

Abstract
Heteromeric nAChRs are pentameric cation channels, composed of combinations of two or three α and three or two β subunits, which play key physiological roles in the central and peripheral nervous systems. The prototypical agonist nicotine acts intracellularly to upregulate many nAChR subtypes, a phenomenon that is thought to contribute to the nicotine dependence of cigarette smokers. The α3β4 subtype has recently been genetically linked to nicotine dependence and lung cancer; however, the mode of action of nicotine on this receptor subtype has been incompletely investigated. Here, using transfected mammalian cells as model system, we characterized the response of the human α3β4 receptor subtype to nicotine and the mechanism of action of the drug. Nicotine, when present at 1 mm concentration, elicited a ∼5-fold increase of cell surface α3β4 and showed a more modest upregulatory effect also at concentrations as low as 10 μM. Upregulation was obtained if nicotine was present during, but not after, pentamer assembly and was caused by increased stability and trafficking of receptors assembled in the presence of the drug. Experimental determinations as well as computational studies of subunit stoichiometry showed that nicotine favors assembly of pentamers with (α3)2(β4)3 stoichiometry; these are less prone than (α3)3(β4)2 receptors to proteasomal degradation and, because of the presence in the β subunit of an endoplasmic reticulum export motif, more efficiently transported to the plasma membrane. Our findings uncover a novel mechanism of nicotine-induced α3β4 nAChR upregulation that may be relevant also for other nAChR subtypes.
Introduction
Nicotine is the primary substance responsible for tobacco addiction, which is mediated by its interaction with neuronal nicotinic acetylcholine receptors (nAChRs). These pentameric ligand-gated receptors, composed of α only (homomeric) or of α and β (heteromeric) subunits, play important roles in the physiology of the central and peripheral nervous systems (Gotti et al., 2009).
One of the most remarkable effects of chronic nicotine exposure is the upregulation of brain nAChRs. Because of the well-known addictive and pathogenic properties of nicotine, many efforts have been dedicated to elucidate the mechanisms by which this upregulation is achieved. Most of the investigations have focused on the α4β2 subtype, which is the most widespread heteromeric nAChR subtype of the CNS (Govind et al., 2009). The general conclusion of the studies is that nicotine's upregulatory effect is the result of its interaction with the intracellular and not the surface-exposed receptor (Corringer et al., 2006; Lester et al., 2009). Among suggested mechanisms are nicotine's ability to assist the assembly of pentamers, to increase their export from the endoplasmic reticulum (ER), and to stabilize the receptors, in particular those with the (α4)2(β2)3 stoichiometry (Nelson et al., 2003; López-Hernández et al., 2004; Darsow et al., 2005; Kuryatov et al., 2005; Sallette et al., 2005; Moroni et al., 2006; Srinivasan et al., 2011). Indeed, α4β2 receptors are present in two alternative stoichiometries: 2α/3β and 3α/2β. Although these two forms both present two nicotine orthosteric binding sites at the two α/β interfaces, the fifth accessory subunit may confer subtle differences to the receptor's pharmacological properties.
Although the α4β2 subtype is the most thoroughly investigated nAChR, there has recently been a surge of interest in α3-containing receptors that, not only mediate ACh-induced fast excitatory ganglionic transmission in the autonomic nervous system and adrenal medulla (De Biasi, 2002), but are also present in selected regions of the brain, where they may influence the behavioral effects of nicotine and some manifestations of nicotine withdrawal (for review, see Picciotto and Kenny, 2013). Indeed, a series of linkage analyses, as well as candidate gene and genome-wide association studies, have recently shown that variants in the human α3-α5-β4 nAChR subunit gene cluster on chromosome 15q24–25.1 are linked to the risk of nicotine dependence and smoking-associated diseases, including lung cancer (Berrettini et al., 2008; Spitz et al., 2008; Thorgeirsson et al., 2008). Two of the clustered nAChR genes (α3 and β4) are significantly overexpressed in small-cell lung carcinoma cells, an aggressive form of lung cancer that is causally related to cigarette smoking (Improgo et al., 2010).
The observations summarized above suggest that the α3β4 subtype carries out a prominent role in nAChR-linked pathophysiological phenomena, and prompted us to carry out an in-depth investigation of the response of this receptor subtype to nicotine. Our purpose was, on the one hand, to characterize the response of the α3β4 subtype to nicotine and, on the other hand, to unravel the basis of the intracellular action of the alkaloid, focusing on aspects that have remained unclear for other receptor subtypes.
Materials and Methods
Materials.
(+/−)[3H]-Epibatidine (specific activity = 70.6 Ci/mmol) and [125I]-α-bungarotoxin (αBgtx; specific activity = 220 Ci/mmol) were purchased from PerkinElmer; CC4 and cytisine were synthesized as described previously (Carbonnelle et al., 2003); MG132 was from Calbiochem. EZ-link NHS-SS-biotin and streptavidin-agarose were from Pierce. All other reagents were from Sigma-Aldrich.
Plasmids.
Human α3-pcDNA3 and β4-pcDNA3 were kind gifts of Dr. Sergio Fucile (University of Rome). pN1EGFP was from Clontech; pCINeoHA-CD3δ and mRFP-KDEL were generously provided by A.M. Weissman (National Institutes of Health) and by Erik L. Snapp (Einstein College of Medicine), respectively. FP22 carrying tdTomato as fluorophore was generated from Venus-22 (Ronchi et al., 2008).
Point mutations were generated using the QuikChangeLightning Site-Directed Mutagenesis Kit (Agilent Technologies). The hβ4-L345M export mutant was generated from β4-pcDNA3, by substituting CTC in position 1033 with ATG: forward primer, 5′-GCTGCCTACCTTCATGTTCATGAAGCGCCCTGGCCCCG-3′; reverse primer, 5′-CGGGGCCAGGGCGCTTCATGAACATGAAGGTAGGCAGC-3′ (mutation in italics). The hα3 W180A nicotinic binding site mutant was generated from α3-pcDNA3, by substituting TG in position 532 with GC: forward primer, 5′-CCATGAAGTTCGGTTCCGCGTCCTAC GATAAGGCG-3′; reverse primer, 5′-CGCCTTATCGTAGGACGCGGAACCGAACTT CATGG-3′ (mutation in italics).
Antibodies.
The α3 and β4 pAbs were produced in male rabbits immunized with the human peptides TRPTSNEGNAQKPRPLYGAELSNLNC and GPDSSPARAFPPSKSCVTKPEATATSPP, respectively, affinity purified, and characterized as previously described (Grady et al., 2009). Anti-GFP was from Abcam, anti-HA from Tebu-bio, and anti-ubiquitin from Biomol.
Cell transfection.
HeLa and SH-SY5Y cells were transfected by the Ca3(PO4)2 method or with the Jet-PEI reagent (Polyplus transfection). Equimolar amounts of α3 and β4 plasmids were used in both cases. Cells were analyzed 24 h after transfection or subjected to further treatments as specified in the figure legends.
Biochemical analyses: SDS-PAGE immunoblotting.
SDS-PAGE and blotting were performed by standard procedures. Primary antibodies were revealed either by secondary peroxidase-conjugated IgG, followed by ECL (PerkinElmer), or by infrared-conjugated IgG IRDye 800CW (LI-COR Bioscience). In the first case, films were digitized, and band intensities were determined with ImageJ software (National Institutes of Health) after calibration with the optical density calibration step table (Stouffer Graphics Arts). In the second case, blots were scanned with the Odyssey CLx Infrared Imaging System (LI-COR Biosciences), and band intensities were determined with Image Studio software (LI-COR Biosciences).
Binding assays.
Cells were collected, rinsed twice with PBS (containing 2 mm PMSF), homogenized in the same buffer using a Potter homogenizer, and then centrifuged for 60 min at 30,000 × g. The supernatant was discarded, and the pellet was washed twice by centrifugation before final suspension in PBS + 2 mm PMSF. The affinity constant (Kd) and the apparent displacement constant (Ki) of nicotinic ligands were determined as previously described (Riganti et al., 2005).
Sucrose gradient centrifugation.
Membrane suspensions, prepared as described in binding assays, were supplemented with Triton X-100 to a final concentration of 2% and incubated for 2 h at 4°C. After elimination of insoluble material by centrifugation, the extracts were analyzed on linear sucrose gradients, as described previously (Riganti et al., 2005). Immobilization of the receptor contained in gradient fractions on microwells coated with affinity-purified anti-β4 or α3 antibodies was done as previously described (Riganti et al., 2005). A preparation of Torpedo californica nicotinic AChR, prelabeled with 125I-α-Bgtx was run in parallel.
Surface biotinylation assay.
Cells were incubated for 30 min at 4°C with 0.3 mg/ml EZ-link NHS-SS-biotin; after quenching of free biotin with 50 mm glycine for 10 min at 4°C, the cells were lysed in 1% SDS lysis buffer. Equal amounts of total protein were incubated overnight at room temperature with streptavidin beads to capture biotinylated proteins. After washing in lysis buffer, biotinylated proteins were eluted from streptavidin beads by boiling in Laemmli sample buffer.
Immunofluorescence.
Cells grown on coverslips were fixed with 4% paraformaldehyde, permeabilized with Triton X-100, and processed for immunofluorescence using Alexa-488/568 secondary antibodies (Invitrogen). z-stack and confocal images were taken with Axiovert 200M Microscope (Carl Zeiss) equipped with Spinning Disk confocal system LCI MLS (PerkinElmer) using the 63× PlanApo lens and the 50 mW solid state diode laser 488/561. Wide-field imaging was done with an Axioplan microscope (Carl Zeiss), using the 63× PlanApo lens. Illustrations were prepared with Adobe Photoshop software.
Statistical analysis.
Data from binding studies and Western blotting were expressed as mean ± SEM and analyzed by Student's t test (single comparisons) or one-way ANOVA followed by post hoc Bonferroni test or by Kruskal–Wallis test followed by Dunn's post hoc test (in the case of multiple comparisons). The accepted level of significance was p < 0.05. All statistical analyses were done with Prism software, version 5 (GraphPad).
Homology modeling and molecular dynamics (MD).
The 3D models of the (α3)2(β4)3 and (α3)3(β4)2 nAChR-LBD (ligand-binding domain) were built using as reference structure the crystallographic coordinates of Aplysia-AChBP (ACh-binding protein) cocrystallized with the full agonist (−)-epibatidine (PDB code 2BYQ) (Hansen et al., 2005). The applied procedure was previously optimized by us to build the α7-nAChR-LBD model (Grazioso et al., 2008). Human α3- and β4-nAChR-LBD amino acid sequences were downloaded from the SWISS-PROT database (codes P32297 and P30926, respectively) and aligned onto the template sequence by means of the LALIGN server (Huang and Miller, 1991). After optimization of the sequence alignment, several models for each of the two alternative stoichiometries (α3)2(β4)3 and (α3)3(β4)2 were generated by means of Modeler9v11 software (Sali and Blundell, 1993). MD simulations were then performed on the best models of each of the two alternative pentamers in three different conditions: receptors carrying the mutated α3 subunit (W148A), wild-type receptors with a molecule of nicotine in each binding site (wild/nicotine), and wild-type receptors without ligand (wild/apo). Nicotine adopted the same binding mode of epibatidine, cocrystallized in the template structure. The amino groups were ionized to better simulate physiological conditions, and the atomic electrostatic charges were calculated at the AM1 level of theory. Then, Generalized Amber Force Field (GAFF) (Wang et al., 2004) parameters were assigned to the ligand by the antechamber module implemented in AMBER9 suite (Case et al.,. 2006). The ff03 (Duan et al., 2003) version of the AMBER force field was used for the protein and the counterions, whereas the TIP3P model (Jorgensen et al., 1983) was used to explicitly represent water molecules.
Before starting the MD simulations, a minimization of the entire ensemble was performed by setting a convergence criterion on the gradient of 0.01 kcal mol−1 Å−1. After equilibration of water shells and counterions for 40 ps at 300 K, MD simulations (at least 10 ns) on the receptors were performed in isothermal-isobaric ensemble, and the root mean square deviation of the Cα atoms of the polypeptide chains was monitored versus the simulation time. In the production runs, the ligand-receptor systems were simulated in periodic boundary conditions. The other parameters were obtained following a previously applied computational protocol model (Grazioso et al., 2008). Once the (α3)2(β4)3 and (α3)3(β4)2 nAChRs models attained geometrical stability, the trajectories were further studied by applying the molecular mechanics generalized-born surface area (MM-GBSA) approach. The last 2 ns of MD simulations allowed the collection of the snapshots that were used for the subsequent MM-GBSA calculations.
MM-GBSA approach.
The MM-GBSA approach uses the outcome of MD simulations of ligand–protein or protein–protein complexes to calculate the binding free energy according to the Gibbs equation (Eq. 1). Therefore, the G terms were calculated from the difference between the subunit complex enthalpy and the enthalpy of each subunit alone, averaged from several MD frames (snapshots; Eq. 2) (Xiong et al., 2007).
In Equation 3, ΔHMM is the average sum of the terms calculated by molecular mechanics (MM) methods, Hbond + Hangle + Htorsion + HvdW + HElectrostatics for the subunit complex, and for each subunit 1 or 2 alone. ΔGpolar and ΔGnonpolar (Eq. 4) are the solvation energy terms, where the first one is obtained from the solution of the Generalized Born approximation, and Gnonpolar is the result of scaling the solvent accessible surface area by an appropriate surface tension, derived from the application of the molsurf module implemented in AMBER9 suite. The TΔS term is the entropic contribution that can be calculated from the normal mode (Kottalam and Case, 1990) analysis.
The HMM, Gpolar, and Gnonpolar terms of Equations 3 and 4 were easily calculated by means of the MMPBSA.py module of AmberTools13.0; the polar desolvation terms (Gpolar) were estimated using the Onuffriev-Bashford-Case model (Onufriev et al., 2004) and the modified Born atomic radii. Surface area was computed applying the LCPO model. SURTEN and SURFOFF values were used to estimate nonpolar contributions to the solvation (Gnonpolar) according to the following:
SURFTEN and SUROFF of Equation 5 were fixed to standard values.
To estimate the entropic contribution with the normal mode analysis (Kottalam and Case, 1990) (nmode module of AMBER9 suite), the unsolvated complexes should be minimized for a maximum number of 1 × 105 cycles to give an energy gradient 1 × 10−4 kcal mol−1Å−1. Because of the extensive computational requirement and the large SD of the results, the normal mode analyses were not performed, assuming that this term could be identical for both nAChR stoichiometries. For details on the application of this method, the reader is referred to Grazioso et al. (2008).
Results
Characterization of α3β4 transfected cells
We first studied the distribution and assembly of α3β4 receptors in human epithelial (HeLa) and neuroblastoma (SH-SY5Y) cells cotransfected with equal amounts of cDNAs specifying the two subunits. In HeLa cells, immunolocalization experiments with anti-α3 and anti-β4 (data not shown) antibodies showed extensive colocalization of the subunits with the ER marker RFP-KDEL (Fig. 1Aa,b), indicating that the majority of the receptors fail to be exported from the ER. A different result was obtained with SH-SY5Y cells. These are neuroblastoma cells that express mRNAs for α3, α5, α7, β2, and β4 subunits. Under our conditions, however, these endogenous receptors could not be detected either by immunofluorescence or by Western blot (data not shown). Confocal analysis detected transfected receptor in neurites, in which the concentration of the ER marker was low (Fig. 1Ac,d), but which were enriched in the plasma membrane (PM) protein FP22. In transfected cells, this protein is transported to the PM, although some is also detectable in the ER (Bulbarelli et al., 2002; Ronchi et al., 2008) (Fig. 1Af).
Figure 1.
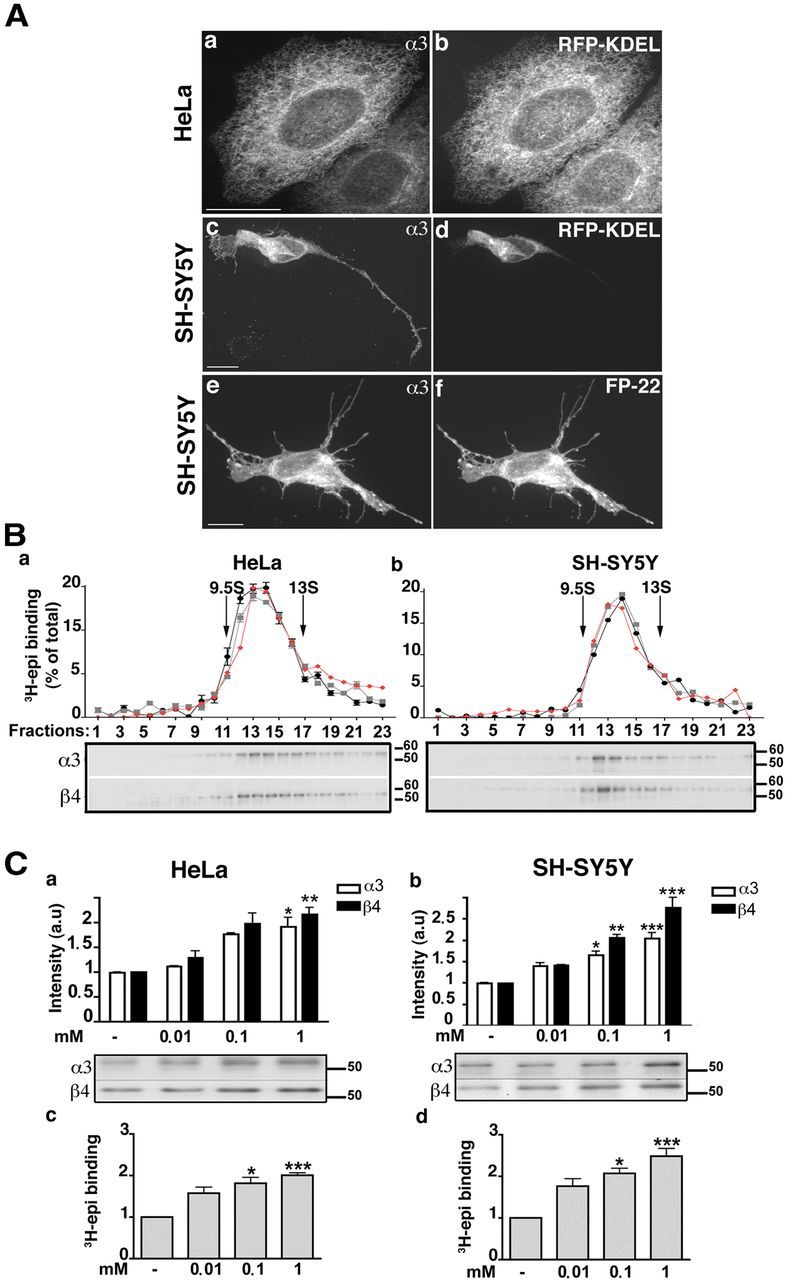
Characterization of α3β4 receptors expressed in HeLa and SH-SY5Y cells in the presence or absence of nicotine. A, Immunofluorescence analysis. HeLa or SH-SY5Y cells cotransfected with α3 and β4 cDNAs and the cDNA of either the ER protein RFP-KDEL or the PM red fluorescent protein FP-22, as indicated, were stained with anti-α3 antibody. Maximum intensity projection of z-stack images are shown. Scale bar, 10 μm. B, Sedimentation analysis of α3β4 receptors expressed in HeLa or SH-SY5Y cells. Aliquots of fractions from sucrose gradients (sedimentation from left to right) were either analyzed for 3H-epibatidine (epi) binding after immobilization on anti-α3- (gray profile) or anti-β4 (black profile) coated microwells, or subjected to Western blot (red profile, and gels below the sedimentation profile). 125I-αBgtx-labeled torpedo AChRs were centrifuged in parallel: single pentameric receptors (9.5S) and receptor dimers (13S) are indicated. C, Chronic treatment with nicotine increases the amount of subunits and of assembled pentamers in HeLa (a,c) and SH-SY5Y (b,d) cells. Transfected cells were incubated for 24 h with the indicated concentrations of nicotine; equal amounts of protein were then analyzed by Western blotting (a,b) or 3H-epibatidine binding (c,d). a, b, the histogram shows the results of blots developed with anti-α3 (white columns) or anti-β4 (black columns) antibodies (n = 5). A representative blot is shown under the quantification panel. c, d, 3H-Epibatidine binding to membrane preparations (n = 6). Immunoblot and binding results are expressed as fold increase over control samples grown in parallel in the absence of nicotine. Asterisks indicate statistical probabilities of treated versus control samples determined by Kruskal–Wallis test followed by Dunn's post hoc test: *p < 0.05; **p < 0.01; ***p < 0.001.
To assess whether the failure of the receptor to reach the PM of HeLa cells is the result of incomplete assembly of the subunits into pentamers, we analyzed the size of the transfected receptors by sucrose gradient sedimentation analysis (Fig. 1Ba). After centrifugation, each fraction was analyzed by Western blotting and by tritiated agonist (3H-epibatidine) binding. For the latter determinations, the receptors were first immobilized on anti-α3 (black circles) or anti-β4 (gray squares) Ab-coated wells. Both the binding of 3H-epibatidine to immunoimmobilized receptors and the Western blot analysis showed that the majority of the detergent solubilized α3β4 receptors sediment as a single species between the Torpedo californica AChR single pentameric receptors (9.5S) and receptor dimers (13S). Importantly, the Western blot (red profile) and agonist-binding profiles (black and gray) showed a close correspondence. Because the agonist binds only to fully assembled pentamers, these results indicate that the vast majority of receptors expressed in the ER is pentameric. Similar results were obtained with transfected SH-SY5Y detergent extracts (Fig. 1Bb). Thus, although receptor subunits correctly assemble both in HeLa and in SH-SY5Y cells, in HeLa cells these pentamers are unable to exit the ER.
Nicotine upregulates α3β4 pentameric receptors
Nicotine binds to various nicotinic receptor subtypes, although with different affinity. In preliminary studies, we performed competition binding experiments to investigate the affinity of nicotine for the human α3β4 transfected subtype and determined an affinity (Ki) of 300 nm. We then treated transfected HeLa and SH-SY5Y cells with nicotine, at concentrations in excess of the estimated Ki, for 24 h. In this and subsequent experiments, nicotine was added to cells 24 h after transfection because concomitant addition with cDNAs leads to reduced transfection efficiency and cell viability.
Western blots showed that nicotine caused a dose-dependent increase in the amount of both α and β subunits in both cell lines (Fig. 1Ca,b). The increase in subunit receptor protein was paralleled by an increase in 3H-epibatidine binding (Fig. 1Cc,d) with a half-maximal concentration (EC50) of 14.8 ± 4 μm in HeLa cells and 16.5 ± 2 μm (EC50 ± SE, n = 3) in SH-SY5Y cells. Treatment with nicotine had no significant effect on the binding affinity for agonist (epibatidine) of the receptor expressed either in HeLa or SH-SY5Y cells (Kd determined on triplicate samples were as follows: HeLa, no nicotine: 0.443 ± 0.09 nm; HeLa + nicotine 1 mm, 24 h: 0.548 ± 0.09 nm; SH-SY5Y, no nicotine: 0.350 ± 0.08 nm; SH-SY5Y + nicotine, 1 mm, 24 h: 0.485 ± 0.09 nm).
Nicotine prevents proteasomal degradation of α3β4 nicotinic receptors if present during their synthesis
Because nicotine has been reported to decrease proteasomal degradation of α7 and α4β2 nAChR subtypes (Rezvani et al., 2007; Govind et al., 2012), we investigated whether this effect underlies the upregulation also of the α3β4 subtype. These and the subsequent experiments were performed only on HeLa cells because transfected SH-SY5Y cells did not tolerate the drug treatments involved.
To analyze the stability of the receptor subunits, transfected cells were treated for 8 h with the protein synthesis inhibitor cycloheximide (CHX), and receptor levels were then assayed by Western blotting, 3H-epibatidine binding, and immunofluorescence. As shown in Figure 2A, Ba, b, ∼50% of the receptors were degraded by the end of the eight CHX chase. The degradation of the receptor was mainly the result of the proteasome, as it was prevented by the proteasome inhibitor MG132 (Fig. 2A,Bc). Although previous work (Christianson and Green, 2004) has demonstrated that unassembled nicotinic subunits are dislocated to the cytosol and degraded by proteasome, these results demonstrate that also ER-retained pentamers are subject to ER-associated degradation (ERAD).
Figure 2.

α3β4 receptors assembled in the presence of nicotine are protected from proteasomal degradation. A, α3β4 receptors are degraded by the proteasome. Transfected HeLa cells were incubated with 50 μg/ml CHX or CHX plus 10 μm MG132 for 8 h. Subunits were analyzed by Western blotting (top and first histogram), and pentamers by 3H-epibatidine binding (bottom histogram): blot, n = 6 (CHX) or 2 (CHX + MG132); 3H-epi binding, n = 5. Values are normalized to sample without drugs. Statistical significance was determined as in Figure 1. B, Nicotine does not stabilize receptors synthesized in its absence. Transfected HeLa cells were fixed (a) or incubated for 8 h with CHX alone (b), with CHX plus MG132 (c), or with CHX plus 1 mm nicotine (d). Wide-field images of cells stained with α3-antibody are shown. e, Transfected cells were either lysed or further incubated for 8 h with CHX alone or CHX plus nicotine and analyzed by Western blot with anti-β4 antibody. C, α3β4 receptors synthesized in the presence of nicotine are less prone to proteasomal degradation. a, Schematic representation of the experiment. Cells were incubated overnight in the presence (2) or absence (1) of 1 mm nicotine and then incubated for an additional 8 h with (4, 6) or without (3, 5) MG132. b, Representative blot of the samples illustrated in a and quantification panel (n = 6). D, The presence of nicotine is not required to stabilize receptors synthesized in its presence. a, Schematic representation of the experiment. Transfected cells were incubated overnight in the absence (1, 2) or in the presence (3, 4) of 1 mm nicotine. Cells were either lysed (1 and 3, respectively) or, after nicotine wash-out, additionally incubated for 8 h with CHX before being lysed (2, 4). b, Lysates were analyzed by Western blot, using anti-α3 antibody followed by IR secondary antibody. Results are normalized to the values before addition of CHX (n = 3). *p < 0.05. **p < 0.01. ns, Not significant.
To assess the effect of nicotine on receptor stability, we first added the drug together with CHX to the 24 h transfected cells and found that, under these conditions, nicotine was without effect (Fig. 2Bd,e). We then investigated whether the alkaloid might stabilize the receptor by acting at an early stage in its biogenesis. To this end, we created a pool of receptors assembled in the presence of nicotine, by treating transfected cells with the drug for 16 h, and then continued the incubation with or without addition of MG132. Cells that were not exposed to nicotine were similarly incubated with or without the proteasomal inhibitor (Fig. 2Ca). As shown in the Western blot analysis and in the corresponding quantification panel (Fig. 2Cb), without nicotine preincubation, the proteasome inhibitor increased the levels of nAChR subunits, in agreement with the data of Figure 2A (Fig. 2Cb, samples 3,4). In contrast, in cells preincubated with nicotine, MG132 had no significant effect, demonstrating a stabilizing effect of nicotine when present during receptor synthesis (Fig. 2Cb, samples 5 and 6).
To confirm that nicotine is effective in stabilizing the receptor if present at early stages of its biogenesis but is ineffective on previously assembled receptors, we compared the degradation, in the absence of nicotine, of receptors that were generated in the presence or absence of the alkaloid (Fig. 2Da). As shown in Figure 2Db, receptor synthesized in the absence of nicotine was reduced to ∼50% of its initial value after 8 h of CHX chase. In contrast, receptors synthesized in the presence of nicotine were more stable during the chase performed in the absence of the alkaloid, maintaining ∼75% of the prechase concentration. Thus, the receptor population generated in the presence of nicotine does not require the subsequent presence of the drug to maintain stability.
It has recently been reported that concentrations of nicotine achieved in the plasma of smokers reduce proteasome activity, thereby increasing total ubiquitinated protein levels (Rezvani et al., 2007). To evaluate whether nicotine affects proteasome activity in HeLa cells, we determined by Western blot the levels of ubiquitinated proteins after incubation with or without nicotine or MG132. Incubation of cells with nicotine was without effect on the levels of ubiquitinated proteins, whereas, as expected, MG132 led to their accumulation (Fig. 3A). We also investigated the effect of nicotine on a specific ERAD substrate, the CD3δ subunit of the T-cell antigen receptor. When expressed in the absence of the other T-cell antigen receptor subunits, CD3δ is ubiquitinated by the ER E3 ubiquitin ligase gp78 (Fang et al., 2001; Ballar et al., 2011) and degraded by the proteasome (Yang et al., 1998). Twenty-four hours after transfection, translation was blocked with CHX, and the cells were incubated with the proteasome inhibitor or with different concentrations of nicotine. As shown and quantified in Figure 3B, degradation of the substrate was prevented only by MG132 (lane 2), whereas nicotine, even at 1 mm, had no effect. These results exclude any direct effect of nicotine on proteasome activity in our cell systems and confirm that its presence during receptor biogenesis has a specific effect on assembled pentamer stability.
Figure 3.
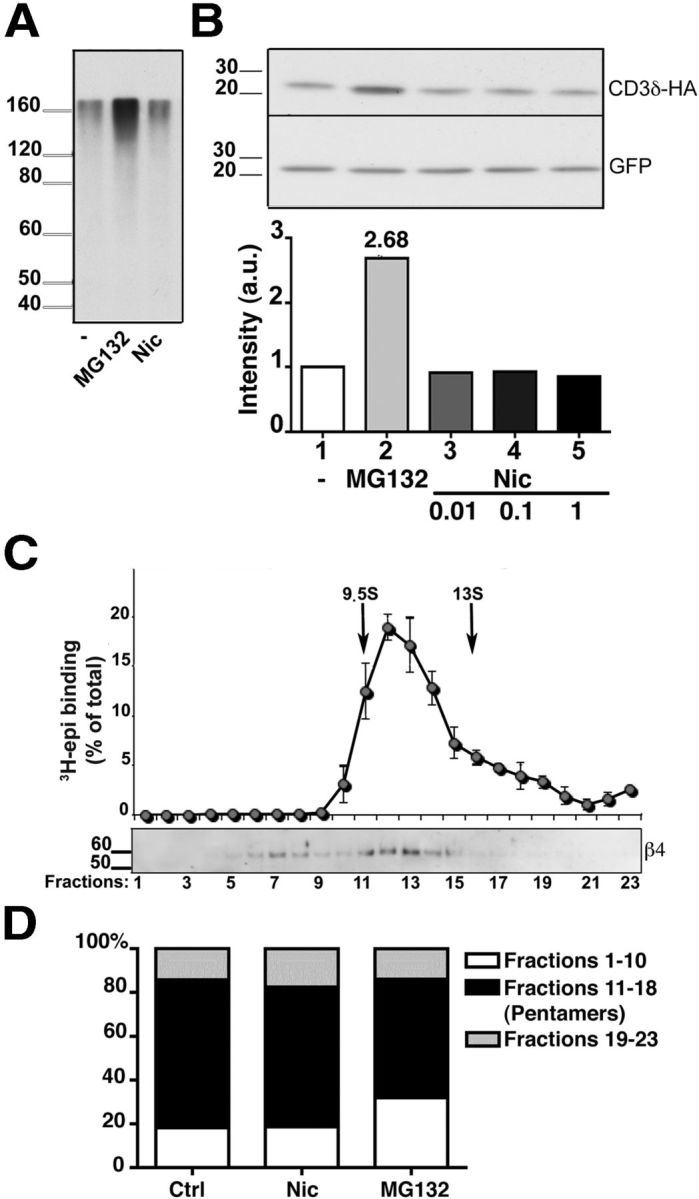
Effect of nicotine on proteasomal activity and on α3β4 receptor assembly. A, B, Nicotine does not impair proteasome activity. A, HeLa cells were incubated with 50 μg/ml of CHX for 3 h in the presence of 10 μm MG132, 1 mm nicotine, or no additional drug, as indicated. The presence of polyubiquitinated proteins was then analyzed by Western blotting with anti-ubiquitin antibodies. B, The degradation of the ERAD substrate CD3δ is not affected by the presence of nicotine. HeLa cells were transfected with HA-tagged CD3δ and GFP cDNAs. After 24 h, cells were incubated for 3 h with 50 μg/ml of CHX either alone, or together with 10 μm MG132, or with the indicated concentration of nicotine. Cell lysates were then analyzed by Western blotting, using anti-HA and anti-GFP antibodies. C, D, α3β4 receptors are assembled efficiently into pentamers. C, Sedimentation analysis of α3β4 receptors from transfected cells treated for 8 h with MG132. Detergent extracts were analyzed as in Figure 1. D, Extent of pentamer assembly in untreated, nicotine-treated (1 mm; 24 h), or MG132-treated (10 μm; 8 h) cells. The histograms represent the percentage of material sedimenting more slowly than pentamers (fractions 1–10), as pentamers (fractions 11–18), and as aggregates (fractions 19–23) detected by Western blot with anti β4 antibodies (n = 4, 3, and 5 for control, nicotine-, and MG132-treated cells, respectively).
Nicotine has at most a modest effect on α3β4 pentamer assembly
A well-known effect of nicotine investigated mainly on the α4β2 subtype is to enhance pentamer assembly. Because unassembled subunits are rapidly degraded by the ERAD process (Christianson and Green, 2004), increased assembly could also have contributed to the α3β4 receptor upregulation that we observed. Sucrose gradient analysis of transfected cells revealed that the material recovered in the more slowly sedimenting fractions, corresponding to unassembled subunits or assembly intermediates, was <20% of the total sample, whether or not the cells were treated with nicotine (Figs. 1 and 3D), confirming the previously reported efficient assembly of this nAChR subtype (Wang et al., 1998; Sallette et al., 2004). To determine the amount of subunits that do not assemble and that are therefore normally degraded by ERAD, we treated cells with MG132. Even under this condition, however, the majority of subunits were assembled in pentamers, although a clear increase in unassembled subunits compared with control cells was observed (Fig. 3C). Nevertheless, the difference between the amounts of unassembled material from MG132-treated cells (31.9% of total expressed receptor) compared with untreated cells (∼18%; Fig. 3D) cannot account by itself for the observed upregulation of α3β4 receptors (2- to 2.5-fold; Fig. 1), which must therefore be mainly attributed to increased stability of the assembled pentamers.
Nicotine increases the proportion of α3β4 receptors at the PM
Up until now, we have described the effect of nicotine on the total cellular levels of α3β4 receptors. To investigate nicotine's effect on receptor surface expression, we devised a protocol in which a burst of receptor synthesis was induced in the presence or absence of the drug. To this aim, we incubated the 24 h transfected cells with CHX for a long enough time to allow degradation of the majority of preexisting receptors (Fig. 4Aa2), obtaining cells with low levels of receptor protein but presumably with high levels of the corresponding mRNAs. We then allowed translation to resume in the presence or absence of nicotine for 22 h (Fig. 4Aa4,3). In agreement with our initial observations (Fig. 1), the receptor was detected only in the ER when synthesis was resumed in the absence of nicotine (Fig. 4Aa3). However, when nicotine was present during α3β4 synthesis, many cells displayed a fluorescent pattern suggestive of partial PM localization (Fig. 4Aa4). As antibodies against the extracellular domain of the receptor suitable for immunofluorescence are not available, we assessed surface expression by colocalization with the cotransfected fluorescent PM protein FP22. A clear colocalization of surface-localized FP22 with the receptor expressed in the presence (Fig. 4Ab4) but not in the absence of nicotine (Fig. 4Ab3) was detected. Surface expression of the receptor was also enhanced at lower concentrations of nicotine (10 and 100 μm), as illustrated in Figure 4C.
Figure 4.
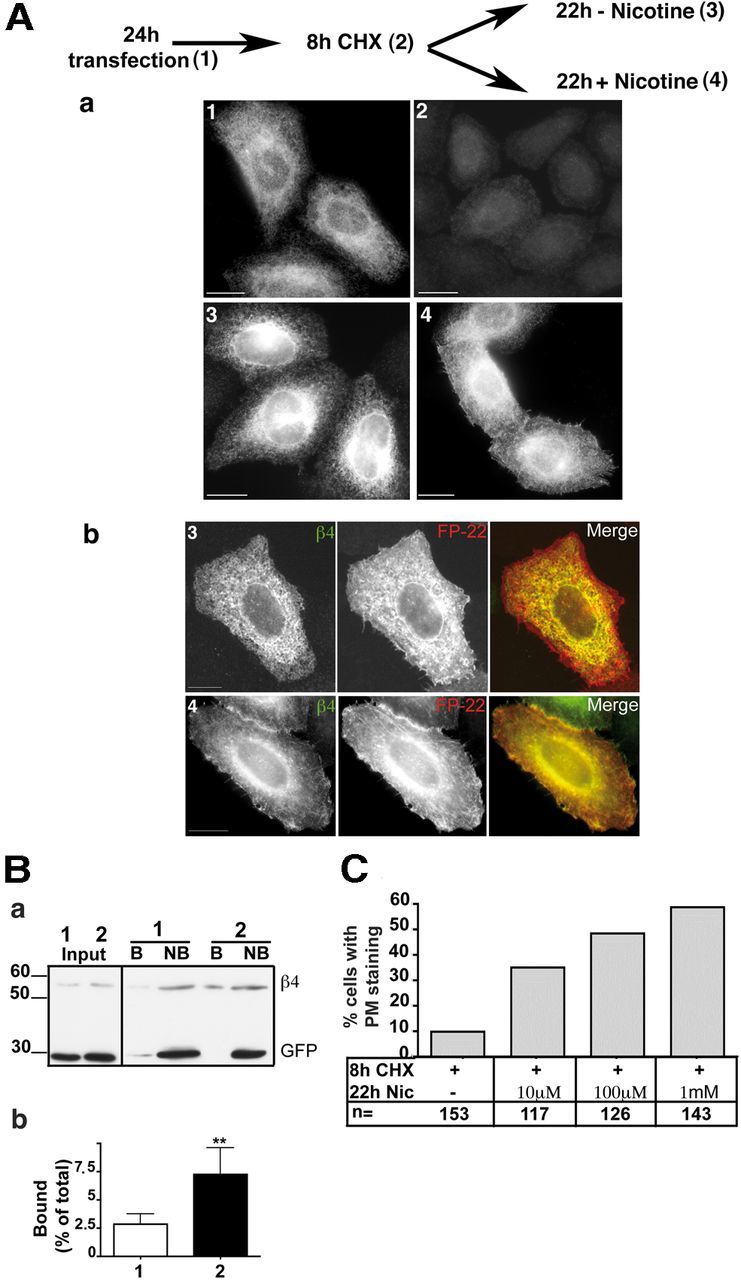
Nicotine facilitates transport of the receptor to the PM. A, Immunofluorescence analysis. a, Transfected HeLa cells were either fixed (1), treated for 8 h with CHX (2), or, after CHX removal, incubated for 22 h in the presence (4) or absence (3) of 1 mm nicotine. Wide-field images of cells stained with anti-β4 antibody are shown. b, HeLa cells, cotransfected with α3, β4, and FP-22 cDNAs, were treated as described for condition 3 and 4, respectively. In cells incubated with nicotine (4), but not in those incubated without the drug (3), colocalization of the receptor with PM-localized FP-22 is visible. Confocal images are shown. Scale bar, 10 μm. B, Surface biotinylation assay. a, HeLa cells, cotransfected with α3, β4, and GFP cDNAs, were treated for 8 h with CHX followed by 22 h without drugs (sample 1) or with 1 mm nicotine (sample 2). Before lysis, cells were surface biotinylated at 4°C. A fraction of the cell lysates was directly analyzed by Western blotting (first two lanes, labeled Input), whereas the remaining part of each sample was incubated with streptavidin beads followed by centrifugation to recover the bound (B) and not-bound (NB) fraction. The input samples correspond to one-fourth of not bound and one-sixteenth of bound samples. b, Quantification of the results (mean ± SEM) (n = 5). **p < 0.01. C, Dose dependency of nicotine-induced α3β4 receptor export to the PM. Transfected cells were incubated for 8 h with CHX and then for 22 h with the indicated concentration of nicotine. Cells were stained with α3 antibody, and the number of cells with PM staining was counted.
We then used cell-surface biotinylation to quantify the proportion of receptors at the cell surface. Cells treated as above were incubated with the cell-impermeant biotinylation reagent EZ-link NHS-SS-biotin, and PM proteins from lysed cells were recovered with streptavidin beads. As shown in Figure 4B, a small fraction of receptor β4 subunits deriving from untreated cells was biotinylated, but this fraction increased almost threefold when nicotine was present during receptor synthesis (4 times the amount of bound vs not-bound sample was loaded on the gel of Fig. 4Ba). The cells of these experiments were cotransfected with GFP as control for the biotinylation assay. As shown in Figure 4Ba, the cotransfected soluble GFP was recovered only in the not-bound fraction, indicating that intracellular proteins were not biotinylated.
The biotinylation experiments indicate that the observed nicotine-induced increase of receptor expression on the cell surface was not only in absolute amount, but also, importantly, in the proportion of surface-exposed receptor. To confirm this conclusion, we tested the effect of proteasomal inhibition, which increases the levels of α3β4 (Figs. 2C and 5A,B) and 3H-epibatidine binding receptors (Fig. 5C), on nAChR PM expression. Although most of these subunits are assembled to form pentamers, (Fig. 3C,D), the resulting receptors were not detected on the PM and remained trapped in the ER (Fig. 5E). In agreement with the immunofluorescence data, the biotinylation assay failed to reveal an increase in the proportion of PM receptors after proteasome inhibition (Fig. 5D). Thus, in addition to enhancing receptor stability, nicotine also facilitates receptor export from the ER.
Figure 5.

Inhibition of the proteasome increases the number of α3β4 receptors but does not promote their exit from the ER. A, Transfected HeLa cells were incubated for 8 h in the presence or absence of 10 μm MG132. The cells were then fixed, permeabilized, and immunostained with anti-α3 antibody. Wide-field images are shown. B, Lysates of cells treated as in A were analyzed by Western blotting. The histogram shows the quantification of three experiments (mean ± SEM) normalized to the amount of β4 subunit detected in the absence of MG132. **p < 0.01. C, 3H-Epibatidine binding to membrane preparations from cells treated as described for A. B, C, Statistical probabilities were determined by unpaired t test. *p < 0.05. D, Transfected HeLa cells, incubated with (sample 2) or without (sample 1) 10 μm MG132, were surface biotinylated and analyzed as described in Figure 4 legend. The input and the not bound samples are one-fifteenth of bound samples. The percentage of surface receptor is indicated below the lanes. E, HeLa cells, cotransfected with α3, β4, and FP-22 cDNAs, treated with MG132 as in A–D, were fixed, permeabilized, and immunostained with the anti-α3 IgG followed by secondary Alexa-488-conjugated secondary antibodies. Maximum intensity projections of z-stacks are shown. Scale bar, 10 μm. Inset shows a 1.3× enlargement.
An ER export motif present in the β4 subunit is required for surface expression of α3β4 receptors in nicotine-treated HeLa cells
A recent paper reported the presence of an export motif (LXM) (Mancias and Goldberg, 2008) in the intracellular loop between the transmembrane domains M3 and M4 of murine α4 and β4 subunits which, by interacting with the COPII component Sec24, increased the recruitment of α4-containing receptors to ERES (Srinivasan et al., 2011). To investigate whether this export motif, present also in the human β4 subunit but absent in the α3 subunit, plays a role in α3β4 receptor trafficking, we mutated the Leu in position 345 to Met (Fig. 6A). The cells were then transfected with the mutated β4 subunit (*) together with the wild-type α3 subunit, subjected to the same CHX-nicotine protocol illustrated in Figure 4A, and finally analyzed by immunofluorescence. As reported in Figure 6C, deletion of the export motif led to the almost complete retention of α3β4 receptors in the ER in the absence as well as in the presence of nicotine. To exclude a possible effect of the mutation on the pharmacological properties of the receptors, we performed saturation binding experiments on WT and mutated receptors under control conditions and after chronic nicotine treatment and found that 3H-epibatidine had the same affinity for the two receptors and that both were equally upregulated by nicotine (Fig. 6B). Thus, although the mutant receptor was still responsive to the stabilizing effect of nicotine, it was crucially dependent on the presence of the export motif for nicotine-enhanced export.
Figure 6.

The LFM export motif is necessary for α3β4 receptor exit from the ER. A, Sequence of intracellular loop of β4 subunit. The export motif (bold) was mutated in β4*, as indicated. B, Unaltered epibatidine binding and nicotine-induced upregulation of α3β4* receptors. The curves illustrate a representative experiment in which ligand concentrations were tested in triplicate. Kd values of 0.443 ± 0.09 and 0.370 ± 0.07 nm for WT and α3β4*, respectively, were obtained by means of nonlinear regression analysis of 3 or 4 independent experiments. C, Nicotine fails to enhance PM localization of export motif-deleted receptor. Transfected cells were incubated for 8 h with CHX and then for 22 h with or without nicotine. Cells were then analyzed by immunofluorescence with anti-β4 antibody.
Membrane-permeant competitive nicotinic ligands upregulate α3β4 receptors and promote their arrival to the PM
We next asked whether the observed effects of nicotine on α3β4 receptors were specific to this agonist or whether other membrane-permeant nicotinic ligands could affect receptor stability and trafficking. We found (Fig. 7A) that a 24 h treatment of transfected HeLa cells with the full agonist cytisine (0.1 and 1 mm) or the very partial agonist CC4 (1 mm) upregulates the α3β4 receptors to a similar extent as nicotine, regardless of the pharmacological properties of the two ligands (Sala et al., 2013). Despite its different pharmacological properties, CC4, like nicotine, was capable of enhancing transport of the α3β4 receptor to the PM (Fig. 7B).
Figure 7.

Membrane-permeant competitive ligands upregulate and enhance trafficking of α3β4 receptors. A, Upregulation of nicotinic receptors in HeLa cells. Transfected cells were incubated for 24 h without drugs, with cytisine, or with CC4, at the indicated concentrations. Samples were analyzed by Western blotting, using anti-α3, and anti-β4 antibodies (top), or by 3H-epi binding. The results are expressed as the fold increase over untreated sample (n = 3). B, CC4 favors the arrival of the α3β4 receptor to the PM. HeLa cells, cotransfected with α3, β4, and FP22 cDNAs, were incubated for 8 h with CHX and then for 22 h in the presence of 1 mm CC4. Wide-field images of cells stained with the anti-β4 antibody are shown. Scale bar, 10 μm. C, Mecamylamine does not upregulate α3β4 receptors. Transfected HeLa cells were further incubated for 24 h without drugs, with 1 mm nicotine, or with 1 mm mecamylamine. Cell lysates were analyzed by Western blotting (top). Bottom, 3H-epi binding to membrane preparations obtained from similarly treated cells. Statistical significance was determined by Kruskal–Wallis test followed by Dunn's post hoc test (n = 4). *p < 0.05. **p < 0.01. ns, Not significant.
Both cytisine and CC4 are competitive ligands that bind to the same site (interface between the α and β subunits) as nicotine; to evaluate whether binding at this specific site is necessary for the upregulatory effect of nicotinic ligands, we tested the effect of mecamylamine, a membrane-permeant noncompetitive antagonist that binds to nicotinic receptors in the lumen of the channel (Papke et al., 2001). As shown in Figure 7C, chronic treatment with this noncompetitive ligand failed to upregulate α3β4 receptors in the transfected cells.
Competitive membrane permeant ligands induce (α3)2(β4)3 stoichiometry
In heteromeric nAChR receptors, two α and two β subunits generate two binding sites for ACh and nicotine at the two α/β subunit interfaces; the fifth, accessory, subunit, which may be either α or β, has no influence on the number of agonist binding sites. Because nicotine must be present during receptor biogenesis to exert its effects, we hypothesized that it might promote the assembly of pentamers with a stoichiometry that could affect both receptor stability and export. We therefore determined the β4 to α3 stoichiometry in untreated and nicotine-treated cells. We first performed preliminary experiments with HeLa cells transfected with an equal ratio of α3 and β4 cDNAs. Western blotting was performed with infrared-conjugated secondary antibodies, and the optimal concentration of lysate proteins to obtain linearity was determined. We then compared the ratio of the β4 to α3 signal in control and nicotine-treated cells. As shown in Figure 8A, after 24 h of treatment with nicotine, the β to α ratio was increased (1.39-fold), indicating that nicotine promoted the formation of pentamers with (α3)2(β4)3 stoichiometry. To be noted, the expected enrichment ratio of a population of receptors with uniform (α3)2(β4)3 stoichiometry versus a population of receptors with exclusive (α3)3(β4)2 stoichiometry is 2.25. We obtained a similar result with cells transfected with the export sequence mutated β4* subunit (Fig. 8B). In contrast, the stoichiometry of receptors generated in the presence of MG132 was unchanged (Fig. 8C).
Figure 8.

Nicotine and CC4 promote the formation of pentameric receptors with two α3 and three β4 subunits. A–C, HeLa cells were transfected with cDNAs coding for α3 and for either the wild-type β4 (A,C) or for the export motif-deleted β4* subunit (B, see Fig. 6A). The transfected cells were incubated for 24 h either with or without 1 mm nicotine (A,B) or for 8 h with or without 10 μM MG132 (C). D, E, HeLa cells, transfected with the wild-type α3 and β4 subunits, were incubated for 8 h with CHX and then for 22 h with or without 1 mm nicotine (D) or 1 mm CC4 (E). For all panels, total cell lysates were analyzed by Western blotting; samples were run in duplicate on the same gel and probed separately with anti-α3 and β4 antibodies followed by incubation with an IR secondary antibody. The mean β4/α3 signal ratio, normalized to the ratio obtained in the absence of treatment, is shown. Statistical significance was evaluated by paired t test: n = 4 for A, B, D, E and = 3 for C. ***p < 0.001.
We then investigated whether the partial agonist CC4, which, like the agonist nicotine, upregulates the receptor and enhances its trafficking, similarly affects subunit stoichiometry. As shown in Figure 8E, receptors assembled in the presence of CC4 had a similarly increased proportion of (α3)2(β4)3 pentamers as found in cells incubated with nicotine with the same experimental protocol (Fig. 8D). Thus, occupancy of the agonist binding site at the α3β4 interface, independently from the pharmacological effect that is elicited, appears to be sufficient to promote (α3)2(β4)3 stoichiometry, with the consequent effects on receptor stability and export from the ER.
Mutation of an α subunit residue required for nicotine binding induces the (α3)2(β4)3 stoichiometry and promotes receptor transport to the PM
To further investigate the basis of ligand-induced stoichiometry modulation, we analyzed the effect of mutating a residue known to be essential for agonist binding (Trp 180 of the α3 subunit) (Xiu et al., 2009). As expected, the mutant receptor (α* W180A) expressed in HeLa cells was incapable of binding epibatidine (Fig. 9B), although it was expressed at similar levels as the wild-type (Fig. 9A) and was correctly assembled into pentamers (Fig. 9C). We then assessed the effect of nicotine on the stability and surface expression of the receptor. As expected, nicotine had no effect on receptor stability (Fig. 9D). However, very surprisingly, we observed a constitutive PM staining of the mutant receptor at a level comparable with that observed when the wild-type receptor was synthesized in the presence of 1 mm nicotine (Fig. 9E; compare with Fig. 4C).
Figure 9.
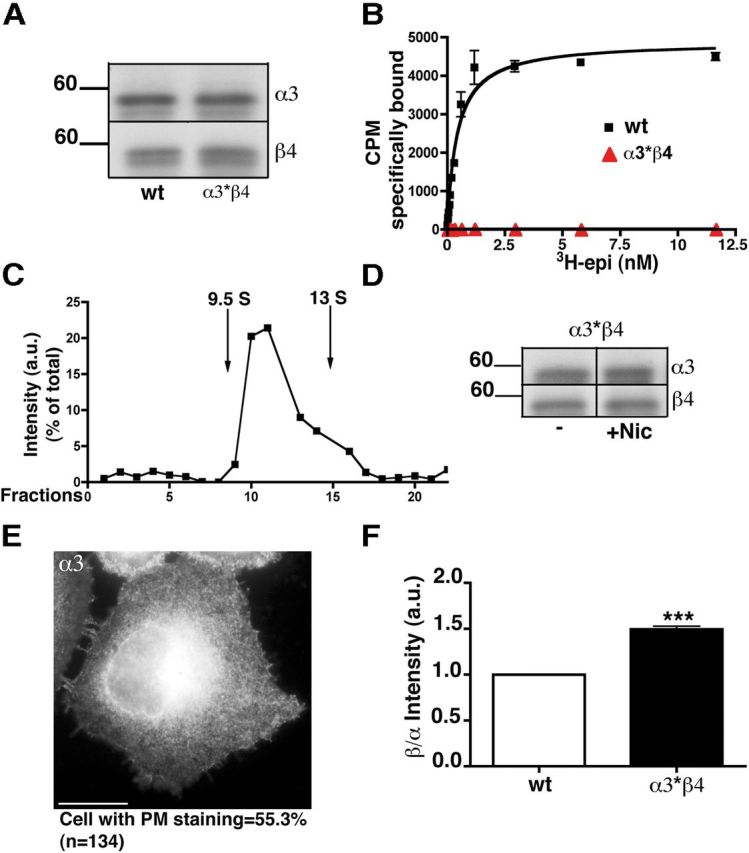
Mutation of an α3 residue essential for agonist binding alters subunit stoichiometry and enhances receptor transport to the PM. A, Equal amounts of protein from cells transfected with wild-type or mutated (α3* containing the W180A mutation) receptors were analyzed with anti-α3 and anti-β4 antibodies. B, Saturation curve of specific 3H-epibatidine binding to membrane preparations from HeLa cells transfected with wild-type or mutant receptors. The binding curves are from a representative experiment, which was repeated three times with superimposable results. C, Sucrose gradient analysis of detergent extracts of HeLa cells transfected with the α3*β4 receptor. Fractions were analyzed by Western blotting with anti-α3 antibodies. D, Lack of effect of nicotine on α3*β4 receptor stability. Transfected cells, incubated for 24 h with or without 1 mm nicotine, were analyzed by Western blotting. E, α3*β4 receptor is detected on the PM. Wide-field image of cells stained with α3 antibody is shown. The percentage of cells showing PM staining is reported below the image. F, Comparison of β4/α3 stoichiometry in wild-type and α3*-containing receptors. Lysates prepared from HeLa cells transfected either with wild-type or α3*-containing receptors were analyzed as in Figure 8. ***p < 0.001.
To evaluate a possible correlation between subunit stoichiometry and increased surface expression of the mutant receptor, we compared the β to α ratio of the mutant and wild-type receptors. In accordance with a role of subunit stoichiometry in promoting export, we found that the (α3)2(β4)3 stoichiometry was favored in the mutant receptors (Fig. 9F). Thus, as in the case of nicotine-treatment, a change in subunit stoichiometry of the mutant receptor correlates with increased transport to the cell surface.
Computational investigations predict that (α3)2(β4)3 stoichiometry is energy-favored in the presence either of nicotine or of mutated α subunit
To elucidate the mechanism underlying the change in stoichiometry induced by both nicotine and the mutated α subunit, we performed computational investigations using MD simulations to estimate the intersubunit binding energy.
The 3D homology models of (α3)2(β4)3 and (α3)3(β4)2 pentamers were produced from the crystallographic coordinates of Aplysia-AChBP (ACh-binding protein) (Hansen et al., 2005) as described in detail in the experimental section. Both pentamers contain two conventional α3(+)/β4(−) orthosteric binding site interfaces and two dissimilar interfaces resulting from the presence of a different fifth subunit (β4 or α3). We refer to these as interfaces I and II in the (α3)2(β4)3 model and interfaces III and IV in the (α3)3(β4)2 model, respectively (Fig. 10A). MD simulations were performed on both the (α3)2(β4)3 and (α3)3(β4)2 nAChR models. The contact strengths between different subunits were estimated by means of the MM-GBSA technique that estimates the binding energy of two different interacting molecules (Gilson and Zhou, 2007). The calculations yielded the ΔEbind, which includes the strength of the internal (including bond, angle, and torsional angle energies), electrostatic, and van der Waals energies as well as the solvation free energy shift (see Materials and Methods). Three different conformational states were simulated: wild-type receptor without ligand (wild/apo), wild-type receptor with a molecule of nicotine in each binding site (wild/nicotine), and receptor carrying the mutated α3 subunit (W148A). As shown Figure 10B, the presence of nicotine or of the α3 mutant strongly increased the binding energy for interface I, although it had much smaller effects on the other interfaces analyzed.
Figure 10.

MD simulations on homology models of α3β4 receptor. A, Schematic representation of α3β4 pentamers with the two stoichiometries: (α3)2(β4)3 on the left and (α3)3(β4)2 on the right. The two conventional α3(+)/β4(−) orthosteric binding sites are depicted. The fifth subunit can be either β4 (on the left, in red) or α3 (on the right in blue), determining two different interfaces in each stoichiometry: interfaces I and II in the (α3)2(β4)3 model and interfaces III and IV in (α3)3(β4)2. B, Predicted binding energy (kcal/mol) by the MM-GBSA method. The results obtained for (α3)2(β4)3 (interfaces I and II) and (α3)3(β4)2 (interfaces III and IV) models are highlighted in red and blue, respectively. Indicated values were obtained from the difference between the subunit complex energy and the sum of energy of each subunit alone. MD simulations were performed for 10 ns. Values from 1 frame every 5 of the last 200 frames (40 frames in total) corresponding to the last 2 ns of the simulation (at which time equilibrium had been reached in all the simulations) were averaged to obtain the indicated values.
These results suggest that nicotine, by binding in its orthosteric site in α/β assembly intermediates, favors the recruitment of β4 as accessory subunit; the results also explain the increased (α3)2(β4)3 stoichiometry in receptors carrying the α3 W148A mutation.
Discussion
In this study, we investigated the previously poorly characterized effects of nicotine on the α3β4 nAChR subtype. Because this receptor assembles efficiently into pentamers also in the absence of nicotine (Wang et al., 1998; Sallette et al., 2004; current study), the work presented here has allowed us to highlight a second effect of the drug on receptor assembly: induction of the assembly of pentamers consisting of two α and three β subunits [(α3)2(β4)3]. We think that our results, which show that enhanced stability and trafficking of the receptor are secondary to this altered β4 to α3 ratio, are of general relevance to the mechanisms of nicotine-induced nAChR upregulation.
Nicotine induces assembly of receptors with (α3)2(β4)3 stoichiometry
It is well known that nicotine, by accessing the ER lumen, promotes the generation of α3β2 and α4β2 pentamers, which assemble slowly in the absence of the drug (Wang et al., 1998; Sallette et al., 2004; Kuryatov et al., 2005; Sallette et al., 2005). Instead, as reported by others (Wang et al., 1998; Sallette et al., 2004) and confirmed here, α3β4 receptors assemble efficiently also in the absence of nicotine. Sucrose gradient analysis of untreated cells revealed a low proportion of assembly intermediates, even when the proteasome was inhibited. Thus, an improvement over a situation in which pentamer formation is already efficient could not explain the more than twofold upregulation of total α3β4 receptors and the threefold increase in the proportion exported to the cell surface. We therefore concluded that nicotine was acting through a mechanism additional to enhancing pentamer formation.
One possibility that we considered was that nicotine could be binding to preassembled pentamers, and that the nicotine-receptor complex was both more stable and more efficiently exported from the ER; however, our results ruled out this hypothesis, as nicotine had no stabilizing effect on preassembled receptors, nor did it increase their trafficking to the cell surface. Thus, nicotine's effect is exerted only when it is present during receptor synthesis/assembly.
We thus hypothesized that the drug, present during the assembly process, might promote assembly of a particular subunit stoichiometry, and that this might be the basis for its observed biological effects. Both the α4β2 (Nelson et al., 2003) and the α3β4 (Krashia et al., 2010) receptor exist in vivo as two populations with different subunit stoichiometry (three α/two β or two α/three β). These two receptor types have the same number of agonist binding sites (two) per pentamer but have distinct pharmacological and functional properties (Moroni et al., 2006; Krashia et al., 2010; Mazzaferro et al., 2011). The two stoichiometries are present also in cell lines expressing α4β2 receptors, and the proportion of (α4)2(β2)3 receptors can be increased by conditions that promote the availability of the β2 subunit or by chronic nicotine treatment (Nelson et al., 2003).
Our quantitative determinations revealed that cells treated with nicotine showed a 30–40% increase in the β to α ratio of receptor subunits, indicating that the agonist promotes assembly of receptors with two α and three β subunits. Although the observation that nicotine alters the subunit stoichiometry in nAChRs is not new, the mechanism of this alteration was previously not clear, and it was generally thought that nicotine selectively stabilizes receptors with the (α4)2(β2)3 stoichiometry (Srinivasan et al., 2011), leading to the enrichment of this pentamer species in the receptor population. In contrast, our results, which show that nicotine is ineffective on preassembled receptors, indicate that the alkaloid acts directly during the assembly process to promote generation of the (α3)2(β4)3 stoichiometry.
This mechanism of action of nicotine is also strongly supported by our MD simulations, which are compatible with the hypothesis that nicotine, by binding in the orthosteric site at α/β interfaces of assembly intermediates, favors the recruitment of β4 as accessory fifth subunit.
Our work also demonstrates that other permeable competitive ligands, independently from their pharmacological effects, favor the (α3)2(β4)3 stoichiometry, whereas an antagonist that binds within the channel is without effect on receptor stability and trafficking. Thus, occupation of the ACh binding site, located at the α/β interface, appears to be sufficient to promote the (α3)2(β4)3 stoichiometry.
Effects of altered subunit stoichiometry on receptor trafficking and stability
Regardless of the mechanism underlying their generation, (α3)2(β4)3 receptors appear to be endowed with two independent properties, which together result in their approximately fivefold upregulation at the cell surface: (1) increased trafficking to the PM and (2) enhanced stability.
1. Our conclusion that nicotine enhances α3β4 trafficking independently from its effect on receptor stability rests on two observations: first, treatment with nicotine resulted in an increase (determined by surface biotinylation) of surface receptors not only in absolute amount but also in proportion to total expressed receptor; second, this proportion was not increased by inhibition of the proteasome, which did, however, increase the total amount of receptor. Interestingly, a mutant (α3W180A), incapable of binding nicotine, showed increased surface expression compared with the wild-type, in parallel with an increased β to α ratio, indicating that the presence of nicotine is not itself required for receptor upregulation at the plasma membrane; rather, competitive ligands appear to act indirectly by inducing a conformational change in the subunits during the early stages of pentamer assembly.
Our work also points to the mechanism by which (α3)2(β4)3 stoichiometry favors export from the ER. Based on the work of Srinivasan et al. (2011), which identified an export motif (LXM) in the intracellular loop of murine α4 and β4 subunits and of human β2, we deleted this motif from human β4 and found that cell surface expression of the mutant receptor was nearly completely abolished both in the presence and absence of nicotine, although total receptor levels were upregulated by the drug to the same extent as the wild-type. This result again disjoins the two phenomena of receptor stabilization and receptor export from the ER and demonstrates that the latter is crucially dependent on the LXM motif. Because the α3 subunit does not have this motif, a receptor with (α3)2(β4)3 stoichiometry, by being enriched 1.5-fold in the export motif compared with those with the three α-two β composition, would be recruited more efficiently to ERES.
2. Whereas the mechanism behind increased export of pentamers with (α3)2(β4)3 stoichiometry is straightforward, it is more difficult at present to understand why pentamers of this composition escape ERAD. Our results clearly rule out a direct effect of nicotine on the proteasome (as reported by Rezvani et al., 2007) and also a stabilizing effect on assembled receptors. We can only imagine that, as in the case of α4β2 receptors, α3β4 receptors with different stoichiometries may behave differently with respect to post-translational modifications, such as phosphorylation (Exley et al., 2006; Wecker et al., 2010) or glycosylation; such differences could affect the interactions of the receptor with the ERAD machinery (Jeanclos et al., 2001; Ficklin et al., 2005; Rezvani et al., 2009).
Pathophysiological implications
Because predicted nicotine concentrations in smokers' brain are well below its EC50 for α3β4 upregulation (Matta et al., 2007), it is generally thought that nicotine-induced upregulation of this receptor subtype does not play a role in the neuroadaptations underlying somatic withdrawal symptoms during nicotine abstinence (Davila-Garcia et al., 2003; Nguyen et al., 2003; De Biasi and Dani, 2011). In agreement, studies on animal models of chronic nicotine exposure have failed to reveal α3β4 upregulation (Davila-Garcia et al., 2003; Nguyen et al., 2003). Nevertheless, in neurons, such as those of the habenula–interpeduncular pathway, expressing many different nAChRs (Grady et al., 2009), subtle changes in specific subtype levels could be masked. Thus, it is possible that a minor increase of this receptor at the cell surface does occur in some brain regions of smokers. Future quantitative, subtype-specific studies will be required to definitely settle this important question.
In addition to a possible role of nicotine-induced α3β4 upregulation in the brain, it is worthwhile to consider extraneuronal tissues, where nicotine may reach much higher concentrations. α3 and β4 transcripts are present in bronchiolar epithelial cells as well as in lung cell lines, and there is increasing evidence that these receptors play crucial roles in signal transduction underlying tumor initiation or growth (Egleton et al., 2008; Improgo et al., 2011). Furthermore, proliferative and prosurvival effects of nicotine in small-cell lung carcinoma have been documented (Schuller, 2009; Improgo et al., 2011). Because nicotine concentration in the sputum of human subjects who have smoked one cigarette (Clunes et al., 2008) is in the range of the EC50 observed by us for α3β4 upregulation, it is tempting to speculate that receptor upregulation may enhance the proliferative and prosurvival effects of nicotine on tumor cells.
In conclusion, we think that the mechanism of action of nicotine on α3β4 receptors revealed here may be relevant to the response to nicotine also of other receptor subtypes. Nicotine-enhanced recruitment of β subunits into pentamers could explain several functional data of altered receptor stability/stoichiometry of the α4β2 subtype after nicotine treatment (Srinivasan et al., 2011; Govind et al., 2012). Future studies will be aimed at unraveling the relevance of the mechanism described here to all nAChRs.
Footnotes
This work was supported by the Union Seventh Framework Programme (Grant HEALTH-F2-2008-202088 “NeuroCypres” project), the Italian PRIN 2009R7WCZS, the European Union Grant Eranet, and the CNR Research Project on Aging as well as CINECA and the Regione Lombardia Award under the LISA initiative (for the availability of high performance computing resources and support). We thank Dr Milena Moretti for help in sucrose gradient experiments; Dr. Matteo Fossati for the tdTomato 22 (FP22) construct; and the Monzino Foundation (Milan, Italy) for its generous gift from Spinning Disk confocal system LCI MLS (Perkin Elmer) and the Fondazione Giancarla Vollaro, Milano.
The authors declare no competing financial interests.
References
- Ballar P, Pabuccuoglu A, Kose FA. Different p97/VCP complexes function in retrotranslocation step of mammalian ER-associated degradation (ERAD) Int J Biochem Cell Biol. 2011;43:613–621. doi: 10.1016/j.biocel.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, Waterworth D, Muglia P, Mooser V. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry. 2008;13:368–373. doi: 10.1038/sj.mp.4002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulbarelli A, Sprocati T, Barberi M, Pedrazzini E, Borgese N. Trafficking of tail-anchored proteins: transport from the endoplasmic reticulum to the plasma membrane and sorting between surface domains in polarised epithelial cells. J Cell Sci. 2002;115:1689–1702. doi: 10.1242/jcs.115.8.1689. [DOI] [PubMed] [Google Scholar]
- Carbonnelle E, Sparatore F, Canu-Boido C, Salvagno C, Baldani-Guerra B, Terstappen G, Zwart R, Vijverberg H, Clementi F, Gotti C. Nitrogen substitution modifies the activity of cytisine on neuronal nicotinic receptor subtypes. Eur J Pharmacol. 2003;471:85–96. doi: 10.1016/S0014-2999(03)01817-X. [DOI] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, et al. AMBER 9. University of California: San Francisco; 2006. [Google Scholar]
- Christianson JC, Green WN. Regulation of nicotinic receptor expression by the ubiquitin-proteasome system. EMBO J. 2004;23:4156–4165. doi: 10.1038/sj.emboj.7600436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clunes LA, Bridges A, Alexis N, Tarran R. In vivo versus in vitro airway surface liquid nicotine levels following cigarette smoke exposure. J Anal Toxicol. 2008;32:201–207. doi: 10.1093/jat/32.3.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corringer PJ, Sallette J, Changeux JP. Nicotine enhances intracellular nicotinic receptor maturation: a novel mechanism of neural plasticity? J Physiol Paris. 2006;99:162–171. doi: 10.1016/j.jphysparis.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Darsow T, Booker TK, Piña-Crespo JC, Heinemann SF. Exocytic trafficking is required for nicotine-induced up-regulation of α 4 β 2 nicotinic acetylcholine receptors. J Biol Chem. 2005;280:18311–18320. doi: 10.1074/jbc.M501157200. [DOI] [PubMed] [Google Scholar]
- Dávila-García MI, Musachio JL, Kellar KJ. Chronic nicotine administration does not increase nicotinic receptors labeled by [125I]epibatidine in adrenal gland, superior cervical ganglia, pineal or retina. J Neurochem. 2003;85:1237–1246. doi: 10.1046/j.1471-4159.2003.01774.x. [DOI] [PubMed] [Google Scholar]
- De Biasi M. Nicotinic mechanisms in the autonomic control of organ systems. J Neurobiol. 2002;53:568–579. doi: 10.1002/neu.10145. [DOI] [PubMed] [Google Scholar]
- De Biasi M, Dani JA. Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci. 2011;34:105–130. doi: 10.1146/annurev-neuro-061010-113734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Egleton RD, Brown KC, Dasgupta P. Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol Sci. 2008;29:151–158. doi: 10.1016/j.tips.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Exley R, Moroni M, Sasdelli F, Houlihan LM, Lukas RJ, Sher E, Zwart R, Bermudez I. Chaperone protein 14-3-3 and protein kinase A increase the relative abundance of low agonist sensitivity human α 4 β 2 nicotinic acetylcholine receptors in Xenopus oocytes. J Neurochem. 2006;98:876–885. doi: 10.1111/j.1471-4159.2006.03915.x. [DOI] [PubMed] [Google Scholar]
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficklin MB, Zhao S, Feng G. Ubiquilin-1 regulates nicotine-induced up-regulation of neuronal nicotinic acetylcholine receptors. J Biol Chem. 2005;280:34088–34095. doi: 10.1074/jbc.M506781200. [DOI] [PubMed] [Google Scholar]
- Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Govind AP, Vezina P, Green WN. Nicotine-induced upregulation of nicotinic receptors: underlying mechanisms and relevance to nicotine addiction. Biochem Pharmacol. 2009;78:756–765. doi: 10.1016/j.bcp.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind AP, Walsh H, Green WN. Nicotine-induced upregulation of native neuronal nicotinic receptors is caused by multiple mechanisms. J Neurosci. 2012;32:2227–2238. doi: 10.1523/JNEUROSCI.5438-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L, Clementi F, Gotti C. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J Neurosci. 2009;29:2272–2282. doi: 10.1523/JNEUROSCI.5121-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grazioso G, Cavalli A, De Amici M, Recanatini M, De Micheli C. Alpha7 nicotinic acetylcholine receptor agonists: prediction of their binding affinity through a molecular mechanics Poisson-Boltzmann surface area approach. J Comput Chem. 2008;29:2593–2602. doi: 10.1002/jcc.21019. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Miller W. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12:337–357. doi: 10.1016/0196-8858(91)90017-D. [DOI] [Google Scholar]
- Improgo MR, Schlichting NA, Cortes RY, Zhao-Shea R, Tapper AR, Gardner PD. ASCL1 regulates the expression of the CHRNA5/A3/B4 lung cancer susceptibility locus. Mol Cancer Res. 2010;8:194–203. doi: 10.1158/1541-7786.MCR-09-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Improgo MR, Tapper AR, Gardner PD. Nicotinic acetylcholine receptor-mediated mechanisms in lung cancer. Biochem Pharmacol. 2011;82:1015–1021. doi: 10.1016/j.bcp.2011.05.020. [DOI] [PubMed] [Google Scholar]
- Jeanclos EM, Lin L, Treuil MW, Rao J, DeCoster MA, Anand R. The chaperone protein 14-3-3eta interacts with the nicotinic acetylcholine receptor α 4 subunit: evidence for a dynamic role in subunit stabilization. J Biol Chem. 2001;276:28281–28290. doi: 10.1074/jbc.M011549200. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein LM. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- Kottalam J, Case DA. Langevin modes of macromolecules: applications to crambin and DNA hexamers. Biopolymers. 1990;29:1409–1421. doi: 10.1002/bip.360291008. [DOI] [PubMed] [Google Scholar]
- Krashia P, Moroni M, Broadbent S, Hofmann G, Kracun S, Beato M, Groot-Kormelink PJ, Sivilotti LG. Human alpha3beta4 neuronal nicotinic receptors show different stoichiometry if they are expressed in Xenopus oocytes or mammalian HEK293 cells. PloS One. 2010;5:e13611. doi: 10.1371/journal.pone.0013611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Luo J, Cooper J, Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human alpha4beta2 acetylcholine receptors. Mol Pharmacol. 2005;68:1839–1851. doi: 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R, Banghart MR, Dougherty DA, Goate AM, Wang JC. Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry: implications for drug discovery. AAPS J. 2009;11:167–177. doi: 10.1208/s12248-009-9090-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Hernández GY, Sánchez-Padilla J, Ortiz-Acevedo A, Lizardi-Ortiz J, Salas-Vincenty J, Rojas LV, Lasalde-Dominicci JA. Nicotine-induced up-regulation and desensitization of alpha4beta2 neuronal nicotinic receptors depend on subunit ratio. J Biol Chem. 2004;279:38007–38015. doi: 10.1074/jbc.M403537200. [DOI] [PubMed] [Google Scholar]
- Mancias JD, Goldberg J. Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J. 2008;27:2918–2928. doi: 10.1038/emboj.2008.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- Mazzaferro S, Benallegue N, Carbone A, Gasparri F, Vijayan R, Biggin PC, Moroni M, Bermudez I. Additional acetylcholine (ACh) binding site at alpha4/alpha4 interface of (alpha4beta2)2alpha4 nicotinic receptor influences agonist sensitivity. J Biol Chem. 2011;286:31043–31054. doi: 10.1074/jbc.M111.262014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. α4β2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70:755–768. doi: 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J. Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;63:332–341. doi: 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Rasmussen BA, Perry DC. Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther. 2003;307:1090–1097. doi: 10.1124/jpet.103.056408. [DOI] [PubMed] [Google Scholar]
- Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- Papke RL, Sanberg PR, Shytle RD. Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. J Pharmacol Exp Ther. 2001;297:646–656. [PubMed] [Google Scholar]
- Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med. 2013;3:a012112. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, Shim D, De Biasi M. Nicotine regulates multiple synaptic proteins by inhibiting proteasomal activity. J Neurosci. 2007;27:10508–10519. doi: 10.1523/JNEUROSCI.3353-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, Pan Y, Dani JA, Lindstrom J, García Gras EA, McIntosh JM, De Biasi M. UBXD4, a UBX-containing protein, regulates the cell surface number and stability of α3-containing nicotinic acetylcholine receptors. J Neurosci. 2009;29:6883–6896. doi: 10.1523/JNEUROSCI.4723-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riganti L, Matteoni C, Di Angelantonio S, Nistri A, Gaimarri A, Sparatore F, Canu-Boido C, Clementi F, Gotti C. Long-term exposure to the new nicotinic antagonist 1,2-bisN-cytisinylethane upregulates nicotinic receptor subtypes of SH-SY5Y human neuroblastoma cells. Br J Pharmacol. 2005;146:1096–1109. doi: 10.1038/sj.bjp.0706434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi P, Colombo S, Francolini M, Borgese N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J Cell Biol. 2008;181:105–118. doi: 10.1083/jcb.200710093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala M, Braida D, Pucci L, Manfredi I, Marks MJ, Wageman CR, Grady SR, Loi B, Fucile S, Fasoli F, Zoli M, Tasso B, Sparatore F, Clementi F, Gotti C. CC4, a dimer of cytisine, is a selective partial agonist at α4β2/α6β2 nAChR with improved selectivity for tobacco smoking cessation. Br J Pharmacol. 2013;168:835–849. doi: 10.1111/j.1476-5381.2012.02204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sallette J, Bohler S, Benoit P, Soudant M, Pons S, Le Novère N, Changeux JP, Corringer PJ. An extracellular protein microdomain controls up-regulation of neuronal nicotinic acetylcholine receptors by nicotine. J Biol Chem. 2004;279:18767–18775. doi: 10.1074/jbc.M308260200. [DOI] [PubMed] [Google Scholar]
- Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, Corringer PJ. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46:595–607. doi: 10.1016/j.neuron.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer. 2009;9:195–205. doi: 10.1038/nrc2590. [DOI] [PubMed] [Google Scholar]
- Spitz MR, Amos CI, Dong Q, Lin J, Wu X. The CHRNA5–A3 region on chromosome 15q24–25.1 is a risk factor both for nicotine dependence and for lung cancer. J Natl Cancer Inst. 2008;100:1552–1556. doi: 10.1093/jnci/djn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Pantoja R, Moss FJ, Mackey ED, Son CD, Miwa J, Lester HA. Nicotine up-regulates α4β2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J Gen Physiol. 2011;137:59–79. doi: 10.1085/jgp.201010532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, Thorlacius S, Gudmundsson J, Jonsson T, Jakobsdottir M, Saemundsdottir J, Olafsdottir O, Gudmundsson LJ, Bjornsdottir G, Kristjansson K, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Nelson ME, Kuryatov A, Olale F, Cooper J, Keyser K, Lindstrom J. Chronic nicotine treatment up-regulates human α3 β2 but not α3 β4 acetylcholine receptors stably transfected in human embryonic kidney cells. J Biol Chem. 1998;273:28721–28732. doi: 10.1074/jbc.273.44.28721. [DOI] [PubMed] [Google Scholar]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Wecker L, Pollock VV, Pacheco MA, Pastoor T. Nicotine-induced up regulation of α4β2 neuronal nicotinic receptors is mediated by the protein kinase C-dependent phosphorylation of α4 subunits. Neuroscience. 2010;171:12–22. doi: 10.1016/j.neuroscience.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Li Y, He H, Zhan CG. Theoretical calculation of the binding free energies for pyruvate dehydrogenase E1 binding with ligands. Bioorg Med Chem Lett. 2007;17:5186–5190. doi: 10.1016/j.bmcl.2007.06.095. [DOI] [PubMed] [Google Scholar]
- Xiu X, Puskar NL, Shanata JA, Lester HA, Dougherty DA. Nicotine binding to brain receptors requires a strong cation-pi interaction. Nature. 2009;458:534–537. doi: 10.1038/nature07768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Omura S, Bonifacino JS, Weissman AM. Novel aspects of degradation of T cell receptor subunits from the endoplasmic reticulum (ER) in T cells: importance of oligosaccharide processing, ubiquitination, and proteasome-dependent removal from ER membranes. J Exp Med. 1998;187:835–846. doi: 10.1084/jem.187.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]