

Abstract
The bed nucleus of the stria terminalis (BNST) exerts a coordinated modulation of the psychoneuroendocrine responses to stress. However, how acute stress impacts on BNST in vivo plasticity is a crucial question that still remains unanswered. Here, neurons from the anterior portion of the BNST (aBNST) were recorded in vivo during and after stimulation of their medial prefrontal cortical (mPFC) afferents. In C57BL/6N mice, a 1 h restraint stress induced a switch from long-term depression (LTD) to long-term potentiation (LTP) in the aBNST after a 10 Hz mPFC stimulation. This switch was independent from glucocorticoid receptor stimulation. Because the endocannabinoid system regulates aBNST activity, we next examined the role of cannabinoid type-1 receptors (CB1-Rs) in these changes. Mutant mice lacking CB1-Rs (CB1−/− mice) displayed a marked deficit in the ability to develop plasticity under control and stress conditions, compared with their wild-type littermates (CB1+/+ mice). This difference was not accounted for by genetic differences in stress sensitivity, as revealed by Fos immunohistochemistry analyses. Local blockade of CB1-Rs in the aBNST and the use of mutant mice bearing a selective deletion of CB1-Rs in cortical glutamatergic neurons indicated that stress-elicited LTP involved CB1-Rs located on aBNST excitatory terminals. These results show that acute stress reverts LTD into LTP in the aBNST and that the endocannabinoid system plays a key role therein.
Keywords: corticosterone, endocannabinoid, in vivo, LTD, LTP
Introduction
Acute and chronic stress alter the ability of cortical excitatory synapses to undergo plasticity (Diamond et al., 2007). Across species, the bed nucleus of the stria terminalis (BNST) has been documented to be a key target of stress (Davis et al., 2010). As an illustration, the anterior portion of the BNST (aBNST), which innervates the paraventricular hypothalamic nucleus (PVN; Radley et al., 2009), has been shown thereby to negatively regulate the hypothalamus-pituitary-adrenal axis (Ulrich-Lai and Herman, 2009). Although the aBNST receives a major input from the medial prefrontal cortex (mPFC; Massi et al., 2008; Radley et al., 2009), it is unknown whether stress impacts the firing and plastic responses of aBNST neurons in response to mPFC stimulation. Accumulating evidence points to a prominent role of the endocannabinoid (eCB) system in controlling neuronal circuits during stress (Hill et al., 2010). Actually, eCBs are released from postsynaptic neurons and act retrogradely on cannabinoid type-1 receptors (CB1-Rs) to control transmitter release and plasticity in many brain structures, including the aBNST (Grueter et al., 2006; Puente et al., 2011). We reported previously that CB1-Rs, which are present on aBNST axon terminals arising from the mPFC, exert a phasic control on cortical afferents to aBNST neurons in vivo (Massi et al., 2008). These results thus open the hypothesis that an acute stress might affect plasticity in the aBNST in response to mPFC stimulation, with the eCB system playing a role therein. Using in vivo electrophysiology, pharmacology, and genetics, we show here that (1) an acute stress reverses aBNST neuronal plasticity from long-term depression (LTD) to long-term potentiation (LTP) in response to mPFC stimulation, and (2) this shift is controlled by CB1-Rs located on aBNST glutamatergic terminals.
Materials and Methods
Animals.
Two-month-old male C57BL/6N mice (Janvier) and 2- 3-month-old male CB1-R wild-type/mutant animals (bred at the Neurocentre Magendie) were housed individually with food/water ad libitum under controlled conditions (22−23°C, 50–55% relative humidity, 12 h light/dark cycle with lights on at 07:00). Constitutive CB1 mutant mice (CB1−/−) or conditional mutant mice lacking CB1-Rs from cortical glutamatergic neurons (hereafter called Glu-CB1−/−), and their respective wild-type littermates were obtained, maintained, and genotyped/re-genotyped as described previously (Monory et al., 2006; Dubreucq et al., 2012). Glu-CB1−/− mice were obtained using the Cre/loxP system by crossing CB1-floxed mice with NEX-Cre mice (Monory et al., 2006). Those animals were in a mixed genetic background with a predominant C57BL/6N contribution. All procedures were conducted in accordance with the European directive 2010-63-EU and with approval from the Bordeaux University Animal Care and Use Committee (N°50120205-A).
Acute restraint stress procedure.
The acute stress procedure, which was performed in the morning, consisted of restraining the animals for 1 h in well ventilated 50 ml plastic tubes. Thereafter, stressed mice (and unstressed, i.e., controls) were immediately anesthetized for in vivo electrophysiology experiments (Fig. 1A,I).
Figure 1.
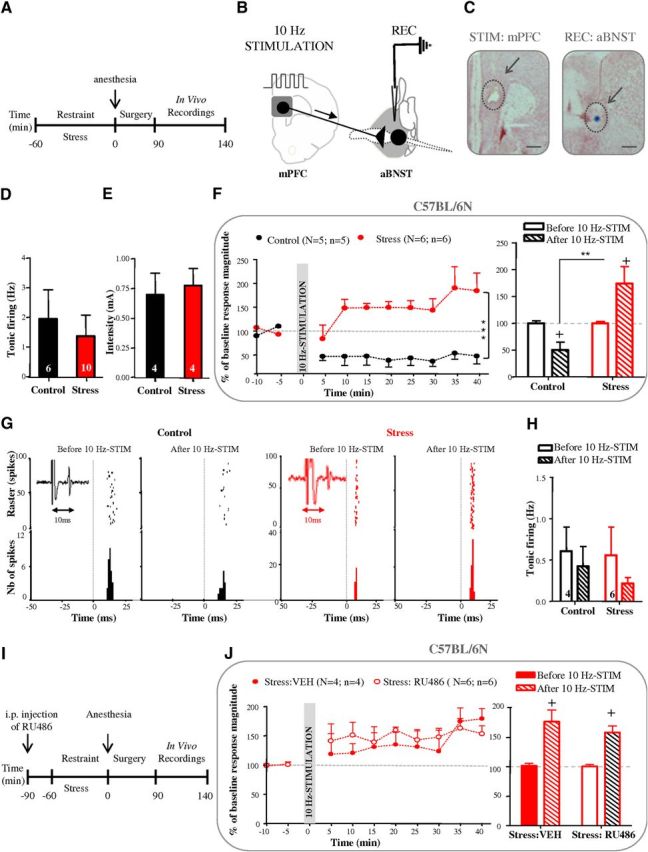
The 10 Hz mPFC stimulation has opposite effects on aBNST plasticity in control and stressed anesthetized mice. A, Time line of experiment. B, Stimulation and recording protocol. C, Histological controls of stimulation (mPFC, lesion at arrow) and recording (aBNST, blue spot at arrow) sites. Scale bar, 1 mm. D, E, Graphs illustrating the effects of stress on basal activity frequency (D) and cortical excitation strength (E). F, Kinetic (left) and quantification (right) of the mean percentage change (±SEM) in mPFC-evoked spike probability, normalized to the baseline, after 10 Hz mPFC stimulation (at t0) in control and stressed mice. G, Typical PSTHs and associated rasters illustrate responses of aBNST neurons before and after 10 Hz mPFC stimulation in control (left) and stressed mice (right). Stimulus at t0 (gray lines). Bin width, 1 ms. Representative electrophysiological traces in insets. H, Quantification of basal firing activity of aBNST neurons (mean ± SEM) before and 35–40 min after 10 Hz mPFC stimulation in control and stressed mice. I, Time line of experiment. J, Kinetic (left) and quantification (right) of the mean percentage change (±SEM) in mPFC-evoked spike probability, normalized to the baseline, after 10 Hz mPFC stimulation (at t0) in stressed mice pretreated with RU486 or vehicle. N = number of mice; n = number of neurons. *indicates significant differences between control and stress groups; + indicates significant differences between baseline (before) and 35–40 min after 10 Hz mPFC stimulation. +p < 0.05, **p < 0.01; ***p < 0.001. Numbers in brackets of histogram bars refer to the number of aBNST neurons recorded. The same nomenclature is used for all figure legends.
Surgery.
Stereotaxic surgery for in vivo electrophysiology experiments was performed under isoflurane anesthesia as previously described (Georges and Aston-Jones, 2002). Stimulation electrodes and recording pipettes were, respectively, inserted into the mPFC (+2.0 mm/bregma, 0.5 mm/midline, and 2.5 mm/brain surface) and the aBNST (+0.2 mm/bregma, 0.8 mm/midline, 3.2–4.2 mm/brain surface).
Electrical stimulation of the mPFC.
Bipolar electrical stimulation of the mPFC was conducted with a concentric electrode (Phymep) and a stimulus isolator (500 μs, 0.2–1-mA; Digitimer). Baseline was recorded for 10 min (2 × 100 pulses; 0.5 Hz) and tetanic stimulation was performed (1 min, 10 Hz). Only one cell per mouse was recorded for neuroplastic responses.
aBNST recordings and pharmacological micro-infusion.
A glass micropipette (tip diameter, 1–2 μm; 10–15-MΩ) filled with a 2% pontamine sky blue solution in 0.5 m sodium acetate was lowered into the aBNST. The extracellular potential was recorded with an Axoclamp-2B amplifier and filtered (300 Hz/0.5 kHz; Georges and Aston-Jones, 2002). Single neuron spikes were collected online (CED1401, SPIKE2; Cambridge Electronic Design). During electrical stimulation of the mPFC, cumulative peristimulus time histograms (PSTHs, 5 ms bin width) of BNST activity were generated for each neuron recorded. For local delivery of 1 μm AM251 (Tocris Bioscience), or its vehicle (artificial CSF containing 5% dimethylsulfoxide (DMSO) and 5% cremophor), double-barrel pipettes (Georges and Aston-Jones, 2002) were used. Five minutes after micro-infusion into the aBNST, the mPFC was electrically stimulated for 1 min at 10 Hz. Post-tetanic evoked responses were recorded for 40 min while stimulating the mPFC (0.5 Hz).
In one series of experiments, stressed mice were injected intraperitoneally 30 min beforehand with vehicle (1.2% DMSO and 5% Tween 80 in water) or 30 mg/kg of the glucocorticoid receptor (GR) antagonist mifepristone (RU486; Sigma-Aldrich).
Histology.
At the end of each recording experiment, the electrode placement was marked with an iontophoretic deposit of pontamine sky blue dye (−20 μA, 30 min). To mark electrical stimulation sites, +50 μA was passed through the stimulation electrode for 2 min. (Fig. 1B,C).
Fos immunohistochemistry procedure.
Ninety minutes following the acute restraint stress, mice were perfused transcardially (4% paraformaldehyde). Sections were incubated (overnight/4°C) with rabbit-anti-Fos polyclonal antibody (1:8000; Calbiochem). Following washes, sections were incubated overnight at 4°C in biotinylated donkey anti-rabbit secondary antibody (1:200; Millipore), followed by an Avidin/Biotin Complex (ABC solution; Vector Laboratories). Staining was revealed after incubation in a diaminobenzidine/nickel solution.
Data analysis.
For in vivo electrophysiological experiments, cumulative PSTHs of aBNST activity were generated during electrical stimulation of the mPFC. Excitatory magnitudes (Rmag values) were normalized for different levels of baseline impulse activity. Rmag values for excitation were calculated according to: Excitation Rmag=(counts in excitatory epoch) − (mean counts per baseline bin × number of bins in excitatory epoch). The cortical excitation strength onto aBNST neurons was determined as the amount of current needed to obtain a 50% spike probability for mPFC-evoked responses (Rmag ranging from 30 to 60). For Fos experiments, blind counts of Fos-immunoreactive neurons in the mPFC, aBNST, and PVN were performed and a density of labeling (number of neurons Fos positive per μm2) was calculated to establish comparisons. Two-group comparisons were achieved using Student's t tests. For multiple comparisons, values were subjected to a two-way ANOVA followed (if significant) by Newman–Keuls post hoc tests.
Results
A 10 Hz stimulation of mPFC neurons induced opposite plastic changes in the aBNST of control and stressed mice
Using anesthetized C57BL/6N mice, we first established that neither the tonic firing activity of aBNST neurons (Fig. 1D) nor the cortical excitation strength onto aBNST neurons (Fig. 1E) was affected by acute stress. We then determined whether a 1 min in vivo activation of the mPFC at a physiologically relevant frequency (Jackson et al., 2001) triggers neuroplastic changes in aBNST neurons (Fig. 1F,G). In control mice, a 10 Hz mPFC stimulation led to a significant decrease in mPFC-evoked spike probability in the aBNST that was evidenced by a 49.5% LTD of the excitatory response 35–40 min thereafter (corresponding to an increase in the percentage failure of evoked responses from 66.9 to 84.9% before and after the 10 Hz stimulation). In contrast, in the stress group, a significant increase in mPFC-evoked spike probability was observed following 10 Hz mPFC stimulation, as illustrated by a +74.3% LTP of the excitatory response measured 35–40 min after tetanic stimulation (corresponding to a decrease in the percentage failure of evoked responses from 54.6% in baseline to 21.3% before and after the 10 Hz stimulation). Thus, 10 Hz mPFC stimulation triggered a significant switch in the polarity of aBNST plasticity, from an LTD in controls to an LTP in stressed mice (F(1,64) = 120.27, p < 0.0001 for the overall influence of stress and F(1,16) = 18.9, p < 0.0005 for the stress × 10 Hz tetanus interaction at times 35–40 min; Figure 1F). On the other hand, the tonic firing activity of aBNST neurons remained unaffected by the 10 Hz mPFC stimulation, in both mouse groups (Fig. 1H). Last, we tested whether stress-induced corticosterone release, through GR stimulation, was involved in the stress-dependent LTP induced by 10 Hz mPFC stimulation. As illustrated in Figure 1I, mice were thus pretreated with vehicle or with the GR antagonist RU486 (30 mg/kg, 30 min beforehand: Fiancette et al., 2010). The 10 Hz mPFC stimulation-elicited LTP in the aBNST of C57BL/6N stressed mice was unaltered by RU486 pretreatment, as illustrated by a significant impact of the 10 Hz mPFC stimulation in both mouse groups (F(1,8) = 36.24, p < 0.001) without any interaction with the mouse condition (control or stressed; Fig. 1J).
CB1-Rs control prefrontal LTD and LTP in aBNST neurons
We first compared Fos responses to acute stress in brain regions of mutant mice lacking CB1-Rs (CB1−/−) and in their wild-type littermates (CB1+/+). We focused on three brain regions thought to play a key role in shaping major stress responses, namely the PVN, the mPFC, and the aBNST (Spencer et al., 2005). Exposure to acute stress resulted in a marked increase in Fos staining in the PVN (F(1,8) = 567, p < 0.0001) and the mPFC (F(1,8) = 34.14, p < 0.001), but not in the aBNST of CB1+/+ and CB1−/− mice (Fig. 2). Moreover, CB1+/+ and CB1−/− mice showed no significant difference in Fos activation in any of the three brain regions (Fig. 2), suggesting that both genotypes displayed a similar response to stress. We next investigated the consequences of stress on the neuroplastic changes in the aBNST of anesthetized CB1+/+ and CB1−/− mice. CB1+/+ mice behaved as C57BL/6N mice (see above) as stress triggered a switch in the polarity of aBNST plasticity responses to the 10 Hz mPFC stimulation (F(1,51) = 95.18, p < 0.0001 for the overall influence of stress and F(1,12) = 18.32, p < 0.005 for the stress × 10 Hz tetanus interaction at times 35–40 min; Figure 3A). Thus, the 10 Hz mPFC stimulation led to LTD in the aBNST of the control group (corresponding to an increase in the percentage failure of evoked responses from 59.38 to 83.75% before and after the 10 Hz stimulation; Fig. 3A) while it promoted LTP in the stress group (corresponding to a decrease in the percentage failure of evoked responses from 60.3 to 38% before and after the 10 Hz stimulation; Fig. 3A). Conversely, the 10 Hz mPFC stimulation failed to trigger a stable depression throughout the 40 min of analysis in control CB1−/− mice or LTP in stressed CB1−/− mice (Fig. 3B). In both genotypes, neither stress nor the 10 Hz mPFC stimulation influenced the tonic firing activity of aBNST neurons and the cortical excitation strength onto aBNST neurons (Fig. 3C–F).
Figure 2.

CB1+/+ and CB1−/− mice displayed a similar response to stress. A–F, Effect of acute stress on Fos-immunoreactive neurons in PVN, mPFC, and aBNST of control and stressed CB1+/+ or CB1−/− mice. A, C, E, Histograms showing the density of Fos-positive neurons after an acute stress in CB1+/+ and CB1−/− mice. PVN and mPFC displayed the expected enhancement of stress-induced cellular activation in CB1+/+ but also in CB1−/− mice. Stress-induced Fos staining was similar in CB1+/+ and CB1−/− mice. B, D, F, Representative micrographs of immunostained sections for Fos of the PVN (B), mPFC (D), and aBNST (F). Note that no stress-induced Fos activation was observed in the aBNST of CB1+/+ or CB1−/− mice (E, F). + indicates significant differences between control and stress groups. +p < 0.05, ++p < 0.01. Scale bars: B, 0.4 mm; D, 0.2 mm; F, 0.6 mm. ac, Anterior commissure; PL, prelimbic cortex; IL, infralimbic cortex; cc, corpus callosum; Ov, oval nucleus; aBNSTv, ventral part of aBNST; dBNSTd, dorsal part of aBNST; 3V, third ventricle. Numbers in brackets of histogram bars refer to the number of mice used.
Figure 3.

Neuroplastic changes in the aBNST depend on CB1-Rs activation. A, B, Kinetic (left) and quantification (right) of the mean percentage change (±SEM) in mPFC-evoked spike probability, normalized to the baseline, after 10 Hz mPFC stimulation (at t0) in control and stressed CB1+/+ or CB1−/− mice. C–F, Graphs illustrating the effects of stress and/or 10 Hz mPFC stimulation on basal activity frequency (C, E, F) and cortical excitation strength (D). See Figure 1 legend for nomenclature.
The CB1-Rs that control the stress-dependent LTP are located on cortical glutamatergic terminals of the aBNST
In keeping with the aforementioned observations, we first tested the hypothesis that the CB1-Rs controlling stress-induced LTP are located in the aBNST (Fig. 4A). Actually, the 10 Hz mPFC stimulation-elicited LTP in the aBNST of C57BL/6N stressed mice was fully blocked by the intra-aBNST infusion (60 nl, 1 μm) of the CB1-R antagonist AM251 (F(1,40) = 41.68, p < 0.0001 and F(1,10) = 8.05, p < 0.05 for the overall influence of the pretreatment and for the pretreatment × 10 Hz tetanus interaction at times 35–40 min; Figure 4B). Notably, intra-aBNST blockade of CB1-Rs decreased the excitatory evoked-responses to below prestimulation levels (corresponding to an increase in the percentage failure of evoked responses from 55.1 to 77.5% before and after the 10 Hz stimulation; Fig. 4B). Because CB1-Rs in the aBNST are located on excitatory axon terminals arising from the mPFC (Massi et al., 2008), we next hypothesized that a selective deletion of CB1Rs in cortical glutamatergic neurons (Glu-CB1−/− mice) might also block 10 Hz mPFC stimulation-elicited LTP in the aBNST. As observed in CB1+/+ mice, stress application to Glu-CB1+/+ mice induced a switch in the polarity of aBNST plasticity responses to the 10 Hz mPFC stimulation (F(1,124) = 60.04, p < 0.0001 for the overall influence of stress and F(1,30) = 14.81, p < 0.001 for the stress × 10 Hz tetanus interaction at times 35–40 min; Figure 4C). On the other hand, the 10 Hz mPFC stimulation evoked only a slight, albeit significant, decrease in the excitatory responses of aBNST neurons in both control and stressed Glu-CB1−/− mice (F(1,18) = 10.47, p < 0.005 for the negative impact of the 10 Hz stimulation at times 35–40 min; Fig. 4D). The analysis of the percentage failures of evoked responses confirmed the aforementioned genotype influence (from 60.1 to 76.6% before and after the 10 Hz stimulation in control Glu-CB1−/− mice, respectively, and from 48.9 to 72.8% before and after the 10 Hz stimulation in stressed Glu-CB1−/− mice, respectively).
Figure 4.

CB1-Rs that control LTD and LTP are located in the aBNST on mPFC terminals. A, Stimulation and recording protocol. B, The CB1-R antagonist AM251 (60 nl, 1 μm) or vehicle was infused into the aBNST before 10 Hz mPFC stimulation in stressed mice. Kinetic (left) and quantification (right) of the mean percentage change (±SEM) in mPFC-evoked spike probability, normalized to the baseline, after 10 Hz mPFC stimulation (at t0) after vehicle (VEH) or AM251 infusion in the aBNST of stressed mice. C, D, Kinetic (left) and quantification (right) of the mean percentage change (±SEM) in mPFC-evoked spike probability, normalized to the baseline, after 10 Hz mPFC stimulation (at t0) in control and stressed Glu-CB1+/+(C) or Glu-CB1−/−(D) mice. See Figure 1 legend for nomenclature.
Discussion
The first aim of this work was to investigate the consequences of acute stress on the mPFC-aBNST pathway. Our data first reveal that acute stress reverses the long-term depressive impact of mPFC stimulation on aBNST neurons onto an LTP. Second, our pharmacological and genetic approaches indicate that the eCB system, through the stimulation of CB1-Rs located on aBNST glutamatergic terminals, plays a key role in that plasticity shift.
The BNST has been associated with stress and negative emotional state (Ulrich-Lai and Herman, 2009), and recent optogenetic findings have revealed how that structure controls aversive and motivational behaviors (Jennings et al., 2013; Kim et al., 2013). Although these recent findings have highlighted the potential role exerted by the BNST in adaptation to aversive stimuli, the impact of acute stress on synaptic plasticity in the BNST, and the role of corticosterone release therein, has been mainly studied in vitro (Grueter et al., 2006; Conrad et al., 2011), while in vivo analyses are still lacking. This paucity of investigations contrasts with the well documented effects of stress on synaptic plasticity in the mPFC (Diamond et al., 2007) and the PVN (Inoue et al., 2013; Wamsteeker Cusulin et al., 2013), i.e., structures that, respectively, project to (mPFC) and to which projects (PVN) the BNST. Interestingly, CB1-Rs in the PVN have been reported to control feedback inhibition of the hypothalamic-pituitary-adrenal axis (Di et al., 2003; Evanson et al., 2010). However, in our hands, stress-induced Fos activation in the PVN was unaltered by CB1 deletion, suggesting that under our conditions, additional neuromodulators (e.g., monoamines) could participate to stress-induced Fos activation in the PVN (Herman et al., 2002; Inoue et al., 2013). Moreover, without any mPFC stimulation, aBNST neurons proved insensitive to stress, an observation in keeping with the weak effect of acute stress on basal firing properties of the neurons. However, using a stimulation protocol of the mPFC that mimics the rate of firing of mPFC neurons during a cognitive task (Jackson et al., 2001), we observed a long-lasting reduction in evoked potentials of aBNST neurons whatever the control (unstressed) mouse group considered, i.e., C57BL/6N, CB1+/+, and Glu-CB1+/+ mice. After acute stress, this aBNST LTD reverted into LTP in C57BL/6N mice, a change which occurred without any change in intrinsic excitability. Because corticosterone release, mostly through GR stimulation, (1) plays a key role in acute stress-elicited changes in synaptic plasticity in cortical and hypothalamic areas (Diamond et al., 2007; Wamsteeker Cusulin et al., 2013), and (2) tunes the activity of the eCB system (Hill et al., 2010, 2011), we examined the functional impact of GR blockade in our experimental setting. The results show that GRs do not mediate the aBNST LTP response to the 10 Hz mPFC stimulation. This indicates the need to investigate the contribution of other likely candidates such as noradrenaline or corticotropin-releasing hormone (Silberman and Winder, 2013).
Through eCB release (Puente et al., 2011), CB1-Rs present on these mPFC glutamatergic terminals control aBNST responses to cortical excitation (Massi et al., 2008). In keeping with these results, we measured through genetic and pharmacological means the contribution of the eCB system to mPFC stimulation-elicited changes in the plasticity of aBNST cells in control and stressed mice. The results gathered with the constitutive (CB1−/−) and conditional (Glu-CB1−/−) mutant mice for the CB1-Rs indicated that the stress-elicited shift in plasticity is fully controlled by CB1-Rs located on glutamatergic terminals. This observation allowed us to rule out the contribution of the CB1-Rs population located on mPFC GABA interneurons that mediates the stress-induced disinhibition of mPFC excitatory transmission (Hill et al., 2011). This statement is reinforced by the efficient impact of the local application of the CB1-Rs antagonist AM251 in the aBNST on stress-induced LTP, hence indicating that stress triggered LTP in the aBNST through CB1-Rs harbored by local glutamatergic terminals. Because the aBNST sends inhibitory projections to the PVN, our study indicates that the aBNST-eCB system might control corticosteroid release during and/or after stress. In our study, all recorded neurons were identified with respect to their anatomical location and their monosynaptic excitatory responses to mPFC stimulation. However, further work will be needed to determine both the identity of these aBNST neurons and the consequences of the aforementioned neuroplastic shift on their efferent projections (Kim et al., 2013).
The present study reveals that CB1-Rs located on aBNST excitatory terminals control the shift in the plasticity response to mPFC stimulation that is elicited by acute stress. One question, which remains to be solved, is the functional impact of such a control. Actually, CB1-Rs located on glutamatergic terminals are involved in the control of the psychoneuroendocrine responses to acute and chronic stress, doing so through different means (Hill et al., 2010; Iremonger et al., 2013). For example, in the PVN, stress-induced corticosteroid secretion may favor eCB release from neurosecretory cells as to stimulate CB1-Rs at glutamatergic terminals and thereby reduce the excitatory drive to maintain rapidly an efficient feedback of the corticotropic axis (Di et al., 2003). Hence, pharmacological experiments have indicated that this stress-induced amplification of excitatory transmission might in turn contribute to increased corticotropic axis activity (Hill et al., 2010). As recently reported in the mPFC (Hill et al., 2011), the present observation thus opens the additional hypothesis that after activation of the mPFC, aBNST CB1-Rs underlie the negative feedback on neuroendocrine consequences of stress (Radley et al., 2009).
Footnotes
This work was supported by grants from Centre National de la Recherche Scientifique, University of Bordeaux and Région Aquitaine.
References
- Conrad KL, Louderback KM, Gessner CP, Winder DG. Stress-induced alterations in anxiety-like behavior and adaptations in plasticity in the bed nucleus of the stria terminalis. Physiol Behav. 2011;104:248–256. doi: 10.1016/j.physbeh.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Miles L, Grillon C. Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology. 2010;35:105–135. doi: 10.1038/npp.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond DM, Campbell AM, Park CR, Halonen J, Zoladz PR. The temporal dynamics model of emotional memory processing: a synthesis on the neurobiological basis of stress-induced amnesia, flashbulb and traumatic memories, and the Yerkes-Dodson law. Neural Plast. 2007;2007:60803. doi: 10.1155/2007/60803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreucq S, Matias I, Cardinal P, Häring M, Lutz B, Marsicano G, Chaouloff F. Genetic dissection of the role of cannabinoid type-1 receptors in the emotional consequences of repeated social stress in mice. Neuropsychopharmacology. 2012;37:1885–1900. doi: 10.1038/npp.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151:4811–4819. doi: 10.1210/en.2010-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiancette JF, Balado E, Piazza PV, Deroche-Gamonet V. Mifepristone and spironolactone differently alter cocaine intravenous self-administration and cocaine-induced locomotion in C57BL/6J mice. Addict Biol. 2010;15:81–87. doi: 10.1111/j.1369-1600.2009.00178.x. [DOI] [PubMed] [Google Scholar]
- Georges F, Aston-Jones G. Activation of ventral tegmental area cells by the bed nucleus of the stria terminalis: a novel excitatory amino acid input to midbrain dopamine neurons. J Neurosci. 2002;22:5173–5187. doi: 10.1523/JNEUROSCI.22-12-05173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Gosnell HB, Olsen CM, Schramm-Sapyta NL, Nekrasova T, Landreth GE, Winder DG. Extracellular-signal regulated kinase 1-dependent metabotropic glutamate receptor 5-induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J Neurosci. 2006;26:3210–3219. doi: 10.1523/JNEUROSCI.0170-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Tasker JG, Ziegler DR, Cullinan WE. Local circuit regulation of paraventricular nucleus stress integration: glutamate-GABA connections. Pharmacol Biochem Behav. 2002;71:457–468. doi: 10.1016/S0091-3057(01)00681-5. [DOI] [PubMed] [Google Scholar]
- Hill MN, Patel S, Campolongo P, Tasker JG, Wotjak CT, Bains JS. Functional interactions between stress and the endocannabinoid system: from synaptic signaling to behavioral output. J Neurosci. 2010;30:14980–14986. doi: 10.1523/JNEUROSCI.4283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Pan B, Fitzgerald ML, Roberts CJ, Lee TT, Karatsoreos IN, Mackie K, Viau V, Pickel VM, McEwen BS, Liu QS, Gorzalka BB, Hillard CJ. Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci. 2011;31:10506–10515. doi: 10.1523/JNEUROSCI.0496-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue W, Baimoukhametova DV, Füzesi T, Cusulin JI, Koblinger K, Whelan PJ, Pittman QJ, Bains JS. Noradrenaline is a stress-associated metaplastic signal at GABA synapses. Nat Neurosci. 2013;16:605–612. doi: 10.1038/nn.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iremonger KJ, Wamsteeker Cusulin JI, Bains JS. Changing the tune: plasticity and adaptation of retrograde signals. Trends Neurosci. 2013;36:471–479. doi: 10.1016/j.tins.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Jackson ME, Frost AS, Moghaddam B. Stimulation of prefrontal cortex at physiologically relevant frequencies inhibits dopamine release in the nucleus accumbens. J Neurochem. 2001;78:920–923. doi: 10.1046/j.1471-4159.2001.00499.x. [DOI] [PubMed] [Google Scholar]
- Jennings JH, Sparta DR, Stamatakis AM, Ung RL, Pleil KE, Kash TL, Stuber GD. Distinct extended amygdala circuits for divergent motivational states. Nature. 2013;496:224–228. doi: 10.1038/nature12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Adhikari A, Lee SY, Marshel JH, Kim CK, Mallory CS, Lo M, Pak S, Mattis J, Lim BK, Malenka RC, Warden MR, Neve R, Tye KM, Deisseroth K. Diverging neural pathways assemble a behavioural state from separable features in anxiety. Nature. 2013;496:219–223. doi: 10.1038/nature12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi L, Elezgarai I, Puente N, Reguero L, Grandes P, Manzoni OJ, Georges F. Cannabinoid receptors in the bed nucleus of the stria terminalis control cortical excitation of midbrain dopamine cells in vivo. J Neurosci. 2008;28:10496–10508. doi: 10.1523/JNEUROSCI.2291-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wölfel B, Dodt HU, Zieglgänsberger W, Wotjak CT, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci. 2011;14:1542–1547. doi: 10.1038/nn.2974. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Gosselink KL, Sawchenko PE. A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J Neurosci. 2009;29:7330–7340. doi: 10.1523/JNEUROSCI.5924-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Winder DG. Emerging role for corticotropin releasing factor signaling in the bed nucleus of the stria terminalis at the intersection of stress and reward. Front Psychiatry. 2013;4:42. doi: 10.3389/fpsyt.2013.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SJ, Buller KM, Day TA. Medial prefrontal cortex control of the paraventricular hypothalamic nucleus response to psychological stress: possible role of the bed nucleus of the stria terminalis. J Comp Neurol. 2005;481:363–376. doi: 10.1002/cne.20376. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsteeker Cusulin JI, Füzesi T, Inoue W, Bains JS. Glucocorticoid feedback uncovers retrograde opioid signaling at hypothalamic synapses. Nat Neurosci. 2013;16:596–604. doi: 10.1038/nn.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]