

Abstract
Small-conductance calcium-activated potassium (SK) channels play an important role in regulating neuronal excitability. While SK channels at the soma have long been known to contribute to the medium afterhyperpolarization (mAHP), recent evidence indicates they also regulate NMDA receptor activation in dendritic spines. Here we investigate the activation of SK channels in spines and dendrites of rat cortical pyramidal neurons during action potentials (APs), and compare this to SK channel activation at the soma. Using confocal calcium imaging, we demonstrate that the inhibition of SK channels with apamin results in a location-dependent increase in calcium influx into dendrites and spines during backpropagating APs (average increase, ∼40%). This effect was occluded by block of R-type voltage-dependent calcium channels (VDCCs), but not by inhibition of N- or P/Q-type VDCCs, or block of calcium release from intracellular stores. During these experiments, we noticed that the calcium indicator (Oregon Green BAPTA-1) blocked the mAHP. Subsequent experiments using low concentrations of EGTA (1 mm) produced the same result, suggesting that somatic SK channels are not tightly colocalized with their calcium source. Consistent with this idea, all known subtypes of VDCCs except R-type were calcium sources for the apamin-sensitive mAHP at the soma. We conclude that SK channels in spines and dendrites of cortical pyramidal neurons regulate calcium influx during backpropagating APs in a distance-dependent manner, and are tightly coupled to R-type VDCCs. In contrast, SK channels activated by APs at the soma of these neurons are weakly coupled to a variety of VDCCs.
Introduction
Calcium (Ca2+) is an important signaling molecule in neurons with diverse actions ranging from regulation of neuronal excitability to gene expression. In addition, calcium plays a critical role in many forms of synaptic plasticity (Alvarez and Sabatini, 2007; Sjöström et al., 2008). One of the main ways in which neurons regulate calcium influx during electrical activity is via calcium-activated potassium channels. While it is well known that these channels regulate excitability by contributing to the action potential (AP) afterhyperpolarization at the soma (Berkefeld et al., 2010), much less is known about their role in regulating dendritic excitability. Calcium influx into dendrites and spines can occur during a range of electrical events, including synaptic input, backpropagating APs (bAPs), and dendritic regenerative events such as calcium and NMDA spikes (Jaffe et al., 1992; Yuste and Denk, 1995; Schiller et al., 1997, 2000; Sabatini et al., 2002). Dendritic calcium-activated potassium channels would be expected to regulate these electrical events, and thereby have a significant impact on neuronal function and plasticity.
Neurons in the CNS express two main types of calcium-activated potassium channels, termed BK and SK channels due to their respective large and small single-channel conductance (Berkefeld et al., 2010). A third variety of calcium-activated potassium channel with intermediate conductance has recently been identified in cerebellar Purkinje neurons (Engbers et al., 2012). At the soma of many neuronal cell types, SK channels are activated by APs and contribute to the medium afterhyperpolarization (mAHP), impacting on AP accommodation, burst firing, and output gain (Schwindt et al., 1988; Faber and Sah, 2002; Womack and Khodakhah, 2003; Womack et al., 2004). The role of SK channels in dendrites is less clear. Dendritic SK channels have been shown to regulate glutamate-evoked dendritic plateau potentials in CA1 pyramidal neurons (Cai et al., 2004) and somatic AP firing in cerebellar Purkinje cells (Womack and Khodakhah, 2003). In addition, SK channels in dendritic spines can constrain NMDA receptor activation and calcium influx during synaptic input (Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007; Faber, 2010), as well as regulate synaptic plasticity (Faber et al., 2005; Lin et al., 2008; Ohtsuki et al., 2012). Whether SK channels in dendrites and spines can be activated by bAPs, and if so, how this impacts on dendritic calcium influx, is unclear.
Here we investigate SK channel activation in spines and dendrites of cortical pyramidal neurons during bAPs and identify their calcium source. Furthermore, we compare dendritic SK channel activation during bAPs with SK channel activation during the mAHP at the soma of the same neurons. We find that SK channels in spines and dendrites of cortical pyramidal neurons regulate calcium influx during bAPs in a location-dependent manner and are tightly coupled to R-type voltage-dependent calcium channels (VDCCs). In contrast, at the soma SK channels activated during the mAHP are weakly coupled to all known VDCCs except R-type VDCCs.
Materials and Methods
Preparation.
Wistar rats (3–5 weeks old) of either sex were anesthetized by inhalation of isoflurane and decapitated according to guidelines approved by the Animal Experimentation Ethics Committee of the Australian National University. Parasagittal brain slices of somatosensory cortex were made using a Vibroslice (Campden) or Leica VT1000S tissue slicer. During slicing, the brain was submerged in ice-cold artificial CSF (ACSF) containing the following: 125 mm NaCl, 25 mm NaHCO3, 15 mm sucrose, 10 mm glucose, 3 mm KCl, 1.25 mm NaH2PO4, 0.5 mm CaCl2, and 6 mm MgCl2, bubbled with 95%O2/5%CO2, pH 7.4. Slices were then transferred to a holding chamber and incubated at 35°C for 30–45 min in standard ACSF containing the following: 125 mm NaCl, 25 mm NaHCO3, 25 mm glucose, 3 mm KCl, 1.25 mm NaH2PO4, 2 mm CaCl2, and 1 mm MgCl2 bubbled with 95%O2/5%CO2, pH 7.4, before being maintained at room temperature. During recordings, slices were perfused a rate of ∼4 ml/min with standard ASCF maintained at 35–36°C.
Electrophysiology.
Somatic whole-cell current-clamp recordings were made from visually identified layer 5 (L5) pyramidal neurons using differential interference contrast optics (Stuart et al., 1993). Recording electrodes were filled with an intracellular solution containing the following: 130 mm K-gluconate, 10 mm HEPES, 10 mm KCl, 4 mm Mg2ATP, 0.3 mm Na2GTP, and 10 mm Na2-Phosphocreatine, pH 7.2 with KOH. In some experiments, EGTA was added to the intracellular solution at a concentration of either 200 μm or 1 mm, as denoted in the Results. Recordings were made using a BVC 700A amplifier in current-clamp mode (Dagan). Voltage traces were filtered at 10 kHz and digitized at 50 kHz using an ITC-18 computer interface (Instrutech). Data acquisition was performed using AxoGraph X software (AxoGraph Scientific) running on an Apple iMac computer. Data were analyzed only from cells with stable (<2 mV change) resting membrane potentials more negative than −60 mV. For some experiments, input resistance was measured using repeated current injections of −20 pA for a duration of 1 s (60–100 sweeps were typically averaged). Rheobase was determined using 200 ms somatic current injections and denoted as the lowest current amplitude that evoked an action potential.
Confocal calcium imaging.
Changes in intracellular calcium concentration were detected with the high-affinity fluorescent calcium indicator Oregon Green BAPTA-1 (OGB-1; 170–200 μm; Kd, 170 nm; Invitrogen) or the lower affinity fluorescent calcium indicator Oregon Green BAPTA-6F (OGB6F; 200 μm; Kd, 3 μm; Invitrogen) using a confocal laser scanning microscope (FV300, Olympus) equipped with a 60× 1.1 numerical aperture water-immersion objective (Olympus). Neurons were loaded with calcium indicator via the recording pipette for 50–60 min before imaging, although loading for up to 120 min was required when imaging dendritic locations greater than ∼150 μm from the soma. Calcium indicators were excited at 488 nm using a solid-state laser (Melles Griot). Baseline fluorescence was used to locate spines projecting from basal dendrites. Line scans for spines were positioned to traverse the spine head and parent dendrite and changes in fluorescence during APs recorded at 500–700 Hz. Typically, 10–20 trials were averaged for each condition. To reduce phototoxicity typically only one spine and parent dendrite was examined per cell. High-speed point scans (filtered at 5 kHz and digitized at 20 kHz) were used during experiments investigating dendritic calcium influx using OGB6F. This was necessary to obtain an acceptable signal-to-noise ratio due to the fast kinetics of this indicator. Point scans (700 Hz) were also used to investigate somatic and dendritic calcium influx during some experiments using SNX 482 (PEPTIDE INSTITUTE, Inc.) at the soma. The change in fluorescence divided by the resting fluorescence (ΔF/F) was measured at the peak by taking the average value within a 10 ms time window. Baseline fluorescence was determined by averaging over a 30 ms time window immediately before AP onset. Only responses with a peak amplitude greater than four times the SD of the baseline noise were included for analysis. In addition, data were excluded if the baseline fluorescence changed >10% during the course of the experiment. Calcium changes during acetylcholine (ACh) applications were recorded using frame scans of the soma at 20 Hz.
Pharmacology.
SK and VDCC antagonists were dissolved in oxygenated ACSF and locally applied using a large-diameter pipette positioned above the brain slice near the region of interest (pipette tip, 20 μm; using <10 mmHg constant pressure). Spines in experiments using VDCC antagonist were located 100–120 μm from the soma. Apamin and calcium channel antagonists were bath applied in experiments on the mAHP and during somatic calcium imaging. Bath application of SK and VDCCs antagonists had no significant impact on the membrane potential or input resistance. In experiments on spines where one drug was tested in the presence of another, a second local application pipette containing both drugs was used. In experiments investigating the role of intracellular stores on spines, cyclopiazonic acid (CPA) was bath applied for 15 min before the commencement of the experiment and apamin locally coapplied with CPA. ACh (100 μm) was locally applied to the soma using brief (10 ms) pulses applied with a Picospritzer III (Parker) to a standard patch pipette (tip 2–3 μm). Apamin, ACh, nifedipine, and NNC 55–0396 were purchased from Sigma-Aldrich; SNX 482 was purchased from the PEPTIDE INSTITUTE, Inc. or Alomone Labs; conotoxin GVIA and agatoxin IVA were purchased from Alomone Labs; conotoxin MVIIC was purchased from the PEPTIDE INSTITUTE, Inc.; and CPA and UCL 1684 were purchased from Tocris Bioscience.
Data analysis.
Fluorescence responses were analyzed using ImageJ, Microsoft Excel, and Axograph X, and were quantified as the ΔF/F. Distances from the soma were determined from confocal image stacks using ImageJ. All electrophysiology was analyzed using AxoGraph X. Statistical differences between datasets were evaluated using one sampled paired or unpaired t test, as appropriate. Correlation analysis was performed using Pearson's coefficient. p < 0.05 was considered significant. All data were tested for normality using the D'Agostino–Pearson omnibus test. All statistical analyses were performed using GraphPad Prism 6.
Results
To investigate the activation of SK channels in spines and dendrites during bAPs, we used confocal fluorescent calcium imaging. Somatic whole-cell patch-clamp recordings were made from L5 pyramidal neurons in brain slices of somatosensory cortex taken from 3- to 5-week-old Wistar rats. Before imaging, neurons were filled with the calcium-sensitive indicator OGB-1 via the recording pipette. We focused on spines protruding from basal dendrites between 50 and 200 μm from the soma. Single APs were evoked by somatic current injections (2–3 nA, 2 ms), and were accompanied by large changes in fluorescence in both spines and dendrites (Fig. 1A). Previous studies indicate that calcium influx into spines and dendrites during bAPs are mediated almost exclusively by VDCCs (Schiller et al., 1998; Sabatini and Svoboda, 2000; Bloodgood and Sabatini, 2007).
Figure 1.

SK channels constrain calcium influx into spines and dendrites during bAPs. A1, Confocal image of a layer 5 pyramidal neuron filled with 200 μm OGB-1. Scale bar, 25 μm. Yellow box indicates a region of interest in a distal basal dendrite. A2, Region of interest at high magnification. Dashed line shows location of line scan. Scale bar, 1 μm. A3, Repeated line scans through the spine and dendrite shown in A2. A single AP evoked after 30 ms (red arrowhead) caused a large increase in fluorescence in both compartments. B, AP-evoked change in fluorescence in a spine (top) and dendrite (bottom) before and after local application of apamin (1 μm). Same spine as in A2. In this and all subsequent figures, the solid line and shaded regions represent the mean ± SEM for an example spine and dendrite. C, Average AP-evoked peak ΔF/F (±SEM) in spines in the absence and presence of apamin (n = 32). D, Average change in AP-evoked fluorescence (±SEM) in spines and dendrites after application of apamin (n = 32). E, F, Change in AP-evoked fluorescence in spines (E) and dendrites (F) after apamin application versus distance from the soma. Lines represent linear fits to OGB-1 data. Pearson's coefficient 0.868 (E) and 0.860 (F). G, H, Average change in AP-evoked fluorescence (±SEM) in spines and dendrites after application of UCL 1684 (G; 1 μm; n = 15) and normal ACSF (H; n = 12). *p < 0.05, **p < 0.01. ns, Not significant.
SK channels constrain calcium influx into spines and dendrites during bAPs
To investigate the role of SK channels in regulating calcium influx into spines and dendrites during bAPs, we applied the SK channel antagonist apamin. Local application of apamin (1 μm in ACSF) caused a significant increase in calcium influx during bAPs in both spines and dendrites (Fig. 1B,C; n = 31). Across all spines, we observed an average increase in fluorescence of ∼40%, which was larger than the increase in the adjacent dendrite (Fig. 1D). The properties of APs recorded at the soma were unaffected by apamin (AP amplitude: ACSF, 100.9 ± 2.4 mV; apamin, 101.3 ± 2.1 mV; p = 0.77; half-width: ACSF, 0.62 ± 0.01 ms; apamin, 0.63 ± 0.03 ms; p = 0.82; n = 5), confirming that the impact of inhibiting SK channels on spine and dendrite calcium dynamics is dendritic in origin. Importantly, the affect of apamin on bAP-evoked calcium influx into spines and dendrites was distance dependent, with apamin having the greatest impact at distal dendritic locations (Fig. 1E,F). As OGB-1 is a high-affinity calcium buffer (Kd, 170 nm) and may perturb SK channel activation, the observed distance dependence could be due to a higher concentration of calcium indicator at proximal locations compared with distal locations. To investigate this possibility, we repeated these experiments with the low-affinity indicator OGB6F (Kd, 3 μm). While we did not have the sensitivity to resolve calcium transients in spines in these experiments, using OGB6F apamin caused a similar distance-dependent increase in dendritic calcium transients during bAPs (Fig. 1F, red symbols). This result suggests that the distance dependence of the impact of apamin on bAP calcium transients is not due to spatial differences in calcium buffering.
In addition, we investigated the impact of the alternative SK channel inhibitor UCL 1684. Local application of UCL 1684 (1 μm in ACSF) led to a similar increase in calcium influx in spines and dendrites during bAPs to that seen during apamin applications (Fig. 1G; n = 15), indicating that apamin is working specifically. To check that the affect of apamin and UCL 1684 were not a result of the local application method, we performed control experiments where ACSF alone was locally applied in the same way as during drug applications. Comparison of fluorescence changes during bAPs at the beginning and end of ACSF applications indicated no significant impact of our local perfusion system on bAP-evoked calcium transients (Fig. 1H; n = 11). Together, these data indicate that SK channels in L5 cortical pyramidal neurons play an important role in regulating calcium influx into basal spines during bAPs, particularly at distal dendritic locations.
R-type calcium channels control SK channel activation during bAPs
We next investigated the calcium sources controlling the activation of SK channels in spines and dendrites during bAPs. Other groups have shown that SK channels in spines during synaptic input are activated by calcium influx through R-type VDCCs (Bloodgood and Sabatini, 2007; Faber, 2010) and/or NMDA receptors (Faber et al., 2005; Ngo-Anh et al., 2005). Given that NMDA receptor activation during bAPs is negligible (Koester and Sakmann, 2000; Sabatini and Svoboda, 2000; Bloodgood and Sabatini, 2007), we examined whether SK channel activation during bAPs also requires calcium influx through R-type VDCCs using the antagonist SNX 482. Local application of SNX 482 (1 μm) caused a significant decrease in the bAP-evoked calcium transient in dendrites, but, interestingly, not in spines (Fig. 2A–C; spines, p = 0.561; dendrites, p = 0.0182; n = 12). There are two possible explanations for this result. The first is that bAPs do not activate R-type VDCCs in spines. The second possibility is that R-type VDCCs are present in spines and activated by bAPs, but the expected impact of blocking these channels on calcium influx is masked by enhanced activation of other VDCCs following removal of the calcium source for SK channel activation. To isolate the impact of SNX 482 on calcium influx from its potential impact on SK channel activation, SNX 482 was applied in the presence of apamin. Under these conditions, SNX 482 caused a significant decrease in bAP-evoked calcium transients in both dendrites and spines (Fig. 2D–F; n = 19). These findings indicate that R-type VDCCs are present in both spines and dendrites. To test whether calcium influx through these channels is required for the activation of SK channels in spines, apamin was applied in the presence of SNX 482. Under these conditions, apamin had no significant impact on bAP-evoked calcium transients in either spines or dendrites (Fig. 2G–I; n = 19). Together, these data indicate that R-type VDCCs in spines and dendrites are activated by bAPs, and that blocking these channels occludes the impact of apamin on bAP-evoked calcium transients.
Figure 2.

R-type VDCCs control SK channel activation during bAPs. A, AP-evoked change in fluorescence in a spine (top) and dendrite (bottom) in control (black) and after local application of SNX 482 (1 μm; blue). B, Average AP-evoked peak ΔF/F for spines before and after the local application of SNX 482 (±SEM; n = 15). C, Average percentage change in AP-evoked fluorescence in SNX 482 compared with control in spines and dendrites (±SEM; n = 15). D, AP-evoked change in fluorescence in a spine (top) and dendrite (bottom) in the presence of apamin (1 μm; red) and after local application of SNX 482 in apamin (blue). E, Average AP-evoked peak ΔF/F for spines before and after the application of SNX 482 in the continued presence of apamin (±SEM; n = 19). F, Average percentage change in AP-evoked fluorescence in SNX 482 plus apamin compared with apamin alone in spines and dendrites (±SEM; n = 19). G, AP-evoked change in fluorescence in a spine (top) and dendrite (bottom) in the presence of SNX 482 (blue) and after local application of apamin in SNX 482 (red). H, Average AP-evoked peak ΔF/F for spines before and after the application of apamin in the continued presence of SNX 482 (±SEM; n = 21). I, Average percentage change in AP-evoked fluorescence in apamin plus SNX 482 compared with SNX 482 alone in spines and dendrites (±SEM; n = 21). *p < 0.05, **p < 0.01. ns, Not significant.
Role of other calcium sources in spines
While these data indicate a critical role of R-type VDCCs in controlling the activation of SK channels in spines and dendrites during bAPs, are they the sole calcium source for SK channel activation? Calcium influx into spines and dendrites during bAPs is mediated by multiple VDCC subtypes (Sabatini and Svoboda, 2000; Bloodgood and Sabatini, 2007). Consistent with this, blocking R-type VDCCs in the presence of apamin only reduced bAP-evoked calcium transients by ∼20% (Fig. 2D–F). To investigate the role of other VDCCs in activating SK channels, we perturbed calcium influx into spines and dendrites by inhibiting N- and P/Q-type VDCCs using local applications of conotoxin MVIIC (10 μm). Blocking N- and P/Q-type VDCCs significantly reduced bAP-evoked calcium influx into spines and dendrites by ∼20% (Fig. 3A–C; spines, n = 10), indicating that N- and P/Q-type VDCCs are also present in basal spines and dendrites of L5 pyramidal neurons. In contrast to SNX 482, however, the application of apamin in the presence of conotoxin MVIIC still led to a significant (∼30%) increase in bAP-evoked calcium transients in both spines and dendrites (Fig. 3D–F; n = 10). These data show that calcium influx through N- and P/Q-type VDCCs in spines and dendrites during bAPs is not required for SK channel activation. Furthermore, they indicate that, despite multiple VDCCs contributing to calcium influx in spines and dendrites during bAPs, SK channel activation is triggered solely by calcium influx through R-type VDCCs.
Figure 3.
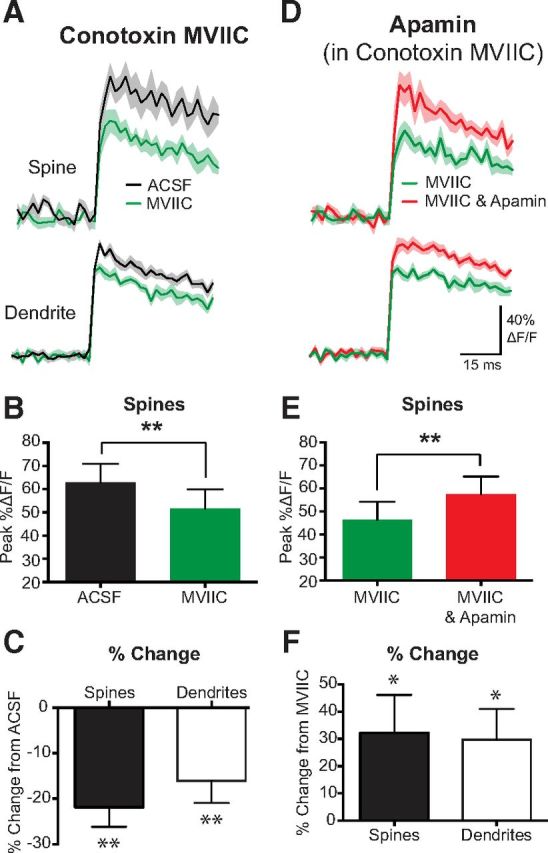
Block of N- and P/Q-type VDCCs does not affect SK channel activation. A, AP-evoked change in fluorescence in a spine (top) and dendrite (bottom) before (black) and after application of conotoxin MVIIC (10 μm; green) to block N- and P/Q-type VDCCs. B, Average AP-evoked peak ΔF/F in spines before and after the application of contoxin MVIIC. C, Average percentage change in AP-evoked fluorescence for spines and dendrites after application of conotoxin MVIIC (±SEM; n = 10). D, AP-evoked change in fluorescence in a spine (top) and dendrite (bottom) in conotoxin MVIIC (green) and after application of apamin (1 μm) in the presence of conotoxin MVIIC (red). E, Average AP-evoked peak ΔF/F in spines before and after the application of apamin in the continued presence of conotoxin MVIIC (±SEM; n = 10). F, Average percentage change in AP-evoked fluorescence after application of apamin in the presence of conotoxin MVIIC (±SEM; n = 10). *p < 0.05, **p < 0.01.
A recent study showed that SK channel activation during EPSPs can be abolished by blocking calcium release from intracellular stores, suggesting a role of calcium-induced calcium release in SK channel activation (Faber, 2010). To investigate the impact of calcium release from intracellular stores on SK channel activation during bAPs, we depleted intracellular calcium stores with the calcium-ATPase inhibitor CPA (30 μm in the bath) (Seidler et al., 1989). Local application of apamin in the presence of CPA caused a similar enhancement of bAP-evoked calcium transients in basal spines and dendrites to that seen under control conditions (compare Figs. 4A–C, 1C, spines; n = 10). These data suggest that intracellular calcium stores are unlikely to be involved in controlling SK channel activation during bAPs. To confirm that CPA had emptied intracellular calcium stores, we locally applied ACh to the soma, which is known to release calcium from intracellular stores leading to a large increase in somatic calcium (Gulledge and Stuart, 2005; Gulledge et al., 2007). The low-affinity calcium indicator OGB6F (200 μm) was included in the recording pipette, and calcium transients in these experiments were evoked by brief (10 ms) applications of ACh (100 μm) in the absence and presence of CPA. As expected, the presence of CPA abolished ACh-induced calcium transients (Fig. 4D,E; n = 4), indicating that CPA was effective in emptying intracellular calcium stores under our experimental conditions.
Figure 4.
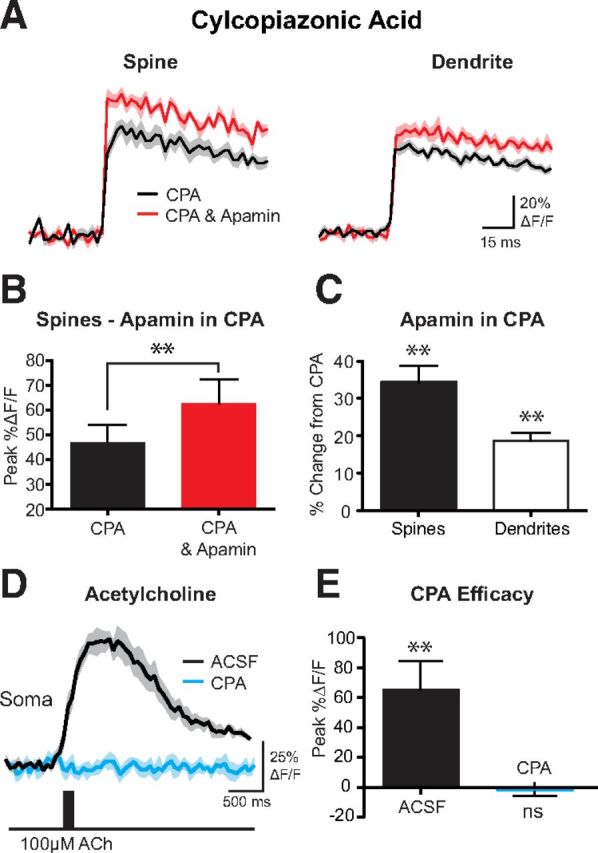
Inhibition of intracellular calcium stores does not affect SK channel activation. A, AP-evoked change in fluorescence in a spine (left) and dendrite (right) in the presence of cyclopiazonic acid (CPA) to deplete intracellular calcium stores (30 μm; black) and after application of apamin (1 μm) in the presence of CPA (red). B, Average AP-evoked peak ΔF/F in spines before and after the application of apamin in the continued presence of CPA (±SEM; n = 10). C, Average percentage change in AP-evoked fluorescence after the application of apamin in the presence of CPA (±SEM; n = 10). D, Change in fluorescence at the soma in response to a 10 ms local application of 100 μm acetylcholine to the soma in normal ACSF (black) and in the presence of CPA (blue). E, Average change in fluorescence during somatic acetylcholine applications in ACSF and CPA (n = 4). **p < 0.01. ns, Not significant.
Somatic SK channels are not tightly coupled to their calcium source
SK channels have long been known to contribute to the generation of the mAHP in L5 pyramidal neurons (Schwindt et al., 1988). We were therefore surprised to find that applications of apamin did not affect the mAHP in our calcium imaging experiments. We hypothesized that this may be the case because OGB-1, which is related to the fast calcium buffer BAPTA, perturbs SK channel activation at the soma through calcium chelation. To test this hypothesis, we quantified changes in amplitude of the mAHP in the absence and presence of OGB-1 in the pipette internal solution. Single APs in these experiments were elicited by 200-ms-long positive current pulses just above rheobase. In the absence of OGB-1 in the internal solution, bath application of apamin (100 nm) led to a significant reduction in the amplitude of the mAHP (Fig. 5A,D; n = 5). Inclusion of OGB-1 (200 μm) in the internal solution, however, led to a time-dependent decrease in the mAHP, with the impact of apamin on the mAHP occluded 30 min after break-in (Fig. 5B,D; n = 5). These data suggest that the coupling of somatic SK channels to their calcium source is weak. To investigate this further, we tested the impact of the slow calcium buffer EGTA on the mAHP. Using the same concentration of EGTA (200 μm), a partial perturbation of the mAHP was observed (n = 6) with full inhibition of the mAHP occurring when the EGTA concentration was increased to 1 mm (Fig. 5C,E; n = 6). The diffusional distance between somatic SK channels and their calcium source can be approximated by: (Neher, 1998). Based on a diffusion coefficient of calcium (DCa) of 220 μm2/s (Neher, 1998), a buffer (EGTA) concentration of 1 mm, and assuming a Kon of EGTA of 1 × 107 M−1 s−1 (Nägerl et al., 2000), we estimate the average distance between somatic SK channels and their calcium source to be greater than ∼150 nm. These data suggest that somatic SK channels are not tightly coupled to their calcium source.
Figure 5.
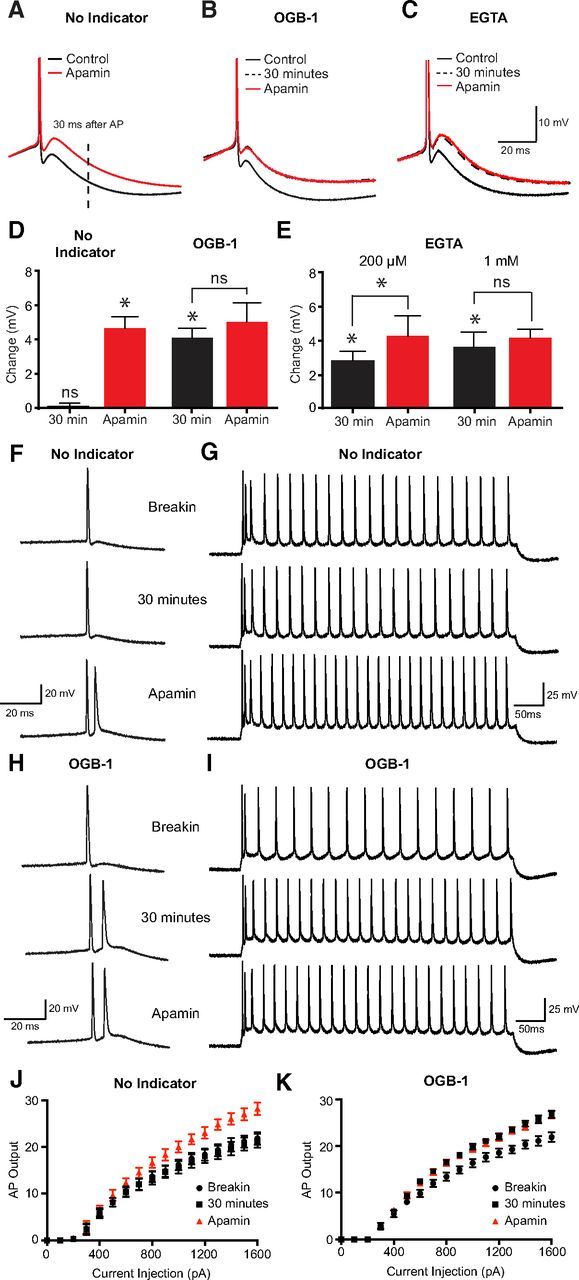
Impact of calcium chelators on the mAHP and action potential firing. A–C, Truncated somatic action potentials evoked by 200 ms somatic current pulses showing the mAHP in control just after initiation of recording (black, solid line), 30 min after recording (black, dashed line) and in the presence of apamin (red, solid line). Recordings were made with no internal calcium indicator in the pipette solution (A), and with OGB-1 (B; 200 μm) or EGTA (C; 1 mm) in the pipette solution. mAHP amplitude was measured 30 ms after AP onset (dashed vertical line in A). D, E, Average change in mAHP amplitude in the absence (black) and presence (red) of apamin in cells with and without OGB-1 in the pipette solution (D) and in cells dialyzed with 200 μm or 1 mm EGTA (E). F, G, Action potential firing in response to a 200 ms current step just above rheobase (F) or in response to a 500 ms current step (G, 1600 pA amplitude) just after break-in (top), after 30 min (middle), and in apamin (bottom) in control. H, I, Action potential firing in response to a 200 ms current step just above rheobase (H) or in response to a 500 ms current step (I, 1600 pA amplitude) just after break-in (top), after 30 min (middle), and in apamin (bottom) in cells dialyzed with 200 μm OGB-1. J, K, Input–output properties during 500 ms somatic current injections in control (J) and in cells dialyzed with 200 μm OGB-1 (K) immediately after break-in (black circles), after 30 min (black squares), and after the addition of apamin (red triangles). *p < 0.05. ns, Not significant.
Impact of OGB-1 on action potential firing
One consequence of the inhibition of the mAHP is a decrease in the interspike interval (Schwindt et al., 1988; Stocker et al., 1999). Consistent with previous findings, the addition of apamin to the extracellular solution caused a significant increase in firing frequency during long current steps (500 ms) and led to burst firing in 7 of 10 neurons during shorter current steps (200 ms) just above rheobase (Fig. 5F,G,J). We next investigated whether dialysis of OGB-1 also leads to similar changes in AP firing frequency and the propensity for burst firing. So as to obtain conditions in the absence of OGB-1 pipettes in these experiments were tip filled with intracellular solution without the indicator and then backfilled with solution containing the dye. Over the first 30 min of the recording, there was a significant change in the f–I relationship with many (8 of 10) regular spiking neurons displaying burst firing after dialysis with OGB-1 (Fig. 5H,I,K). Subsequent addition of apamin to the extracellular solution in these experiments led to no additional effect on AP firing, indicating that SK channels were blocked (Fig. 5H,I,K). Rheobase and input resistance were unchanged by the dialysis of OGB-1 or the addition of apamin (Table 1). An increase in the incidence of burst firing was also observed after dialysis with 1 mm EGTA (three of six neurons). These data indicate that the inclusion of low concentrations of exogenous calcium buffers, such as fluorescent calcium indicators, can significantly alter the firing properties of L5 pyramidal neurons due to the weak coupling between somatic SK channels and their calcium source.
Table 1.
Impact of apamin and OGB-1 on rheobase and input resistance
| Break-in | 30 min | Apamin | N | |
|---|---|---|---|---|
| No indicator | ||||
| Rheobase (pA) | 324 ± 32.31 | 323 ± 35.30 | 319 ± 35.51 | 10 |
| RN (MΩ) | 27 ± 1.71 | 27 ± 1.63 | 26 ± 1.60 | 10 |
| OGB-1 | ||||
| Rheobase (pA) | 269 ± 12.69 | 265 ± 16.20 | 265 ± 14.92 | 10 |
| RN (MΩ) | 29 ± 1.37 | 28 ± 1.51 | 27 ± 2.13 | 10 |
Data are given as the mean ± SEM, unless otherwise stated. RN, Input resistance.
The calcium source for SK channel activation during the mAHP
We next investigated the calcium source for the activation of SK channels at the soma during the mAHP. The observation that low concentrations of OGB-1 (200 μm) or EGTA (1 mm) were sufficient to abolish the mAHP (Fig. 5) suggests that somatic SK channels are not tightly coupled to their calcium source. As a result, one might expect that calcium influx through multiple VDCCs can activate SK channels during the mAHP. To investigate this, we bath applied antagonists to different VDCC subtypes and monitored their impact on the apamin-sensitive mAHP. We found that antagonists to all known VDCC subtypes except R-type VDCCs caused a reduction in the amplitude of the mAHP (Fig. 6). To determine whether an additional calcium source other than N-, P/Q-, L-, and T-type VDCCs contributes to SK channel activation during the mAHP, we applied a cocktail of antagonists to these VDCCs. Under these conditions, the addition of apamin had no significant affect on the mAHP (Fig. 6F,G; n = 6), indicating that the activation of SK channels during the mAHP can be completely perturbed by blocking N-, P/Q-, L-, and T-type VDCCs.
Figure 6.

The calcium source for the mAHP in cortical pyramidal neurons. A–F, Truncated somatic action potentials evoked by 200 ms somatic current pulses showing the mAHP in control (black, solid line), after bath application of VDCC blockers (colored dashed line) and in the presence of apamin (red, solid line). A, Impact of the R-type VDCC blocker SNX 482 (300 nm; n = 5). B, Impact of the N-type VDCC blocker conotoxin GVIA (1 μm; n = 6). C, Impact of the L-type VDCC blocker nifedipine (15 μm; n = 6). D, Impact of the T-type VDCC blocker NNC 55–0396 (20 μm, n = 5). E, Impact of the P/Q-type VDCC blocker agatoxin IVA (300 nm; n = 6). F, Impact of a cocktail of N-, L-, T-, and P/Q-type VDCC blockers (n = 4). G, Average change in mAHP amplitude (±SEM) relative to control under the different recording conditions (mAHP measured 30 ms after AP onset). *p < 0.05, **p < 0.01. ns, Not significant.
One likely explanation for the absence of a contribution of R-type VDCCs to the amplitude of the mAHP is that R-type VDCCs are not expressed at the soma of layer 5 pyramidal neurons. To investigate this, somatic and dendritic calcium imaging was performed in the absence and presence of SNX 482. There was no difference in calcium influx at the soma in the two conditions, whereas a ∼20% decrease in calcium influx was observed in basal dendrites ∼100 μm from the soma in the presence of SNX 482 (Fig. 7; n = 5). These data suggest that R-type VDCCs are not expressed at the soma but, as also shown in Figure 2, are expressed in basal dendrites, and indicates that the absence of a role of R-type VDCCs in the generation of the mAHP (Fig. 6) is due to a lack of expression of these channels at the soma.
Figure 7.

R-type VDCCs are not expressed at the soma of L5 pyramidal neurons. A, Layer 5 pyramidal neuron filled with OGB-1. The black and yellow boxes indicate somatic and dendritic regions of interest for point scans, the results of which are shown in B. Scale bar, 35 μm. B, Point scans of regions shown in A during a single AP before and after wash-in of SNX 482. Solid line and shaded regions represent the mean ± SEM. C, Average peak ΔF/F for the soma and dendrite before and after wash-in of SNX 482 (±SEM; n = 5). D, Average percentage change in fluorescence transients after wash-in of SNX 482 for the soma and dendrite (±SEM; n = 5). **p < 0.01, ns, Not significant.
Together, these findings demonstrate that SK channels mediating the apamin-sensitive component of the mAHP can be activated by calcium influx through a variety of VDCC subtypes, consistent with the idea that they are not tightly coupled to their calcium source. Furthermore, in contrast to SK channels in spines and dendrites, which are coupled solely to R-type VDCCs, all known VDCC subtypes except R type are involved in the activation of the SK channels mediating the mAHP at the soma of cortical L5 pyramidal neurons.
Discussion
The experiments presented here provide the first observations of SK channel activation in spines and dendrites of cortical pyramidal neurons during bAPs. We find that during bAP SK channels regulate calcium influx into spines and dendrites in a distance-dependent manner, with a greater impact at distal dendritic locations. Furthermore, we show that R-type VDCCs exhibit tight and specific control of SK channel activation in spines during bAPs. In contrast, coupling of SK channels at the soma to VDCCs is much less specific, with all known VDCCs, except R-type channels, playing a role in SK activation during the mAHP.
SK channel activation in dendrites and spines during action potentials
The observed increase in bAP-evoked calcium influx during SK channel block is presumably due to enhanced activation of VDCCs in spines and dendrites following an increase in amplitude or broadening of bAPs. Consistent with this idea, it has recently been observed that SK channels can control bAP amplitude in cerebellar Purkinje neurons (Ohtsuki et al., 2012). The observation that dendritic SK channels can influence bAPs is surprising given that apamin had no impact on the somatic AP waveform, but may be due to the tighter coupling of SK channels in spines and dendrites to their calcium source, speeding their activation compared with SK channels at the soma. Previous studies indicate that SK channels can activate within a millisecond during rapid changes in intracellular calcium at room temperature (Xia et al., 1998), and would be expected to activate even faster at physiological temperatures. In addition, one might expect SK channels to have a greater impact on bAPs due to their increased duration compared with somatic APs (Stuart et al., 1997). Consistent with this idea, the impact of SK channels on bAP-evoked calcium transients was greatest at distal basal dendritic locations where bAP duration is longest (Kampa and Stuart, 2006; but see Antic, 2003). The distance-dependent impact of apamin on bAP-evoked calcium transients could also be due to differences in the expression of SK or R-type calcium channels. Finally, we observed that blocking SK channels caused a greater increase in bAP-evoked calcium influx in spines compared with dendrites. While this effect may also be due to differences in the expression of SK or R-type calcium channels, the larger surface-to-volume ratio of spines compared with dendrites is also likely to contribute (Sabatini et al., 2002).
What possible function might SK channels in spines and dendrites serve when activated by bAPs? The capacity of SK channels in spines and dendrites to constrain the amplitude and/or width of bAPs would be expected to influence NMDA receptor activation during EPSP—AP pairing. This effect would be greatest at distal dendritic locations, where NMDA receptor activation during synaptic events is most pronounced (Branco and Häusser, 2011). Given that changes in synaptic strength during spike timing-dependent plasticity (STDP) are dependent on NMDA receptor activation (Markram et al., 1997), the impact of SK channels on bAP time course may play a role in setting the STDP time window (Froemke et al., 2005; Letzkus et al., 2006), possibly increasing the fidelity of coincidence detection during STDP, particularly at distal dendritic locations.
As SK channels play an important role in regulating dendritic calcium dynamics, modulation of these channels would be expected to modify neuronal excitability and synaptic plasticity. Consistent with this idea, downregulation of SK channels following activation of M1 muscarinic or β-adrenoceptor receptors in CA1 pyramidal (Buchanan et al., 2010; Giessel and Sabatini, 2010) and lateral amygdala neurons (Faber et al., 2008), respectively, increases synaptic strength. Conversely, changes in synaptic strength during synaptic plasticity have been shown to be associated with changes in SK channel function (Lin et al., 2008; Ohtsuki et al., 2012). Given the specific coupling of R-type VDCCs to SK channels in spines as shown here and previously (Bloodgood and Sabatini, 2007), modulation of R-type VDCCs following activation of D2 dopamine receptors (Higley and Sabatini, 2010) could provide another mechanism in which SK channel activation in spines may be regulated. Recent evidence indicates that the inhibition of individual dendritic spines can modulate spine calcium influx during bAPs in a selective manner (Chiu et al., 2013). These data suggest that SK channel modulation in individual spines could selectively influence calcium influx into only those spines during bAPs, although this effect may be dominated by the progressive recruitment of SK channels distributed along the entire length of a dendritic branch. Consistent with this idea, the impact of SK channel activation on bAP calcium influx increased with distance from the soma (Fig. 1E,F).
Calcium sources for SK channel activation in dendritic spines
While there is evidence that R-type VDCCs control SK channel activation in spines during EPSP-like events evoked by glutamate uncaging (Bloodgood and Sabatini, 2007), other studies have suggested that calcium influx through NMDA receptors contributes to SK channel activation during EPSPs (Faber et al., 2005; Ngo-Anh et al., 2005; Faber, 2010). Consistent with this latter idea, NMDA receptors and SK channels are colocalized within the postsynaptic density in CA1 pyramidal neurons (Lin et al., 2008). Because SK channels in spines modulate NMDA receptor activation (Faber et al., 2005; Ngo-Anh et al., 2005; Faber, 2010), which provides the main calcium source during synaptic activation (Kovalchuk et al., 2000; Sabatini et al., 2002), identifying the calcium source driving SK channel activation in spines during EPSPs is complicated. This complication does not exist in our experiments as NMDA receptor activation during bAPs is negligible (Koester and Sakmann, 2000; Sabatini and Svoboda, 2000; Bloodgood and Sabatini, 2007). During bAPs, we find that the inhibition of solely R-type VDCCs is sufficient to block SK channel activation. Moreover, the inhibition of N- and P/Q-type VDCCs, which are expressed in spines along with R-type VDCCs and led to similar calcium influx during bAPs, did not influence SK channel activation. This indicates tight and specific coupling between R-type VDCCs and SK channels within the spine head. This conclusion is similar to that made previously during EPSP-like events evoked by glutamate uncaging in spines from CA1 pyramidal neurons (Bloodgood and Sabatini, 2007). These data suggest that R-type VDCCs and SK channels in the spine head are coupled in “nanodomains,” consistent with previous observations showing that only high concentrations of the fast, high-affinity calcium buffer BAPTA are able to interfere with SK channel activation in spines during EPSPs (Ngo-Anh et al., 2005).
Calcium sources for SK channel activation during the mAHP
SK channels contribute to the mAHP in many neuronal cell types, including L5 pyramidal neurons (Schwindt et al., 1988; Sah and McLachlan, 1991; Faber and Sah, 2002; Womack and Khodakhah, 2003), although this is controversial in CA1 pyramidal neurons (Stocker et al., 1999; Gu et al., 2008). The ability of low concentrations of both fast (OGB-1) and slow (EGTA) calcium buffers to inhibit the mAHP in L5 cortical pyramidal neurons suggests that the calcium influx driving activation of somatic SK channels is working within a microdomain rather than a nanodomain (Neher, 1998; Augustine et al., 2003; Eggermann et al., 2012), with a coupling distance greater than ∼150 nm. Consistent with this idea, we show that the SK channel-dependent component of the mAHP in L5 neurons is controlled by all known VDCC subtypes except R-type VDCCs, which are not expressed at the soma. The coupling between somatic SK channels and their calcium source in L5 neurons differs from that in other cell types, where the calcium source for SK channel activation during the mAHP has been linked to specific VDCC subtypes. For example, in midbrain dopaminergic neuron activation of SK channels during the mAHP is solely dependent on T-type VDCCs (Wolfart and Roeper, 2002), whereas only L-type channels are coupled to somatic SK channels in hippocampal pyramidal neurons (Marrion and Tavalin, 1998). The reasons for this difference between neuronal cell types, and why it is that SK channels are weakly coupled to multiple calcium sources in L5 neurons is unclear. Finally, it is worth noting that our observation that low concentrations of the calcium indicator OGB-1 blocks the mAHP, increasing firing rate and promoting burst firing, raises the concern that the use of high-affinity calcium indicators to investigate network activity (Garaschuk et al., 2006) may inadvertently influence neuronal excitability and thereby network dynamics.
In conclusion, we show that SK channels in spines and dendrites are activated by bAPs and act to constrain dendritic and spine calcium influx in a distance-dependent manner. This effect of SK channels would be expected to influence STDP, particularly at distal dendritic locations. Furthermore, we provide evidence that SK channels in spines and dendrites are strongly coupled to their calcium source, forming nanodomains with R-type VDCCs. In contrast, SK channels at the soma of L5 cortical pyramidal neurons are weakly coupled to multiple calcium sources, forming microdomains with all known VDCCs except R-type channels. These findings provide evidence for heterogeneous and location-dependent coupling of SK channels to VDCCs within the same neuronal cell type. Such exquisite compartmentalization exemplifies the contrasting role calcium plays in regulating neuronal excitability at different cellular locations even within the same neuron.
Footnotes
This work was supported by the Australian National Health and Medical Research Council and the John Curtin School of Medical Research. We are grateful to Dr. Jens-Karl Eilers, Dr. Jean-Didier Breton, and Tobias Bock for many helpful discussions; and Dr. Kaori Ikeda for comments on the manuscript.
The authors declare no competing financial interests.
References
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Antic SD. Action potentials in basal and oblique dendrites of rat neocortical pyramidal neurons. J Physiol. 2003;550:35–50. doi: 10.1113/jphysiol.2002.033746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/S0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Berkefeld H, Fakler B, Schulte U. Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev. 2010;90:1437–1459. doi: 10.1152/physrev.00049.2009. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Branco T, Häusser M. Synaptic integration gradients in single cortical pyramidal cell dendrites. Neuron. 2011;69:885–892. doi: 10.1016/j.neuron.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan KA, Petrovic MM, Chamberlain SE, Marrion NV, Mellor JR. Facilitation of long-term potentiation by muscarinic M(1) receptors is mediated by inhibition of SK channels. Neuron. 2010;68:948–963. doi: 10.1016/j.neuron.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Kao JP, Tang CM, Thompson SM. Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron. 2004;44:351–364. doi: 10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis-Davies GC, Higley MJ. Compartmentalization of GABAergic inhibition by dendritic spines. Science. 2013;340:759–762. doi: 10.1126/science.1234274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca(2) channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci. 2012;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engbers JD, Anderson D, Asmara H, Rehak R, Mehaffey WH, Hameed S, McKay BE, Kruskic M, Zamponi GW, Turner RW. Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc Natl Acad Sci U S A. 2012;109:2601–2606. doi: 10.1073/pnas.1115024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES. Functional interplay between NMDA receptors, SK channels and voltage-gated Ca2+ channels regulates synaptic excitability in the medial prefrontal cortex. J Physiol. 2010;588:1281–1292. doi: 10.1113/jphysiol.2009.185645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Power JM, Sedlak PL, Crane JW, Sah P. Modulation of SK channel trafficking by beta adrenoceptors enhances excitatory synaptic transmission and plasticity in the amygdala. J Neurosci. 2008;28:10803–10813. doi: 10.1523/JNEUROSCI.1796-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Poo MM, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Milos RI, Konnerth A. Targeted bulk-loading of fluorescent indicators for two-photon brain imaging in vivo. Nat Protoc. 2006;1:380–386. doi: 10.1038/nprot.2006.58. [DOI] [PubMed] [Google Scholar]
- Giessel AJ, Sabatini BL. M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron. 2010;68:936–947. doi: 10.1016/j.neuron.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Hu H, Vervaeke K, Storm JF. SK (KCa2) channels do not control somatic excitability in CA1 pyramidal neurons but can be activated by dendritic excitatory synapses and regulate their impact. J Neurophysiol. 2008;100:2589–2604. doi: 10.1152/jn.90433.2008. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ. Heterogeneity of phasic cholinergic signaling in neocortical neurons. J Neurophysiol. 2007;97:2215–2229. doi: 10.1152/jn.00493.2006. [DOI] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- Kampa BM, Stuart GJ. Calcium spikes in basal dendrites of layer 5 pyramidal neurons during action potential bursts. J Neurosci. 2006;26:7424–7432. doi: 10.1523/JNEUROSCI.3062-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol. 2000;529:625–646. doi: 10.1111/j.1469-7793.2000.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J Neurosci. 2000;20:1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Kampa BM, Stuart GJ. Learning rules for spike timing-dependent plasticity depend on dendritic synapse location. J Neurosci. 2006;26:10420–10429. doi: 10.1523/JNEUROSCI.2650-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Luján R, Watanabe M, Adelman JP, Maylie J. SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat Neurosci. 2008;11:170–177. doi: 10.1038/nn2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Novo D, Mody I, Vergara JL. Binding kinetics of calbindin-D28k determined by flash photolysis of caged Ca2+ Biophys J. 2000;79:3009–3018. doi: 10.1016/S0006-3495(00)76537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/S0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Ohtsuki G, Piochon C, Adelman JP, Hansel C. SK2 channel modulation contributes to compartment-specific dendritic plasticity in cerebellar Purkinje cells. Neuron. 2012;75:108–120. doi: 10.1016/j.neuron.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Svoboda K. Analysis of calcium channels in single spines using optical fluctuation analysis. Nature. 2000;408:589–593. doi: 10.1038/35046076. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca(2+) ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/S0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Sah P, McLachlan EM. Ca(2+)-activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: a role for Ca(2+)-activated Ca2+ release. Neuron. 1991;7:257–264. doi: 10.1016/0896-6273(91)90264-Z. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol. 1997;505:605–616. doi: 10.1111/j.1469-7793.1997.605ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Clapham DE. NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nat Neurosci. 1998;1:114–118. doi: 10.1038/363. [DOI] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Stafstrom CE, Chubb MC, Crill WE. Multiple potassium conductances and their functions in neurons from cat sensorimotor cortex in vitro. J Neurophysiol. 1988;59:424–449. doi: 10.1152/jn.1988.59.2.424. [DOI] [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflugers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- Stuart G, Schiller J, Sakmann B. Action potential initiation and propagation in rat neocortical pyramidal neurons. J Physiol. 1997;505:617–632. doi: 10.1111/j.1469-7793.1997.617ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfart J, Roeper J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci. 2002;22:3404–3413. doi: 10.1523/JNEUROSCI.22-09-03404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J Neurosci. 2003;23:2600–2607. doi: 10.1523/JNEUROSCI.23-07-02600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Chevez C, Khodakhah K. Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci. 2004;24:8818–8822. doi: 10.1523/JNEUROSCI.2915-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]