

Abstract
Mutations within the central region of prion protein (PrP) have been shown to be associated with severe neurotoxic activity similar to that observed with Dpl, a PrP-like protein. To further investigate this neurotoxic effect, we generated lines of transgenic (Tg) mice expressing three different chimeric PrP-Dpl proteins. Chi1 (amino acids 1–57 of Dpl replaced by amino acids 1–125 of PrP) and Chi2 (amino acids 1–66 of Dpl replaced by amino acids 1–134 of PrP) abrogated the pathogenicity of Dpl indicating that the presence of a N-terminal domain of PrP (23–134) reduced the toxicity of Dpl, as reported. However, when the amino acids 1–24 of Dpl were replaced by amino acids 1–124 of PrP, Chi3 Tg mice, which express the chimeric protein at a very low level, start developing ataxia at the age of 5–7 weeks. This phenotype was not counteracted by a single copy of full-length-PrPc but rather by its overexpression, indicating the strong toxicity of the chimeric protein Chi3. Chi3 Tg mice exhibit severe cerebellar atrophy with a significant loss of granule cells. We concluded that aa25 to aa57 of Dpl, which are not present in Chi1 and Chi2 constructs, confer toxicity to the protein. We tested this possibility by using the 25–57 Dpl peptide in primary culture of mouse embryo cortical neurons and found a significant neurotoxic effect. This finding identifies a protein domain that plays a role in mediating Dpl-related toxicity.
Introduction
Prion diseases consist of a group of fatal neurodegenerative disorders characterized by the transition of the cellular prion protein (PrPc) into an aberrant protein conformer, PrPSc. This form of PrP is the hallmark of prion infection (Prusiner, 1982). However, many experiments have shown that PrPSc may not be the proximate cause of neuronal dysfunction and neurodegeneration (Solomon et al., 2010). Toxicity and infectivity are distinct properties that do not necessarily coincide (Hill and Collinge, 2003; Mallucci et al., 2003; Solomon et al., 2010; Sandberg et al., 2011). Several PrP mutants that cause neurodegeneration in the absence of infectious prion propagation have been studied in transgenic (Tg) mouse models (Aguzzi et al., 2008). Notably, a category of mutations within the central region of PrP is found to be associated with severe neurotoxic activity: mice expressing truncated PrPc mutants develop specific phenotypes, such as massive degeneration of cerebellar granule cells (GCs) for mutant Δ105–125 (Li et al., 2007) or widespread axomyelinic degeneration for mutant Δ94–134 (Baumann et al., 2007). It has been concluded that the absence of these segments, called central domains (CDs), which lie within the highly conserved but unstructured N-terminal region, is responsible for induced neurodegeneration characterized by ataxia. This degenerative phenotype is reminiscent of the phenotype associated with a PrP-like protein, namely Doppel (Dpl) (Moore et al., 1999). In PrP-null mice, ectopic neuronal expression of Dpl leads to ataxia. The C-terminal region of Dpl and PrP has a very similar folding pattern with a globular three-helix region (Mo et al., 2001), therefore Dpl resembles the neurotoxic PrP mutants cited above, lacking the amino proximal region.
To understand the dual function of PrP (neurotoxicity vs neuroprotection), Baumann et al. (2009) created Tg mice expressing a fusion protein consisting of the CD of PrP inserted within the Dpl sequence. These Tg mice failed to develop a Dpl-mediated typical neurological disorder indicating that the presence of this N-terminal portion of PrPc reduces the toxicity of Dpl. Here we report a divergent result. Three Tg lines of mice expressing chimeric Dpl-PrP proteins were generated with different localizations of the PrP-Dpl junction site and length of the Dpl segment. Two Tg lines, Chi1 and Chi2, did not develop any clinical sign. In contrast, the third Tg line, Chi3 mice, starts developing ataxia at a very young age. Moreover, in contrast with previously published Tg mice (Sakaguchi et al., 1996; Shmerling et al., 1998; Rossi et al., 2001), this phenotype could only be counteracted by PrPc overexpression, indicating the strong toxicity of the chimeric protein Chi3. Anatomo-pathological analysis of the cerebellum confirmed the phenotype of Chi3 mice. These data led to conclude that the N-terminal portion of PrP is not sufficient to protect from Dpl neurotoxicity. The different phenotypes observed for Chi1, Chi2, and Chi 3 mice suggested that Dpl neurotoxicity could be contained within aa25 to aa57. We tested this hypothesis by investigating in vitro the toxicity of this protein segment (peptide D25–57) in mouse embryonic cortical neurons.
Materials and Methods
Animals.
Tg and WT mice were anesthetized (200 mg/kg ketamine and 25 mg/kg xylazine, i.p.) and perfused transcardiacally with either 4% paraformaldehyde in 0.1 m phosphate buffer (PB) or a mixture of 2% paraformaldehyde and 2% glutaraldehyde in 0.1 m PB. All mice were maintained under specific pathogen-free conditions. Mice of both sexes have been used in all experiments.
Ethics statement.
All experiments with Tg and non-Tg lines of mice were conducted according to national and international laws for laboratory animal welfare and experimentation (EEC Council Directive 2010/63/EU, September 2010). Experiments were performed under the supervision of J.-Y.C. (agreement 38 05 17) in the animal care facilities approved by the Direction des Services Vétérinaires de l'Isère (A 38 516 10006).
Construction of the transgenes.
The coding region of murine Prnp and Prnd genes has been cloned previously (Locht et al., 1986; Moore et al., 1999) and was used as the template to generate the different PrP and Dpl fragments. The construction of the Chi1 fragment has already been described (Erlich et al., 2008). To create the Chi2 fragment, murine Prnp and Prnd ORFs were used as the template to obtain two PCR fragments (PrP2 and Dpl2) with primer set P1 (5′-CGATGTCCAGCTTCCTGCTCATGGCGCTCC-3′) and P2 (5′-ACGCGTGCAGATCAGTCATCATGGCGAACCTTGGCTACTGG-3′) and primer set P3 (5′-GGAGCGCCATGAGCAGGAAGCTGGACATCG-3′) and P4 (5′-ACGCGTCCCTTACTTCACAATGAACCAAACGAAACCCAGC-3′), respectively. To create the Chi3 fragment, we used primer set P5 (5′-TATGCCCCTTGCTTTAAGGCCCCCCACTAC-3′) and P2 and primer set P6 (5′-GTAGTGGGGGGCCTTAAAGCAAGGGGCATA-3′) and P4 to amplify, respectively, the PrP and Dpl fragments (PrP3 and Dpl3). To generate the chimeric fragments Chi2 and Chi3, the two initial PCR products (PrP2 and Dpl2 for Chi2 and PrP3 and Dpl3 for Chi3) were then fused in a third PCR with the flanking primers P2 and P4. These products were then cloned into the pCR2.1-Topo vector (Invitrogen). The 3F4 epitope (L109M/V112M) was introduced by using the QuikChange site-directed mutagenesis kit (Stratagene, Agilent Technologies) according to the manufacturer's instructions. All mutations were verified by sequencing. A 692 bp fragment was then amplified using the primers P7 (5′-GGAGGGTGGAACACCGGTGGAAGCCGGTAT-3) and P8 (5′-GGAAGGCCTCC CTCACTTCACAATGAACCA-3′). The 5′ primer is homologous to the region from positions 184 to 213 of the murine Prnp cDNA (GenBank accession number M13685) and possessed an Age1 restriction site. The 3′ primer is complementary to the 3′ end of the murine ORF [starting at nucleotide 876 and includes 15 nt complementary to the murine Dpl sequence (nucleotides 36749 to 36763; GenBank accession number U29187). Another PCR was performed using P9 (5′-TGGTTCATTGTGAAGTGAGGGAGGCCTTCC-3′) and P10 (5′-GGCTGTTTTCCAG GGCGCCATCCCC-3′) as the 5′ and 3′ primers and half-genomic vector (pHGPrP) (Fischer et al., 1996) containing a single Nar1 restriction site (pHGPrPmutNarI) (Erlich et al., 2008) as the template, to amplify part of the 3′ murine Prnp untranslated region (UTR). For each construct (Chi1, Chi2, and Chi3), both amplified fragments (Chi1 ORF, Chi2 ORF, Chi3 ORF, and the murine 3′ UTR) were fused in a third PCR with the flanking primers P7 and P10. These products were digested with Age1/Nar1 and ligated into the Age1/Nar1 site of pHGPrPmutNarI. The presence of the new inserts was confirmed by sequencing of the ORF.
Generation, identification, and maintenance of Tg mice.
The transgene fragments containing chimera coding sequences were cut out from pHGPrP vector with NotI and SalI. Pronuclear injection was performed into oocytes of hybrid C57BL6/SJL mice. Tg mice were generated by microinjecting the purified transgene fragments into the pronucleus of Prnp+/+ single-cell embryos (C56BL/6 × DBA/2 F1 hybrids) and implanted into pseudo-pregnant female mice. Founder mice were identified by genomic PCR with transgene-specific primers (P11, 5′-GCGGCGCCATCCCCAAACAGTGGCTTC-3′; P12, 5′-CCTAGGAGCCTTCATCAAGCAAGGCCGG-3′), producing an ∼770 bp fragment for the Chi1 and Chi3 transgenes. Chi2 mice were identified with a second set of primers (P13, 5′-AACTATTGGCAGTTCCCTGATGGG-3′; P14, 5′-CTCTGGGTACAAGTCAGGGGATACACC-3′). Founder mice were bred with C57BL/6J mice to produce F1 hemizygous mice for expression analysis. In general, Tg mice were maintained as hemizygotes by backcrossing to the C57BL/6J strain for five to six generations. Tg founders were then serially crossed with C57BL/6J ZrchI Prnp0/0 mice. All Tg lines were maintained in the Prnp+/+ or Prnp0/0 allelotype. Chi3 mice were also maintained in the Prnp+/0 allelotype. To test for the presence of Prnp alleles, additional PCR was performed using primers P15 (5′-TCATGCCCATTTCCAAATTCCACTACA-3′), P16 (5′-GGGTAGCGGTACATGTTTTCACGGTAG-3′), and P17 (5′-ACGCGTCACCTTAATATGCGAAGTGGA-3′). A fragment of 566 bp indicates the presence of the Prnp0/0 allele, whereas a product of 779 bp is detected for the Prnp+/+ allele. Heterozygous mice display both amplicons. Chi3Prnp+/+ mice were crossed with tga20 mice with a pure C57Bl6J background and maintained on tga20+/−Prnp+/−. The presence of tga20 allele was detected by PCR using primers P16 and P18 (5′-ACTCTTCATTTTAGGCCCCACGTATCA-3′), producing an ∼1066 bp fragment. The transgene copy number was calculated by dot blot assay on genomic DNA.
Anti-PrP and anti-Dpl antibodies.
Anti-PrP monoclonal antibodies (mAbs) were gifts from Jacques Grassi (CEA, Gif sur Yvette, France). SAF32 recognizes amino acids 59–89 in the N-terminal domain of murine PrP and thus can also detect the chimeric protein (dilution, 1:5000). Mouse mAb 3F4 (Kascsak et al., 1987) raised against human PrP 109–112 was diluted 1:5000 for Western blot. Rabbit polyclonal anti-Dpl antibody was obtained by immunizing rabbits with recombinant full-length Dpl [expression and purification of Dpl has been described in previous work (Erlich et al., 2008)]. Sera were tested by Western blotting using total protein extracts from mouse testis for their ability to detect cellular Dpl (dilution, 1:7500).
Western blot.
Brain homogenates (10% w/v) were prepared in a lysis buffer containing 150 mm NaCl, 0.5% Triton X-100, 0.5% sodium deoxycholate, 50 mm Tris-HCl, pH 7.5, 2 mm EDTA, and 1 μm of proteases inhibitors pepstatin and leupeptin (both from Roche Diagnostics). After 10 min of centrifugation at 3000 × g, the supernatant was collected, and its total protein concentration was determined by the bicinchoninic protein assay (Uptima; Interchim). For PrPc and chimera detection, the equivalent of 30 μg of total protein was mixed with reducing Laemmli buffer, boiled at 100°C for 5 min, and subjected to 12% SDS-PAGE. The proteins were transferred (400 mA for 60 min) onto a nitrocellulose membrane (Trans-Blot Transfer Medium, 0.2 μm; Bio-Rad). The membrane was then rinsed in PBS for 5 min and blocked with 3% BSA in PBS–0.1% Tween 20. The next day, the membrane was rinsed two times for 10 min in PBS–0.1% Tween 20 and incubated 1 h at room temperature with the appropriate primary antibody diluted in PBS–0.1% Tween 20. After washing, the membrane was incubated with the secondary antibody [goat anti-mouse HRP conjugate or goat anti-rabbit HRP conjugate (both 1:10,000; Jackson ImmunoResearch Laboratories)] for 60 min. Blots were developed by enhanced chemiluminescence (ECL; GE Healthcare) and exposed on X-ray film (Hyperfilm ECL; GE Healthcare) or analyzed with the ChemiDoc XRS system (Bio-Rad). Commercially available monoclonal anti-actin (1:10,000; Sigma-Aldrich) antibody was used for controls. For deglycosylation, 50 μg of denatured total protein from brain lysates was incubated at 37°C for 4 h with 500 U of PNGase F (New England Biolabs), according to the manufacturer's instructions, in the presence of protease inhibitors leupeptin and pepstatin (10 μg/ml). Reaction was stopped by adding reducing Laemmli buffer, and samples were analyzed by Western blotting as described above. Quantification of Western blots was performed using ImageJ densitometry software.
Flotation assays.
Appropriate brain homogenates were extracted for 2 h on ice in cold lysis buffer (150 mm NaCl, 25 mm Tris–HCl, pH 7.4, 5 mm EDTA, 1% Triton X-100; total protein, 1 mg in 1 ml). Extracts were mixed with 2 vol (2 ml) of 60% Optiprep (Sigma-Aldrich) to reach a final concentration of 40%. All lysates were loaded at the bottom of ultracentrifuge tubes (Sorvall). An Optiprep step gradient in a buffer containing 150 mm NaCl, 25 mm Tris–HCl, pH 7.4, and 5 mm EDTA was then overlaid onto the lysate (6 ml of 30% Optiprep and 2.5 ml of 5% Optiprep). Tubes were centrifuged for 24 h at 4°C in a T-641 rotor (Sorvall) at 100,000 × g. Fractions (1 ml) were collected from the top of the tube. The equivalent of 300 μl of each fraction was treated with 3 vol of methanol (900 μl), 1 vol of chloroform (300 μl), and 3 vol of H20 and centrifuged for 5 min at 4°C at 10,000 × g. Aqueous upper phase was eliminated, and proteins were precipitated with 3 vol of methanol and centrifuged as above. The pellet was dried, resuspended in reducing Laemmli buffer, and processed for immunoblotting and visualization with anti-PrP SAF32 and 3F4 antibodies.
Behavioral assessment.
All mice were examined twice weekly for behavioral abnormalities, such as trembling gait, disorientation, kyphosis, rough hair coat, and paresis that could rapidly progress to severe ataxia. The onset of ataxia is defined by the clear appearance of trembling. For ethical reasons, the mice were killed when the following neurological symptoms were observed: feet clasping when lifted, paresis of the hind legs, and prostration. Motor performance was evaluated with a rotating rod apparatus (Accelerating Model; Ugo Basile Biological Research Apparatus). Mice were placed on a rotating rod with a speed slowly accelerating from 4 to 40 rpm. Maximum speed was reached after 240 s. We recorded the latencies at which the animal fell off the rod. The median latency to fall was tested three times in a row with an interval of 30–60 min between trials. Mice were tested from 12 to 25 weeks of age on a weekly or biweekly basis.
Histology and immunohistochemistry.
Sagittal paraffin sections were cut from paraformaldehyde-fixed cerebellum of mice. Sections were selected at 10 equidistant levels along the transverse dimension of the cerebellum, mounted on slides, and submitted to Mason's trichrome staining (Kascsak et al., 1987) and to a classical immunoperoxidase staining protocol for detection of calcium binding protein (CaBP) in Purkinje cells (PCs), glial fibrillary acidic protein (GFAP) in astrocytes, and NeuN in granule cells. Briefly, the deparaffinized sections were incubated overnight at 4°C in the primary antibody (anti-CaBP, 1:300, Sigma-Aldrich; anti-NeuN, 1:1000, Millipore; anti-GFAP, 1:300, Sigma-Aldrich) solution of 0.1 m PBS containing 0.3% Triton X-100 after rinsing for 45 min in 3% normal horse serum (NHS) in PBS containing 0.5% Triton. The sections were then rinsed in PBS before incubation in secondary antibodies (biotinylated horse anti-mouse 1/200 (CaBP, GFAP) and 1/400 (NeuN) diluted in PBS containing 0.3% NHS. After rinsing in PBS, the secondary antibodies were revealed for 1 h with streptavidin–horseradish peroxidase complexes (Vector Laboratories) and subsequent reaction in diaminobenzidine (2 min). The sections were dehydrated in ethanol and mounted in Eukitt.
Morphometry in Mason's trichrome-stained and immunostained sections.
The length of the Purkinje cell layer (PCL) was measured in each of the 10 Mason's trichrome-stained cerebellar sections from each mouse using ImageJ software. Statistical comparison between the mean lengths measured in each animal was achieved using a Student's t test (p < 0.01, p < 0.05). The number of CaBP-positive PCs per millimeter of PCL was counted in each of the 10 CaBP immunostained sections from each mouse using ImageJ. Statistical comparison between the mean PC number/millimeter of PCL ratios measured in each animal was achieved using a Student's t test (p < 0.01). The surface of the internal granular layer (IGL) and of NeuN immunoperoxidase-stained area, which mostly concerned granule cell nuclei in the IGL, were measured using ImageJ to estimate the density of granule cells by the ratio between the NeuN immunostained IGL area and the IGL area in each of the 10 NeuN immunostained sections from each mouse. Both mean IGL surfaces and mean granule cell densities were statistically compared between the different mice using a Student's t test (p < 0.05).
Morphometry in Toluidine blue-stained semithin sections.
Sagittal vibratome sections were cut in the glutaraldehyde/paraformaldehyde-fixed cerebellum and postfixed in osmium tetroxide before classical processing for flat embedding in Araldite. Four sagittal semithin sections were cut from the whole cerebellum of each mouse in the right and left hemispheres (n = 1 section/hemisphere) and in the vermis (n = 2 sections). These sections were stained with Toluidin blue, and the length of the PCL was measured using ImageJ. A mean PC number per millimeter of PCL was estimated in the hemispheres and vermis of each mouse by counting the total number of PCs in each section. In addition, the perimeter and surface of each vermal section were measured using ImageJ. The mean values of PC number per millimeter (p ≤ 0.01) and of cerebellar vermis perimeter and surface (p ≤ 0.01–0.05) were compared between each mouse using a Student's t test.
Transmission electron microscopy.
Four blocks were randomly sampled in the cortex and deep nuclei in each of the four Araldite-embedded vibratome sections of cerebellar hemispheres and vermis of mice. Ultrathin sections were cut from each bloc and examined with a transmission electron microscope (Hitachi 7500). In each mouse, 23–38.104 μm2 were observed throughout the cerebellar cortex (14–20.104 μm2) and deep nuclei (8–22.104 μm2), and the ultrastructural content of 120–230 PC somata was examined.
Primary neuron culture and toxicity assay.
Cortices from C57Bl6 × 129/Pas E17.5 mice embryos were isolated and chemically and mechanically dissociated as described previously (Dotti et al., 1988). Two hemispheres produced a cell suspension plated on poly-l-lysine-coated P96 wells (BD Biosciences) in DMEM plus 10% FBS, replaced with Neurobasal medium supplemented with B-27 (Invitrogen) about 90 min later. After 3 d in vitro (DIV), 3 μm cytosine β-d-arabinofuranoside (Sigma-Aldrich) was added in fresh medium to prevent proliferation of non-neuronal cells. At 8 DIV, the indicated concentrations of peptide in H2O were added in fresh medium. After an additional 3 DIV, survival of cortical neurons was tested by adding 10 μl of the cell proliferation reagent CCK-8 to each well, according to the manufacturer's recommendations (Dojindo Molecular Technologies). The optical density at 450 nm (reference, 650 nm) was measured 0.5, 1, 2, and 4 h later, using a microplate reader (PHERAstar; BMG Labtech). Background-subtracted results were plotted in Prism5 (GraphPad Software) and analyzed using a two-way ANOVA test followed by Bonferroni's post hoc tests.
Peptide.
The murine sequence of the Dpl protein fragment D25–57, KARGIKHRFKWNRKVLPSSGGQITEARVAENRPG, and its control scrambled peptide, VGPKAWKTSLGKFRQVGRPESRNHIKAARIENRG, were purchased from Genosphere Biotechnologies. These peptides were dissolved in water at a concentration of 10 mm, distributed in 20 μl aliquots, and stored at −20°C until used.
Statistics.
Statistical analysis was performed using Prism 4 (GraphPad Software). Results are shown as the means ± SEM. Nonparametric Mann–Whitney tests were used to compare statistical differences between any two groups. For more than two groups, a one-way ANOVA was used, followed by Bonferroni's post hoc comparisons or Tukey's post hoc comparisons.
Results
Characterization of Tg mice expressing chimeric PrP-Dpl proteins
To study the effect of PrP on Dpl toxicity in the brain, we created three chimeric PrP-Dpl constructs. The first two, Chi1 and Chi2, consisted in the N terminus of PrP comprising, respectively, codons 1–125 and 1–135 fused to the C terminus of Dpl comprising, respectively, codons 58–179 and 66–179. These particular junctions were chosen because secondary structure comparisons and hydrophobic predictions suggested that the resulting chimeric proteins would resemble wild-type PrPc, Chi1 having its first β strand from Dpl and Chi2 from PrP (Fig. 1). In contrast, we designed a third construct (Chi3) to have the full-length Dpl (25–179) fused to the same N terminus of PrP as Chi1 (Fig. 1).
Figure 1.

Schematic representation of WT PrP, Dpl, and chimeric PrP/Dpl mutants (Chi1, Chi2, and Chi3). Sequences are hatched for PrP and white for Dpl. SP, Secretory signal peptide, cleaved during synthesis; OR, octarepeats, five repeats of eight amino acids; α 1–3 and α′1–3, α-helices 1, 2, and 3 of the globular C-terminal domain of PrP and Dpl, respectively; β1–2 and β′1–2, β sheets 1 and 2 of PrP and Dpl, respectively; GPI, sequence cleaved after attachment of the glycosylphosphatidyl inositol anchor; 3F4, epitopes recognized by the mAb 3F4 (white arrowhead). For chimeric constructs, the junctions between PrP and Dpl are indicated by a black arrowhead.
To overcome the simultaneous detection of the chimeric and endogenous PrP proteins in mice expressing endogenous Prnp, we inserted two amino acid substitutions from human PrP, L109M and V112M, which are included in the epitope recognized by 3F4 anti-PrP mAb (Kascsak et al., 1987). Four Chi1, one Chi2, and one Chi3 founder mice were found to carry the transgenes. The Chi1, Chi2, and Chi3 Tg lines selected for additional studies harbored, respectively, 15, 10, and 15 copies per haploid genome. Tg mice were maintained as hemizygotes by serial backcrossing to the C57BL/6J strain and were then successively intercrossed with nonataxic PrPc-deficient Zürich-I mice [Prnp0/0 (Büeler et al., 1992)] previously bred on the C57BL6/J background (Loeuillet et al., 2010). In this way, offsprings carrying a single allele of the Chi transgene associated or not with the endogenous Prnp gene (Chi+/− and Prnp+/+, Chi+/− and Prnp+/0, Chi+/−and Prnp0/0) were obtained. Because we have only used heterozygous Chi Tg mice, Chi+/− Tg mice will be referred as Chi mice for convenience.
Expression of the chimeric proteins was analyzed by Western blotting. The mAb 3F4 (Kascsak et al., 1987) allowed detection of protein bands between 30 and 43 kDa in the brain tissue homogenates from Chi1, Chi2, and Chi3 mice (Fig. 2A). The analysis indicates significant expression of Chi1, 10–15 times higher than Chi2 and 20–25 times higher than Chi3. Chi3 showed a lower electrophoretic mobility than Chi1 and Chi2, as a result of the additional 33 aa present in the Dpl portion of Chi3. The expression of each chimeric protein was similar, whatever the Prnp+/+ or Prnp0/0 background, suggesting that removal of the WT Prnp alleles did not downregulate or upregulate the expression of the Chi proteins in the brain of Chi Tg mice (Fig. 2A). To quantify the expression of the chimeric proteins against the WT and tga20 mice, a mouse line overexpressing PrPc at about 6–10 times the WT level (Fischer et al., 1996), we performed Western blots using the SAF32 mAb (Kascsak et al., 1987). This mAb is directed against amino acids 79–92 of the N-terminal portion of mouse PrPc. As shown in Figure 2B, the expression of Chi1 was much higher than that of PrPc in WT mice, almost at the same level as PrPc in tga20 mice, whereas Chi2 and Chi3 expression levels were 5- to 10-fold lower than that of PrPc in WT mice. As Chi proteins are detected as several bands, we also investigated the chimeric proteins after PNGase-F treatment. As shown in Figure 2C, the Chi proteins detected with the 3F4 mAb was reduced to a single band of lower molecular weight, indicating that these proteins were N-glycosylated. Both Dpl and PrPc have been found to associate with membrane microdomains enriched in cholesterol and sphingolipids known as detergent-resistant membrane or lipid rafts (Shyng et al., 1995; Taylor and Hooper, 2006; Caputo et al., 2010). To investigate the localization of the chimeric proteins, we performed flotation assays on OptiPrep gradients using cold Triton X-100-treated brain extracts from Tg mice. As shown in Figure 2D, chimeric proteins were detected in low-density fractions and displayed buoyancy similar to that of PrPc, whose cellular localization in raft is well established (Sunyach et al., 2003; Chen et al., 2009). The molecular weight and the migration profile of Chi1, Chi2, and Chi3 protein in the raft are similar to what is observed in whole-brain extract (Fig. 2A,B). Together, these data indicated that the chimeric proteins are properly expressed, correctly processed to the cell surface, and localized in lipid rafts.
Figure 2.
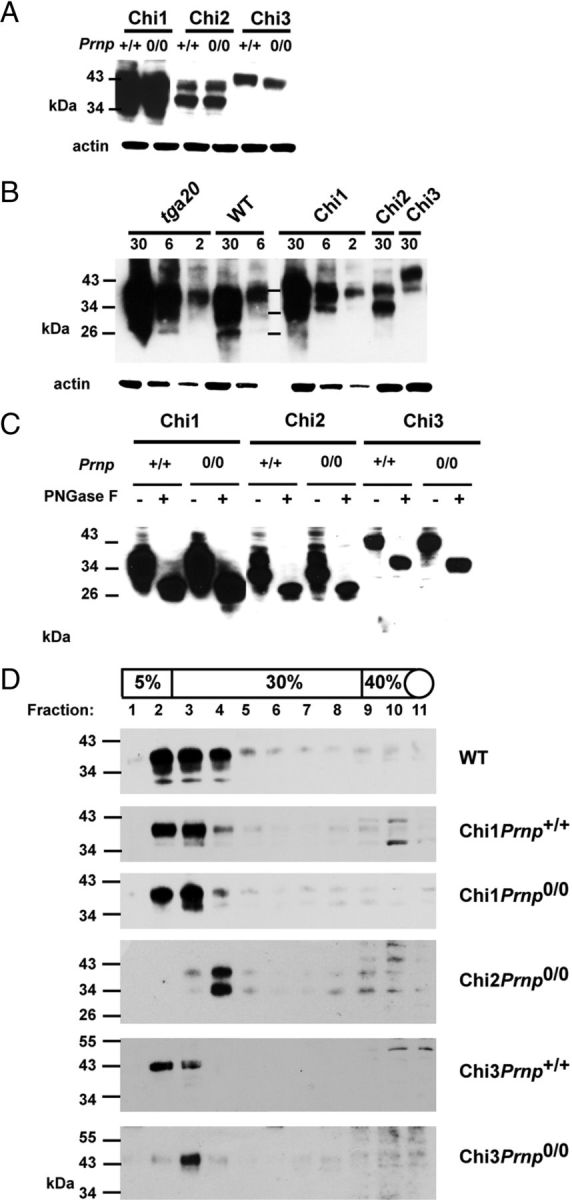
Expression of Tg proteins. A, The expression levels of each chimeric protein are independent of the Prnp status (Prnp+/+ or Prnp0/0). Thirty micrograms of protein from crude brains homogenate were loaded. B, Expression of chimeric protein in brain compared with that of PrP in WT and tga20 mice. Chi3 expression is lower than that of WT PrP. Amounts of protein (in micrograms) loaded for each line are indicated on the top of the gel. C, The glycosylation pattern of full-length Chi proteins is similar in Chi Tg mice expressed on mouse Prnp+/+and Prnp0/0 background. Thirty micrograms of protein from total brain homogenates are subjected (+) or not (−) to PNGase F treatment. D, Analyses of density gradient of detergent-resistant membranes from brains of WT, Chi1, Chi2, and Chi3 mice (on Prnp+/+ and Prnp0/0 background). Western blots were performed with mAb SAF32 (B, D; ChiPrnp0/0 Tg mice) or mAb 3F4 (A, C, D; ChiPrnp+/+ Tg mice). Molecular size markers (in kilodaltons) are indicated on the left.
Ataxia in Chi3 mice
The three Chi lines were maintained on the Prnp+/+, Prnp+/0, and Prnp0/0 allelotype and monitored for abnormal behavior. The hemizygous Chi1 and Chi2 mice did not elicit any clinical syndrome on the Prnp+/+ or Prnp0/0 background, up to 2 years. These results confirmed the previous data obtained (Yoshikawa et al., 2008; Baumann et al., 2009) indicating that the presence of N-terminal portions ([1–125] for Chi1 and [1–135] for Chi2) of PrP suppress the toxicity of Dpl (Aguzzi et al., 2008). However, although the Chi3 protein was expressed at a very low level in the heterozygous Tg mice on the Prnp+/+ background, these mice developed early motor deficits (47 ± 11 d) that progressed with age. The earliest symptoms were coarse tremor followed by a progressive inability to stride on cage grids. The mice then developed paresis of the hind legs, which progressed to full paraplegia (Fig. 3A). Ultimately (324 ± 99 d), they were severely handicapped in their movements and were killed. These data indicated that, contrary to Chi1 and Chi2, Chi3 protein retained the neurotoxic property of Dpl.
Figure 3.
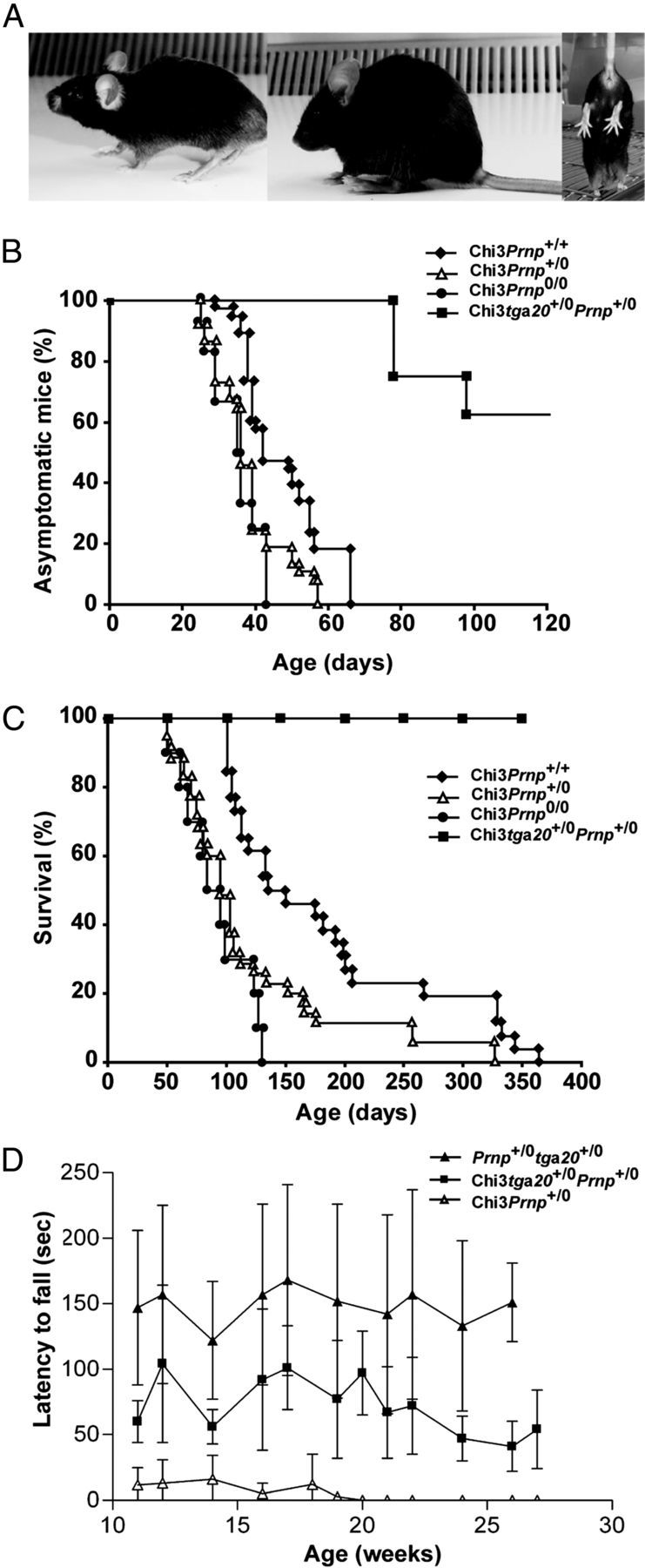
Phenotypic analysis of Chi3 mice. A, Typical clinical phenotypes at the terminal stage: ataxia (left), kyphosis (middle), and clasping when held head down by the tail (right). B, Chi3 mice were maintained on the Prnp+/+ (n = 38), Prnp+/0 (n = 37), Prnp0/0 (n = 12), and Prnp+/0tga20+/− (n = 8) allelotypes and monitored for the onset of the ataxia. Chi3Prnp+/0 and Chi3Prnp0/0 mice start showing coarse tremor slightly more rapidly than Chi3Prnp+/+ (p ≤ 0.001, Mann–Whitney test), whereas most Chi3tga20+/−Prnp+/0 remain asymptomatic. C, The mice were killed when the following neurological symptoms were observed: feet clasping when lifted, paresis of the hind legs, and prostration. The survival of mice is dependent of Prnp status, with Chi3Prnp+/0 and Chi3Prnp+/+ mice surviving longer than Chi3Prnp0/0 (p ≤ 0.001, Mann–Whitney test), whereas Chi3tga20+/−Prnp+/0 mice have a longer life expectancy (≥400d). D, Rotarod testing in acceleration mode of Chi3Prnp+/0 (n = 5), Chi3tga20+/−Prnp+/0 (n = 5) and tga20+/−Prnp+/0 (n = 5) mice. Overexpression of WT PrPc improves rotarod performance in Chi3 mice (Chi3tga20+/−Prnp+/0, p < 0.0001). This improvement still differs from tga20+/−Prnp+/0 control mice. Chi3Prnp+/0 mice continue to deteriorate, not being able to stay on the rod from 19 weeks of age. Significant differences were tested using one-way ANOVA with Tukey's post hoc comparison.
Impact of PrP expression on ataxia in Chi3 mice
The PrP knock-out mice with ectopic neuronal expression of Dpl exhibit spontaneous ataxia that can be rescued by introduction of a WT Prnp allele (Nishida et al., 1999; Moore et al., 2001; Rossi et al., 2001). Therefore, we developed mice on the following genotypes: (1) Chi3Prnp+/+, (2) Chi3Prnp+/0, (3) Chi3Prnp0/0, (4) Chi3tga20+/−Prnp+/0, and (5) tga20+/−Prnp+/0. The age of onset of ataxia was compared between the various Prnp background Tg mice (Fig. 3B). Trembling gait appeared at 35 d in Chi3Prnp0/0 mice (n = 12) and at 38 and 47 d, respectively, in Chi3Prnp+/0 (n = 37) and Chi3Prnp+/+ (n = 38) mice. The onset of ataxia was observed in all these mice within 65 d. In contrast, ∼40% of mice bred on tga20 background (n = 8) started to show slight clinical signs at 80 d (Fig. 3B). These data showed that the suppression of two alleles of Prnp shortened significantly the onset of ataxia phenotype, whereas it appeared to be partially rescued when PrPc is overexpressed on tga20 background (p ≤ 0.001 for each comparison, Mann–Whitney test). Furthermore, mice showed significantly faster progressive symptoms of ataxia, i.e., 91 d for Chi3Prnp0/0 mice compared with 122 d for Chi3Prnp+/0 and 182 d for Chi3 Prnp+/+. Survival was significantly shorter than for Chi3Prnp+/+ mice (p ≤ 0.001; Fig. 3C), whereas none of the Chi3 tga20+/−Prnp+/0 mice progressed to a severe ataxia (up to 386 d).
To further investigate the ataxia in Chi3 mice linked to different Prnp backgrounds, we studied the locomotor function, by measuring the latency to fall from an accelerating Rotarod device (Fig. 3D). The mice tested were between 12 and 28 weeks old. The 12-week-old Chi3Prnp+/0 mice, which presented clinical signs, could not stay on the rod for longer than 13 s (n = 5), whereas Chi3 mice overexpressing PrP (Chi3tga20+/−Prnp+/0) fell after 104 s (n = 5). The control mice (tga20+/−Prnp+/0) that did not express Chi3 performed better with an average time of 157 s (n = 5). These data showed that Chi3tga20+/−Prnp+/0 performed better than Chi3Prnp+/0 (p < 0.0001, ANOVA followed by Tukey post hoc tests), but not as well as control mice (tga20+/−Prnp+/0, p < 0.0001). The performances of Chi3Prnp+/0 diminished rapidly over time since they could not stay on the rod when older than 19 weeks. In contrast, the Chi3tga20+/−Prnp+/0 mice fell with a constant latency up to 26 weeks of age. These results suggested that the severity of the ataxic phenotype displayed by Chi3Prnp+/0 mice was partially reversed by overexpression of WT PrPc.
Neuropathological changes
Histological examination of either Mason's trichrome- or Toluidin blue-stained sections of Chi3 cerebellar tissue did not disclose evident structural abnormalities. Nevertheless, the size of the cerebellum of the 3-month-old ataxic Chi3Prnp+/+ (Fig. 4B) was obviously reduced compared with that of the non-Tg littermate of the same age (Fig. 4A). This reduction was also observed in Chi3Prnp+/0 (Fig. 4C) and Chi3Prnp0/0 (Fig. 4D) mice from 3 to 13.5 months of age. This phenotype was not observed in Chi1 mice, which are not ataxic (Fig. 4H,G). On tga20+/− background, the Chi3 cerebellum (Fig. 4F) was not reduced compared with the control tga20+/− mice (Fig. 4E). This observation was confirmed by morphometrical analyses that revealed a significant reduction (60%) of the area of the cerebellum of these Chi3Prnp+/+mice (p < 0.01, Student's t test), whereas no difference was observed between Chi3tga20+/− and tga20+/− mice (Fig. 5A).
Figure 4.

Histological studies. Toluidine blue (A–F) and Mason's trichrome (G, H) histological staining of sagittal sections in the cerebellum of 3-month-old WT mice (A), Chi3Prnp+/+ mice (B), 2-month-old Chi3Prnp+/0 mice (C), 3-month-old Chi3Prnp0/0 mice (D), tga20+/− mice (E), Chi3tga20+/− mice (F), 8-month-old WT mice (G), and 11-month-old Chi1Prnp+/+ mice (H). Scale bar, 500 μm.
Figure 5.

A, Mean area of the median cerebellar vermis in 3- to 8-month-old WT mice (n = 2), 3- to 13.5-month-old Chi3Prnp+/+ mice (n = 4), 3-month-old tga20+/− mice (n = 2), and Chi3tga20+/− mice (n = 2) (*p < 0.01). B, Mean number of PCs per millimeter of PCL in Toluidin blue-stained semithin sections of the median cerebellar vermis of 3- to 8 month-old WT mice (n = 2), 3- to 13.5-month-old Chi3Prnp+/+ mice (n = 4), 3-month-old tga20+/− mice (n = 2), and 3-month-old Chi3tga20+/− mice (n = 2) (*p < 0.01). C, Mean density of NeuN immunolabeled GCs in the IGL in sagittal paraffin sections of the median cerebellar vermis of 3- to 8-month-old WT (n = 2) and 3- to 13.5-month-old Chi3Prnp+/+ (n = 4) mice (*p < 0.05).
The impact of the expression of PrP-Dpl chimeras on the PC population was investigated by specific CaBP immunohistochemical staining of these neurons in the cerebellar cortex of the Tg mice. In the vermis of the cerebellum, CaBP-positive PCs did not exhibit any obvious alteration of their somato-dendritic morphology in the cerebellar molecular layer of Chi3Prnp+/+, Chi1Prnp+/+, and WT mice (Fig. 6A–C). Nevertheless, in the Chi3 mice (Fig. 6B), the PCs appeared to be kept closer together in the PCL than in the WT (Fig. 6A) and in the Chi1Prnp+/+ (Fig. 6C) mice. Quantifying the number of PCs per millimeter of PCL length in Toluidin blue-stained semithin sections showed that the linear density of PCs was significantly greater in Chi3 mice than in WT mice (Fig. 5B) (p < 0.01, Student's t test). This was observed at all ages of Chi3 mice, whatever their Prnp genetic background. However, in Chi3 mice overexpressing Prnp (Chi3tga20+/−Prnp+/0), such an increase of PC density was not observed, compared with the nonchimeric tga20+/− (tga20+/−Prnp+/0) mice as shown in Figure 5B.
Figure 6.

Calcium-binding protein immunoperoxidase staining of the Purkinje cells in the cerebellum of 8-month-old WT (A) and Chi3Prnp+/+ (B) mice and 11-month-old Chi1Prnp+/+ mice (C). NeuN immunohistochemical staining of the GCs in the median cerebellar vermis of 8-month-old WT (D) and Chi3Prnp+/+ (E) mice and 11-month-old Chi1Prnp+/+ mice (F). Scale bars, 50 μm.
The granule cell population was analyzed using NeuN immunohistochemical staining in the cerebellar cortex of all Tg mice. As opposed to the Purkinje cells, the granule cell density was lower in the IGL of the Chi3 mice than in the IGL of the WT and Chi1Prnp+/+ mice (Fig. 6D—F). Indeed, cell quantification showed that the mean density of the NeuN-stained GC was significantly reduced in the Chi3 mice compared with WT mice (Fig. 5C) (p < 0.05, Student's t test). This was observed throughout the cerebellum as early as 3 months up to 13.5 months of age, whatever the Prnp background of the mice. The reaction of astrocytic glia to PrP-Dpl expression was analyzed using GFAP immunohistochemistry in the cerebellum of all Tg mice. In the cerebellar cortex of the WT (Fig. 7A,D) and Chi1 (Fig. 7C,F) mice, GFAP-positive astrocytes were stained throughout the PCL and IGL whereas parallel processes of Bergman glia were faintly stained in the molecular layer (ML). In contrast, in the cerebellum of the Chi3 mice, an important astrocytic gliosis was observed, particularly prominent in the PCL and IGL as well as in Bergman glia in the ML (Fig. 7B,E).
Figure 7.

Immunohistochemical staining of GFAP in the cerebellum of WT, Chi3, and Chi1 mice. A, B, D, E, Vermal sections and cortex of the dorsal side of the lobule IXa of the cerebellum of 8-month-old WT (A, D), 8.5-month-old Chi3Prnp+/+ (B, E), and 11-month-old Chi1Prnp+/+ (C, F) mice. GFAP overexpression signals reactive astrogliosis in Chi3 mice compared with WT and Chi1Prnp+/+. Scale bars: A–C, 50 μm; D–F, 200 μm.
The ultrastructure of the cerebellar cortex was then investigated by transmission electron microscopy in the Chi3 mice. In the IGL of these mice, abnormal wrapping of glial profiles was frequent (Fig. 8A), confirming the astrocytic reaction evidenced by GFAP immunohistochemistry. However, the mossy fiber–granule cell glomerules displayed a normal aspect and composition (Fig. 8A). More spectacular abnormalities were observed within the myelinated PC axons, which contained piles of tubules (Fig. 8A) or autolysosomal profiles. In some cases, groups of degenerated GCs displayed electron-opaque content of various sizes (Fig. 8B). The PCs of the Chi3 mice presented several somatic as well as dendritic abnormalities. Autophagic-like phagophores born from trans saccules of Golgi dictyosomes were frequently found in the PC soma and main dendrites, which also contained stacked endoplasmic reticulum profiles (Fig. 8C) and numerous dense bodies distinct from the classical lipofuchsin bodies. Stacked membranes (Fig. 8D) and phagophores as well as putative autolysosomes were also found in large and tertiary PC dendrites. Although we did not quantify PC spines and their presynaptic partners, we observed PC spines embedded in the surrounding glia, free of any presynaptic innervation (Fig. 8E). In the deep cerebellar nuclei, myelinated axons displayed similar abnormal structures such as stacked tubules and autophagolysosomes as observed in the IGL. However, we did not find any alterations of the PC terminals synapsing symmetrically on the dendrites of the deep cerebellar neurons (Fig. 8F). As we did not observe any loss of PCs in the cerebellar cortex of the Chi3 mice compared with the WT mice, the ultrastructural abnormalities observed in these cells result from the loss of GCs, a main afferent input of the cerebellar cortex, which likely underlies the ataxic phenotype of the Chi3 mice.
Figure 8.

Ultrastructure analysis. A, B, Ultrastructure of the internal granular layer of Chi3 mice. A, A mossy fiber terminal (mf) makes asymmetric synapses (arrowheads) with GC dendrites (GD) and is surrounded by abnormal wraps of glial profiles (g). A myelinated PC-like axon contains an abnormal crystalloid stacking of tubular profiles (*). B, Steps of GC degeneration from shrunken cell body with cytoplasmic electron-opaque profiles (arrows) to overall electron-opacity of the neurons (*). See the normal-like GC on the right side. C–F, Ultrastructure of abnormal profiles in PC somata and dendrites of Chi3 mice. C, Autophagic-like phagophore (*) forming from the trans saccules of a Golgi (Go) apparatus dictyosome wrapping in a PC body. Note also the abnormal stacking of endoplasmic reticulum (white asterisks) and a dense body (arrow). D, A main PC dendrite (PCD) displays abnormal stacking of endoplasmic reticulum (*). Arrowheads indicate asymmetric synapses between presynaptic parallel fiber boutons (p) and postsynaptic PC dendritic spines (s). E, A PC dendritic spine (white s) is completely surrounded by glia (G) and displays a postsynaptic density (arrow) free of a presynaptic partner. Another PC spine (s) is innervated (arrowheads) by a presynaptic parallel fiber bouton (p). F, PC axon terminals (Pa) make symmetric synapses (arrowheads) on dendrites of a deep nuclear neuron (DCN). Scale bars: A, E, 500 nm; B, C, D, F, 2 μm.
D25–57 peptide toxicity
It has been described that PrPc can suffer a proteolytic cleavage at amino acids 110/111 or 111/112, leading to the production of the C-terminal fragment C1 (Mangé et al., 2004). The ectopic expression of Dpl in brain neurons is known to be the primary cause of ataxia (Moore et al., 2001; Rossi et al., 2001). Since the C1 cleavage site is present in Chi3 construct, we tested for the release of a C1 equivalent Dpl fragment from Chi3 protein, which could result in neurotoxicity. Because the cleavage would occur within the linear 3F4 epitope, the putative Dpl fragment can only be recognized by an anti-Dpl antibody. Thus, Western blot analysis of PNGase-F-treated samples was performed using a rabbit polyclonal antibody directed against the full-length Dpl protein (Fig. 9). Brain homogenate from Dpl-overexpressing Prnpngsk/ngsk mice and from WT mice as well as WT testis homogenate were added as controls. As expected, a Dpl-specific signal could not be observed in the brain sample of WT mice (Fig. 9). Testis control showed a heterodisperse signal between 29 and 37 kDa. Subsequent to deglycosylation, this signal was reduced to a major unglycosylated band at 17 kDa, indicating that Dpl is heavily glycosylated, as described previously (Moore et al., 1999; Al Bersaoui et al., 2005). Additionally, a minor band at ∼20 kDa, presumably representing the monoglycosylated form of Dpl, was observed. Cerebral tissues from Chi3 mice on the three different Prnp backgrounds revealed a fragment of similar size. This Dpl form in Chi3 is indicative of a minor proteolytic cleavage of the chimeric protein. This form is also observed in Prnpngsk/ngsk mice, which are known to develop ataxia. Thus, the cleaved form of Dpl might explain the toxicity of the chimeric protein, but only partially because (1) the ataxia in Prnpngsk/ngsk mice developed much later in age than in Chi3 mice and (2) the Chi3 mice on a tga20+/− background did not develop severe ataxia.
Figure 9.

Glycosylation pattern of Chi3 brain proteins compared with that of WT mice and Prnpngsk/ngsk mice. T, Testis extract from WT mice. Chi3 mice are expressed on mouse Prnp+/+, Prnp0/0, and on tga20+/− background with (+) or without (−) PNGase F pretreatment. Thirty micrograms of protein from total brain homogenates are subjected to PNGase F treatment as indicated and analyzed by Western blot revealed with rabbit anti-Dpl polyclonal antibody.
The obvious ataxic phenotype in Chi3 Tg mice associated to the very low expression level of Chi3 protein in the Tg mice, compared with the innocuousness of the very high expression level of the Chi1 protein, led us to hypothesize that ataxia could result from the 33 aa of the N-terminal portion ([25–57]) of Dpl. Indeed the Chi1 and Chi3 proteins carry an identical N-terminal segment of PrP protein ([1–125]) but differ only by the length of their Dpl part, which is, respectively, [58–179] and [25–179]. Therefore, the neurotoxicity of the Dpl peptide D25–57 was investigated in vitro. Primary cultures of cortical neurons from embryonic 17.5 d WT mice were exposed for 48 h to 40, 80, and 160 μm of the peptide. Neuronal viability was monitored by measuring the reduction of the mitochondrial activity using the cell proliferation reagent CCK-8 assay. Treatment with D25–57, but not with the control scrambled peptide, induced a significant decrease of neuron viability in a concentration-dependent manner (p < 0.01; Fig. 10).
Figure 10.

Neurotoxic effect of D25–57 doppel peptide on primary cortical neurons from embryonic 17.5 d WT mice. Neurons were chronically exposed to the indicated concentrations of D25–57 peptide for 48 h at 37°C. Neuronal viability was monitored by using the cell proliferation reagent CCK-8 assay. The neurotoxicity of 160 μm P25–57 peptide was statistically significant after a 48 h incubation time compared with 40 and 80 μm and with the control scrambled peptide. Significant differences (p < 0.01) were tested using a two-way ANOVA Bonferroni's post hoc test.
Discussion
Spontaneous neurodegenerative diseases have never been observed in Tg mice expressing PrP-Dpl chimeric proteins. For the first time, we report ataxia with cerebellar lesion in such a Tg mouse line. We have developed and characterized three lines of Tg mice expressing chimeric PrP-Dpl proteins that contain the CD of PrP. Our results confirm that fusion of either the N terminus of PrP [1–135] or [1–125] to, respectively, the [66–179] or [58–179] C-terminus part of Dpl fails to provoke neurological disorder in Tg Chi2 and Chi1 mice (Yoshikawa et al., 2008; Baumann et al., 2009). Unexpectedly, the third chimeric protein (Chi3) composed of the N terminus of PrP [1–124] linked to Dpl [25–179] is neurotoxic in Tg mice, resulting in GC loss. Although a single line of Chi3 Tg mice was obtained, it is extremely unlikely that the ataxia is not directly linked to the presence of the Chi3 transgene, (1) because ataxia is the common phenotype observed in Dpl-expressing Tg mice (Sakaguchi et al., 1996; Moore et al., 2001; Rossi et al., 2001) and (2) because the ataxic phenotype is rescued by PrPc overexpression as observed with some other Dpl Tg mice.
Histological analyses of brain from Tg mice expressing a low level of Chi3 protein show severe cerebellar atrophy with reduction of the PCL, whereas the number of PCs remains unchanged. However, ultrastructural data indicate abnormalities in PCs, suggesting autophagy, whereas GC degeneration is likely to be responsible for the ataxic phenotype. It is noteworthy that Chi1 mice expressing a high level of the same N-terminal part of PrP grafted to Dpl [58–179] do not show any obvious lesions. In Prnp0/0 mice, Dpl toxicity specifically induces a loss of PCs only at an advanced age (Sakaguchi et al., 1996; Moore et al., 2001; Rossi et al., 2001). In Chi3 mice, we observe a severe atrophy at a very early age of the cerebellum without a significant reduction of PCs. The use of the half genomic vector to construct Chi1, Chi2, and Chi3 transgenes may explain this discrepancy since this vector is known to exclude the expression in PCs (Shmerling et al., 1998; Karapetyan et al., 2009). PCs that lie at the interface between the internal granular layer and the molecular layer possess a wealth of synaptic connections with GCs. Hence, Chi3 expression in GCs, even at a low level, is able to compromise both GC and PC functions leading to ataxia.
Striking features of our result are the greatly enhanced toxicity of Chi3 compared with Dpl Tg mice [Ngsk Prnp-deficient mice or Prnpngsk/ngsk mice (Sakaguchi et al., 1996)] and the requirement for much higher levels of WT PrP to partially rescue the Chi3 phenotype (Nishida et al., 1999). Indeed, even though Prnpngsk/ngsk mice express the Dpl transgene at approximately the same level as Chi3 mice, they exhibit ataxia and PC loss later in life, i.e., at 24 weeks of age (Heitz et al., 2007), compared with 5-week-old for GC loss in Chi3 Tg mice. Furthermore, whereas ataxia in Prnpngsk/ngsk is rescued by a single copy of Prnp, we show that the toxicity of Chi3 protein can only be partially inhibited when Prnp is overexpressed by fourfold to fivefold. It is interesting to note that this phenotype in Chi3 mice is also reminiscent of those described in mice expressing PrP harboring Δ32–121, Δ32–134, Δ105–125, and Δ94–134 deletions, which resulted in cerebellar atrophy and GC degeneration (Shmerling et al., 1998; Baumann et al., 2007; Li et al., 2007).
Since the only difference between the proteins encoded by Chi1 and Chi3 transgenes is the presence or absence of residues 23–57 from Dpl, we suggest that this segment of Dpl may represent an important toxicity-determining region. The dose-dependent neurotoxic effect of D25–57 in primary mouse embryonic cortical neurons supports this hypothesis. The peptide concentration used might seem to be important, but a short peptide does not reflect a full-length protein, which can be more toxic because of its proper conformation and its natural cellular environment. It would be interesting to investigate how this region might play a role in the toxic activity of Dpl and whether the mechanisms involved is similar to 23–31 PrP toxicity as discussed by Harris and colleagues (Solomon et al., 2010; Westergard et al., 2011). This could provide insights into understanding the molecular mechanisms involved in the pathogenesis of prion disease and more globally of neurodegenerative diseases.
Footnotes
We thank C. Weissmann for providing Prnp0/0 and tga20 mice and pHGPrP construct. We thank J. Grassi for providing SAF32. We are grateful to the Plateforme de Haute Technologie Animale and F. Blanquet-Grossard for contribution to experimental mouse studies and to G. Gavazzi for providing the rotarod. We thank S. Kamche for assistance with mice.
References
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol. 2008;3:11–40. doi: 10.1146/annurev.pathmechdis.3.121806.154326. [DOI] [PubMed] [Google Scholar]
- Al Bersaoui R, Robert I, Lutz Y, Blanc F, Sommermeyer-Leroux G, Shibaguchi H, Aunis D, Fuchs JP. Purkinje-cell degeneration in prion protein-deficient mice is associated with a cerebellum-specific Doppel protein species signature. FEBS Lett. 2005;579:2715–2721. doi: 10.1016/j.febslet.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rülicke T, Bürkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26:538–547. doi: 10.1038/sj.emboj.7601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann F, Pahnke J, Radovanovic I, Rülicke T, Bremer J, Tolnay M, Aguzzi A. Functionally relevant domains of the prion protein identified in vivo. PLoS One. 2009;4:e6707. doi: 10.1371/journal.pone.0006707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell- surface PrP protein [see comments] Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Caputo A, Sarnataro D, Campana V, Costanzo M, Negro A, Sorgato MC, Zurzolo C. Doppel and PrPC co-immunoprecipitate in detergent-resistant membrane domains of epithelial FRT cells. Biochem J. 2010;425:341–351. doi: 10.1042/BJ20091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Jen A, Warley A, Lawrence MJ, Quinn PJ, Morris RJ. Isolation at physiological temperature of detergent-resistant membranes with properties expected of lipid rafts: the influence of buffer composition. Biochem J. 2009;417:525–533. doi: 10.1042/BJ20081385. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich P, Cesbron JY, Lemaire-Vieille C, Curt A, Andrieu JP, Schoehn G, Jamin M, Gagnon J. PrP N-terminal domain triggers PrP(Sc)-like aggregation of Dpl. Biochem Biophys Res Commun. 2008;365:478–483. doi: 10.1016/j.bbrc.2007.10.202. [DOI] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Heitz S, Lutz Y, Rodeau JL, Zanjani H, Gautheron V, Bombarde G, Richard F, Fuchs JP, Vogel MW, Mariani J, Bailly Y. BAX contributes to Doppel-induced apoptosis of prion-protein-deficient Purkinje cells. Dev Neurobiol. 2007;67:670–686. doi: 10.1002/dneu.20366. [DOI] [PubMed] [Google Scholar]
- Hill AF, Collinge J. Subclinical prion infection. Trends Microbiol. 2003;11:578–584. doi: 10.1016/j.tim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Karapetyan YE, Saá P, Mahal SP, Sferrazza GF, Sherman A, Salès N, Weissmann C, Lasmézas CI. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One. 2009;4:e5730. doi: 10.1371/journal.pone.0005730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locht C, Chesebro B, Race R, Keith JM. Molecular cloning and complete sequence of prion protein cDNA from mouse brain infected with the scrapie agent. Proc Natl Acad Sci U S A. 1986;83:6372–6376. doi: 10.1073/pnas.83.17.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeuillet C, Lemaire-Vieille C, Naquet P, Cesbron-Delauw MF, Gagnon J, Cesbron JY. Prion replication in the hematopoietic compartment is not required for neuroinvasion in scrapie mouse model. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013166. pii:e13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Mangé A, Béranger F, Peoc'h K, Onodera T, Frobert Y, Lehmann S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol Cell. 2004;96:125–132. doi: 10.1016/j.biolcel.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Mo H, Moore RC, Cohen FE, Westaway D, Prusiner SB, Wright PE, Dyson HJ. Two different neurodegenerative diseases caused by proteins with similar structures. Proc Natl Acad Sci U S A. 2001;98:2352–2357. doi: 10.1073/pnas.051627998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol. 1999;292:797–817. doi: 10.1006/jmbi.1999.3108. [DOI] [PubMed] [Google Scholar]
- Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D, DeArmond SJ, Tremblay P. Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci U S A. 2001;98:15288–15293. doi: 10.1073/pnas.251550798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel SP, Nakaoke R, Atarashi R, Houtani T, Torchia M, Sakaguchi S, DeArmond SJ, Prusiner SB, Katamine S. A mouse prion protein transgene rescues mice deficient for the prion protein gene from Purkinje cell degeneration and demyelination. Lab Invest. 1999;79:689–697. [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Rossi D, Cozzio A, Flechsig E, Klein MA, Rülicke T, Aguzzi A, Weissmann C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001;20:694–702. doi: 10.1093/emboj/20.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y, Houtani T, Shirabe S, Okada H, Hasegawa S, Miyamoto T, Noda T. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature. 1996;380:528–531. doi: 10.1038/380528a0. [DOI] [PubMed] [Google Scholar]
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011;470:540–542. doi: 10.1038/nature09768. [DOI] [PubMed] [Google Scholar]
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Moulder KL, Lesko A, Harris DA. The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J Biol Chem. 1995;270:14793–14800. doi: 10.1074/jbc.270.24.14793. [DOI] [PubMed] [Google Scholar]
- Solomon IH, Schepker JA, Harris DA. Prion neurotoxicity: insights from prion protein mutants. Curr Issues Mol Biol. 2010;12:51–61. [PMC free article] [PubMed] [Google Scholar]
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003;22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Hooper NM. The prion protein and lipid rafts. Mol Membr Biol. 2006;23:89–99. doi: 10.1080/09687860500449994. [DOI] [PubMed] [Google Scholar]
- Westergard L, Turnbaugh JA, Harris DA. A nine amino acid domain is essential for mutant prion protein toxicity. J Neurosci. 2011;31:14005–14017. doi: 10.1523/JNEUROSCI.1243-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa D, Yamaguchi N, Ishibashi D, Yamanaka H, Okimura N, Yamaguchi Y, Mori T, Miyata H, Shigematsu K, Katamine S, Sakaguchi S. Dominant-negative effects of the N-terminal half of prion protein on neurotoxicity of prion protein-like protein/doppel in mice. J Biol Chem. 2008;283:24202–24211. doi: 10.1074/jbc.M804212200. [DOI] [PMC free article] [PubMed] [Google Scholar]