporated brains were fixed in 4% paraformaldehyde (PFA) at 4°C overnight or until they sank to the bottom, cryoprotected, and cryosectioned coronally at 16 μm.
Immunocytochemistry and histological analysis.
Immunocytochemistry was performed as previously described (Barnabé-Heider et al., 2005). Briefly, sections were fixed in 4% PFA for 10 min, and then permeabilized and blocked with 5% bovine serum albumin (BSA) and 0.3% Triton-X in PBS for 1 h. Primary antibodies were diluted in the same permeabilization and blocking buffer and incubated either at 4°C overnight or room temperature for 3 h. Secondary antibodies and Hoechst were diluted in PBS. Sections were washed three times for 5 min with PBS between each step. For FoxP2 immunostaining, an antigen retrieval step was added postfixation that involved boiling slides at 92°C in sodium citrate buffer, pH 6, for 20 min. For cultures, 4-well chambers were fixed for 10 min in PFA and then permeabilized with 0.2% NP40 in PBS. Cells were then blocked with 0.5% BSA and 6% normal goat serum or normal donkey serum diluted in PBS. Primary antibodies were diluted in 0.25% BSA and 3% normal goat serum or normal donkey serum in PBS and incubated overnight. Secondary antibodies were diluted in the same blocking solution as primary antibodies and incubated for 1 h. Hoechst was diluted in PBS. The wells were washed with PBS in between each step. The primary antibodies used were mouse anti-GFP (1:1000; Invitrogen), rabbit anti-GFP (1:5000; Abcam), chicken anti-GFP (1:2000; Abcam), mouse anti-β-III-tubulin (1:1000; Covance), rabbit anti-Pax6 (1:1000; Covance), mouse anti-Pax6 (1:100; Development Studies Hybridoma Bank, The University of Iowa), rabbit anti-Tbr2 (1:1000; Abcam), mouse anti-Satb2 (1:400; Abcam), rabbit anti-cleaved caspase 3 (1:200; Millipore Bioscience Research Reagents), mouse anti-Flag (1:1000; Sigma), goat anti-FoxP2 (1:100; Santa Cruz Biotechnology sc-21069). rat anti-Ctip2 (1:400; Abcam), mouse anti-HuD (1:100; Life Tech), and rabbit anti-Tbr1 (1:1000; Abcam). The secondary antibodies used were Alexa Fluor 555-, Alexa Fluor 488-, and Alexa Fluor 647-conjugated goat or donkey antibodies to mouse, rabbit, or goat IgG (1:1000 for 488/555, 1:500 for 647; Invitrogen). Nuclear staining was performed with Hoechst 33258 (Sigma). In some cases, immunostaining was performed using the MOM Kit (Vector Laboratories), as per manufacturer's instructions.
Western blot analysis.
Western blots were performed as described previously (Barnabé-Heider et al., 2005). Briefly, tissue or cells were harvested and incubated for 30 min at 4°C in radioimmunoprecipitation assay lysis buffer supplemented with 1 mm PMSF, 1 mm Na3VO4, 10 μg/ml aprotenin, and 10 μg/ml leupeptin. Lysate was then centrifuged at 13,000 rpm for 10 min and the supernatant was collected. Protein concentration was measured with the BCA Protein Assay Kit (Thermo Scientific). For most experiments, 50–100 μg of lysate was loaded onto each lane of a 10% SDS gel. Proteins were then transferred to nitrocellulose membranes (Bio-Rad) at 250 mA at 4°C overnight (Bio-Rad). Membranes were blocked with 5% skim milk in Tris-buffered saline—Tween 20 (TBST) for 1 h, and incubated with primary antibodies diluted in 5% skim milk in TBST at 4°C overnight. Secondary antibodies were also diluted in 5% skim milk in TBST and incubated for 1 h at room temperature. Three 5 min washes were applied between every step. To develop the blots, membranes were incubated in either ECL or ECL-plus (GE Healthcare) for 5 min and then exposed for varying periods of time. The primary antibodies used were rabbit anti-FoxP2 (Ab #1) (1:1000; Abcam), rabbit anti-FoxP2 (Ab #2) (1:1000; Sigma), goat anti-FoxP2 (Ab #3) (1:1000; Santa Cruz Biotechnology), mouse anti-Flag (1:5000; Sigma), mouse anti-c-myc (1:1000; Santa Cruz Biotechnology), and rabbit anti-ERK1 K23 (1:5000; Santa Cruz Biotechnology). Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG (1:5000; Boehringer Mannheim).
Flow cytometry.
E13 littermate embryos were harvested from crosses between Sox2-EGFP and wild-type B6 mice. EGFP-positive and -negative animals were separated, and cortical cells were harvested as described for the culture experiments, dissociated to single cells in 1% BSA, and sorted for EGFP expression on a MoFlo fluorescent-activated cell sorter (Dako) with viable cells identified by propidium iodide exclusion as previously described (Biernaskie et al., 2009). Gates were set using cells isolated from the EGFP-negative cortices.
HEK293 experiments.
HEK293 cells were cotransfected with mouse FoxP1, FoxP2, or FoxP4 cDNA and, for the knockdown experiments, with the negative control shRNA or shRNAs against mouse FoxP2. Briefly, cells were plated in 2 ml of DMEM (Lonza) supplemented with 10% fetal bovine serum, 2 μm l-glutamine, (Cambrex Biosciences), and 1% penicillin-streptomycin (Invitrogen) in 6-well polystyrene multiwell plates (BD Falcon). The next day, 500 μl of Opti-MEM (Invitrogen) and 5 μl of Lipofectamine 2000 (Invitrogen) were mixed with the DNA and incubated at room temperature for 30 min. The medium was replaced with 1.5 ml Opti-MEM, and 500 μl of the transfection reaction mixture was added. The medium was replaced the next day with DMEM, and cells were harvested 1 or 2 d later for analysis.
Reverse transcription-PCR.
Cortices were dissected from embryos and directly subjected to RNA isolation or plated and cultured as described above for the indicated number of days before RNA isolation. Trizol (Invitrogen) was used to isolate RNA according to the manufacturer's protocol. RNA was treated with DNase (Fermentas) to remove any contaminating genomic DNA. Reverse transcription was performed using RevertAid H Minus M-MuLV Reverse Transcriptase (Fermentas) primed with random hexamers, according to the manufacturer's instructions. All reactions were subjected to the following PCR protocol: 94°C for 2 min, 35 cycles of 94°C for 15 s, 58°C for 30 s, and 72°C for 30 s, and a final elongation at 72°C for 2 min. Products were then resolved on a 2% agarose gel.
Microscopy and quantification.
For quantification of cell culture experiments, depending on the transfection efficiency, 44–820 cells per condition per experiment were counted and analyzed using the Zeiss Axioplan2 upright microscope equipped with fluorescence optics. In clonal studies, 28–210 clones were analyzed per condition per experiment. Clones were defined as groups of EGFP-positive cells in close proximity to each other that were well separated from any other EGFP-positive clusters, but which were not part of distinct unlabeled clusters. Digital image acquisition was performed with Northern Eclipse software (Empix) using a Sony XC-75CE CCD video camera. For quantification of tissue sections, cortical sections were chosen that showed a similar anatomical distribution and amount of EGFP-positive cells for comparison. Brains were sectioned at 16 μm at E16/17; three to four brain sections at the same anatomical level per embryo were analyzed using a Zeiss Pascal confocal microscope and the manufacturer's software. A mean of two single optical sections taken with a 40× objective were computed for each image. A total of 3–4 sections was analyzed per embryo by taking up to three 8–10 mm pictures to cover the electroporated ventricular zone (VZ), subventricular zone (SVZ), and cortical plate (CP) of each coronal section with a 40× objective and comparing them with equivalent sections in littermate control counterparts. For some experiments, sections were instead analyzed using a similar approach with an Olympus IX81 inverted fluorescence microscope equipped with a Hamamatsu C9100-13 back-thinned EM-CCD camera and Yokogawa CSU X1 spinning disk confocal scan head (with Spectral Aurora Borealis upgrade) using Volocity (PerkinElmer) software. Signals were considered to be positive when they were more intense than background labeling, and showed the predicted cellular localization. For measurement of cell location in electroporated cortices, image acquisition was performed using Northern Eclipse Software (Empix) with a Sony-XC-75CE CCD video camera. Ventricular, subventricular, and CP layers were delineated using Hoechst staining. For quantification of FoxP2 overexpression, confocal images were taken as described above. ImageJ was used to quantify immunofluorescent intensity as described in a published study (Burgess et al., 2010). Briefly, the corrected fluorescence was obtained by subtracting the integrated density by the measured area multiplied by the mean fluorescence background reading, which was determined as an average of three background readings taken of the region adjacent to the cells of interest. Approximately 50 cells from three electroporated embryos each for human versus mouse FoxP2 were measured and the mean fluorescence determined.
Statistics.
All data were expressed as the mean ± SEM, and were tested for statistical significance with two-tailed unpaired Student's t tests unless otherwise indicated, in which case they were analyzed with a Student–Neuman–Keuls post hoc ANOVA. Differences were considered significant if p < 0.05.
Results
FoxP2 is expressed in neural precursors in the embryonic murine cortex
To determine whether FoxP2 is expressed in neural precursors of the embryonic cortex, as suggested by previous in situ hybridization studies (Shu et al., 2001; Ferland et al., 2003; www.genepaint.org; set ID EH3230), we initially analyzed expression of FoxP2 mRNA. Reverse transcription(RT)-PCR of RNA isolated from the murine cortex at E12 demonstrated that FoxP2 mRNA is expressed at this time point (Fig. 1A), when the cortex is predominantly comprised of precursors. To ask whether the protein is also expressed, we performed Western blot analysis. To do this, we first characterized three different FoxP2 antibodies for their specificity. HEK293 cells were transfected with myc- or flag-tagged expression constructs for FoxP1, FoxP2, and FoxP4, all of which are expressed in the embryonic cortex (Shu et al., 2001; Lu et al., 2002). Western blots of these lysates were then probed with three different FoxP2 antibodies and with antibodies for flag or myc, as relevant. This analysis (Fig. 1B) demonstrated that all three FoxP2 antibodies recognized overexpressed FoxP2, but that two of them also recognized other FoxP family members. We therefore used the third FoxP2-specific antibody to probe Western blots of the embryonic cortex; FoxP2 protein was present at E11, when the cortex does not contain neurons or glial cells, and this expression was maintained throughout embryogenesis, into early postnatal life (Fig. 1C).
To ask whether FoxP2 is expressed in cortical precursors, as suggested by its early embryonic expression, we performed two additional experiments. First, we analyzed E12 cortical precursor cultures which, when plated, are almost entirely comprised of proliferating radial precursors that go on to generate transit-amplifying intermediate progenitors and newly born neurons. RT-PCR and Western blot analysis demonstrated that FoxP2 mRNA and protein were both expressed in these cultures 1 d postplating (Fig. 1A,C). Second, we used flow cytometry to isolate neural precursors from the embryonic cortex, taking advantage of mice where EGFP is knocked-in to the Sox2 locus (Ellis et al., 2004). In these mice, embryonic radial precursors and intermediate progenitors can be sorted from other cells by virtue of their expression of EGFP (Hutton and Pevny, 2011). RT-PCR and Western blot analysis of Sox2:EGFP-positive cells isolated from the E13 cortex demonstrated that cortical precursors expressed both FoxP2 mRNA and FoxP2 protein (Fig. 1A,C).
Two classes of precursors are present in the embryonic cortex, radial glial precursors, and intermediate progenitors. To ask which of these two populations express FoxP2, we immunostained the E12/13 cortex with the FoxP2-specific antibody. At this stage, low levels of nuclear FoxP2 were present in most Pax6-positive radial precursors (Fig. 1D). Tbr2-positive intermediate progenitors also expressed nuclear FoxP2, although this expression was more heterogeneous, with some cells expressing low levels and some higher levels (Fig. 1E). Similar results were obtained with E12 precursors cultured for 1 d; the most robust levels of nuclear FoxP2 were observed in Tbr2-positive intermediate progenitors, and lower levels were seen in Tbr2-negative radial precursors (Fig. 1F).
FoxP2 knockdown decreases neurogenesis in the embryonic cortex
To ask about a potential function for FoxP2 in these neural precursors, we generated two shRNAs to knock it down. Western blot analysis of HEK293 cells cotransfected with a mouse FoxP2 overexpression vector and each of the FoxP2 shRNAs showed that these shRNAs robustly decreased FoxP2 protein levels (Fig. 1G). In contrast, these FoxP2 shRNAs had no effect on the levels of FoxP1 or FoxP4 in similar cotransfection experiments (Fig. 1G), as predicted by their sequences (FoxP2 shRNA #1 had 13/21 mismatches with FoxP1 mRNA and 12/21 mismatches with FoxP4 mRNA while FoxP2 shRNA #2 had 5/21 and 8/21 mismatches with FoxP1 and FoxP4 mRNAs, respectively). To ask whether these shRNAs were similarly efficacious in cortical precursors, we cotransfected either FoxP2 shRNA #2 or a control shRNA together with a plasmid-expressing EGFP into E12 precursor cultures and immunostained these cultures 3 d later with the FoxP2-specific antibody. Quantification of three independent experiments demonstrated that the number of EGFP-positive cells that expressed bright FoxP2 (as seen in Fig. 1F) was significantly decreased (control shRNA, 8.0 ± 1.7%, FoxP2 shRNA, 1.5 ± 0.3%; p < 0.05; n = 3 independent experiments).
Having demonstrated their efficacy, we used these two FoxP2 shRNAs to ask about the biological role of FoxP2 in embryonic cortical precursors. To do this, we electroporated E13/14 cortices with plasmids encoding nuclear EGFP and one of the two shRNAs; this manipulation electroporates embryonic cortical radial precursors, many of which will generate neurons that migrate out of the precursor regions of the cortex (VZ/SVZ) into the CP region. Some of these radial precursors will also generate intermediate progenitors that will then divide and differentiate into neurons. Analysis of cortices at E16/17, 3 d following electroporation, revealed that both of the FoxP2 shRNAs altered the location of EGFP-positive cells relative to a control shRNA (Fig. 2A,B). In particular, they caused a significant reduction in the percentage of EGFP-positive cells, presumably newly born neurons, that were present in the CP, and a significant increase in the percentage that remained in the VZ/SVZ (Fig. 2A,B).
Figure 2.

FoxP2 knockdown decreases neurogenesis in the embryonic cortex in vivo. A, Fluorescence micrographs of coronal sections through E16/17 cortices that were coelectroporated at E13/14 with nuclear EGFP and control shRNA (Control) or FoxP2 shRNA #2 (shFoxP2 #2) and immunostained for EGFP (green) and Tbr1 (red; blue is Hoechst 33258 nuclear counterstain). Tbr1 staining was used to mark the border between the intermediate zone (IZ) and the CP. In each set the left side shows the merged images of Tbr1 and Hoechst 33258, and the right side shows the EGFP immunostaining. The top white line demarcates the border between the CP and IZ, and the bottom white line the border between IZ and the VZ/SVZ. Scale bar, 100 μm. B, Quantification of sections similar to those in A for the proportion of EGFP-positive cells in the VZ/SVZ (left pair), IZ (middle pair), and CP (right pair). ***p < 0.001; n = 5 each for control and FoxP2 shRNA#1; n = 4 each for control and FoxP2 shRNA#2. C, Confocal micrographs of coronal E16/17 cortical sections immunostained for Satb2 (red) and Pax6 (middle, green) or Tbr2 (right, green). Scale bar, 50 μm. D, Higher magnification confocal images of sections similar to those shown in C. Arrows denote cells that are positive for Satb2 (red) and arrowheads denote cells that are positive for Pax6 (green, top) or Tbr2 (green, bottom; right side shows the merges). Note the absence of colocalization between Satb2-positive cells and Pax6-positive or Tbr2-positive cells. Scale bar, 20 μm. E, Confocal micrographs of coronal cortical sections from E16/17 brains 3 d after electroporation with EGFP and control shRNA (Control) or FoxP2 shRNA #2 (shFoxP2), immunostained for EGFP (green) and Satb2 (red). The white lines demarcate the different regions of the embryonic cortex. Scale bar, 50 μm. F, High-magnification confocal images of sections similar to those in E. The bottom shows Satb2 (red) alone, and the top shows Satb2 plus EGFP (green). Arrows denote EGFP-positive Satb2-positive cells. Scale bar, 20 μm. G, Quantification of sections similar to those in E and F for the percentage of EGFP-positive cells that are also positive for Satb2 or HuD, as indicated, 3 d after electroporation with control shRNA or with FoxP2 shRNA#1 or FoxP2 shRNA#2. FoxP2 shRNA#2 was somewhat less efficacious than FoxP2 shRNA#1 in this regard. *p < 0.05; **p < 0.01; ***p < 0.001; n = 3–5 embryos for each of the shRNAs. Error bars indicate SEM.
We asked whether this change in cellular distribution could be due to enhanced cell death by immunostaining sections for EGFP and the apoptotic marker cleaved caspase-3 (CC3) 2 and 3 d postelectroporation. Numbers were similar in control and experimental brains at both time points, averaging ∼0–3 cells per section (at 3 d, p > 0.05 for the comparisons between control vs FoxP2 shRNA#1 and control vs FoxP2 shRNA#2, n = 3 embryos each, five sections per embryo). We then asked whether the changes were due to perturbed neurogenesis by immunostaining sections for the transcription factor Satb2. Satb2 is a marker for one subpopulation of cortical neurons (Alcamo et al., 2008), and, consistent with this, we found that at E16/17, no Satb2-positive cells coexpressed the intermediate progenitor marker Tbr2 or the radial precursor marker Pax6 (Fig. 2C,D). Moreover, virtually all of the electroporated, EGFP-positive neurons in the CP expressed Satb2 when cortices were electroporated at E13/14 (Fig. 2E,F). Quantification of electroporated cortical sections immunostained with Satb2 demonstrated that FoxP2 knockdown caused a decrease in the number of neurons that were generated (Fig. 2G), consistent with the decrease in EGFP-positive cells in the CP (Fig. 2B). A similar decrease was observed when sections were immunostained for the panneuronal marker HuD (Fig. 2G).
To ask whether cortical precursors were also altered by FoxP2 knockdown, we immunostained sections for Pax6 and Tbr2, markers for radial precursors and intermediate progenitors, respectively, 3 d following electroporation of the two different FoxP2 shRNAs. This analysis demonstrated that FoxP2 knockdown caused an increase in the proportion of EGFP-positive, Pax6-positive radial precursors (Fig. 3A,B) and a decrease in the proportion of EGFP-positive, Tbr2-positive intermediate progenitors (Fig. 3C,D).
Figure 3.
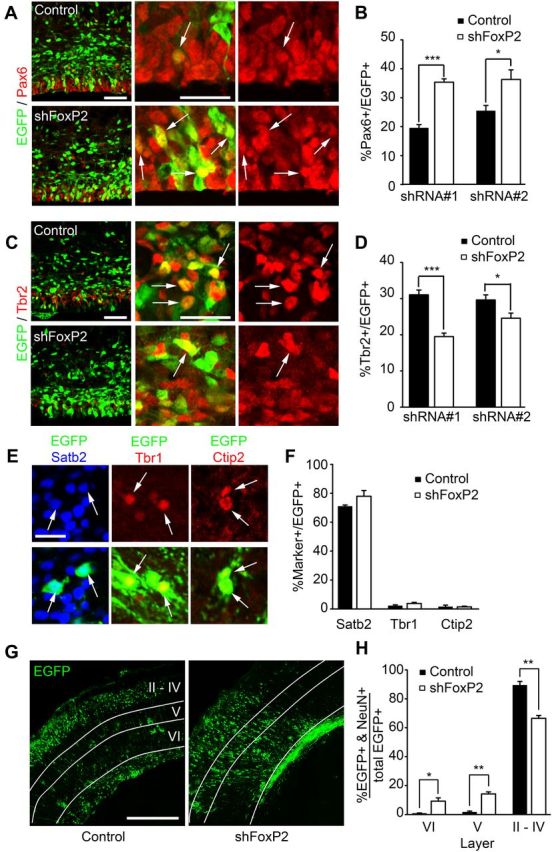
FoxP2 knockdown increases radial precursors at the expense of intermediate progenitors in the embryonic cortex. A–D, Murine cortices were electroporated at E13/14 with EGFP and control shRNA (Control) or FoxP2 shRNAs #1 or #2 (shFoxP2) and analyzed 3 d later. A, Confocal micrographs of coronal cortical sections from electroporated brains immunostained for EGFP (green) and Pax6 (red). Left, Low-magnification images of the VZ/SVZ of cortices electroporated with control shRNA (top) or FoxP2 shRNA #2 (bottom). Middle and right, High-magnification images showing EGFP-positive cells that are positive (arrows) for Pax6, with the middle panels showing the merges and the right panels Pax6 immunostaining only. Scale bar, 50 μm. B, Quantification of sections similar to those in A for the percentage of EGFP-positive cells that are also positive for Pax6. *p < 0.05; ***p < 0.001; n = 4 or 5 embryos each. C, Confocal micrographs of coronal cortical sections from electroporated brains immunostained for EGFP (green) and Tbr2 (red). Left, Low-magnification images of the VZ/SVZ of cortices electroporated with control shRNA (top) or FoxP2 shRNA #2 (bottom). Middle and right, High-magnification images showing EGFP-positive cells that are positive (arrows) for Tbr2, with the middle showing the merges and the right Tbr2 immunostaining only. Scale bar, 50 μm. D, Quantification of sections similar to those in C for the percentage of EGFP-positive cells that are also positive for Tbr2. *p < 0.05; ***p < 0.001; n = 4 or 5 embryos each. E–H, Murine cortices were electroporated at E13/14 with a PB transposon-based EGFP reporter and control shRNA (Control) or FoxP2 shRNA #2 and analyzed at P3. E, Confocal micrographs of coronal cortical sections electroporated with control shRNA (Satb2 and Ctip2) or FoxP2 shRNA#2 (Tbr1) and immunostained for EGFP (green) and Satb2 (left, blue), Tbr1 (middle, red), or Ctip2 (right, red; the bottom shows the merges). The Satb2 images are from the superficial cortical layers. Arrows denote double positive cells. Scale bar, 20 μm. F, Quantification of sections similar to those in E for the percentage of EGFP-positive cells that are also positive for Satb2, Tbr1, and Ctip2 (p > 0.05; n = 3 animals each). G, Confocal micrographs of coronal cortical sections from electroporated P3 brains immunostained for EGFP. The white lines distinguish the various cortical layers, in part as identified by immunostaining for Ctip2 (data not shown), which labels layer V cortical neurons. Scale bar, 400 μm. H, Quantification of sections similar to those in G for the percentage of EGFP-positive cells in the different cortical layers that also expressed NeuN. *p < 0.05; **p < 0.01; n = 3 animals each. Error bars indicate SEM.
These results indicate that the transition from radial precursors to intermediate progenitors and neurons is perturbed by FoxP2 knockdown. To ask whether this perturbation has long-lasting consequences for cortical neurogenesis, we used the PB transposon. In this system, a plasmid encoding the PB transposase is cotransfected with a plasmid-encoding EGFP flanked by inverted terminal repeats, and when the transposase is expressed, this leads to genomic integration of the flanked EGFP sequence (Nagy et al., 2011), thereby allowing long-term EGFP expression in the progeny of electroporated radial precursors. We coelectroporated the transiently expressed FoxP2 shRNA together with plasmids encoding PB transposase and the EGFP reporter at E13/14, and analyzed cortical neurons at P3. This analysis demonstrated that ∼80% of EGFP-expressing progeny of electroporated precursors were Satb2-positive neurons, and that transient knockdown of FoxP2 during embryogenesis did not cause long-term alterations in this number (Fig. 3E,F). Immunostaining for two other cortical neuron markers, Tbr1 and Ctip2, demonstrated that only 1–5% of the EGFP-positive cells expressed either of these markers, and that these were unaltered by FoxP2 knockdown (Fig. 3E,F). However, while the decrease in neuronal number that was observed at E16/17 was lost by P3, potentially due to the transient nature of the FoxP2 knockdown, the localization of these neurons was aberrant (Fig. 3G). In particular, in contrast to controls, in cortices electroporated with the FoxP2 shRNA, ∼20% of the NeuN-positive, EGFP-positive neurons were located in layers V and VI (Fig. 3G,H), and most of these mislocalized neurons expressed Satb2. This analysis also demonstrated that significantly more transfected Satb2-positive neurons were located in the deep cortical layers following FoxP2 knockdown (Fig. 3G,H).
These data indicate that FoxP2 knockdown during embryogenesis perturbs neurogenesis. To ensure that these results are not due to off-target effects of the shRNA, we performed rescue experiments with human FoxP2, which is not targeted by the shRNAs. Specifically, E13/14 cortices were electroporated with plasmids encoding EGFP and FoxP2 shRNA #1 ± a human FoxP2 overexpression vector. Analysis of these cortices 3 d later confirmed that coincident human FoxP2 expression largely reversed the decrease in EGFP-positive cells in the CP and the increase in the VZ/SVZ caused by FoxP2 knockdown (Fig. 4A,B). Similarly, immunostaining demonstrated that coincident human FoxP2 expression completely reversed the decrease in Tbr2-positive intermediate progenitors and Satb2-positive neurons and the increase in Pax6-positive radial precursors seen with FoxP2 knockdown (Fig. 4C–E).
Figure 4.

FoxP2 knockdown increases radial precursors at the expense of intermediate progenitors and neurons, and this is reversed by coincident expression of human FoxP2. A–E, Murine cortices were electroporated at E13/14 with an EGFP plasmid and control shRNA plus pCMV-Tag4A empty vector (shCon/EV), FoxP2 shRNA #1 plus pCMV-Tag4A empty vector (shFoxP2/EV), or FoxP2 shRNA #1 plus human FoxP2 in the pCMV-Tag4A backbone (shFoxP2/hFoxP2) and analyzed 3 d later. A, Fluorescence micrographs of coronal cortical sections showing EGFP-positive cells (green). The white lines denote the different regions of the cortex. Scale bar, 100 μm. B, Quantification of sections similar to those in A for the number of EGFP-positive cells in the different cortical regions. *p < 0.05; **p < 0.01; ***p < 0.001, ANOVA with Student–Newman–Keuls post hoc analysis; n = 3–7 embryos each. C–E, Quantification of the percentage of EGFP-positive cells that were also positive for Pax6 (C), Tbr2 (D), or Satb2 (E) in cortical sections similar to those in A that were immunostained for EGFP and one of these three markers. *p < 0.05; **p < 0.01; ***p < 0.001, ANOVA with Student–Newman–Keuls post hoc analysis; n = 3–5 embryos each. F, E12 cortical precursors were cotransfected with EGFP and control shRNA (Control) or FoxP2 shRNA #2 (shFoxP2 #2) and quantified for the percentage of transfected, Ki67-positive (left two bars) or CC3-positive (right two bars) cells after 3 d. **p < 0.01; n = 3 independent experiments. G–I, E12 cortical precursors were cotransfected with PB-EGFP and PBase, and control shRNA plus empty vector (shCon/EV), FoxP2 shRNA plus pCMV-Tag4A empty vector (shFoxP2/EV), or FoxP2 shRNA plus a plasmid encoding hFoxP2 in the pCMV-Tag4A backbone (shFoxP2/hFoxP2). G, Fluorescence micrographs of cortical precursor cultures cotransfected with plasmids encoding PB-EGFP and PBase and either control shRNA (top) or FoxP2 shRNA#2 (bottom), immunostained 4 d later for EGFP (green). Scale bar, 50 μm. H, Quantification of clone size in experiments similar to those shown in G. Results are pooled data from three independent experiments. I, Quantification of the percentage of transfected, EGFP-positive clones that contained only a single cell (left three bars) or nine or more cells (right three bars). The graphs show pooled data from three independent experiments. *p < 0.05; **p < 0.01, ANOVA with Student–Newman–Keuls post hoc analysis. J–L, E12 cortical precursors were cotransfected with PB-EGFP and PBase, and control shRNA plus empty vector (shCon/EV), or FoxP2 shRNA plus pCMV-Tag4A empty vector (shFoxP2/EV), control shRNA plus a plasmid encoding hFoxP2 in the pCMV-Tag4A backbone (shCon/hFoxP2), or FoxP2 shRNA plus hFoxP2 (shFoxP2/hFoxP2). J, Fluorescence micrographs of control transfected cells immunostained 4 d after transfection for EGFP (green) and Tbr2 (top, red) or β-III-tubulin (bottom, red). Cells were counterstained with Hoechst 33258 (blue). Right, Shows the merges. Scale bar, 20 μm. K, L, Quantification of the percentage of transfected, Tbr2-positive (K) or β-III-tubulin-positive (L) cells in experiments similar to those shown in J. Both graphs show pooled data from three independent experiments. *p < 0.05; **p < 0.01, ANOVA with Student–Newman–Keuls post hoc analysis. Error bars indicate SEM.
FoxP2 knockdown inhibits the genesis of neurogenic intermediate progenitors
These data indicate that FoxP2 knockdown delays the progression from radial precursors to intermediate progenitors and neurons in vivo. To characterize this phenotype further, we knocked down FoxP2 in culture; radial precursors were isolated from the E12 cortex, and upon plating were cotransfected with a control shRNA or with FoxP2 shRNA #2 plus an EGFP expression construct. Initially we characterized these cultures for apoptosis and proliferation. Immunostaining for CC3 demonstrated that FoxP2 knockdown had no significant effect on the low proportion of CC3-positive apoptotic cells (Fig. 4F). In contrast, immunostaining for the proliferation marker Ki67 demonstrated that FoxP2 knockdown caused a significant increase from ∼60 to 80% in the proportion of proliferating cells (Fig. 4F).
To ask whether this increase in proliferation reflected an increase in self-renewing radial precursors, as suggested by the in vivo knockdown data, we performed clonal analysis using the PB transposon. Specifically, E12 precursors were cotransfected with FoxP2 shRNA #2 plus or minus a human FoxP2 overexpression construct together with plasmids encoding PB transposase and the PB EGFP reporter construct, under conditions where only a small number of cells were transfected. We then analyzed the size and composition of EGFP-positive clones in these cultures (Fig. 4G). This analysis showed that FoxP2 knockdown caused a significant increase in the size of the EGFP-positive clones (Fig. 4H), with a decrease in single cell clones, and an increase of almost threefold in the proportion of clones containing nine or more cells (Fig. 4I). Moreover, only a very small number of control clones contained >17 cells, but ∼10% of the FoxP2 knockdown clones were 17 cells or larger (Fig. 4H). Importantly, coexpression of human FoxP2 reversed this increase in clone size (Fig. 4H,I). We also performed an additional control for the single cell clonal analysis, transfecting cells with a control shRNA together with human FoxP2. This control group was statistically similar to the FoxP2 shRNA plus human FoxP2 group (p > 0.05) and was significantly higher than the FoxP2 shRNA plus empty vector group (p < 0.01; n = 3 independent experiments for both comparisons).
We also asked about clone composition by immunostaining these cultures for β-III-tubulin and Tbr2 (Fig. 4J). FoxP2 knockdown increased by ∼30% the proportion of EGFP-positive clones that did not contain β-III-tubulin or Tbr2-positive cells, and this increase in radial precursor clones was significantly reversed by coincident expression of human FoxP2 (p < 0.01 for both comparisons, n = 3 independent experiments). Moreover, quantification showed that the FoxP2 but not control shRNA decreased the genesis of intermediate progenitors and neurons, and that these decreases were also reversed by coincident overexpression of human FoxP2 (Fig. 4K,L). Together, these data indicate that FoxP2 knockdown in culture enhanced radial precursors at the expense of intermediate progenitors and neurons. Moreover, the increased proliferation observed in culture following FoxP2 knockdown (Fig. 4F) suggests that the increase in radial precursors observed following FoxP2 knockdown in vivo (Fig. 3B) may also be due, in part, to enhanced proliferation.
Ectopic expression of human but not mouse FoxP2 promotes the radial precursor to intermediate progenitor transition in the mouse cortex
These data indicate that FoxP2 knockdown decreases genesis of intermediate progenitors from radial precursors. Since a number of groups have suggested that evolutionary changes in mammalian FoxP2 might be important (Enard et al., 2002; Krause et al., 2007; Konopka et al., 2009), we asked whether mouse and human FoxP2 proteins had similar or different activities when ectopically expressed in radial precursors. To do this, we used flag-tagged expression constructs for mouse and human FoxP2 (Konopka et al., 2009) that were identical except that one encoded mouse and the other human FoxP2 coding sequences (Fig. 5A). We verified that these constructs were expressed at similar levels by transfecting them into HEK293 cells, and probing equal amounts of protein on Western blots with either the FoxP2 antibody or with an antibody for the flag tag (Fig. 5B).
Figure 5.
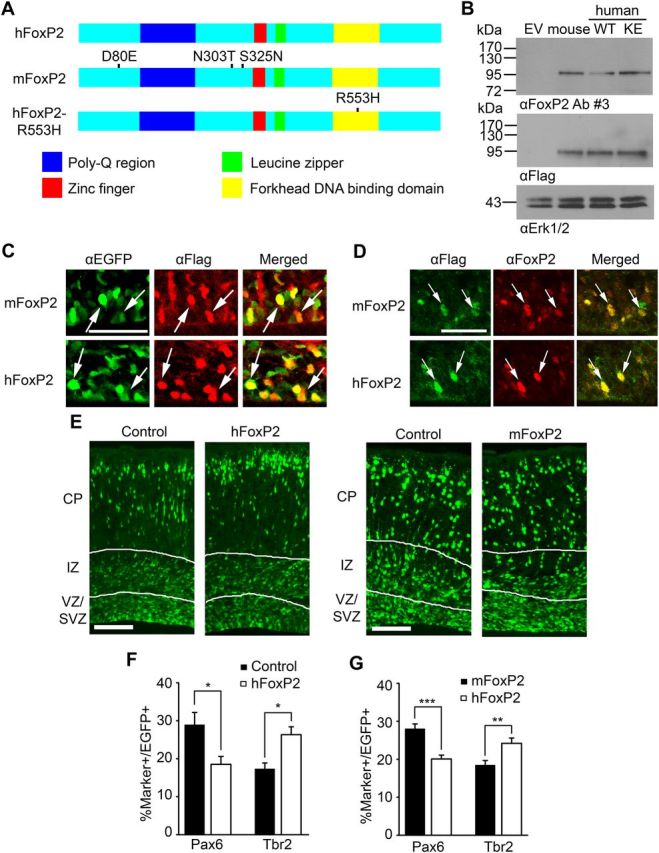
Ectopic expression of human but not murine FoxP2 in the murine embryonic cortex enhances the genesis of intermediate progenitors and neurons. A, Schematics of the domain structure of human FoxP2 (hFoxP2), mouse FoxP2 (mFoxP2), and human FoxP2 carrying the KE mutation in the DNA binding domain (hFoxP2-R553H). Both of the human FoxP2 proteins are 715 aa in length while mouse FoxP2 is 714. All amino acid substitutions indicated in the diagram are with reference to human FoxP2. B, Western blot analysis of lysates of HEK293 cells transfected with pCMV-Tag4A empty vector (EV) or flag-tagged expression constructs for mouse FoxP2, wild-type human FoxP2 (WT), or human FoxP2 carrying the R553H KE family mutation (KE), all in the pCMV-Tag4A backbone, probed with antibodies against FoxP2 or the flag tag. Blots were also probed with an antibody against Erk as a control for equal protein loading. C–G, Murine cortices were electroporated at E13/14 with a nuclear EGFP expression construct and either the empty vector (Control), or the flag-tagged human FoxP2 (hFoxP2) or mouse FoxP2 (mFoxP2) expression constructs, and analyzed 3 d later. C, High-magnification confocal micrographs of coronal cortical sections through the VZ/SVZ of electroporated brains immunostained for EGFP (green) and Flag (red; right side show the merges) 3 d after electroporation. Arrows indicate EGFP-positive cells that were also positive for the flag tag. Scale bar, 50 μm. D, High-magnification confocal micrographs of coronal cortical sections through the VZ/SVZ of electroporated brains immunostained for Flag (green) and FoxP2 (red; right side shows the merges) 3 d after electroporation. Arrows indicate flag-positive cells that were also positive for FoxP2. Scale bar, 50 μm. E, Fluorescence micrographs of coronal sections of electroporated brains 3 d after electroporation showing EGFP-positive cells (green). The white lines demarcate the different cortical regions. Scale bar, 100 μm. F, Quantification of the percentage of double-labeled cells in sections similar to those in E that were immunostained for EGFP and Pax6 or Tbr2 3 d following electroporation with the empty vector (Control) or the expression construct for human FoxP2 (hFoxP2). *p < 0.05; n = 4 embryos each. G, Quantification of the percentage of double-labeled cells in sections similar to those in E that were immunostained for EGFP and Pax6 or Tbr2 3 d following electroporation with EGFP and the expression construct for either mouse FoxP2 (mFoxP2) or human FoxP2 (hFoxP2). **p < 0.01; ***p < 0.001; n = 7 embryos each. Error bars indicate SEM.
We then introduced these constructs into cortical precursors in vivo, electroporating them into the E13/14 cortex along with a nuclear EGFP expression plasmid. Three days later we demonstrated that both constructs were expressed by staining cortical sections either for the flag tag present on the tagged proteins or for FoxP2 itself. Flag-tagged murine and human FoxP2 were both detectably expressed in EGFP-positive cells (Fig. 5C). Moreover, image analysis of sections immunostained for FoxP2 (Fig. 5D) demonstrated that mouse and human FoxP2 were expressed at similar levels in electroporated precursors (mouse FoxP2 levels were set to 1.0; mouse FoxP2, 1.0 ± 0.28, human FoxP2, 1.0 ± 0.17; p = 0.99, n = 3 embryos each). Since both proteins were expressed in electroporated precursors, we asked whether they affected cortical precursor biology. Immunostaining for EGFP (Fig. 5E) demonstrated that the distribution of transfected cells was similar in cortices electroporated with the control vector and mouse FoxP2 (p > 0.05; n = 3 embryos each), but that electroporation with human FoxP2 caused a small but significant decrease in the proportion of electroporated cells in the VZ/SVZ relative to controls (control vector, 31.6 ± 2.6, human FoxP2, 26.0 ± 1.0; p < 0.05, n = 4 embryos each). To ask whether this reflected a change in precursor composition, we immunostained these sections for EGFP and Pax6 or Tbr2. Quantification demonstrated that human FoxP2 overexpression decreased the proportion of Pax6-positive radial precursors and increased the proportion of Tbr2-positive intermediate progenitors (Fig. 5F) relative to controls. In contrast, mouse FoxP2 had no effect on the proportions of these two precursor types (for Pax6-positive cells, control vector, 20.3 ± 0.8%, mouse FoxP2, 20.0 ± 3.4%, p = 0.94; for Tbr2-positive cells, control vector, 16.8 ± 0.8%, mouse FoxP2, 17.1 ± 1.8%, p = 0.87; n = 3 embryos each).
To more definitively establish these differences between human and mouse FoxP2 proteins, we performed similar studies electroporating different embryos in the same mothers with human versus mouse FoxP2 constructs. We then analyzed these cortices 3 d later by immunostaining for Pax6 and Tbr2. Quantification showed that human and mouse FoxP2 overexpression had significantly different effects, with human FoxP2 causing a decrease in Pax6-positive radial precursors and an increase in the percentage of Tbr2-positive intermediate progenitors relative to mouse FoxP2 (Fig. 5G).
Since human FoxP2 increased the number of neurogenic intermediate progenitors, we asked whether it also altered the number or phenotype of cortical neurons. To ask about neuron number, we analyzed cortices 3 d postelectroporation at E16/17. Immunostaining for EGFP and HuD demonstrated a small but significant increase in the proportion of EGFP-positive cortical neurons (Fig. 6A). A similar small but significant increase was observed when sections were immunostained for Satb2 (control vector, 42.3 ± 4.8, human FoxP2, 52.6 ± 4.8; p = 0.03, two-tailed paired Student's t test, n = 4 embryos each). To ask about neuronal phenotypes, we analyzed cortices postnatally, electroporating embryos at E13/14 with plasmids encoding EGFP and human FoxP2, and immunostaining them for different neuronal markers at P3 (Fig. 6B,C). Quantification showed that human FoxP2 did not significantly affect the proportion of Satb2-positive neurons at this longer time point nor did it affect the small number of Ctip2 or Tbr1-positive neurons (Fig. 6D).
Figure 6.

Ectopic expression of human but not mouse FoxP2 promotes genesis of intermediate progenitors and neurons. A–D, Murine cortices were electroporated at E13/14 with EGFP and either the pCMV-Tag4A empty vector (Control) or a human FoxP2 expression construct in the same vector (hFoxP2), and analyzed at E16/17 (A) or at P3 (B–D). A, Confocal micrographs of coronal cortical sections of E16/17 electroporated brains immunostained for EGFP (green) and HuD (red). The graph on the right shows quantification of similar sections for the percentage of EGFP-positive cells that express HuD. *p < 0.05, two-tailed paired Student's t test; n = 4 embryos each. Scale bar, 100 μm. B, Confocal micrographs of coronal cortical sections of electroporated brains at P3 immunostained for EGFP (green), Tbr1 (red), and Satb2 (blue). Scale bar, 400 μm. C, Confocal micrographs of coronal cortical sections of electroporated brains at P3 immunostained for EGFP (green) and Ctip2 (red). Scale bar, 400 μm. D, Quantification of sections similar to those in B and C for the percentage of EGFP-positive cells that were also positive for Satb2 (left two bars), Tbr1 (middle two bars), or Ctip2 (right two bars). p > 0.05; n = 3 animals each. E–H, E12 cortical precursor cultures were cotransfected with an EGFP plasmid and expression constructs for mouse FoxP2 (mFoxP2), wild-type human FoxP2 (hFoxP2), or human FoxP2 carrying the KE mutation (hFoxP2-R553H or KE FoxP2). As a control, cultures were cotransfected with the empty vector (Control). E, Quantification of the sizes of transfected EGFP-positive clones. F, Quantification of the percentage of transfected EGFP-positive clones that contain only a single cell, normalized to the empty vector control cultures. G, H, Quantification of the percentage of transfected, Tbr2-positive (G) or β-III-tubulin-positive (H) cells, normalized to the empty vector control cultures. *p < 0.05; **p < 0.01; ***p < 0.001, ANOVA with Student–Newman–Keuls post hoc analysis; n = 3 or 4 experiments for each. Error bars indicate SEM.
To further explore the possibility that human, but not mouse, FoxP2 enhances the transition from radial precursors to intermediate progenitors when ectopically expressed in the mouse, we performed similar experiments in culture, cotransfecting mouse versus human FoxP2 constructs together with an EGFP expression plasmid into cultured radial precursors. Clonal analysis 4 d later showed that human FoxP2 caused a decrease in clone size, significantly increasing the proportion of one cell clones relative to either mouse FoxP2 or the empty vector (Fig. 6E,F). Consistent with this, immunostaining showed that human, but not mouse FoxP2 increased the proportion of Tbr2-positive intermediate progenitors (Fig. 6G) and β-III-tubulin-positive neurons (Fig. 6H) that were generated in these cultures. Thus, human FoxP2 apparently acts as a gain-of-function protein in murine cortical precursors, acting to promote the radial precursor to intermediate progenitor transition and enhance neurogenesis.
The human FoxP2 KE mutation acts like a dominant-inhibitory FoxP2 protein
Since our data indicate that even the small differences between human and murine FoxP2 proteins can alter its ability to regulate neural precursor development, we asked about a mutant FoxP2 protein that has been implicated in aberrant vocalization. To do this, we examined a FoxP2 mutation from the KE family (Lai et al., 2001) using a flag-tagged expression construct identical to that used for the human versus mouse overexpression studies. This mutation results in a FoxP2 protein carrying a single amino acid switch in the forkhead DNA binding domain (Fig. 5A). Cotransfection into HEK293 cells demonstrated that the KE mutant was expressed at levels similar to the normal human FoxP2 protein (Fig. 5B). We then cotransfected cultured cortical precursors with plasmids encoding nuclear-localized EGFP and the wild-type or mutant human FoxP2 or murine FoxP2, and immunostained them 1 d later for EGFP and the flag tag. This analysis (Fig. 7A) demonstrated that all three proteins were expressed, but that their localization differed. In particular, the normal murine and human FoxP2 proteins were limited to nuclei, colocalizing closely with nuclear EGFP (Fig. 7A). In contrast, the human KE mutant was present in both the cytoplasm and nucleus (Fig. 7A). Quantification showed that >60% of the flag-positive cells transfected with the KE mutant expressed detectable FoxP2 in the cytoplasm (Fig. 7B).
Figure 7.

The KE family FoxP2 mutation acts as a dominant-negative with regard to embryonic cortical neurogenesis. A, Fluorescence micrographs of E12 cortical precursor cultures cotransfected with a nuclear EGFP plasmid and expression constructs for mouse FoxP2 (mFoxP2; left), wild-type human FoxP2 (hFoxP2; left center), or human FoxP2 carrying the KE mutation (hFoxP2-R553H; right center and right), immunostained 1 d later for EGFP (green) and Flag (red). Cells were counterstained with Hoechst 33258 (blue). Dotted lines mark nuclear borders as indicated by Hoechst staining. Note the presence of cytoplasmic Flag-tagged hFoxP2-R553H. Scale bar, 20 μm. B, Quantification of the percentage of transfected cells in experiments as in A where Flag immunoreactivity was detectable in the cytoplasm. ***p < 0.001, ANOVA with Student–-Newman–Keuls post hoc analysis; n = 3 independent experiments. C–E, E13/14 murine cortices were electroporated with EGFP and pCMV-Tag4A empty vector (Control) or an expression construct for hFoxP2-R553H in the same vector. C, Fluorescence micrographs of coronal cortical sections 3 d postelectroporation, showing EGFP-positive cells (green). The white lines demarcate the different cortical regions. Scale bar, 100 μm. D, Quantification of sections similar to those in C for the number of EGFP-positive cells in the VZ/SVZ (left pair), IZ (middle pair), and CP (right pair). **p < 0.01; n = 7 animals for the control and 6 for hFoxP2-R553H. E, Quantification of sections similar to those in C for the percentage of EGFP-positive cells that were positive for Pax6 (left two bars), Tbr2 (middle two bars), or Satb2 (right two bars) 3 d after electroporation. *p < 0.05; ***p < 0.001; n = 6 for control and 5 for hFoxP2-R553H. Error bars indicate SEM.
These experiments indicate that the mutant human FoxP2 protein is expressed at readily detectable levels in cortical precursors, but that it is aberrantly localized. These data are consistent with previous reports expressing it in cell lines (Vernes et al., 2006; Mizutani et al., 2007), which showed that the KE mutant protein was present in the cytoplasm and that it even caused aberrant localization of wild-type FoxP2 protein in the cytoplasm. We therefore electroporated the E13/14 cortex with expression plasmids for the KE mutant protein and EGFP, and analyzed precursor development 3 d later. Immunostaining for EGFP demonstrated that location of transfected cells within the cortex was perturbed by expression of the mutant FoxP2, with significantly more cells present in the VZ/SVZ and significantly fewer in the CP relative to the empty vector (Fig. 7C,D). Coincident with this change in location, there was an increase in the proportion of Pax6-positive radial precursors and a decrease in Tbr2-positive intermediate progenitors (Fig. 7E). There was also a trend toward a decrease in Satb2-positive neurons, but this did not reach significance (Fig. 7E). Thus, in contrast to human FoxP2, the KE mutant FoxP2 protein decreased the radial precursor to intermediate progenitor transition thereby phenocopying the FoxP2 knockdown phenotype, suggesting that it was functioning as a dominant-inhibitory protein.
To further explore the possibility that the KE mutant FoxP2 has a different phenotype from normal human FoxP2 protein, we overexpressed it in cultured cortical precursors for 4 d. Consistent with the idea that the KE mutant protein inhibited cortical precursor differentiation, clonal analysis demonstrated that it increased clone size relative to human and murine FoxP2 proteins (Fig. 6E,F). Moreover, immunostaining demonstrated that there were only approximately half as many Tbr2-positive intermediate progenitors (Fig. 6G) and β-III-tubulin-positive neurons (Fig. 6H) generated from cells that expressed the mutant human FoxP2 compared with normal human FoxP2. Overexpression of the KE mutant also caused a decrease in neurons relative to the empty vector and murine FoxP2 overexpression (Fig. 6H), thereby phenocopying the FoxP2 knockdown data (Fig. 4H). Together with the in vivo studies, these data provide support for the idea that the human KE mutant acts as a dominant-inhibitory FoxP2 protein.
Discussion
Our results suggest a novel role for the FoxP2 transcription factor in developing neural precursors that may have implications for evolution of a larger mammalian cortex. In this regard, our findings support three major conclusions. First, our data indicate that FoxP2 is expressed in developing cortical precursors, and that when it is knocked down, this perturbs the genesis of intermediate progenitors and neurons from multipotent radial precursors. Second, our data indicate that human and mouse FoxP2 proteins differ with regard to their ability to regulate the genesis of intermediate progenitors when ectopically expressed. When mouse FoxP2 is overexpressed either in vivo or in culture, this has no effect on cell genesis, indicating that mouse FoxP2 is not sufficient to promote the genesis of intermediate progenitors, and suggesting that it is normally not limiting for neural precursors. However, when human FoxP2 is overexpressed in the same way, this enhances the genesis of intermediate progenitors and neurons, indicating that human FoxP2 acts as a gain-of-function protein relative to murine FoxP2. This is an intriguing result, given the high homology between these two orthologs. Third, our data indicate that the KE mutant FoxP2, which acts as an autosomal dominant gene in humans, functions as a dominant-inhibitory protein with regard to neurogenesis. Specifically, we show that, as seen previously in cell lines, this mutant FoxP2 is localized to both the nucleus and cytoplasm in cortical precursors, and that it phenocopies FoxP2 knockdown when overexpressed, causing increased radial precursors and decreased intermediate progenitors and neurons. Together, these findings suggest a novel proneurogenic role for FoxP2 and, from a broader context, suggest that genetically defined changes in proteins like FoxP2 may contribute to the evolution of the human cortex.
The FoxP2 gene has garnered significant interest because it is mutated in a monogenic syndrome causing speech and language dysfunction (Lai et al., 2001). This relationship has led to speculation that small changes in FoxP2 sequence are causally related to the evolution of human speech (Enard et al., 2002; Zhang et al., 2002; Vernes et al., 2007). In support of this idea, human FoxP2 has transcriptional targets that are distinct from mouse and even chimpanzee FoxP2 (Vernes et al., 2007, 2011; Konopka et al., 2009), and corticobasal circuitry is functionally different in a mouse where two of the human amino acid changes are knocked-in to the mouse FoxP2 allele (Enard et al., 2009; Reimers-Kipping et al., 2011). Thus, the few amino acid changes between the different FoxP2 proteins are clearly important and are likely somehow involved in functional neural differences between species. In this regard, though, it is still unclear whether FoxP2 is only important for speech and language and/or whether it plays a broader role in neural function.
Previous studies on FoxP2 have used the songbird (Haesler et al., 2007) and the mouse (Shu et al., 2005; French et al., 2007; Fujita et al., 2008) as models for understanding how this gene might regulate vocalization. Work in both models has shown that FoxP2 is expressed in neurons, and that depletion of FoxP2 leads to changes in vocalization (Shu et al., 2005; French et al., 2007; Fujita et al., 2008) and neuronal morphology (Vernes et al., 2011). In this regard, knock-out mice and mice homozygous for mutant human FoxP2 alleles are developmentally delayed, die within several weeks of birth, display perturbed ultrasonic vocalizations, and have smaller brains with a particularly small cerebellum. While these neural phenotypes are thought to be due to perturbed neuronal connectivity, the results presented here suggest that they are, in part, due to aberrant neurogenesis. Strong additional support for this conclusion comes from a recent study that placed FoxP2 and FoxP4 together in a proneurogenic pathway (Rousso et al., 2012). This study showed that Foxp2 is initially expressed throughout the neuroepithelium, and that inactivation of FoxP2 and FoxP4 function led to neuroepithelial disorganization and enhanced maintenance of neural precursors, a result consistent with the findings reported here.
How then does FoxP2 regulate neurogenesis? During cortical development, neurons are generated either directly from radial glial precursors or indirectly via intermediate progenitors. Intriguingly, the number of intermediate progenitors correlates with cortical expansion (Martinez-Ceredeño et al., 2006) and the intermediate progenitor population is enhanced in humans relative to rodents (Hansen et al., 2010). Moreover, deletion of genes required for intermediate progenitors results in decreased cortical thickness and neuron number (Farkas et al., 2008; Sessa et al., 2008). In this regard, our knockdown data argue that FoxP2 promotes neurogenesis in part by enhancing the genesis of intermediate progenitors from radial precursors. Intriguingly, our expression data also show that while FoxP2 is expressed at low levels in radial precursors, its expression is robustly increased in at least a subset of intermediate progenitors. Thus, one interpretation of our data is that FoxP2 levels normally increase as part of a pro-intermediate progenitor gene expression program, and that when we knock it down in radial precursors, this prevents this induction and inhibits the transition. However, our data showing that overexpression of mouse FoxP2 in these same radial precursors does not cause them to prematurely generate intermediate progenitors argues that increased FoxP2 is necessary but not sufficient for this transition.
What are the molecular mechanisms that allow FoxP2 to regulate the radial precursor to intermediate progenitor transition? First, in the recent Rousso et al. (2012) paper, the authors show that ectopic expression of FoxP2 downregulates expression of N-cadherin, a key component of apical adherens junctions. Since radial precursors, but not intermediate progenitors, are part of the apical epithelium, then perhaps a FoxP2-mediated delamination from this epithelium is a key part of the transition between these two cell types. Second, FoxP2 can directly inhibit transcription of the DISC1 gene (Walker et al., 2012), and suppression of DISC1 causes developing neural precursors to stop proliferating and to differentiate (Mao et al., 2009). Third, FoxP2 regulates transcription of the autism-associated gene CNTNAP2, which is expressed in neural precursors, and regulates neuronal development (Peñagarikano et al., 2011). Thus, FoxP2 may regulate these and other downstream targets to promote neurogenesis.
One intriguing result presented here is that human FoxP2 acts as a gain-of-function protein within the murine context. These data imply that the four amino acid changes that distinguish the human and mouse proteins enhance the proneurogenic actions of FoxP2. While we do not yet know which amino acid change accounts for this difference, the amino acid switch at position 324 of mouse FoxP2 (homologous to position 325 of human FoxP2) introduces a consensus protein kinase C (PKC) phosphorylation site into the human FoxP2 protein. In this regard, we recently showed that atypical PKCs regulate cortical neurogenesis (Wang et al., 2012), and PKCs are activated in neural precursors by neurotransmitters, peptide ligands, and growth factors (Canoll et al., 1996; Ma et al., 2000; Hansel et al., 2001; Song and Ghosh, 2004). Moreover, this particular amino acid substitution is one of two that have been knocked-in to the mouse FoxP2 gene, and shown to result in alterations in corticobasal ganglia circuits (Enard et al., 2009; Reimer et al., 2011). Regardless of the underlying mechanism, these findings indicate that FoxP2 evolved an enhanced proneurogenic function coincident with evolution of a larger cortex. Since recent work suggests that an increase in intermediate progenitors was important for evolution of a larger human cortex (Noctor et al., 2007; Pontious et al., 2008), then this raises the possibility that a more active FoxP2 was perhaps somehow involved in facilitating this evolution.
One final conclusion arising from these studies is that the KE mutant FoxP2 acts as a dominant-inhibitory protein with regard to neurogenesis, consistent with the autosomal dominant genetics of the KE family (Fisher et al., 1998). How might it do this? We report here that the KE mutant FoxP2, which is mutated at a single amino acid in the forkhead DNA binding domain, localizes to both the nucleus and cytoplasm of cortical precursors. A similar cytoplasmic localization was seen with this mutant in cell lines (Vernes et al., 2006; Mizutani et al., 2007), and cytoplasmic aggregates of this protein were observed in neurons when it was knocked-in to the murine FoxP2 locus (Fujita et al., 2008). Since the KE mutant FoxP2 can modify the localization of wild-type FoxP2 (Mizutani et al., 2007), then we propose that it acts as a dominant-inhibitory protein by binding to the endogenous protein and sequestering it in the cytoplasm. It may also mediate this dominant-inhibition function in part by inhibiting appropriate DNA binding and transcriptional activation within the nucleus.
In summary, our findings support a novel role for FoxP2 in developmental neurogenesis, and indicate that human FoxP2 acquired an enhanced ability to promote neurogenesis coincident with the evolution of a larger human cortex. While our findings do not preclude a later role for FoxP2 in neuronal development, we propose that perturbations in the numbers or timing of cortical neuron genesis would ultimately cause aberrant neural circuit formation, thereby potentially providing a neural template for the deficits in speech and language seen in individuals with FoxP2 mutations.
Footnotes
This work was supported by Canadian Institutes of Health Research (CIHR) Grant MOP 13958 from the CIHR to F.D.M. and D.R.K. F.D.M. is an Howard Hughes Medical Institute International Research Scholar, and F.D.M. and D.R.K. are CRC Chairs. D.T. was supported by a CIHR CGS-D scholarship, and J.V. was supported by the CIHR Mark Bisby Fellowship. We particularly thank Dr. Steve Scherer for the thoughtful discussions that led to this work; Dr. Matthias Groszer for his valuable intellectual input and supplying tissue; Dr. Edward Morrissey for the kind gift of the FoxP1 and FoxP4 constructs; Dr. Genevieve Konopka for the pCMV-Tag4A, hFoxP2, and hFoxP2-R553H constructs; Dr. Zea Borok for the pCMV-Tag2B-FoxP2 construct; Dr. Larysa Pevny for the Sox2:EGFP mice; Sarah Burns for her superb technical assistance; and members of the Kaplan–Miller laboratories for their input and assistance.
References
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, McConnell SK. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–377. doi: 10.1016/j.neuron.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Wasylnka JA, Fernandes KJ, Porsche C, Sendtner M, Kaplan DR, Miller FD. Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron. 2005;48:253–265. doi: 10.1016/j.neuron.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Biernaskie J, Paris M, Morozova O, Fagan BM, Marra M, Pevny L, Miller FD. SKPs derive from hair follicle precursors and exhibit properties of adult dermal stem cells. Cell Stem Cell. 2009;5:610–623. doi: 10.1016/j.stem.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbé JC, Lorca T, Castro A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A. 2010;107:12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canoll PD, Musacchio JM, Hardy R, Reynolds R, Marchionni MA, Salzer JL. GGF/neuregulin is a neuronal signal that promotes the proliferation and survival and inhibits the differentiation of oligodendrocyte progenitors. Neuron. 1996;17:229–243. doi: 10.1016/s0896-6273(00)80155-5. [DOI] [PubMed] [Google Scholar]
- Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S, McMahon A, Rao M, Pevny L. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci. 2004;26:148–165. doi: 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- Enard W, Przeworski M, Fisher SE, Lai CS, Wiebe V, Kitano T, Monaco AP, Pääbo S. Molecular evolution of FOXP2, a gene involved in speech and language. Nature. 2002;418:869–872. doi: 10.1038/nature01025. [DOI] [PubMed] [Google Scholar]
- Enard W, Gehre S, Hammerschmidt K, Hölter SM, Blass T, Somel M, Brückner MK, Schreiweis C, Winter C, Sohr R, Becker L, Wiebe V, Nickel B, Giger T, Müller U, Groszer M, Adler T, Aguilar A, Bolle I, Calzada-Wack J, et al. A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell. 2009;137:961–971. doi: 10.1016/j.cell.2009.03.041. [DOI] [PubMed] [Google Scholar]
- Farkas LM, Haffner C, Giger T, Khaitovich P, Nowick K, Birchmeier C, Pääbo S, Huttner WB. Insulinoma-associated 1 has a panneurogenic role and promotes the generation and expansion of basal progenitors in the developing mouse neocortex. Neuron. 2008;60:40–55. doi: 10.1016/j.neuron.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- Feuk L, Marshall CR, Wintle RF, Scherer SW. Structural variants: changing the landscape of chromosomes and design of disease studies. Hum Mol Genet 15 Spec No. 2006;1:R57–R66. doi: 10.1093/hmg/ddl057. [DOI] [PubMed] [Google Scholar]
- Fisher SE, Vargha-Khadem F, Watkins KE, Monaco AP, Pembrey ME. Localisation of a gene implicated in a severe speech and language disorder. Nat Genet. 1998;18:168–170. doi: 10.1038/ng0298-168. [DOI] [PubMed] [Google Scholar]
- French CA, Groszer M, Preece C, Coupe AM, Rajewsky K, Fisher SE. Generation of mice with a conditional Foxp2 null allele. Genesis. 2007;45:440–446. doi: 10.1002/dvg.20305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita E, Tanabe Y, Shiota A, Ueda M, Suwa K, Momoi MY, Momoi T. Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells. Proc Natl Acad Sci U S A. 2008;105:3117–3122. doi: 10.1073/pnas.0712298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier AS, Furstoss O, Araki T, Chan R, Neel BG, Kaplan DR, Miller FD. Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron. 2007;54:245–262. doi: 10.1016/j.neuron.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haesler S, Rochefort C, Georgi B, Licznerski P, Osten P, Scharff C. Incomplete and inaccurate vocal imitation after knockdown of FoxP2 in songbird basal ganglia nucleus Area X. PLoS Biol. 2007;5:e321. doi: 10.1371/journal.pbio.0050321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel DE, Eipper BA, Ronnett GV. Neuropeptide Y functions as a neuroproliferative factor. Nature. 2001;410:940–944. doi: 10.1038/35073601. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464:554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- Hutton SR, Pevny LH. SOX2 expression levels distinguish between neural progenitor populations of the developing dorsal telencephalon. Dev Biol. 2011;352:40–47. doi: 10.1016/j.ydbio.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Konopka G, Bomar JM, Winden K, Coppola G, Jonsson ZO, Gao F, Peng S, Preuss TM, Wohlschlegel JA, Geschwind DH. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature. 2009;462:213–217. doi: 10.1038/nature08549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause J, Lalueza-Fox C, Orlando L, Enard W, Green RE, Burbano HA, Hublin JJ, Hänni C, Fortea J, de la Rasilla M, Bertranpetit J, Rosas A, Pääbo S. The derived FOXP2 variant of modern humans was shared with Neandertals. Curr Biol. 2007;17:1908–1912. doi: 10.1016/j.cub.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–523. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- Li S, Weidenfeld J, Morrisey EE. Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol Cell Biol. 2004a;24:809–822. doi: 10.1128/MCB.24.2.809-822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhou D, Lu MM, Morrisey EE. Advanced cardiac morphogenesis does not require heart tube fusion. Science. 2004b;305:1619–1622. doi: 10.1126/science.1098674. [DOI] [PubMed] [Google Scholar]
- Lu MM, Li S, Yang H, Morrisey EE. Foxp4: a novel member of the Foxp subfamily of winged-helix genes co-expressed with Foxp1 and Foxp2 in pulmonary and gut tissues. Mech Dev. 2002;119(Suppl 1):S197–S202. doi: 10.1016/s0925-4773(03)00116-3. [DOI] [PubMed] [Google Scholar]
- Ma W, Maric D, Li BS, Hu Q, Andreadis JD, Grant GM, Liu QY, Shaffer KM, Chang YH, Zhang L, Pancrazio JJ, Pant HC, Stenger DA, Barker JL. Aceytlcholine stimulates cortical precursor cell proliferation in vitro via muscarinic receptor activation and MAP kinase phosphorylation. Eur J Neurosci. 2000;12:1227–1240. doi: 10.1046/j.1460-9568.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cerdeño V, Noctor SC, Kriegstein AR. The role of intermediate progenitor cells in the evolutionary expansion of the cerebral cortex. Cereb Cortex. 2006;16(Suppl 1):i152–i161. doi: 10.1093/cercor/bhk017. [DOI] [PubMed] [Google Scholar]
- Mizutani A, Matsuzaki A, Momoi MY, Fujita E, Tanabe Y, Momoi T. Intracellular distribution of a speech/language disorder associated FOXP2 mutant. Biochem Biophys Res Commun. 2007;353:869–874. doi: 10.1016/j.bbrc.2006.12.130. [DOI] [PubMed] [Google Scholar]
- Nagy K, Sung HK, Zhang P, Laflamme S, Vincent P, Agha-Mohammadi S, Woltjen K, Monetti C, Michael IP, Smith LC, Nagy A. Induced pluripotent stem cell lines derived from equine fibroblasts. Stem Cell Rev. 2011;7:693–702. doi: 10.1007/s12015-011-9239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Kriegstein AR. Contribution of intermediate progenitor cells to cortical histogenesis. Arch Neurol. 2007;64:639–642. doi: 10.1001/archneur.64.5.639. [DOI] [PubMed] [Google Scholar]
- Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontious A, Kowalczyk T, Englund C, Hevner RF. Role of intermediate progenitor cells in cerebral cortex development. Dev Neurosci. 2008;30:24–32. doi: 10.1159/000109848. [DOI] [PubMed] [Google Scholar]
- Reimers-Kipping S, Hevers W, Paabo S, Enard W. Humanized Foxp2 specifically affects cortico-basal ganglia circuits. Neuroscience. 2011;175:75–84. doi: 10.1016/j.neuroscience.2010.11.042. [DOI] [PubMed] [Google Scholar]
- Rousso DL, Gaber ZB, Wellik D, Morrisey EE, Novitch BG. Coordinated actions of the forkhead protein Foxp1 and Hox proteins in the columnar organization of spinal motor neurons. Neuron. 2008;59:226–240. doi: 10.1016/j.neuron.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousso DL, Pearson CA, Gaber ZB, Miquelajauregui A, Li S, Portera-Cailliau C, Morrisey EE, Novitch BG. Foxp-mediated suppression of N-cadherin regulates neuroepithelial character and progenitor maintenance in the CNS. Neuron. 2012;74:314–330. doi: 10.1016/j.neuron.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Mao CA, Hadjantonakis AK, Klein WH, Broccoli V. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron. 2008;60:56–69. doi: 10.1016/j.neuron.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W, Yang H, Zhang L, Lu MM, Morrisey EE. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. 2001;276:27488–27497. doi: 10.1074/jbc.M100636200. [DOI] [PubMed] [Google Scholar]
- Shu W, Cho JY, Jiang Y, Zhang M, Weisz D, Elder GA, Schmeidler J, De Gasperi R, Sosa MA, Rabidou D, Santucci AC, Perl D, Morrisey E, Buxbaum JD. Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. Proc Natl Acad Sci U S A. 2005;102:9643–9648. doi: 10.1073/pnas.0503739102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W, Lu MM, Zhang Y, Tucker PW, Zhou D, Morrisey EE. Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development. 2007;134:1991–2000. doi: 10.1242/dev.02846. [DOI] [PubMed] [Google Scholar]
- Song MR, Ghosh A. FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat Neurosci. 2004;7:229–235. doi: 10.1038/nn1192. [DOI] [PubMed] [Google Scholar]
- Vernes SC, Nicod J, Elahi FM, Coventry JA, Kenny N, Coupe AM, Bird LE, Davies KE, Fisher SE. Functional genetic analysis of mutations implicated in a human speech and language disorder. Hum Mol Genet. 2006;15:3154–3167. doi: 10.1093/hmg/ddl392. [DOI] [PubMed] [Google Scholar]
- Vernes SC, Spiteri E, Nicod J, Groszer M, Taylor JM, Davies KE, Geschwind DH, Fisher SE. High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. Am J Hum Genet. 2007;81:1232–1250. doi: 10.1086/522238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernes SC, Oliver PL, Spiteri E, Lockstone HE, Puliyadi R, Taylor JM, Ho J, Mombereau C, Brewer A, Lowy E, Nicod J, Groszer M, Baban D, Sahgal N, Cazier JB, Ragoussis J, Davies KE, Geschwind DH, Fisher SE. Foxp2 regulates gene networks implicated in neurite outgrowth in the developing brain. PLoS Genet. 2011;7:e1002145. doi: 10.1371/journal.pgen.1002145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RM, Hill AE, Newman AC, Hamilton G, Torrance HS, Anderson SM, Ogawa F, Derizioti P, Nicod J, Vernes SC, Fisher SE, Thomson PA, Porteous DJ, Evans KL. The DISC1 promoter: characterisation and regulation by FOXP2. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds111. [DOI] [PubMed] [Google Scholar]
- Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE, Tucker PW. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131:4477–4487. doi: 10.1242/dev.01287. [DOI] [PubMed] [Google Scholar]
- Wang J, Weaver IC, Gauthier-Fisher A, Wang H, He L, Yeomans J, Wondisford F, Kaplan DR, Miller FD. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell. 2010;18:114–125. doi: 10.1016/j.devcel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Wang J, Gallagher D, DeVito LM, Cancino GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell. 2012;11:23–35. doi: 10.1016/j.stem.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Zhang J, Webb DM, Podlaha O. Accelerated protein evolution and origins of human-specific features: Foxp2 as an example. Genetics. 2002;162:1825–1835. doi: 10.1093/genetics/162.4.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Zhong Q, Minoo P, Li C, Ann DK, Frenkel B, Morrisey EE, Crandall ED, Borok Z. Foxp2 inhibits Nkx2.1-mediated transcription of SP-C via interactions with the Nkx2.1 homeodomain. Am J Respir Cell Mol Biol. 2008;38:750–758. doi: 10.1165/rcmb.2007-0350OC. [DOI] [PMC free article] [PubMed] [Google Scholar]