

Abstract
Regulated exocytosis in neurosecretory cells relies on the timely fusion of secretory granules (SGs) with the plasma membrane. Secretagogue stimulation leads to an enlargement of the cell footprint (surface area in contact with the coverslip), an effect previously attributed to exocytic fusion of SGs with the plasma membrane. Using total internal reflection fluorescence microscopy, we reveal the formation of filopodia-like structures in bovine chromaffin and PC12 cells driving the footprint expansion, suggesting the involvement of cortical actin network remodeling in this process. Using exocytosis-incompetent PC12 cells, we demonstrate that footprint enlargement is largely independent of SG fusion, suggesting that vesicular exocytic fusion plays a relatively minor role in filopodial expansion. The footprint periphery, including filopodia, undergoes extensive F-actin remodeling, an effect abolished by the actomyosin inhibitors cytochalasin D and blebbistatin. Imaging of both Lifeact-GFP and the SG marker protein neuropeptide Y-mCherry reveals that SGs actively translocate along newly forming actin tracks before undergoing fusion. Together, these data demonstrate that neurosecretory cells regulate the number of SGs undergoing exocytosis during sustained stimulation by controlling vesicular mobilization and translocation to the plasma membrane through actin remodeling. Such remodeling facilitates the de novo formation of fusion sites.
Introduction
Chromaffin cells are archetypical neurosecretory cells of the adrenal medulla that undergo regulated Ca2+-dependent exocytosis in which secretory granules (SGs) containing catecholamines fuse with the plasma membrane at specific release sites. Fusion of predocked or primed SGs from the release-ready pool provides the initial burst of catecholamine release (Neher and Zucker, 1993; Parsons et al., 1995; Gillis and Chow, 1997; Sørensen, 2004). However, sustained stimulation requires further mobilization of SGs from the reserve pool. SGs involved in this secondary release phase are required to navigate through the cortical actin network, a dense mesh of actin filaments underlying the plasma membrane, before docking and fusion (Neher and Zucker, 1993; Trifaró, 1999; Stevens et al., 2011).
Recent studies have shown that the cortical actin network plays an active role in SG exocytosis, in addition to serving as a barrier to passive SG diffusion (Trifaró et al., 2008; Wen et al., 2011). F-actin anchors SGs and provides tracks for their directed motion toward fusion sites (Giner et al., 2005). Molecular motors associated with F-actin, such as myosins (Malacombe et al., 2006), are involved in SG tethering (Tomatis et al., 2013), transport (Rose et al., 2002, 2003), and the control of fusion kinetics and fusion pore size (Neco et al., 2004; Neco et al., 2008). The cortical actin network has also been shown to undergo large-scale reorganization, including partial depolymerization, in response to stimulation, not only in chromaffin cells (Aunis and Bader, 1988; Vitale et al., 1995) but also in adipocytes (Kanzaki and Pessin, 2001), pancreatic β-cells (Nevins and Thurmond, 2003), alveolar type II cells (Rose et al., 1999), and sea urchin eggs (Yonemura and Mabuchi, 1987). Secretagogue stimulation can change cell surface morphology in two ways. First, the addition of vesicular membrane to the plasma membrane can promote enlargement of the area in contact with solid support (the footprint) in cultured neurons and neurosecretory cells (Neves and Lagnado, 1999; Llobet et al., 2003). Second, the morphology can change through actin remodeling. Stimulation induces rapid sprouting of filopodia in PC12 cells (Manivannan and Terakawa, 1994) and enhances neurite outgrowth in neurons (Kobayashi et al., 1997; Kimura et al., 1998). Importantly, neurite outgrowth from nerve terminals has been shown to provide de novo functional release sites in vivo and in vitro (Zakharenko et al., 1999; de Paiva et al., 1999), raising the possibility that activity-driven changes in the cortical cytoskeleton could also play a key role in defining de novo release sites in stimulated neurosecretory cells.
In this study, we establish that secretagogue stimulation promotes an enlargement of the footprint surface area in neuroendocrine cells by a mechanism largely independent of functional exocytosis. These activity-dependent structural changes are driven by reorganization of the cortical cytoskeleton and lead to the generation of new fusion sites for SGs in the extending area of the footprint. Our data reveal a novel type of plasticity in neuroendocrine cells that contributes to sustained stimulation by providing new actin tracks capable of capturing, guiding, and enabling fusion of SGs.
Materials and Methods
Cell culture and reagents.
Chromaffin cells were isolated from bovine adrenal glands, as described previously (Meunier et al., 2002; Meunier et al., 2005), and maintained in DMEM supplemented with 10% serum supreme, 2.5 μg/ml fungizone, 50 μg/ml gentamycin, and 10 mm HEPES on 0.1 mg/ml poly-d-lysine-coated culture dishes (MatTek). Cells were transfected using electroporation (Basic Nucleofector Kit for primary neurons [Amaxa] and Program X-001 [Lonza]) and cultured at 37°C in 5% CO2 for at least 24 h before experimentation. PC12 cells were routinely cultured as described previously (Osborne et al., 2008). Munc18–1/2 double knock-down PC12 (DKD-PC12) cells were provided by Shuzo Sugita (University of Toronto, Toronto, Ontario, Canada) and maintained as described previously (Malintan et al., 2009; Martin et al., 2013). PC12 and DKD-PC12 cells were transfected using Lipofectamine LTX (Invitrogen) according to the manufacturer's instructions. After 24 hours, transfected cells were replated onto poly-d-lysine-coated glass-bottomed culture dishes (MatTek) or coverslips and processed for imaging 72 hours after transfection.
VAMP2-pHluorin (Miesenböck et al., 1998) was provided by James Rothman (Yale University, New Haven, CT), Lifeact-GFP and Lifeact-RFP (Riedl et al., 2008) were provided by Roland Wedlich Soldner (MPI Biochemistry, Martinsried, Germany), glycosyl phosphatidyl inositol (GPI)-anchored green GFP (GFP-GPI) was a gift from Kai Simons (MPI-CBG, Dresden, Germany), and pCMV-Munc18–1-emGFP was a gift from Shuzo Sugita (University of Toronto; Arunachalam et al., 2008). pCMV-neuropeptide Y-mCherry (NPY-mCherry) was generated from pCMV-NPY-emerald GFP provided by S. Sugita. In brief, mCherry was amplified from pmCherry-C1 (Takara) by PCR using the following primers: forward, 5′-ATCGATAAGCTCATGGTGGCAAGGGCGAG-3′; and reverse, 5′-CAAGTAAAACCTCTACAAATGTGGTATGGC-3′. It was then subcloned into pCMV-NPY-emerald GFP using ClaI and BamHI, replacing emerald GFP (Tomatis et al., 2013).
Electron microscopy.
Bovine chromaffin cells seeded on poly-d-lysine-coated dishes were washed in Buffer A containing the following (in mm): 145 NaCl, 5 KCl, 1.2 Na2HPO4, 20 HEPES-NaOH, pH 7.4, containing 5.6 mm d-glucose, and were then treated with or without 2 mm BaCl2 for 5–10 min. Cells were fixed with 2.5% glutaraldehyde containing 1 mg/ml ruthenium red to define the cell surface. Fixed cells were contrasted with 1% osmium tetroxide and 4% uranyl acetate, before dehydration and flat embedding in LX-112 resin. Sections (∼55 nm) were cut using an ultramicrotome (UC64; Leica) parallel to the cell footprint and sections within 250 nm of the base of the cell were imaged using a transmission electron microscope (model 1011; JEOL) equipped with a Morada cooled CCD camera. All images were processed using Adobe Photoshop CS3 and figures compiled with Adobe Illustrator CS3.
Total internal reflection fluorescence microscopy.
Transfected cells on glass-bottomed culture dishes (MatTek) were visualized with a total internal reflection fluorescence (TIRF) microscope (Marianas, SDC Everest; Intelligent Imaging Innovations) fitted with a 100× oil-immersion objective (numerical aperture = 1.46, Carl Zeiss Pty Ltd) using an EMCCD camera (QuantEM 512sc) and Slidebook software (version 4.2.0.27/28). Cells were bathed in Buffer A before the addition of 2 mm BaCl2 or 100 μm nicotine + 2 mm CaCl2. Time-lapse videos were captured at 2 frames/s for the indicated period. When cytoskeletal inhibitors were used, these were preincubated with the cells for 20 min before imaging.
Footprint area was measured using the masking function (Otsu mask) of Slidebook software (versions 4.2.0.27/28). The density (number per square micrometer) of SGs in NPY-mCherry- and GFP-GPI-transfected cells was measured using Imaris (version 7.3.1; Bitplane) spot and surface features. The “Spots” function was used to determine SG numbers (from NPY-mCherry-positive structures) and the “Surface” function was used to measure the footprint area (visible area stained by GFP-GPI). To quantify the density of SGs, the average density in 10 cells per time point was measured. Particle tracking of NPY-mCherry-labeled SGs was performed on previously acquired TIRF images using the spot-tracking function of Imaris. SGs were tracked for 3 min after Ba2+ stimulation. The spot diameter was 0.40 μm and SGs were selected based on local contrast thresholding after background subtraction. Tracking was performed using the built-in autoregressive motion model with maximum displacement of 1 μm per time step and a gap size of three frames. Mean square displacement was calculated for each vesicle over incrementing time intervals and then averaged (Steyer and Almers, 1999).
Fusion event analysis.
The fusion of NPY-mCherry-positive SGs with the plasma membrane of the footprint was scored based on two separate metrics. NPY-mCherry-labeled SG fusion was assessed by the disappearance of a labeled SG and a transient local increase in fluorescence intensity allowing fNPY-mCherry release. For VAMP2-pHluorin experiments, fusion events were determined by first measuring the average integrated intensity change for 10 fusions and then using the FISUM (integrated fluorescence intensity) change to estimate the total number of fusion events by dividing through the average for one fusion. For fusion time-lapse analysis, the FISUM change was compared from one frame to the next and fusion events were defined if the FISUM difference was >70% of the calculated average fusion event change. To enhance detection of VAMP2-pHluorin fusion events, we created difference images of subsequent frames and localized individual fusion events using the ImageJ “find maxima” function. Comparisons of the results from both approaches were found to be in good agreement.
Statistical analysis.
All experiments were repeated in at least three different cultures and the number of cultures analyzed is indicated in the figure legends. Data analysis was performed using unpaired, two-tailed Student's t tests unless otherwise stated and data were considered significant at p < 0.05. Values are expressed as mean ± SEM.
Results
Activity-dependent increase of footprint surface area in neurosecretory cells
We used time-lapse TIRF microscopy and imaged both bovine chromaffin and PC12 cell footprints (i.e., the area of the plasma membrane in close contact with the coverslip) before and during secretagogue stimulation. To visualize the footprint membrane, cells were transfected with the plasma membrane marker GFP-GPI (Hiscox et al., 2002), which diffuses readily across the entire plasma membrane and therefore provides an even distribution of fluorescence across the footprint membrane. To elicit a strong stimulation, we initially used Ba2+ (2 mm; Burgoyne et al., 1989; Heldman et al., 1989). Ba2+ stimulation led to a marked increase in footprint area in both chromaffin and PC12 cells (Fig. 1A–D), in good agreement with previous studies (Llobet et al., 2003). The average change in footprint area measured 400 s after stimulation was 50 ± 10% for bovine chromaffin cells and 100 ± 15% for PC12 cells (Fig. 1D). Vehicle treatment (Buffer A alone) did not affect the size of the footprint. Importantly, examination of the microscopy time-lapse videos revealed filopodia-like structures in the newly added areas (Fig. 1A,B, arrowheads), suggesting that plasma membrane expansion is an active process. Interestingly, when observed under electron microscopy, Ba2+-stimulated chromaffin cell footprints displayed a large number of filopodia-like structures protruding from the cell periphery that were largely absent in unstimulated control cells (Fig. 1E). We quantified this by measuring the length of the membrane outline and counting the number of membrane-enclosed objects surrounding the cells before and after stimulation. In both cases, we found a significant increase 5 min after Ba2+ stimulation (Fig. 1E).
Figure 1.
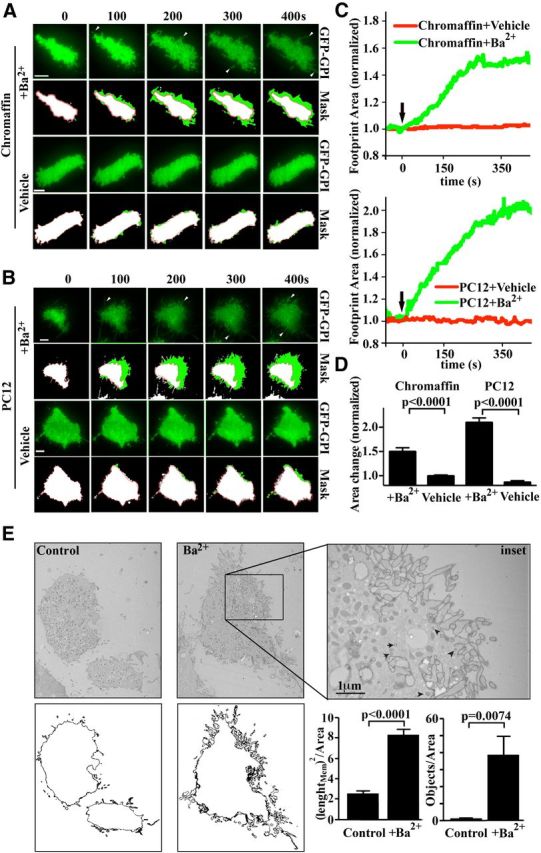
Activity-dependent filopodial extension drives the increase in footprint surface area in neurosecretory cells. A, B, Bovine chromaffin (A) and PC12 (B) cells expressing GFP-GPI were examined by TIRF microscopy, imaged at 2 Hz and stimulated with Ba2+ (2 mm) or vehicle treated as indicated. Top, Time-lapse of the GFP-GPI fluorescence in the footprint. Bottom, GPI-GFP footprint area (green) with an overlay of the pretreatment area (white with red outline). Scale bars, 5 μm. Arrowheads indicate outgrowing filopodia. C, Changes in surface area over time were plotted as indicated. D, Average change in footprint area (normalized to initial area before treatment) for stimulated chromaffin and PC12 cells and vehicle-treated controls 400 s after stimulation (n = 6/10/8/5). E, Bovine chromaffin cells were treated with vehicle or 2 mm BaCl2 for 8 min and the region of the cell containing the cell footprint was analyzed by electron microscopy. Note the numerous filopodia emerging from the footprint in stimulated cells. The inset shows a region of membrane extension containing SGs (arrowheads). Proximity to the plasma membrane is confirmed by the profiles of clathrin-coated pits containing ruthenium red stain, indicative of continuity with the plasma membrane (arrows). Bottom, Outlines of the cells in the top. Left: Ratio of outline length to visible surface before and stimulation. Right: Counts of objects outside the cell surface in unstimulated and stimulated conditions (n = 6).
Major discrepancy between footprint enlargement and estimated incorporated vesicular membrane
Because previous studies have suggested that incorporation of vesicular membrane into the plasma membrane accounts for footprint membrane expansion (Llobet et al., 2003), we analyzed the contribution of exocytosis (i.e., the fusion of SGs with the plasma membrane) to the footprint enlargement. To assess the number of SG fusion events contributing to the footprint area, we used two different approaches. First, we analyzed the fusion of SGs containing an expressed, fluorescently tagged secretory protein, NPY. NPY-mCherry-positive SG fusion events were scored based on a spike in fluorescence intensity and cloud-like dispersal of NPY-mCherry fluorescence (Fig. 2A) and the immediate disappearance of a SG, as described previously (Steyer and Almers, 1999; Allersma et al., 2004; Martin et al., 2013). In this method, the intensity spike stems from the diffusion of released NPY-mCherry molecules into closer proximity to the total internal reflection plane, where the evanescent wave intensity is higher (Allersma et al., 2004). Second, we validated our findings after the expression of VAMP2-pHluorin, in which the transmembrane v-SNARE VAMP2 is luminally tagged with the fluorescent pH reporter protein pHluorin (Miesenböck et al., 1998). pHluorin fluorescence is quenched in the acidic environment of the SG lumen. Upon fusion of the SG with the cell surface, neutralization of the luminal pH unquenches pHluorin, which becomes fluorescent (Miesenböck et al., 1998). As expected, increased pHluorin fluorescence coincided with the disappearance of NPY-mCherry-labeled SGs both spatially and temporally (Fig. 2B; Wen et al., 2011). We then compared this approach with our assay based on fusion events of NPY-mCherry-positive SGs and found very similar time-lapse fusion characteristics (Fig. 2C), thus validating our approach. To determine the proportion of SGs that were actually labeled with fluorescent proteins, we immunostained PC12 cells expressing VAMP-pHluorin for GFP and for endogenous VAMP2 and found that 89.9 ± 1.3% of VAMP2 labeled SGs were also positive for GFP. We also analyzed the colocalization of NPY-mCherry and VAMP2-pHluorin in SGs. The proportion of NPY-mCherry-positive SGs that also contained VAMP2-pHluorin was 80.95 ± 0.03% (Fig. 2D). Conversely, 70.17 ± 0.04% of VAMP2-pHluorin-positive SGs contained NPY-mCherry (Fig. 2D). Having estimated how much membrane surface was incorporated after fusion of SGs with the plasma membrane, we calculated the degree to which SG membrane fusion accounted for the change in footprint area. We compared the estimated footprint growth based on our estimation of the number of SG fusion events with the previously measured increase in the GFP-GPI-labeled footprint surface in both chromaffin and PC12 cells (Fig. 2E). Assuming an average SG is a sphere with a diameter of 300 nm (Plattner et al., 1997; Allersma et al., 2004; Grabner et al., 2005), the membrane surface area added to the footprint area after fusion will be ∼0.28 μm2/SG. In chromaffin cells, this level of membrane incorporation should result in an increase in footprint area of 19%, resulting from an average of 70 fusions. This is significantly lower than the 50% increase in footprint area that we observed by direct measurement of GFP-GPI (Fig. 2E). In PC12 cells, the estimated increase was 14%, resulting from an average of 60 fusion events. This is much lower than the 100% increase observed by direct measurement (Fig. 2E). Together, these data reveal a significant discrepancy between the increase in footprint area when measured directly relative to the estimated increase predicted through direct measurement of the number of fusion events. However, it is important to note that not all SGs fully fuse with the plasma membrane (Ceccarelli et al., 1973; Chow et al., 1992; de Toledo et al., 1993) and, conversely, not all SGs fusing with the plasma membrane in an activity-dependent manner are NPY-mCherry positive. Therefore, this assessment provides an estimate rather than a direct representation of the membrane area that is added to the footprint through SG fusion.
Figure 2.
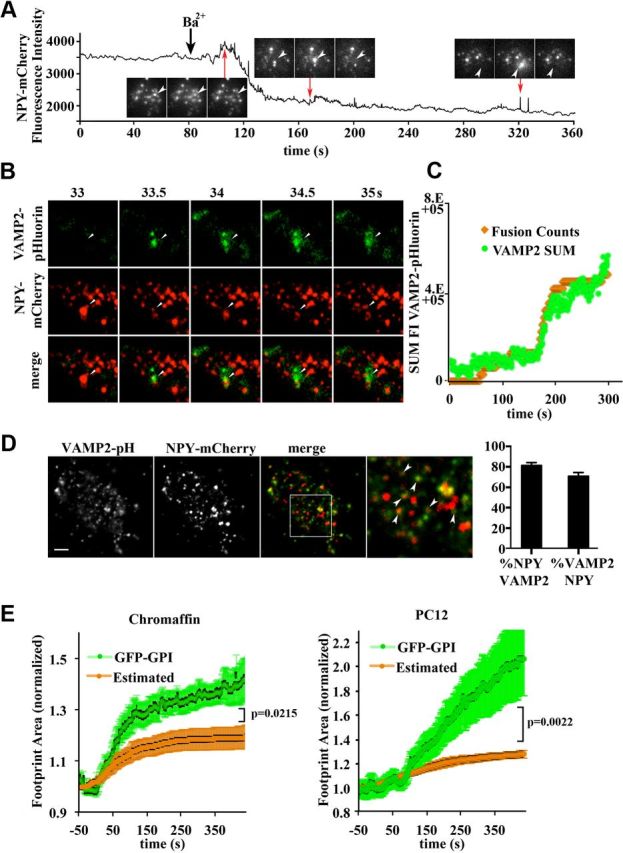
Vesicular fusion does not account for the entire footprint enlargement. SG granule fusion was measured in bovine chromaffin cells expressing NPY-mCherry. Cells were examined by TIRF microscopy and imaged at 2 Hz before and for 7 min after stimulation (black arrow) with Ba2+ (2 mm). A, Individual fusion events were defined as the disappearance of NPY-positive SGs, followed by a cloud-like dispersal of NPY-mCherry to the extracellular space, corresponding to a spike in overall NPY-mCherry intensity. B, SG fusion was measured in bovine chromaffin cells expressing NPY-mCherry and VAMP2-pHluorin. Cells were examined by TIRF microscopy and imaged at 2 Hz before and for 7 min after stimulation with Ba2+ (2 mm). C, Validation of the NPY-mCherry assay described in A by comparison with VAMP2-pHluorin overall fluorescence intensity change. To count fusion events with VAMP2-pHluorin, we determined the average fluorescence intensity change due to alkalinization of the pHluorin moiety during a fusion event by calculating the difference in fluorescence intensity between two adjacent frames before and during a fusion event and averaging over a number of events. We then assigned this value to each individual fusion event detected using the NPY-mCherry assay (orange). Note that these results do not significantly differ from the direct VAMP2-pHluorin fluorescence measurement (green). D, Chromaffin and PC12 cells were cotransfected with NPY-mCherry and VAMP2-pHluorin and analyzed by TIRF microscopy. Arrowheads in the inset indicate vesicles where both markers colocalize. The graph depicts the percentage of VAMP2-pHluorin-positive vesicles that also contain NPY-mCherry and vice versa (n = 7). E, Chromaffin and PC12 cells cotransfected with GFP-GPI and NPY-mCherry and stimulated with Ba2+ (2 mm). The extent of footprint enlargement based on the actual GFP-GPI measurements (green) was compared with the estimated membrane increase based on the measured number of SG fusion events (NPY-mCherry assay) multiplied by the predicted membrane surface area of a 300 nm SG (orange; n = 4–6). Note that the actual footprint increase is significantly larger than that of the estimate based on fusion alone.
Footprint area change is not dependent on SG exocytosis
To investigate directly the contribution of SG fusion to the increase in footprint area, we next tested whether footprint growth could occur in the absence of SG fusion events. Munc18 interacts with the t-SNARE, syntaxin-1a, and the SNARE complex and is required for SG fusion (Shen et al., 2007; Tareste et al., 2008). In the absence of Munc18, SG fusion and neurosecretion are severely impaired (Verhage et al., 2000; de Vries et al., 2000; Fisher et al., 2001; Voets et al., 2001; Medine et al., 2007; Shen et al., 2007; Malintan et al., 2009). To examine the role of SG fusion in footprint area enlargement, we used Munc18–1/2 double knock-down PC12 (DKD-PC12) cells (Han et al., 2009). We have shown previously using NPY-mCherry and TIRF microscopy that fusion of SGs is severely reduced in DKD-PC12 cells and that this can be fully rescued by reexpression of Munc18–1 (Martin et al., 2013). To measure the number of fusion events and changes in footprint size, we imaged control PC12 cells or DKD-PC12 cells cotransfected with NPY-mCherry and GFP-GPI, as well as DKD-PC12 cells in which Munc18–1 was reexpressed in response to 2 mm Ba2+. Consistent with our previous studies (Martin et al., 2013), both control PC12 cells and DKD-PC12 cells reexpressing Munc18–1 underwent a high and comparable number of fusion events, whereas DKD-PC12 cells were fusion incompetent (Fig. 3A). Importantly, however, the increase in footprint area was statistically indistinguishable between the three groups (Fig. 3B,C). This result demonstrates that the increase in the footprint area is largely independent of exocytic SG fusion.
Figure 3.

Footprint expansion does not depend on regulated exocytosis. NPY-mCherry was expressed in PC12 cells, DKD-PC12 cells, and DKD-PC12 cotransfected with Munc18–1-GFP. A, Cells were imaged at 2 Hz by TIRF microscopy before and for 7 min after 2 mm Ba2+ stimulation and fusion numbers were counted and normalized to footprint area. Note the rescue of exocytosis elicited by Munc18–1-GFP reexpression in DKD-PC12 cells. B, GFP-GPI was expressed in DKD-PC12 cells and DKD-PC12 cotransfected with (untagged) Munc18–1. Cells were examined by TIRF microscopy and imaged at 2 Hz before and during Ba2+ (2 mm) stimulation. Top, Time-lapse of the GFP-GPI fluorescence in the footprint. Bottom, GPI-GFP footprint area (green) with an overlay of the prestimulation area (white with red outline). Scale bars, 5 μm. C, Footprint area change for GFP-GPI-transfected DKD PC12 cells expressing empty vector or Munc18–1 and wild-type (wt)-PC12 cells after Ba2+ (2 mm) stimulation (400 s). Note that all three populations exhibit similar increases in footprint area. The numbers in the bars indicate the number of cultures analyzed.
Disrupting actin polymerization and myosin II function reduces footprint growth
Because the activity-dependent footprint growth implicated filopodial extensions (Fig. 1), we examined the contribution of the cortical actin network by inhibiting actin polymerization using cytochalasin D (Brenner and Korn, 1979) or myosin II activity using blebbistatin (Brenner and Korn, 1979; Straight et al., 2003). Bovine chromaffin cells and PC12 cells were cotransfected with GFP-GPI and NPY-mCherry and treated with either blebbistatin (10 μm) or cytochalasin D (2 μm) for 20 min before stimulation with 2 mm Ba2+ (Fig. 4A–D). Cells were imaged by TIRF microscopy before and during stimulation (Fig. 4A,C) and the number of fusion events and changes in footprint size were quantified (Fig. 4B,D). Although the number of fusion events was not significantly affected, we observed a significant block in footprint expansion in both cell types after treatment with either cytochalasin D or blebbistatin (Fig. 4B,D). Notably, the small increase in GFP-GPI-positive footprint size observed upon disruption of the cortical actin network was consistent with the amount of membrane added through SG fusion (Fig. 4B,D).
Figure 4.
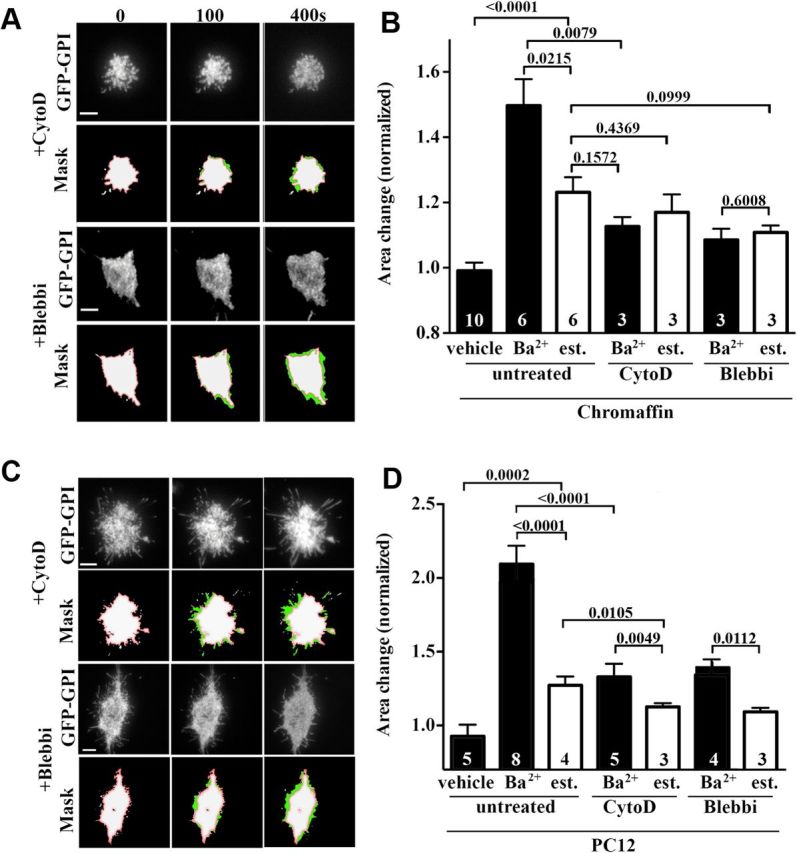
Interfering with actin polymerization and myosin II function reduces footprint enlargement. Bovine chromaffin (A, B) and PC12 (C, D) cells coexpressing GFP-GPI and NPY-mCherry were treated with cytochalasin D (CytoD) or blebbistatin (Blebbi) for 20 min before examination by time-lapse TIRF microscopy. Cells were then stimulated with Ba2+ and imaged at 2 Hz. A, B, Time-lapse profile of the GFP-GPI fluorescence in the footprint of chromaffin (A) or PC12 (B) cells treated as indicated. Bottom, GPI-GFP footprint area (green) with an overlay of the prestimulation area (white with red outline). Scale bars, 5 μm. B, D, Footprint area change under indicated conditions 400 s after Ba2+ stimulation or vehicle treatment for chromaffin (B) and PC12 cells (D) based on either the actual GFP-GPI measurements (black bars) or the corresponding estimated (est.) footprint increase based on fusion events (white bars). Note that for both blebbistatin and cytochalasin D treatments in chromaffin cells, the measured and estimated footprint area changes do not differ significantly. The numbers inside the bars indicate the numbers of cultures analyzed.
Newly added membrane area contains high levels of actin remodeling
To further investigate the role of the cytoskeleton in footprint expansion we next quantified the amount of cortical actin remodeling after Ba2+ stimulation. PC12 cells were cotransfected with GFP-GPI and Lifeact-RFP and dual-color TIRF microscopy was performed before and during stimulation (Fig. 5A). We used the fluorescence intensity ratio of Lifeact-RFP:GFP-GPI to measure the dynamic changes in F-actin distribution occurring in response to stimulation in various regions of the footprint (Fig. 5A,B). This analysis revealed that the ratio decreased in the center of the footprint after stimulation, indicating partial depolymerization of the cortical actin network (Fig. 5C), as described previously (Aunis and Bader, 1988; Vitale et al., 1995). In contrast, we found that the Lifeact-RFP:GFP-GPI ratio at the periphery of the footprint increased significantly, indicating high levels of actin polymerization, which is consistent with growth and remodeling of the footprint area.
Figure 5.
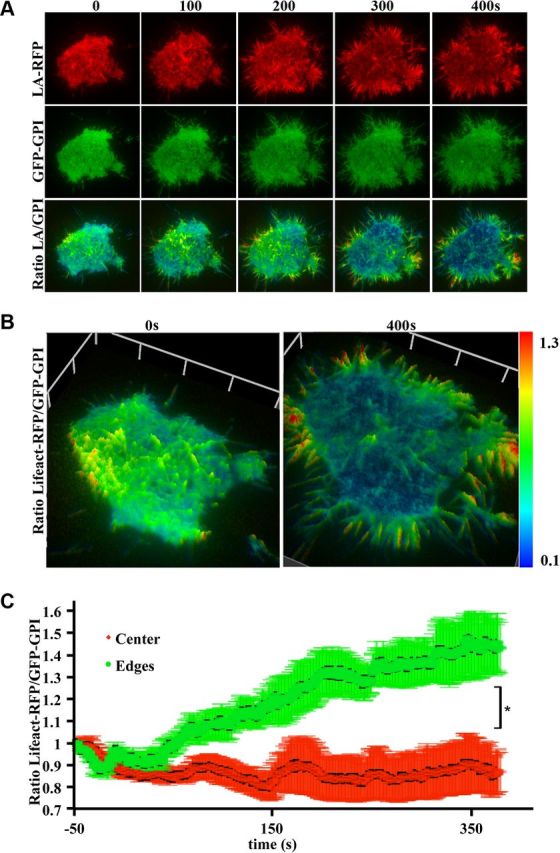
Newly added footprint area contains high levels of F-actin. A, PC12 cells cotransfected with Lifeact-RFP (LA-RFP; red) and GFP-GPI (green) were examined by TIRF microscopy and imaged at 2 Hz before and for 7 min after stimulation with Ba2+ (2 mm). Bottom, Ratio of Lifeact-RFP:GFP-GPI fluorescence in pseudocolor to indicate changes in F-actin concentration. B, 3D surface intensity plot of Lifeact-RFP:GFP-GPI ratio before and 400 s after stimulation, highlighting the increased F-actin concentration (red) at the edge of the footprint and the decrease in F-actin in the center of the footprint. C, Quantification of Lifeact-RFP and GFP-GPI ratios using regions of interest from the center or the expanding edge of PC12 cell footprints (n = 3). After stimulation, the Lifeact-RFP:GFP-GPI ratio in the initial footprint area decreases while simultaneously increasing in the added footprint area.
Secretory granules move into the newly added area and undergo fusion
We next investigated whether the actin-driven increase in the footprint surface area was able to provide additional SG fusion sites. Consistent with a functional role for the increased footprint, we observed that SGs colonized the newly added membrane area (Fig. 6A). Further analysis revealed that the SG density (number of granules per unit area in the total internal reflection plane) remained constant during stimulation (Fig. 6A). Lifeact-GFP expression in bovine chromaffin cells revealed F-actin at the protruding edges of the growing footprint and also showed that NPY-mCherry-positive SGs moved bidirectionally along these newly formed actin structures (Fig. 6B). Furthermore, not only did NPY-mCherry-positive SGs actively move into the added area (Fig. 6B,C), but their mobility also did not differ significantly from that in the original footprint area (Fig. 6C–E). We then investigated whether SGs undergo regulated exocytosis in the newly added area. By analyzing TIRF microscopy time-lapse movies of bovine chromaffin cells expressing Lifeact-GFP and NPY-mCherry, we were able to detect fusion events in the added area (Fig. 6F). The frequency of these events was lower than that found in the original area at the onset of stimulation (Fig. 6G) because the majority of primed vesicles readily fuse upon stimulation. However, when considering the fusion events that occurred in both the original and newly added areas 120 s after stimulation, the values were not significantly different (0.120 ± 0.011/μm2 in the initial area and 0.077 ± 0.018/μm2 in the newly added area; Fig. 6G).
Figure 6.
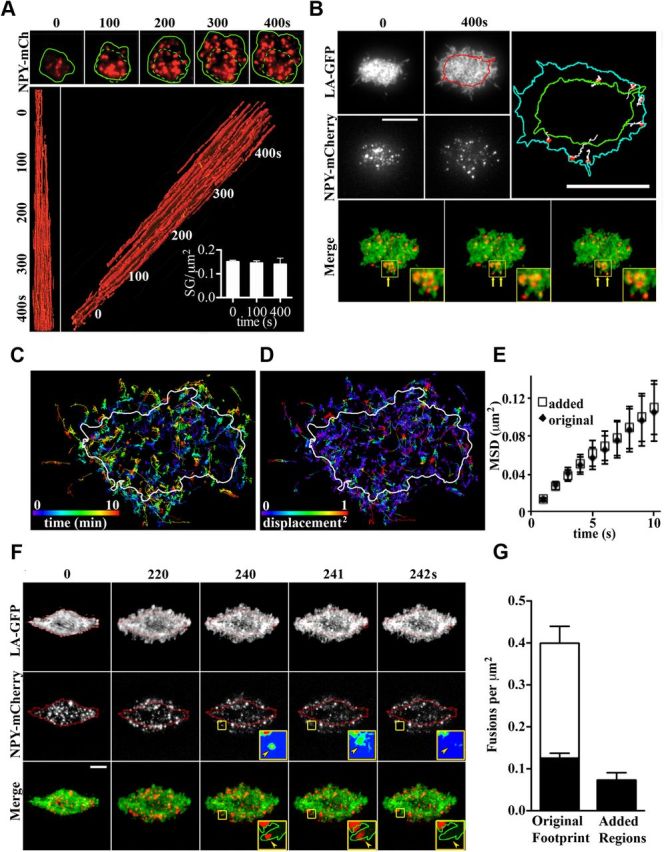
SGs move into the newly added footprint area. A, Time-lapse of PC12 cells cotransfected with NPY-mCherry(NPY-Ch) and GFP-GPI, imaged at 2 Hz before and for 7 min after stimulation with Ba2+ (2 mm). Top, NPY-mCherry-positive SGs appear outside the initial area (green dashed line) and are evenly distributed across the entire footprint (green outline). Bottom and side, x-y-t reconstruction of NPY-mCherry-positive SGs, with new SGs appearing in both the initial and the newly added area after stimulation. Note that the SG density (number of granules per unit area) remains constant over time (small inset) in all areas. B, Time-lapse TIRF microscopy of a bovine chromaffin cell cotransfected with Lifeact-GFP (LA-GFP) and NPY-mCherry showing the distribution of F-actin and SGs before and 400 s after stimulation. Right, Trajectories of SGs in the added footprint membrane area show that they are entering the newly forming area that also contains Lifeact-GFP positive structures (bottom). Scale bar, 5 μm. C,D, Maps of NPY-mCherry positive SGs trajectories in a Ba2+-stimulated chromaffin cell color coded for time after stimulation (C) and square displacement (D), as indicated. The white outline indicates the footprint area before stimulation. E, Mean square displacement (MSD) analysis of SGs in the original footprint and added areas. Note that there is no significant difference in mobility between either SG populations. F, SGs undergo regulated exocytosis in the newly added area. Time-lapse profile of a bovine chromaffin cell transfected with Lifeact-GFP (LA-GFP) and NPY-mCherry (red outline of the initial footprint area) stimulated with Ba2+ at t = 0 and imaged at 2 Hz. Arrowheads depict a SG entering an added area and undergoing fusion (pseudocolored in the inset). In the merged insets, the GFP channel is replaced with an outline of the limit of the footprint. Scale bar, 5 μm. G, Quantification of fusion events per area in both the original footprint and the added area. During footprint expansion, starting 120 s after stimulation, the number of fusions/area in the new area does not differ significantly from those in the initial footprint. Scale bar, 5 μm.
Discussion
Using a combination of pharmacological and genetic manipulations, we have shown that secretagogue stimulation of neurosecretory cells induces an enlargement of the footprint area driven by actin remodeling and filopodial outgrowth. Our results demonstrate that activity-dependent footprint expansion relies predominantly on significant cortical actin remodeling. Indeed, we demonstrate that footprint growth proceeds in the absence of regulated exocytosis but is abolished upon pharmacological inhibition of myosin II activity or actin polymerization. Our data therefore highlight a novel form of plasticity by demonstrating that secretagogue stimulation promotes actin remodeling and filopodia formation, which in turn drive an increase in footprint surface area, culminating in the formation of new SG release sites within this growth zone.
An increase in footprint size has been reported previously to occur seconds after stimulation as a result of SG fusion events and the resulting incorporation of vesicular membrane into the plasma membrane (Llobet et al., 2003). However, longer stimulation periods, we observed chromaffin and PC12 cell footprint enlargement that was independent of exocytic SG fusion. Indeed, using exocytosis-incompetent DKD-PC12 cells, we demonstrated that these morphological changes occur in much the same way in the absence of fusion events. Importantly, minor footprint enlargement was correlated with the number of fusion events when actin polymerization or myosin II activity was inhibited. We conclude that SG fusion lead to a small footprint increase (<10% of the original footprint size) immediately following stimulation, but that at later stages (>120 s), this effect is completely overshadowed by actin-driven remodeling (50% for chromaffin cells and 100% for PC12 cells) leading to a significant enlargement of the footprint area.
Actin is a well-known modulator of regulated exocytosis in neurons (Cingolani and Goda, 2008) and neurosecretory cells (Trifaró et al., 2008). Furthermore, activity-dependent neuronal actin remodeling has been implicated in neurite formation, extension, and branching and in the de novo formation of synapses. High concentrations of actin can be found in dendritic spines, the formation of which is also initiated by filopodia formation (Fifková and Delay, 1982; Matus et al., 1982; Landis et al., 1988; Dailey and Smith, 1996; Ziv and Smith, 1996; Fiala et al., 1998; Matus, 2000; Cingolani and Goda, 2008). Our data show that neurosecretory cells can undergo stimulation-induced morphological changes resembling the filopodia-based neurite extension that occurs during neuronal plasticity. Sustained neuronal stimulation has been related to neurite extension and the formation of new synapses (Fifková, 1985; Korobova and Svitkina, 2010) and our results demonstrate that neurosecretory cells are also capable of undergoing activity-dependent filopodial growth that leads to the creation of new functional release sites.
The functional importance of actin and myosin in exocytosis has become increasingly recognized in recent years. Cortical actin network remodeling plays a crucial role in regulated exocytosis in a range of non-neurosecretory cell types, including SG exocytosis in starfish eggs (Puppo et al., 2008), alveolar epithelial type II cells (Rose et al., 1999), neutrophils (Mitchell et al., 2008), and oocytes (Yonemura and Mabuchi, 1987; Runft et al., 2002; Horner and Wolfner, 2008), in addition to insulin-stimulated GLUT4 translocation in adipocytes (Kanzaki and Pessin, 2001; Kanzaki, 2006). In some cases, a morphological correlate has been reported between cortical actin rearrangement and the activity of small GTPases such as Cdc42 (Nevins and Thurmond, 2003) or Ral1 (Xie et al., 2013). In pancreatic β-cells, Ccdc42-dependent F-actin remodeling occurs during the regulated exocytosis of insulin-containing granules and facilitates filopodia formation through binding to the exocyst complex (Sugihara et al., 2002). In neuroendocrine cells, Cdc42 is activated during exocytosis and recruits N-WASP, leading to enhanced actin polymerization (Gasman et al., 2004). Both N-WASP and Arp2/3 regulate actin polymerization during GLUT4 translocation (Jiang et al., 2002) and during natural killer cell immunological synapse formation (Vyas et al., 2001; Wulfing et al., 2003; Brown et al., 2011). In neurosecretory cells, Cdc42-mediated actin reorganization promotes the mobilization and recruitment of SGs to the plasma membrane after inhibition of PI3-kinase delta (Wen et al., 2011), whereas in neurons, inhibition of PI3-kinase has also been shown to cause actin/myosin II-dependent elongation of filopodia (Tornieri et al., 2006) and to promote a biphasic effect on neuroexocytosis (Milosevic et al., 2005). Together, these data highlight the important and complex role of the cortical actomyosin network, in conjunction with its regulatory elements, in facilitating regulated exocytosis across a range of cell types.
Our findings clearly suggest a role for myosin II in controlling the expansion of the actin footprint, suggesting that the actomyosin II complex helps to establish these new membrane areas. Myosin II has previously been implicated in neuroexocytosis, with inhibition or expression of an inactive mutant myosin II being shown to impair SG mobility (Neco et al., 2002; Neco et al., 2004; Berberian et al., 2009). Myosin II inhibition alters the position of SGs, increasing their distance from the plasma membrane and slowing fusion kinetics (Villanueva et al., 2012). It also affects catecholamine release directly by controlling the size and duration of the fusion pore (Berberian et al., 2009) and has been implicated in the disruption of F-actin, an effect deemed auxiliary to facilitate full fusion (Doreian et al., 2008; Doreian et al., 2009). Our data contribute to this emerging field by demonstrating that myosin II-dependent actin remodeling drives the footprint enlargement elicited by stimulation in neurosecretory cells. Inhibition of actin polymerization with cytochalasin D was sufficient to stop footprint growth, highlighting the driving role of the actin cytoskeleton in these morphological changes. Myosin II inhibition with blebbistatin produced an identical effect, pointing to a dynamic role for the actomyosin II complex in controlling the response to sustained stimulation.
After stimulation, a significant remodeling of F-actin drives the enlargement of the footprint. Based on the ratio of Lifeact-RFP and GFP-GPI signals obtained with TIRF imaging, we found that the growing edges of the footprint, including the filopodia, contain high amounts of F-actin. Importantly, this occurred in parallel to the cortical F-actin depolymerization at the center of the footprint, as described previously (Vitale et al., 1995). Indeed, polymerization in the expanding footprint allows SGs to move into and undergo fusion in these areas, whereas partial F-actin depolymerization in the original footprint area facilitates exocytosis of SGs (Vitale et al., 1991; Trifaró et al., 2008). Our findings that SGs can often be found at the tips of filopodia could indicate that their presence actually facilitates F-actin outgrowth and filopodia formation in a myosin II-dependent manner (Taunton, 2001).
Our data reveal that, by modifying their surface morphology, neurosecretory cells can regulate the number of SGs undergoing vesicular mobilization, translocation to the plasma membrane, and exocytic fusion during sustained stimulation conditions. This actin-driven plasticity creates new tracks and anchoring points for SGs en route to the plasma membrane. The ability of SGs to nucleate actin could facilitate the development of de novo fusion sites in those added areas, thereby contributing to maintaining release rates under sustained stimulation conditions. We have recently shown that the density of SGs detected in the TIRF zone remains constant after stimulation (Tomatis et al., 2013). The expanding footprint therefore helps to regulate the number of SGs available at the plasma membrane, a feature shared by the sprouting of more nerve terminals in vivo (de Paiva et al., 1999) and in cultured neurons (Zakharenko et al., 1999).
In conclusion, we have shown that neurosecretory cells increase their surface in response to secretagogue stimulation by promoting filopodia-driven footprint enlargement. These activity-dependent morphological changes elicited by the interplay between actin and myosin II lead to the establishment of new release sites that contribute to the sustained response to secretagogue stimulation.
Footnotes
This work was supported by the National Health and Medical Research Council of Australia (project grant to F.A.M. and a senior research fellowship [Grant 569596] to F.A.M.) and the Australian Research Council (Linkage Infrastructure, Equipment, and Facilities Grant LE0882864 to F.A.M.). We thank Rowan Tweedale for critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Allersma MW, Wang L, Axelrod D, Holz RW. Visualization of regulated exocytosis with a granule-membrane probe using total internal reflection microscopy. Mol Biol Cell. 2004;15:4658–4668. doi: 10.1091/mbc.E04-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunachalam L, Han L, Tassew NG, He Y, Wang L, Xie L, Fujita Y, Kwan E, Davletov B, Monnier PP, Gaisano HY, Sugita S. Munc18–1 is critical for plasma membrane localization of syntaxin1 but not of SNAP-25 in PC12 cells. Mol Biol Cell. 2008;19:722–734. doi: 10.1091/mbc.E07-07-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aunis D, Bader MF. The cytoskeleton as a barrier to exocytosis in secretory cells. J Exp Biol. 1988;139:253–266. doi: 10.1242/jeb.139.1.253. [DOI] [PubMed] [Google Scholar]
- Berberian K, Torres AJ, Fang Q, Kisler K, Lindau M. F-actin and myosin II accelerate catecholamine release from chromaffin granules. J Neurosci. 2009;29:863–870. doi: 10.1523/JNEUROSCI.2818-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner SL, Korn ED. Substoichiometric concentrations of cytochalasin D inhibit actin polymerization: additional evidence for an F-actin treadmill. J Biol Chem. 1979;254:9982–9985. [PubMed] [Google Scholar]
- Brown AC, Oddos S, Dobbie IM, Alakoskela JM, Parton RM, Eissmann P, Neil MA, Dunsby C, French PM, Davis I, Davis DM. Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy. PLoS Biol. 2011;9:e1001152. doi: 10.1371/journal.pbio.1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Morgan A, O'Sullivan AJ. The control of cytoskeletal actin and exocytosis in intact and permeabilized adrenal chromaffin cells: role of calcium and protein kinase C. Cell Signal. 1989;1:323–334. doi: 10.1016/0898-6568(89)90051-X. [DOI] [PubMed] [Google Scholar]
- Ceccarelli B, Hurlbut WP, Mauro A. Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J Cell Biol. 1973;57:499–524. doi: 10.1083/jcb.57.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow RH, von Rüden L, Neher E. Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature. 1992;356:60–63. doi: 10.1038/356060a0. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Smith SJ. The dynamics of dendritic structure in developing hippocampal slices. J Neurosci. 1996;16:2983–2994. doi: 10.1523/JNEUROSCI.16-09-02983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paiva A, Meunier FA, Molgó J, Aoki KR, Dolly JO. Functional repair of motor endplates after botulinum neurotoxin type A poisoning: biphasic switch of synaptic activity between nerve sprouts and their parent terminals. Proc Natl Acad Sci U S A. 1999;96:3200–3205. doi: 10.1073/pnas.96.6.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Toledo GA, Fernández-Chacon R, Fernández JM. Release of secretory products during transient vesicle fusion. Nature. 1993;363:554–558. doi: 10.1038/363554a0. [DOI] [PubMed] [Google Scholar]
- de Vries KJ, Geijtenbeek A, Brian EC, de Graan PN, Ghijsen WE, Verhage M. Dynamics of munc18–1 phosphorylation/dephosphorylation in rat brain nerve terminals. Eur J Neurosci. 2000;12:385–390. doi: 10.1046/j.1460-9568.2000.00931.x. [DOI] [PubMed] [Google Scholar]
- Doreian BW, Fulop TG, Smith CB. Myosin II activation and actin reorganization regulate the mode of quantal exocytosis in mouse adrenal chromaffin cells. J Neurosci. 2008;28:4470–4478. doi: 10.1523/JNEUROSCI.0008-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doreian BW, Fulop TG, Meklemburg RL, Smith CB. Cortical F-actin, the exocytic mode, and neuropeptide release in mouse chromaffin cells is regulated by myristoylated alanine-rich C-kinase substrate and myosin II. Mol Biol Cell. 2009;20:3142–3154. doi: 10.1091/mbc.E09-03-0197. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifková E. Actin in the nervous system. Brain Res Rev. 1985;9:187–215. doi: 10.1016/0165-0173(85)90012-8. [DOI] [PubMed] [Google Scholar]
- Fifková E, Delay RJ. Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. J Cell Biol. 1982;95:345–350. doi: 10.1083/jcb.95.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RJ, Pevsner J, Burgoyne RD. Control of fusion pore dynamics during exocytosis by Munc18. Science. 2001;291:875–878. doi: 10.1126/science.291.5505.875. [DOI] [PubMed] [Google Scholar]
- Gasman S, Chasserot-Golaz S, Malacombe M, Way M, Bader MF. Regulated exocytosis in neuroendocrine cells: a role for subplasmalemmal Cdc42/N-WASP-induced actin filaments. Mol Biol Cell. 2004;15:520–531. doi: 10.1091/mbc.E03-06-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis KD, Chow RH. Kinetics of exocytosis in adrenal chromaffin cells. Semin Cell Dev Biol. 1997;8:133–140. doi: 10.1006/scdb.1996.0132. [DOI] [PubMed] [Google Scholar]
- Giner D, Neco P, Francés Mdel M, López I, Viniegra S, Gutiérrez LM. Real-time dynamics of the F-actin cytoskeleton during secretion from chromaffin cells. J Cell Sci. 2005;118:2871–2880. doi: 10.1242/jcs.02419. [DOI] [PubMed] [Google Scholar]
- Grabner CP, Price SD, Lysakowski A, Fox AP. Mouse chromaffin cells have two populations of dense core vesicles. J Neurophysiol. 2005;94:2093–2104. doi: 10.1152/jn.00316.2005. [DOI] [PubMed] [Google Scholar]
- Han L, Jiang T, Han GA, Malintan NT, Xie L, Wang L, Tse FW, Gaisano HY, Collins BM, Meunier FA, Sugita S. Rescue of Munc18–1 and -2 double knockdown reveals the essential functions of interaction between Munc18 and closed syntaxin in PC12 cells. Mol Biol Cell. 2009;20:4962–4975. doi: 10.1091/mbc.E09-08-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldman E, Levine M, Raveh L, Pollard HB. Barium ions enter chromaffin cells via voltage-dependent calcium channels and induce secretion by a mechanism independent of calcium. J Biol Chem. 1989;264:7914–7920. [PubMed] [Google Scholar]
- Hiscox S, Hallett MB, Morgan BP, van den Berg CW. GPI-anchored GFP signals Ca2+ but is homogeneously distributed on the cell surface. Biochem Biophys Res Commun. 2002;293:714–721. doi: 10.1016/S0006-291X(02)00280-2. [DOI] [PubMed] [Google Scholar]
- Horner VL, Wolfner MF. Transitioning from egg to embryo: Triggers and mechanisms of egg activation. Dev Dyn. 2008;237:527–544. doi: 10.1002/dvdy.21454. [DOI] [PubMed] [Google Scholar]
- Jiang ZY, Chawla A, Bose A, Way M, Czech MP. A phosphatidylinositol 3-kinase-independent insulin signaling pathway to N-WASP/Arp2/3/F-actin required for GLUT4 glucose transporter recycling. J Biol Chem. 2002;277:509–515. doi: 10.1074/jbc.M108280200. [DOI] [PubMed] [Google Scholar]
- Kanzaki M. Insulin receptor signals regulating GLUT4 translocation and actin dynamics. Endocr J. 2006;53:267–293. doi: 10.1507/endocrj.KR-65. [DOI] [PubMed] [Google Scholar]
- Kanzaki M, Pessin JE. Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J Biol Chem. 2001;276:42436–42444. doi: 10.1074/jbc.M108297200. [DOI] [PubMed] [Google Scholar]
- Kimura K, Yanagida Y, Haruyama T, Kobatake E, Aizawa M. Electrically induced neurite outgrowth of PC12 cells on the electrode surface. Med Biol Eng Comput. 1998;36:493–498. doi: 10.1007/BF02523221. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Nagata S, Kita Y, Nakatsu N, Ihara S, Kaibuchi K, Kuroda S, Ui M, Iba H, Konishi H, Kikkawa U, Saitoh I, Fukui Y. Expression of a constitutively active phosphatidylinositol 3-kinase induces process formation in rat PC12 cells: use of Cre/loxP recombination system. J Biol Chem. 1997;272:16089–16092. doi: 10.1074/jbc.272.26.16089. [DOI] [PubMed] [Google Scholar]
- Korobova F, Svitkina T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell. 2010;21:165–176. doi: 10.1091/mbc.E09-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis DM, Hall AK, Weinstein LA, Reese TS. The organization of cytoplasm at the presynaptic active zone of a central nervous system synapse. Neuron. 1988;1:201–209. doi: 10.1016/0896-6273(88)90140-7. [DOI] [PubMed] [Google Scholar]
- Llobet A, Beaumont V, Lagnado L. Real-time measurement of exocytosis and endocytosis using interference of light. Neuron. 2003;40:1075–1086. doi: 10.1016/S0896-6273(03)00765-7. [DOI] [PubMed] [Google Scholar]
- Malacombe M, Bader MF, Gasman S. Exocytosis in neuroendocrine cells: new tasks for actin. Biochim Biophys Acta. 2006;1763:1175–1183. doi: 10.1016/j.bbamcr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Malintan NT, Nguyen TH, Han L, Latham CF, Osborne SL, Wen PJ, Lim SJ, Sugita S, Collins BM, Meunier FA. Abrogating Munc18–1-SNARE complex interaction has limited impact on exocytosis in PC12 cells. J Biol Chem. 2009;284:21637–21646. doi: 10.1074/jbc.M109.013508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manivannan S, Terakawa S. Rapid sprouting of filopodia in nerve terminals of chromaffin cells, PC12 cells, and dorsal root neurons induced by electrical stimulation. J Neurosci. 1994;14:5917–5928. doi: 10.1523/JNEUROSCI.14-10-05917.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Tomatis VM, Papadopulos A, Christie MP, Malintan NT, Gormal RS, Sugita S, Martin JL, Collins BM, Meunier FA. The Munc18–1 domain 3a loop is essential for neuroexocytosis but not for syntaxin-1A transport to the plasma membrane. J Cell Sci. 2013;126:2353–2360. doi: 10.1242/jcs.126813. [DOI] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medine CN, Rickman C, Chamberlain LH, Duncan RR. Munc18–1 prevents the formation of ectopic SNARE complexes in living cells. J Cell Sci. 2007;120:4407–4415. doi: 10.1242/jcs.020230. [DOI] [PubMed] [Google Scholar]
- Meunier FA, Feng ZP, Molgó J, Zamponi GW, Schiavo G. Glycerotoxin from Glycera convoluta stimulates neurosecretion by up-regulating N-type Ca2+ channel activity. EMBO J. 2002;21:6733–6743. doi: 10.1093/emboj/cdf677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier FA, Osborne SL, Hammond GR, Cooke FT, Parker PJ, Domin J, Schiavo G. Phosphatidylinositol 3-kinase C2alpha is essential for ATP-dependent priming of neurosecretory granule exocytosis. Mol Biol Cell. 2005;16:4841–4851. doi: 10.1091/mbc.E05-02-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Sørensen JB, Lang T, Krauss M, Nagy G, Haucke V, Jahn R, Neher E. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J Neurosci. 2005;25:2557–2565. doi: 10.1523/JNEUROSCI.3761-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell T, Lo A, Logan MR, Lacy P, Eitzen G. Primary granule exocytosis in human neutrophils is regulated by Rac-dependent actin remodeling. Am J Physiol Cell Physiol. 2008;295:C1354–1365. doi: 10.1152/ajpcell.00239.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neco P, Gil A, Del Mar Francés M, Viniegra S, Gutiérrez LM. The role of myosin in vesicle transport during bovine chromaffin cell secretion. Biochem J. 2002;368:405–413. doi: 10.1042/BJ20021090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neco P, Giner D, Viniegra S, Borges R, Villarroel A, Gutiérrez LM. New roles of myosin II during vesicle transport and fusion in chromaffin cells. J Biol Chem. 2004;279:27450–27457. doi: 10.1074/jbc.M311462200. [DOI] [PubMed] [Google Scholar]
- Neco P, Fernández-Peruchena C, Navas S, Gutiérrez LM, de Toledo GA, Alés E. Myosin II contributes to fusion pore expansion during exocytosis. J Biol Chem. 2008;283:10949–10957. doi: 10.1074/jbc.M709058200. [DOI] [PubMed] [Google Scholar]
- Neher E, Zucker RS. Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron. 1993;10:21–30. doi: 10.1016/0896-6273(93)90238-M. [DOI] [PubMed] [Google Scholar]
- Neves G, Lagnado L. The kinetics of exocytosis and endocytosis in the synaptic terminal of goldfish retinal bipolar cells. J Physiol. 1999;515:181–202. doi: 10.1111/j.1469-7793.1999.181ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins AK, Thurmond DC. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol. 2003;285:C698–710. doi: 10.1152/ajpcell.00093.2003. [DOI] [PubMed] [Google Scholar]
- Osborne SL, Wen PJ, Boucheron C, Nguyen HN, Hayakawa M, Kaizawa H, Parker PJ, Vitale N, Meunier FA. PIKfyve negatively regulates exocytosis in neurosecretory cells. J Biol Chem. 2008;283:2804–2813. doi: 10.1074/jbc.M704856200. [DOI] [PubMed] [Google Scholar]
- Parsons TD, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytic burst, and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15:1085–1096. doi: 10.1016/0896-6273(95)90097-7. [DOI] [PubMed] [Google Scholar]
- Plattner H, Artalejo AR, Neher E. Ultrastructural organization of bovine chromaffin cell cortex-analysis by cryofixation and morphometry of aspects pertinent to exocytosis. J Cell Biol. 1997;139:1709–1717. doi: 10.1083/jcb.139.7.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppo A, Chun JT, Gragnaniello G, Garante E, Santella L. Alteration of the cortical actin cytoskeleton deregulates Ca2+ signaling, monospermic fertilization, and sperm entry. PloS One. 2008;3:e3588. doi: 10.1371/journal.pone.0003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose F, Kürth-Landwehr C, Sibelius U, Reuner KH, Aktories K, Seeger W, Grimminger F. Role of actin depolymerization in the surfactant secretory response of alveolar epithelial type II cells. Am J Respir Crit Care Med. 1999;159:206–212. doi: 10.1164/ajrccm.159.1.9801106. [DOI] [PubMed] [Google Scholar]
- Rosé SD, Lejen T, Casaletti L, Larson RE, Pene TD, Trifaró JM. Molecular motors involved in chromaffin cell secretion. Ann N Y Acad Sci. 2002;971:222–231. doi: 10.1111/j.1749-6632.2002.tb04466.x. [DOI] [PubMed] [Google Scholar]
- Rosé SD, Lejen T, Casaletti L, Larson RE, Pene TD, Trifaró JM. Myosins II and V in chromaffin cells: myosin V is a chromaffin vesicle molecular motor involved in secretion. J Neurochem. 2003;85:287–298. doi: 10.1046/j.1471-4159.2003.01649.x. [DOI] [PubMed] [Google Scholar]
- Runft LL, Jaffe LA, Mehlmann LM. Egg activation at fertilization: where it all begins. Dev Biol. 2002;245:237–254. doi: 10.1006/dbio.2002.0600. [DOI] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128:183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Sørensen JB. Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflugers Arch. 2004;448:347–362. doi: 10.1007/s00424-004-1247-8. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Schirra C, Becherer U, Rettig J. Vesicle pools: lessons from adrenal chromaffin cells. Front Synapt Neurosci. 2011:3. doi: 10.3389/fnsyn.2011.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer JA, Almers W. Tracking single secretory granules in live chromaffin cells by evanescent-field fluorescence microscopy. Biophys J. 1999;76:2262–2271. doi: 10.1016/S0006-3495(99)77382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Sugihara K, Asano S, Tanaka K, Iwamatsu A, Okawa K, Ohta Y. The exocyst complex binds the small GTPase RalA to mediate filopodia formation. Nat Cell Biol. 2002;4:73–78. doi: 10.1038/ncb720. [DOI] [PubMed] [Google Scholar]
- Tareste D, Shen J, Melia TJ, Rothman JE. SNAREpin/Munc18 promotes adhesion and fusion of large vesicles to giant membranes. Proc Natl Acad Sci U S A. 2008;105:2380–2385. doi: 10.1073/pnas.0712125105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton J. Actin filament nucleation by endosomes, lysosomes and secretory vesicles. Curr Opin Cell Biol. 2001;13:85–91. doi: 10.1016/S0955-0674(00)00178-2. [DOI] [PubMed] [Google Scholar]
- Tomatis VM, Papadopulos A, Malintan NT, Martin S, Wallis T, Gormal RS, Kendrick-Jones J, Buss F, Meunier FA. Myosin VI small insert isoform maintains exocytosis by tethering secretory granules to the cortical actin. J Cell Biol. 2013;200:301–320. doi: 10.1083/jcb.201204092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornieri K, Welshhans K, Geddis MS, Rehder V. Control of neurite outgrowth and growth cone motility by phosphatidylinositol-3-kinase. Cell Motil Cytoskeleton. 2006;63:173–192. doi: 10.1002/cm.20115. [DOI] [PubMed] [Google Scholar]
- Trifaró JM. Scinderin and cortical F-actin are components of the secretory machinery. Can J Physiol Pharmacol. 1999;77:660–671. doi: 10.1139/y99-074. [DOI] [PubMed] [Google Scholar]
- Trifaró JM, Gasman S, Gutiérrez LM. Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol (Oxf) 2008;192:165–172. doi: 10.1111/j.1748-1716.2007.01808.x. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Villanueva J, Torres V, Torregrosa-Hetland CJ, Garcia-Martinez V, López-Font I, Viniegra S, Gutiérrez LM. F-actin-myosin II inhibitors affect chromaffin granule plasma membrane distance and fusion kinetics by retraction of the cytoskeletal cortex. J Mol Neurosci. 2012;48:328–338. doi: 10.1007/s12031-012-9800-y. [DOI] [PubMed] [Google Scholar]
- Vitale ML, Rodríguez Del Castillo A, Tchakarov L, Trifaró JM. Cortical filamentous actin disassembly and scinderin redistribution during chromaffin cell stimulation precede exocytosis, a phenomenon not exhibited by gelsolin. J Cell Biol. 1991;113:1057–1067. doi: 10.1083/jcb.113.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale ML, Seward EP, Trifaró JM. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron. 1995;14:353–363. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Südhof TC, Neher E, Verhage M. Munc18–1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/S0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Vyas YM, Mehta KM, Morgan M, Maniar H, Butros L, Jung S, Burkhardt JK, Dupont B. Spatial organization of signal transduction molecules in the NK cell immune synapses during MHC class I-regulated noncytolytic and cytolytic interactions. J Immunol. 2001;167:4358–4367. doi: 10.4049/jimmunol.167.8.4358. [DOI] [PubMed] [Google Scholar]
- Wen PJ, Osborne SL, Zanin M, Low PC, Wang HT, Schoenwaelder SM, Jackson SP, Wedlich-Söldner R, Vanhaesebroeck B, Keating DJ, Meunier FA. Phosphatidylinositol(4,5)bisphosphate coordinates actin-mediated mobilization and translocation of secretory vesicles to the plasma membrane of chromaffin cells. Nat Commun. 2011;2:491. doi: 10.1038/ncomms1500. [DOI] [PubMed] [Google Scholar]
- Wulfing C, Purtic B, Klem J, Schatzle JD. Stepwise cytoskeletal polarization as a series of checkpoints in innate but not adaptive cytolytic killing. Proc Natl Acad Sci U S A. 2003;100:7767–7772. doi: 10.1073/pnas.1336920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang Y, Liang T, Dolai S, Xie H, Parsaud L, López JA, He Y, Chidambaram S, Lam PP, James DE, Sugita S, Gaisano HY. RalA GTPase tethers insulin granules to L- and R-type calcium channels through binding alpha2 delta-1 subunit. Traffic. 2013;14:428–439. doi: 10.1111/tra.12047. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Mabuchi I. Wave of cortical actin polymerization in the sea urchin egg. Cell Motil Cytoskeleton. 1987;7:46–53. doi: 10.1002/cm.970070107. [DOI] [PubMed] [Google Scholar]
- Zakharenko S, Chang S, O'Donoghue M, Popov SV. Neurotransmitter secretion along growing nerve processes: comparison with synaptic vesicle exocytosis. J Cell Biol. 1999;144:507–518. doi: 10.1083/jcb.144.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/S0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]