

Abstract
Pain is the most common symptom of bone cancer. TGF-β, a major bone-derived growth factor, is largely released by osteoclast bone resorption during the progression of bone cancer and contributes to proliferation, angiogenesis, immunosuppression, invasion, and metastasis. Here, we further show that TGF-β1 is critical for bone cancer-induced pain sensitization. We found that, after the progression of bone cancer, TGF-β1 was highly expressed in tumor-bearing bone, and the expression of its receptors, TGFβRI and TGFβRII, was significantly increased in the DRG in a rat model of bone cancer pain that is based on intratibia inoculation of Walker 256 mammary gland carcinoma cells. The blockade of TGF-β receptors by the TGFβRI antagonist SD-208 robustly suppressed bone cancer-induced thermal hyperalgesia on post-tumor day 14 (PTD 14). Peripheral injection of TGF-β1 directly induced thermal hyperalgesia in intact rats and wide-type mice, but not in Trpv1−/− mice. Whole-cell patch-clamp recordings from DRG neurons showed that transient receptor potential vanilloid (TRPV1) sensitivity was significantly enhanced on PTD 14. Extracellular application of TGF-β1 significantly potentiated TRPV1 currents and increased [Ca2+]i in DRG neurons. Pharmacological studies revealed that the TGF-β1 sensitization of TRPV1 and the induction of thermal hyperalgesia required the TGF-βR-mediated Smad-independent PKCε and TGF-β activating kinase 1-p38 pathways. These findings suggest that TGF-β1 signaling contributes to bone cancer pain via the upregulation and sensitization of TRPV1 in primary sensory neurons and that therapeutic targeting of TGF-β1 may ameliorate the bone cancer pain in advanced cancer.
Keywords: bone cancer pain, dorsal root ganglion, patch clamp, thermal hyperalgesia, TGF-β, TRPV1
Introduction
Bone cancer pain is one of the most severe types of chronic pain. Abnormal activity in primary sensory neurons is implicated in this pain process. Tumor-derived products have been shown to sensitize and injure primary sensory neurons (Hamamoto et al., 2008). Thus, successful treatment of bone cancer pain will need to address the abnormal sensitization of peripheral nociceptors.
TGF-β is a major bone-derived growth factor that is secreted in a latent form by bone cells and stored in the bone matrix (Dallas et al., 2002). Numerous studies have shown the importance of TGF-β for the development of bone metastases. In advanced bone cancer, excessive levels of TGF-β promote tumor growth, invasion, and metastasis (Massagué, 2008). Tumors in bone secrete a multitude of factors, including osteoblastic factors and osteolytic factors. TGF-β is released and activated from the mineralized bone matrix by osteoclastic bone resorption and further induces the production of osteolytic factors by tumors. The reciprocal interaction between cancer cells and the bone microenvironment drives a feedforward cycle that increases both tumor growth and bone destruction (Mundy, 2002; Juárez and Guise, 2011). Blocking TGF-β to interrupt this vicious cycle between cancer and bone has been considered a promising strategy for the treatment of bone metastasis. Preclinical studies show that TGF-β inhibitors are effective at treating and preventing tumor cell invasiveness and bone metastases as well as at increasing bone mass (Bandyopadhyay et al., 2006; Juárez and Guise, 2011). In this study, we further investigated whether and how TGF-β in the periphery regulates metastatic bone cancer-evoked hyperalgesia in a rat model of bone cancer pain induced by intratibia inoculation of Walker 256 mammary gland carcinoma cells.
A recent report indicates that TGF-β is involved in inflammatory pain signaling by increasing Cdk5-dependent transient receptor potential vanilloid 1 (TRPV1) phosphorylation (Utreras et al., 2012). TRPV1 is a nonselective cation channel that is expressed by nociceptors and can be activated by noxious heat (>43°C), extracellular protons, various lipids, and capsaicin (Caterina et al., 1997). TRPV1 expression increased in the DRG after cancer infiltration and contributed to thermal hyperalgesia (Niiyama et al., 2007; Pan et al., 2010; Han et al., 2012). Selective blockade of TRPV1 results in a significant attenuation of nociceptive behaviors in bone cancer pain models (Ghilardi et al., 2005; Niiyama et al., 2009; Kawamata et al., 2010). Therefore, in the present study, we used in vitro and in vivo approaches to study the role of TGF-β1 in bone cancer pain by sensitizing TRPV1. Our results indicate that TGF-β1 released from tumor-inoculated bone tissue may increase the excitability of DRG neurons by sensitizing TRPV1 via the TGF-β activating kinase 1 (TAK1)/p38 MAPK and PKC signaling pathways, leading to hyperalgesia in animals with bone cancer.
Materials and Methods
Animals.
Female Wistar rats weighing 150–180 g were obtained from the Shanghai Experimental Animal Center of the Chinese Academy of Sciences and were housed under a 12:12 light-dark cycle with food and water ad libitum. Knock-out mice lacking Trpv1 (Trpv1−/−) and C57BL/6 background WT control mice (both sexes, 8–10 weeks) were purchased from The Jackson Laboratory and bred in the Animal Center of Fudan University. All animal experiments were approved by the Shanghai Animal Care and Use Committee and followed the policies issued by the International Association for the Study of Pain on the use of laboratory animal (Zimmermann, 1983). All the following behavioral testing, quantification of Western blot, and immunohistochemical experiments described herein were performed by experimenters that were blind with respect to the treatments.
Model of bone cancer pain.
Tumor cells were extracted from ascetic fluid of rats that were administered Walker 256 rat mammary gland carcinoma cells. Suspensions of 1 × 108/ml tumor cells in PBS were prepared. The surgery was conducted as previously described (Duan et al., 2012). Briefly, rats were anesthetized with sodium pentobarbital (50 mg/kg i.p.). The right leg was shaved, and the skin was disinfected with iodine tincture and 75% ethanol. Superficial incisions were made in the skin overlying the patella. A 23-gauge needle was inserted at the site of the intercondylar eminence of the tibia and was then replaced with a 10 μl microinjection syringe containing a 4 μl suspension of tumor cells. The contents of the syringe were slowly injected into the right tibia cavity. To prevent leakage of cells outside the bone, the injection site was sealed with bone wax. Then, the skin wound was closed. For the sham group (control), 4 μl of PBS was injected instead of carcinoma cells into the tibia. At the end of the experiment, radiological, postmortem, and histological evaluations were performed. Rats that showed no obvious tumor growth and bone destruction after inoculation of tumor cells were excluded from the experiments.
Drugs and administration.
TGF-β1 was purchased from Peprotech and was dissolved in a citrate buffer (50 μg/ml) and then diluted in sterile 0.01 m PBS with 2 mg/ml BSA at 5 μg/ml to create a stock solution. The TGFβRI kinase inhibitor SD-208, the ERK MAPK inhibitors PD98059 and U0126, the PKC inhibitor bisindolylmaleimide VII (BIM), capsaicin, and the TRPV1 receptor inhibitor capsazepine were purchased from Sigma. The p38 MAPK inhibitor SB203580 and the JNK MAPK inhibitor SP600125 were purchased from Merck KGaA. The TAK1 inhibitor (5Z)-7-oxozeaenol was purchased from Tocris Bioscience. The drugs were dissolved in DMSO or normal saline as stock solutions. All of the stock solutions were stored at −20°C or −80°C until use. Working concentrations of the drugs were prepared on the day of the experiment from the stock solutions. The drug dosages were selected based on previous reports and our preliminary studies. In the whole-cell patch-clamp recordings, TGF-β1 (10 ng/ml) was applied continuously for 20 min, capsaicin challenges (1 μm) were applied for 3 s and TRPV1 currents were recorded. The inhibitors were applied to the chamber 30 min before and during the TGF-β1 perfusion. In the calcium imaging experiments, capsaicin (1 μm) was administered 5 times, with a 90 s interval between trials. TGF-β1 (10 ng/ml) was perfused 20 min before and during the capsaicin challenges. SD-208 was applied 15 min before and during the TGF-β1 perfusion. In the behavioral experiments, SD-208 was delivered into local deep tissue around the tumor bone or the spinal space via a lumbar puncture performed with a 30-gauge needle between the L5 and L6 vertebrae (Duan et al., 2012). Capsazepine, SB203580, and BIM were delivered via intraplantar injection 20 min before TGF-β1 (intraplantar).
Preparation of DRG neurons.
Rats were anesthetized with isoflurane and then rapidly decapitated. The DRGs from spinal L3–L5 segments were removed and immediately transferred onto DMEM (Invitrogen) on ice. The ganglia were minced with fine spring scissors and treated with collagenase (type IA, 2.67 mg/ml, Sigma) and trypsin (type I, 1 mg/ml, Sigma) in DMEM at 37°C for 35 min. After washing with a standard external solution, the ganglia were then gently triturated using fine fire-polished Pasteur pipettes. The isolated DRG neurons were plated onto glass coverslips in 3.5 cm culture dishes and incubated with a standard external solution containing (in mm) as follows: 150 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4, at room temperature for >2 h.
Patch-clamp recordings.
Whole-cell patch-clamp recordings of DRG neurons were performed at room temperature (23 ± 1°C) using an Axonpatch 200B amplifier (Molecular Devices). All of the recordings were performed in small-diameter (15–25 μm) DRG neurons and were made 2–8 h after plating, with resting membrane potentials more negative than −50 mV. Microelectrodes (N51A borosilicate glass, Sutter Instruments) with a resistance of 2–6 mΩ were pulled using a P97 puller (Sutter Instruments). The pipette solution contained (in mm) as follows: 140 KCl, 1 MgCl2, 0.5 CaCl2, 5 EGTA, 3 Na2ATP, 0.2 NaGTP, and 10 HEPES, pH 7.2. Seals (1–10 GΩ) between the electrode and the cells were established. After the whole-cell configuration was established, the cell membrane capacitance and series resistance were compensated (>80%). Leak currents were subtracted using the online P/4 protocol. The data were sampled at 10 kHz and low-passed at 2 kHz. Capsaicin-induced TRPV1 currents were recorded in voltage-clamp mode, with the membrane potential held at −70 mV. Action potentials were elicited by injecting a depolarizing current (50 pA, 400 ms). Drugs were applied using a DVD-8VC superfusion application system (ALA Scientific Instruments). The perfusion tube (inner diameter, 100 μm) was located ∼200 μm from the recorded cell, with a rapid solution exchange of 20 ms. Only one recording was performed on each dish to ensure that data were not obtained from cells that had been inadvertently exposed to other test treatments. Two or three cells were studies per animal to ensure the data from different animals. Data acquisition and analysis were performed with Clampfit 10.2 (Molecular Devices) and/or Origin 7.5 (Microcal Software) software.
Calcium imaging.
The acutely isolated DRG neurons were loaded with 1 μm fura-2 AM (DoJinDo Laboratories) for 1 h and were then washed and incubated with standard external solution. The entire process was protected from light. The neurons were observed on an inverted microscope (Olympus IX51) with a 40× UV flour oil-immersion objective lens. The fluorescence of the individual neurons was recorded by a cooled CCD camera (Hamamatsu), with a 1 Hz alternating wavelength, time scanning with excitation wavelengths of 340 and 380 nm and an emission wavelength of 510 nm (monochromators; Till Polychrome IV). Images were captured every 1.5 s. Digitized images were acquired and analyzed by SimplePCI (Compix). The ratio of the fluorescence at the two excitation wavelengths was represented to estimate the changes in the [Ca2+]i.
Behavioral tests.
Animals were habituated to the testing environment daily for at least 3 d before baseline testing. Thermal hyperalgesia was assessed by measuring the paw withdrawal latency latencies (PWLs) in response to a radiant heat source. Animals were placed individually into Plexiglas chambers on an elevated glass platform and allowed to acclimate for 30 min. A radiant heat source (IITC/Life Science Instruments) was applied from underneath the platform to the glabrous surface of the paw through the glass plate. The heat source was turned off when the animal lifted its foot, allowing for measurement of the time from the onset of radiant heat application to the withdrawal of the hindpaw. This time was defined as the PWL. The heat was maintained at a constant intensity, which produced a stable PWL of 10–12 s in naive animals. A 20 s cutoff was used to prevent tissue damage in the absence of a response.
Bone radiological detection.
Rats were placed on a clear Plexiglas plane and exposed to an x-ray source under sodium pentobarbital anesthesia. Using the Digital Radiographer System (Philips, BV Pulsera C-arm), radiographs were taken from both hind limbs of naive, sham, and bone cancer rats.
Bone histology.
Rats were anesthetized with an overdose of urethane (1.5 g/kg, i.p.) and transcardially perfused with 300 ml of 0.9% normal saline followed by 300 ml of 4% paraformaldehyde. Bilateral tibia bones were removed and decalcified in decalcifying solution for 24 h. The bones were rinsed and dehydrated and then embedded in paraffin, cut into 7 μm cross sections using a rotary microtome (Reichert-Jung 820, Cambridge Instruments), and stained with hematoxylin and eosin to visualize the extent of tumor infiltration and bone destruction.
Immunohistochemistry/immunocytochemistry.
Animals were given an overdose of urethane and were then transcardially perfused with normal saline followed by 4% paraformaldehyde in 0.1 m phosphate buffer (pH 7.4, 4°C). DRGs (L3–L5 segments) were removed and postfixed in the same fixative for 2 h at 4°C and then immersed in a 10–30% gradient of sucrose in phosphate buffer for 24–48 h at 4°C for cryoprotection. DRG sections (15 μm) were cut on with a cryostat (Leica 1900, Leica) and mounted onto gelatin-coated slides for immunofluorescence. Control and treated DRG sections were mounted on the same slides and processed under the same conditions.
The slides were blocked after blocking with 10% donkey serum in 0.01 m PBS, pH 7.4, with and 0.3% Triton X-100 for 2 h at room temperature and incubated for 36 h at 4°C; the slides were incubated with goat anti-TRPV1 (1:200; Santa Cruz Biotechnology) primary antibody in PBS with 1% normal donkey serum and 0.3% Triton X-100 for 36 h at 4°C. After three 15 min rinses in PBS, the slides were incubated followed by incubation within rhodamine red-X-conjugated donkey anti-goat IgG (1:200; Jackson ImmunoResearch Laboratories) for 2 h at room temperature. For TRPV1/TGFβRI /TGFβRII or TRPV1/CGRP/NF-200 double immunofluorescence, the slides were incubated in a mixture of goat anti-TRPV1 (1:200) and rabbit anti-TGFβRI (1:200, Santa Cruz Biotechnology), rabbit anti-TGFβRII (1:200, Santa Cruz Biotechnology), mouse anti-CGRP (calcitonin gene-related peptide, a marker for peptide-containing DRG neurons, 1:3000, Sigma), or mouse anti-NF-200 (a marker for DRG neurons with myelinated fibers, 1:3000, Sigma) primary antibodies for 36 h at 4°C. The slides were then incubated with a mixture of rhodamine red-X-conjugated donkey anti-goat IgG (1:200) and FITC-conjugated donkey anti-rabbit IgG (1:200; Jackson ImmunoResearch Laboratories) secondary antibodies for 2 h at room temperature. All of the slides were coverslipped with 50% glycerin in 0.01 m PBS and then examined with an Olympus FV1000 confocal laser scanning microscope (Olympus). Images were acquired using FV10-ASW software. For the quantification of the immunoreactive signals, every 20th section was picked from a series of consecutive DRG sections, and four sections were counted for each DRG. The numbers of immunopositive cells were expressed as percentages of the total neuronal profiles in the same section. The specificities of the immunostaining were verified by observing no immunostaining after omitting the primary antibodies, which resulted in the disappearance of the immunostaining signals. The specificities of the primary antibodies were verified by a preabsorption experiment. Sections were first incubated overnight with a mixture of TRPV1, TGFβRI, or TGFβRII primary antibody and the corresponding blocking peptide (5:1 blocking peptide: primary antibody), followed by incubation with a secondary antibody. The immunostaining signals were abolished after absorption.
For PKCε visualization, DRG neurons on coverslips were treated with TGF-β1 (10 ng/ml) or the TFGβRI antagonist SD-208 (1 μm). TGF-β1 was added for 30 min. SD-208 was added 30 min before TGF-β1. Negative controls were treated alike but without the addition of any pharmaceutical reagents. The DRG neurons were rapidly fixed with 4% paraformaldehyde for 10 min at room temperature. After blocking with 10% donkey serum and 0.3% Triton X-100 for 1 h, the neurons were incubated overnight at 4°C with rabbit anti-pPKCε Ser729 (1:200, Santa Cruz Biotechnology) and goat anti-TRPV1 (1:200,Santa Cruz Biotechnology). The cultured neurons were then incubated for 1 h at room temperature with FITC-conjugated donkey anti-rabbit IgG and rhodamine red-X-conjugated donkey anti-goat IgG. To quantify PKCε translocation, 100 randomly selected cells were evaluated in each optic field. All counting was performed by the same observer, who was blinded to the experimental groups. All treatments were repeated with DRG neurons from four rats. The criteria for determining PKCε membrane translocation were based on previous reports (Cesare et al., 1999; Zhu et al., 2007). Briefly, a 25 μm line was drawn across the cell body to measure the florescence intensity along the line. If the intensity for a particular cell was higher in the cytoplasm than in the membrane, the cell was designated −1. Conversely, if the intensity in the distal membrane of a cell was high with two peak values, the cell was designated +1.
Western blots.
The L3–L5 DRGs and tibia (near the metaphysic) from bone cancer and control rats were collected and then homogenized in the lysis buffer (12.5 μl/mg tissue) containing a mixture of protease inhibitors (Roche) and PMSF (Sigma). The protein concentrations of the lysate were measured using a BCA Protein Assay kit (Pierce Biotechnology). Equal amounts of protein samples were loaded and separated in 10% SDS-PAGE gel and then transferred to PVDF membranes (Millipore). After 2 h, the membranes were blocked in blocking with 5% nonfat dry milk for 2 h at room temperature, and membranes were incubated overnight at 4°C with rabbit anti-TGF-β1 (1:1000, Sigma), rabbit anti-TGFβRI (1:1000, Santa Cruz Biotechnology), rabbit anti-TGFβRII (1:1000, Santa Cruz Biotechnology), or rabbit anti-p-p38 (1:500, Cell Signaling Technology) primary antibodies. The blots were then incubated with the secondary antibody, followed by HRP-conjugated goat anti-rabbit IgG conjugated with HRP secondary antibody (1:3000, Santa Cruz Biotechnology) for 2 h at room temperature. The blots were probed with GAPDH antibody as a loading control. Signals were finally detected using enhanced chemiluminescence (Pierce), and the bands were visualized with the ChemiDoc XRs system (Bio-Rad). All Western blot analysis was performed at least 3 times, and consistent results were obtained. Bio-Rad Image Analysis System was then used to measure the integrated optic density of the bands.
Statistical analysis.
The data are presented as the mean ± SEM. Student's t tests (for comparisons of two groups) or one-way ANOVAs (or two-way ANOVA, for multiple group comparisons) followed by post hoc Student-Newmann-Keuls tests were used to identify significant differences. In all cases, p < 0.05 was considered statistically significant.
Results
TGF-β1 and TGFβRs are involved in bone cancer pain
After intratibia inoculation with Walker 256 mammary gland carcinoma cells, thermal hyperalgesia developed within days 6–8, peaked at day 14, and persisted for at least 20 d in the ipsilateral hindpaw (two-way ANOVA, treatment: F(1,13) = 46.308, p < 0.001; treatment × time: F(10,117) = 2.466, p = 0.009) (Fig. 1A). Thus, in this study, we selected day 14 post-tumor inoculation to perform the following experiments. The radiographs showed signs of radiolucent lesions in the tibia close to the injection site, especially at the proximal epiphysis, on post-tumor day 14 (PTD 14) (Fig. 1B). The postmortem evaluation showed robust tumor growth in the ipsilateral but not in the contralateral tibia (Fig. 1C) or other bones (data not shown). The hematoxylin and eosin-stained tibia sections showed both tumor growth and bone destruction with medullary bone loss on PTD 14 (Fig. 1D).
Figure 1.
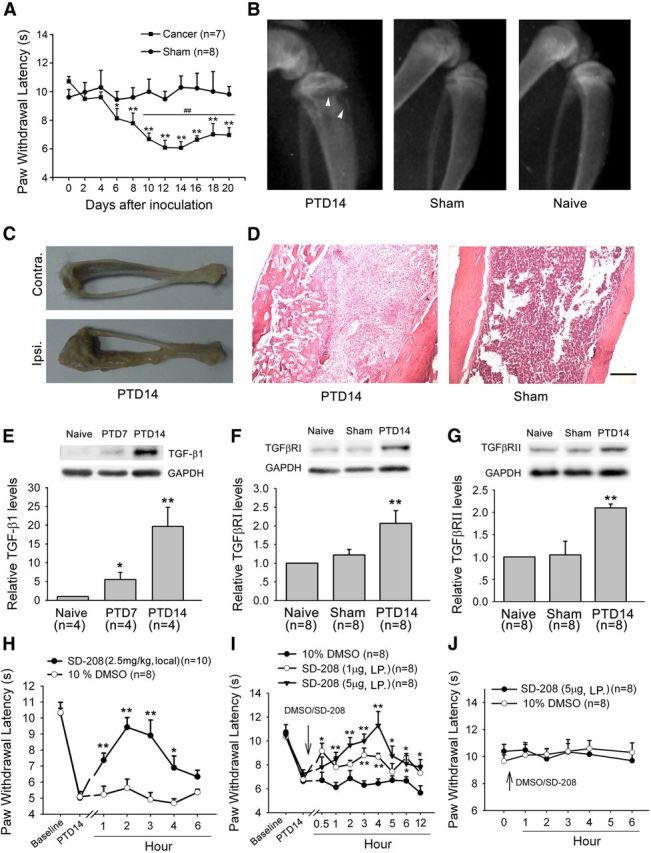
Involvement of bone-derived TGF-β1 and its receptors TGFβRI and TGFβRII in bone cancer pain. A, Intratibia inoculation with Walker 256 mammary gland carcinoma cells induces significant thermal hyperalgesia in the hindpaw ipsilateral to the tumor-bearing limb. *p < 0.05 versus basal (before tumor inoculation). **p < 0.01 versus basal (before tumor inoculation). ##p < 0.01 versus sham control. B, Radiographs represent robust radiolucent lesions (arrowhead) of the tibia on PTD 14. C, Photograph of a rat tibia on PTD 14 with obvious tumor growth in the ipsilateral but not contralateral tibia. D, Histopathological sections (hematoxylin and eosin stain) show that the bone marrow was largely replaced by invading tumor cells with medullary bone loss and tibial bone destruction on PTD 14. Scale bar, 500 μm. E, Western blot analysis reveals a progressive increase in the level of TGF-β1 in the affected bone after tumor inoculation. F, G, Western blot analysis reveals a significant increase in the level of TGFβRI and TGFβRII in L3–L5 DRGs ipsilateral to the tumor-bearing bone on PTD 14. *p < 0.05 versus naive. **p < 0.01 versus naive. H, Local injection (around tumor bone) of SD-208 (2.5 mg/kg) attenuated bone cancer-induced thermal hyperalgesia on PTD 14. I, Lumbar puncture injection of SD-208 (1 and 5 μg) significantly reduces bone cancer-induced thermal hyperalgesia on PTD 14. J, Lumbar puncture injection of SD-208 does not alter the paw withdrawal response to radiant heat stimulation in naive rats. *p < 0.05 versus vehicle (DMSO). **p < 0.01 versus vehicle (DMSO).
Tumor growth and bone destruction lead to the production of TGF-β (Mundy, 2002; Juárez and Guise, 2011). We therefore examined TGF-β1 expression in tumor-bearing bone. As shown in Figure 1E, the level of TGF-β1 protein robustly increased in the advanced cancer rats (one-way ANOVA, F(3,9) = 9.656, p = 0.006).
TGF-β mediates its biological effect through TGF-β receptor type I (TGFβRI) and the TGFβRII transmembrane serine/threonine receptor (Derynck and Zhang, 2003). We measured TGFβRI and TGFβRII protein levels in L3–L5 DRGs ipsilateral to the affected bone in cancer rats. Western blot analysis showed that there was significant upregulation of both TGFβRI and TGFβRII on PTD 14 (one-way ANOVA, TGFβRI: F(2,21) = 6.95, p = 0.005; TGFβRII: F(2,21) = 11.461, p < 0.001) (Fig. 1F,G).
To address whether TGFβRs are involved in bone cancer-induced thermal hyperalgesia, we observed the effects of local injection (around tumor bone) of the TFGβR antagonist, SD-208, in the PTD14 rats. When SD-208 (2.5 mg/kg) was injected into deep tissue around tumor bone, bone cancer-induced thermal hyperalgesia was significantly inhibited (two-way ANOVA, treatment: F(1,16) = 9.157, p < 0.001) (Fig. 1H). Consistently, the hyperalgesia was also suppressed by lumbar puncture (LP) injection of SD-208 (1 or 5 μg/20 μl) (Fig. 1I). It has been reported that drugs can be predominantly delivered into DRG neurons by direct lumbar puncture (Vulchanova et al., 2010; Duan et al., 2012). LP injection of SD-208 (5 μg/20 μl) had no effect on PWL in naive rats (Fig. 1J).
TGF-β mediates bone cancer pain by sensitization of TRPV1
The tumor-induced osteoclastic process results in TGF-β activation and local environment acidification (Griffiths, 1991), which can activate TRPV1 at normal body temperatures (Tominaga et al., 1998). Therefore, we examined TRPV1 expression and colocalization with TGF-βRs in primary sensory neurons. A significant increase in the number of TRPV1-positive neurons was consistently detected in L3–L5 DRGs ipsilateral to the tumor-inoculated bone on PTD 14 (Fig. 2A). Double immunofluorescence revealed that TRPV1 was predominantly expressed in isolectin B4 (IB4)-positive and CGRP-positive DRG neurons. At PTD 14, TRPV1-positive signals were significantly increased in both IB4- and CGRP-positive neurons. Few NF-200-positive neurons were TRPV1-positive in both the sham and bone cancer rats (Fig. 2B). Moreover, we observed that either TGFβRI or TGFβRII was heavily colocalized with TRPV1 in the DRG neurons; ∼90% TRPV1-positive neurons expressed TGF βRI or TGFβRII (Fig. 3A). To address whether increased peripheral TGF-β1 is involved in the bone cancer-induced upregulation of TRPV1 and TGF-βRs in the DRG, we examined the effects of directly intraplantar TGF-β1 on TRPV1 and TGFβRI levels in the L3–L5 DRGs. As shown in Figure 3B, a robust increase in either TRPV1- or TGFβRI-positive neurons was detected at 4 and 24 h after intraplantar injection of TGF-β1 (250 ng/50 μl). These results provided morphological evidence for the functional interaction between TGFβ and TRPV1.
Figure 2.

Expression of TRPV1 in the DRG. A, Immunohistochemistry reveals that the number of TRPV1-positive cells increases in L3, L4, and L5 DRGs on PTD 14. B, Double immunofluorescence reveals the expression of TRPV1 in IB4-, CGRP-, and NF-200-positive neurons in the DRGs from sham and PTD 14 rats. **p < 0.01 versus sham.
Figure 3.
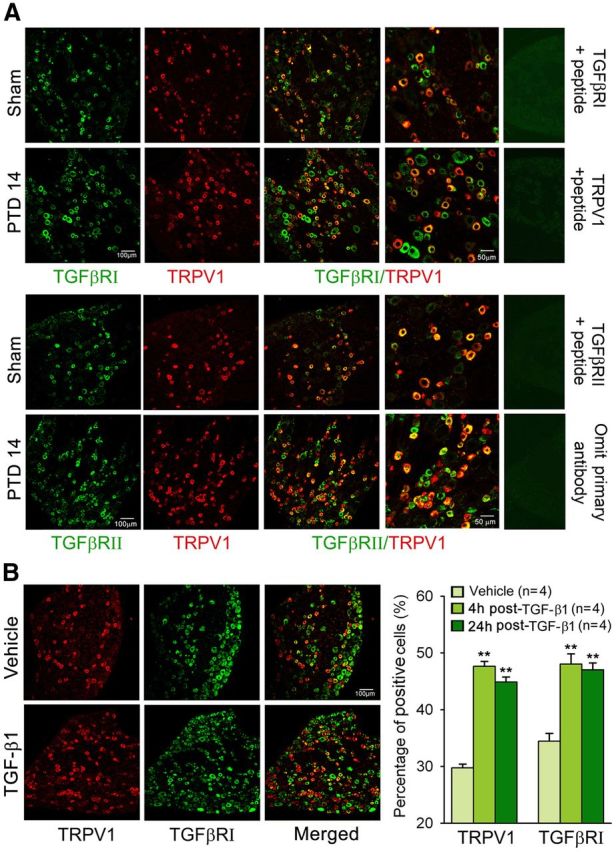
Expression of TGFβRs (TGFβRI and TGFβRII) and TRPV1 in the DRG. A, Double immunofluorescence reveals colocalization of TRPV1 with TGFβRI and TGFβRII in DRGs from naive and PTD 14 rats. B, Immunohistochemistry reveals a significant increase in the number of TRPV1- and TGFβRI-positive neurons in L3–L5 DRGs both 4 and 24 h after intraplantar injection of TGF-β1. **p < 0.01 versus vehicle.
Given that TRPV1 was predominantly expressed in small-diameter IB4-positive and CGRP-positive DRG neurons, whole-cell patch recordings were performed in acutely isolated small-diameter (<25 μm) DRG neurons. Consistent with our previous studies (Zhang et al., 2007), capsaicin (0.5–5 μm) induced an inward current in a dose-dependent manner in ∼70% of the recorded neurons (Fig. 4A,B). After bone cancer development, the percentage of DRG neurons that were responsive to capsaicin was increased to >90%, and the dose–response curve of capsaicin currents was left-shifted on PTD 14 (Fig. 4B). Successive applications of 1 μm capsaicin (3 s, interval of 60 s) produced a progressive decline or desensitization in the responses in the neurons from both normal and bone cancer rats (Fig. 4C,D). The amplitude of the TRPV1 currents was much greater, and the rate of desensitization was much less in DRG neurons from PTD 14 rats than those from control rats (Fig. 4C–F).
Figure 4.

Dynamic changes in TRPV1 in DRG neurons after tumor inoculation. A, Representative recordings of different doses of capsaicin-induced TRPV1 currents. The competitive antagonist of capsaicin, capsazepine, completely blocked the current. Capsaicin was perfused for 3 s for each dose. B, Capsaicin dose–response curves were left-shifted in the DRG neurons from PTD 14 rats compared with those from sham rats. C, D, Representative current traces of successive applications of capsaicin-induced TRPV1 currents in individual DRG neurons from naive (C) and PTD 14 rats (D). E, A plot of the normalized mean (±SEM) amplitude of TRPV1 currents evoked by sequential applications of capsaicin (1 μm) in DRG neurons from controls (pooled naive and sham) and PTD 14 rats. F, TRPV1 current density (pA/pF) is significantly increased in DRG neurons from PTD 14 rats. *p < 0.05, versus controls. **p < 0.01 versus controls. #p < 0.05 versus the previous application.
Next, we examined whether peripheral TGF-β can directly produce thermal hyperalgesia. As shown in Figure 5A, intraplantar injection of TGF-β1 (250 ng/50 μl) produced a robust thermal hyperalgesia in naive rats. Interestingly, this TGF-β1-induced hyperalgesia was completely abolished by the TRPV1 antagonist, capsazepine (1.5 μg/50 μl) (Fig. 5A). A two-way ANOVA revealed significant differences in the treatment groups (F(3,28) = 26.796, p < 0.001) and in the interaction between the treatment and time (F(24,224) = 2.531, p = 0.002). This result was further confirmed in Trpv1 knock-out mice. The TGF-β1 (intraplantar 25 ng/5 μl)-induced thermal hyperalgesia was observed only in wild-type (C57BL/6) mice and not in Trpv1−/− mice (Fig. 5B). Consistent with the behavioral hypersensitivity, whole-cell current-clamp recordings showed that TGF-β1 (10 ng/ml) greatly increased the action potential firing frequencies of small-diameter DRG neurons (paired t test, t(0.05,6.357), p < 0.001) (Fig. 5C,D).
Figure 5.
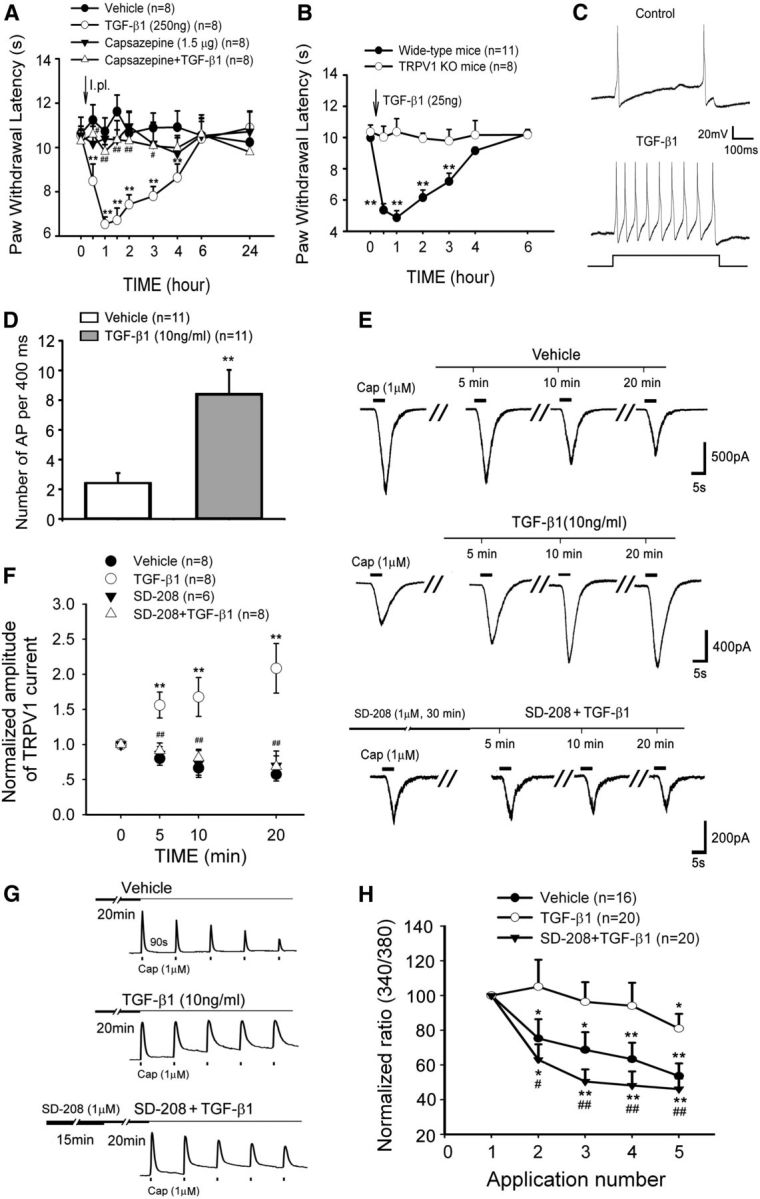
TGF-β1 evokes nociceptive behavior and potentiates action potential firing and TRPV1 function in small DRG neurons. A, An intraplantar injection of TGF-β1 acutely induced thermal hyperalgesia in naive rats. Preadministration (intraplantar) of the TRPV1 antagonist capsazepine 20 min before TGF-β1 completely prevents TGF-β1-induced thermal hyperalgesia. **p < 0.01 versus vehicle. #p < 0.05 versus TGF-β1 treatment. ##p < 0.01 versus TGF-β1 treatment. B, An intraplantar injection of TGF-β1 acutely induced thermal hyperalgesia in wide-type mice but did not in TRPV1 knock-out mice. **p < 0.01 versus wide-type. C, D, TGF-β1 robustly enhances the firing frequency of evoked action potentials obtained by a 50 pA (400 ms) current injection in DRG neurons. **p < 0.01 versus vehicle. E, Representative current traces from individual DRG neurons challenged by multiple capsaicin (Cap) applications during the 20 min treatment with vehicle, TGF-β1, or pretreatment with the TGFβRI kinase inhibitor SD-208 before TGF-β1. F, Summary data (mean ± SEM) of normalized TRPV1 currents indicate that TGF-β1 sensitizes TRPV1 currents in DRG neurons but can be blocked by pretreatment with SD-208. **p < 0.01 versus vehicle control. ##p < 0.01 versus TGF-β1 treatment. G, Representative traces of calcium imaging from individual DRG neurons challenged by five capsaicin (Cap) applications. H, Summary data (mean ± SEM) of normalized capsaicin-induced increases in [Ca2+]i reveal that TGF-β1 rescues TRPV1 desensitization through TGFβRI. *p < 0.05 versus first application of capsaicin. **p < 0.01 versus first application of capsaicin. #p < 0.05 versus TGF-β1 treatment. ##p < 0.01 versus TGF-β1 treatment.
To determine the effects of TGF-β1 on TRPV1 activity, 1 μm capsaicin-induced TRPV1 currents were recorded in the presence of TGF-β1 (10 ng/ml). As shown in Figure 5, E and F, TGF-β1 progressively potentiated TRPV1 currents during a 20 min perfusion in small-diameter DRG neurons. Pretreatment with SD-208 (1 μm), a selective TFGβRI antagonist, for 30 min prevented the TGF-β1-induced potentiation of TRPV1 currents completely. Analogously, the repetitive capsaicin-induced desensitization in the responses of [Ca2+]i increases by capsaicin was also antagonized by exogenous TGF-β1. This effect was abolished by the TFGβRI antagonist SD-208 (Fig. 5G,H).
TGF-β1-induced sensitization of TRPV1 is mediated by Smad-independent PKC and TAK1/p38
TGF-β can alter cell behavior through the activation of Smad-dependent and Smad-independent pathways. To address whether the Smad-dependent pathway is involved in TRPV1 sensitization, the Smad3 inhibitor SIS3 was used to block the phosphorylation of Smad3 and the interaction of Smad3 with Smad4. As shown in Figure 6, the TGF-β1-induced potentiation of TRPV1 currents was not blocked by SIS3 (10 μm).
Figure 6.
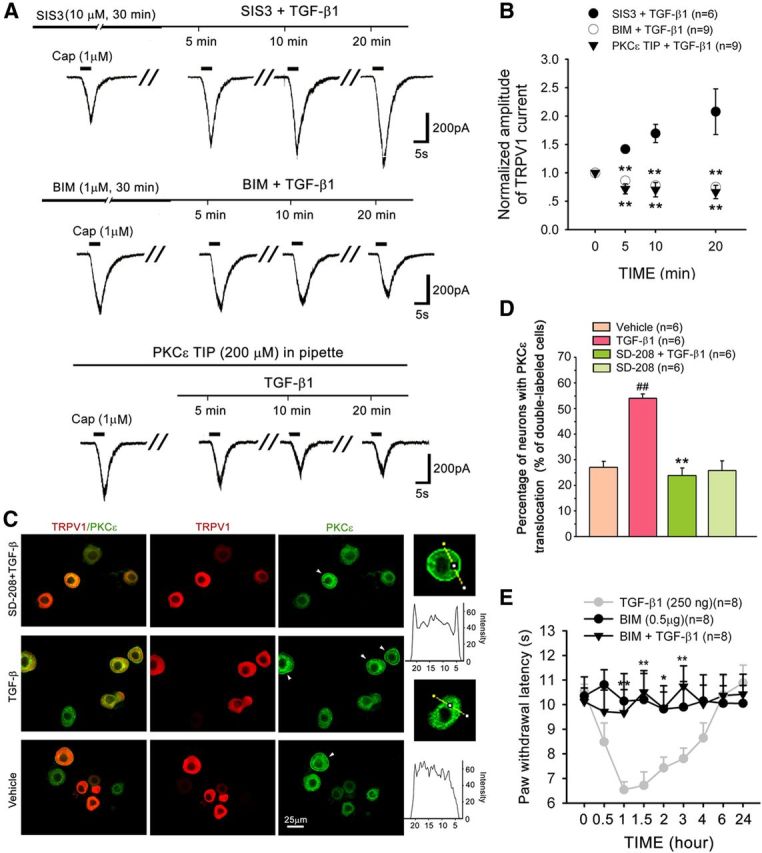
TGF-β1 sensitizes TRPV1 through PKCε activation in DRG neurons. A, B, The addition of the PKCε translocation inhibitor peptide (PKCε TIP) to the recording pipette or pretreatment with the PKC inhibitor BIM, but not the Smad 3 inhibitor SIS3, completely prevents the TGF-β1-induced enhancement of TRPV1 currents in DRG neurons. **p < 0.01 versus TGF-β1 treatment. C, D, Double staining of PKCε and TRPV1 in dissociated DRG neurons treated by vehicle, TGF-β1 and SD-208 plus TGF-β1. In most vehicle control cells, PKCε is found in the cytoplasm, whereas TRPV1 is present in the cytoplasm and on the membrane. After incubation in TGF-β1 for 30 min, PKCε translocated to the plasma membrane (arrowhead), and the percentage of neurons demonstrating PKCε membrane translocation was significantly increased. Pretreatment with SD-208 for 30 min completely prevented the TGF-β1-induced PKCε membrane translocation. ##p < 0.01 versus vehicle. **p < 0.01 versus TGF-β1 treatment. E, TGF-β1-induced thermal hyperalgesia is blocked by an intraplantar injection of the PKC inhibitor BIM 20 min before TGF-β1. *p < 0.05 versus TGF-β1 treatment. **p < 0.01 versus TGF-β1 treatment.
Several non–Smad-signaling pathways have been linked to the TGFβRI/TGFβRII complex. PKC has been reported to be activated by a wide variety of growth factors, including TGF-β 1 (Mulsow et al., 2005; Nagaraj and Datta, 2010), and is strongly implicated in TRPV1 sensitization (Numazaki et al., 2002; Bhave et al., 2003; Zhang et al., 2007). We therefore assessed whether PKC is involved in the sensitization of TRPV1 by TGF-β1 in small DRG neurons. Although the PKC inhibitor BIM (1 μm) did not affect TRPV1 currents per se, it completely abrogated the TGF-β1-induced enhancement of TRPV1 currents (Fig. 6A,B).
Several PKC isoforms (δ, βΙ, βΙΙ, ε, and ξ) are present in rat DRG neurons (Cesare et al., 1999). Among the different PKC isoforms, PKCε is coexpressed with TRPV1 in DRG neurons and is phosphorylated after inflammation (Zhou et al., 2003). Translocation of PKCε to the cell membrane triggers TRPV1 phosphorylation and sensitization (Numazaki et al., 2002). Our results showed that the addition of PKCε translocation inhibitor peptide (200 μm) to the recording pipette completely abolished the TGF-β-induced TRPV1 sensitization (Fig. 6A,B). TGF-β also caused a significant translocation of PKCε immunoreactivity to the plasma membrane in DRG neurons, especially in TRPV1-positive neurons (Fig. 6C,D). Preincubation with the TFGβRI antagonist SD-208 (1 μm) for 30 min greatly diminished TGF-β-induced translocation (one-way ANOVA, F(3,20) = 21.838, p < 0.001) (Fig. 6C,D). Moreover, behavioral testing showed that an intraplantar injection of the PKC inhibitor BIM (0.5 μg) blocked peripheral TGF-β1-induced thermal hyperalgesia. BIM (0.5 μg/50 μl) per se did not change basal PWL in response to noxious heat stimulation (Fig. 6E).
TGF-β can also elicit cellular responses through TAK1, a member of the MAPKKK family. TAK1 is involved in the activation of several MAP kinases (MAPK), including ERK, p38, and JNK (Massagué, 2008; Zhang, 2009; Nagaraj and Datta, 2010; Mu et al., 2012). Thus, we investigated the role of the TAK1-MAPK pathways in the TGF-β1-induced sensitization of TRPV1. As shown in Figure 7, TGF-β1-induced potentiation of TRPV1 currents was completely blocked by the TAK1 inhibitor (5Z)-7-oxozeaenol (1 μm). Furthermore, blockade of p38 activation by SB203580 (10 μm) also abolished TGF-β1-induced TRPV1 sensitization. In contrast, neither the JNK inhibitor SP600125 (10 μm) nor the MEK (upstream of ERK1/2) inhibitors PD98059 (10 μm) and U0126 (5 μm) prevented the TGF-β1-induced sensitization of TRPV1 (Fig. 7A,B). Accordingly, an intraplantar injection of TGF-β1 induced behavioral hypersensitivity, which was blocked by a preinjection of the p38 inhibitor SB203580 (10 μg) (Fig. 7C).
Figure 7.
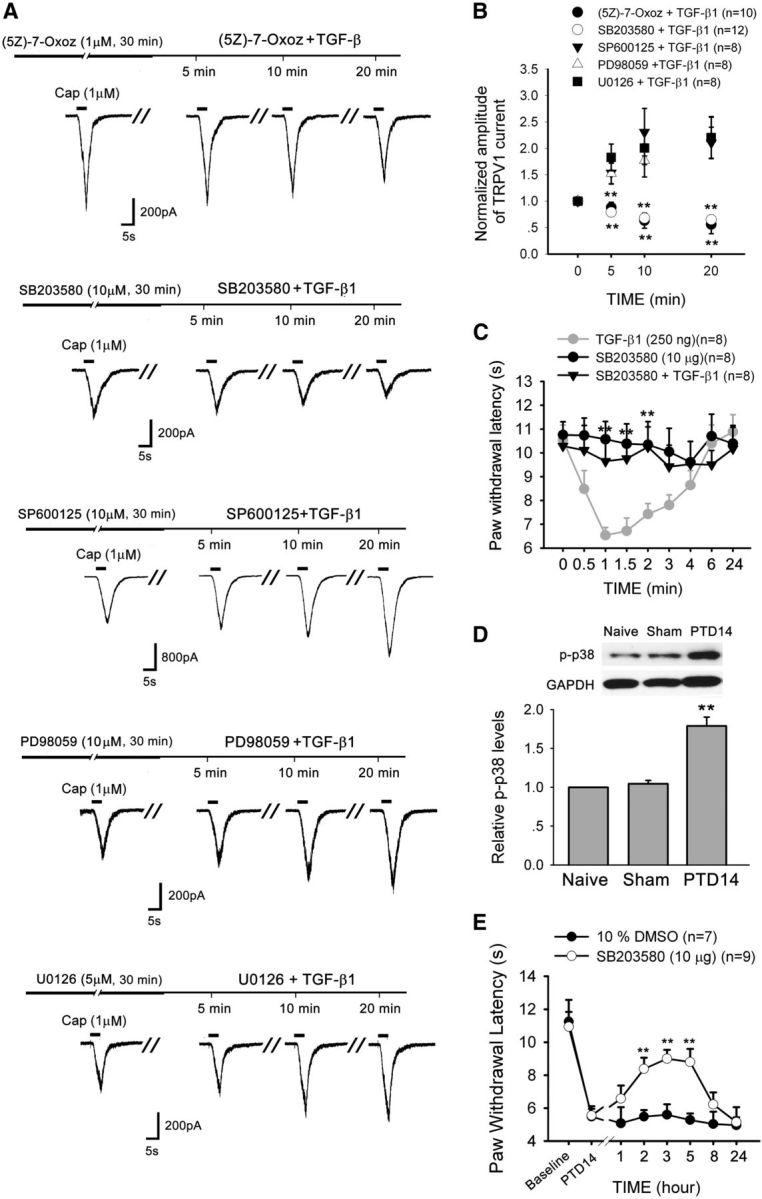
TGF-β1 sensitizes TRPV1 through activation of the TAK1-p38 pathway in DRG neurons. A, B, The TGF-β1-induced enhancement of TRPV1 currents in DRG neurons is prevented by the TAK1 inhibitor (5Z)-7-oxozeaenol or p38 inhibitor SB203580 but not by the JNK inhibitor SP600125 or the MEK inhibitors PD98059 and U0126. **p < 0.01 versus TGF-β1 treatment. C, TGF-β1-induced thermal hyperalgesia is blocked by an intraplantar injection of the p38 inhibitor SB203580. **p < 0.01 versus TGF-β1 treatment. D, Western blot analysis reveals a significant increase in the level of p-p38 in the ipsilateral L3–L5 DRGs on PTD 14. **p < 0.01 versus naive and sham. E, Lumbar puncture injection of p38 inhibitor SB203580 significantly reduces bone cancer-induced thermal hyperalgesia on PTD 14. **p < 0.01 versus vehicle.
The involvement of p38 in bone cancer pain was further confirmed. Western blot analysis showed that there was significant upregulation of p-p38 level in affected DRGs on PTD 14 (one-way ANOVA, F(2,12) = 40.99, p < 0.001) (Fig. 7D). On PTD 14 when the thermal hyperalgesia was steadily developed, LP injection of the p38 inhibitor, SB203580 (10 μg/20 μl), significantly reversed existing thermal hyperalgesia in the hindpaw that was ipsilateral to the affected bone (Fig. 7E).
Discussion
The TGF-β superfamily includes activins, BMPs, growth and differentiation factors, and TGF-βs (Lantero et al., 2012). The TGF-βs family contains three related mammalian isoforms: TGF-β1, TGF-β2, and TGF-β3 (Janssens et al., 2005). All three isoforms are detected in bone, but the most abundant in bone is TGF-β1 (Hering et al., 2001). It has been shown that increased expression of TGF-β1 is often correlated with the aggressiveness and grade/stage of a tumor, as well as a high incidence of metastasis (Muraoka et al., 2002; Massagué, 2008). Blocking TGF-β1 or its signaling inhibits tumor cell invasiveness in vitro and metastatic ability in vivo (Ananth et al., 1999; Rowland-Goldsmith et al., 2002). A recent study demonstrated that, in bone metastases mice, the osteolytic lesions were significantly decreased after 4 weeks of treatment with the TGFβR-I inhibitor, SD-208 (Mohammad et al., 2011). In the present study, we further showed that excessive levels of TGF-β1 in advanced-stage bone tumors also contributed to bone cancer pain. Thus, TGF-β1 and its signaling effectors and modulators may be promising molecular targets of novel therapeutic agents for both the treatment of bone metastases and the management of bone cancer pain.
TGF-β1 contributes to bone cancer-induced thermal hyperalgesia
The most common symptom of bone cancer is pain. The severity of the pain is closely correlated with the extent of bone destruction (Adami, 1997). TGF-β, which plays a prominent role in breast cancer progression and bone metastasis, is a central mediator that drives a feedforward cycle of tumor growth in bone (Mundy, 2002; Juárez and Guise, 2011). Correspondingly, in a rat model of bone cancer, we found that TGF-β1 was highly expressed in the tumor-inoculated bone and that this increased expression was associated with tumor growth and bone destruction. A concomitant increase in the expression of its receptors, TGFβRI and TGFβRII, in the DRG and robust thermal hyperalgesia in the affected hindpaw were observed. In particular, the TGFβRI antagonist, SD-208, significantly suppressed bone cancer-induced thermal hyperalgesia, indicating a role for TGF-β1 in bone cancer pain.
TGF-β has been implicated in pain signaling, but its precise role is not completely clear. For instance, Echeverry et al. (2009) showed that TGF-β1 was implicated in the negative modulation of neuropathic pain through neuroprotective effects, inhibiting the activation of spinal microglia and astrocytes. Studies from the Hurle group using BMP and activin membrane-bound inhibitor (BAMBI)-KO mice (Bambi−/− mice) implicated the TGF-β superfamily members in the modulation of chronic and acute pain perception through the transcriptional regulation of genes that encode endogenous opioids (Tramullas et al., 2010; Lantero et al., 2012). In contrast, activin A, another member of the TGF-β superfamily, was reported to cause acute thermal hyperalgesia by sensitizing peripheral TRPV1 (Zhu et al., 2007) and increasing the expression in DRG neurons of CGRP, a peptide marker of the major subset of peripheral nociceptors that contribute to the development of hyperalgesia in pathological pain (Xu et al., 2005); TGF-β1 and TGF-β2 increased the levels of substance P (SP) (Lantero et al., 2012), which can sensitize TRPV1 and produce thermal hyperalgesia via the NK-1 receptor (Zhang et al., 2007). TGF-β superfamily members, BMP2, BMP4, BMP6, and BMP7, also induce a strong increase in the expression of CGRP and SP in cultured embryonic DRG neurons (Ai et al., 1999). A recent study also showed that a deficiency in TGF-β signaling in Tgfb1−/− mice, or mice in which the transforming growth factor-β receptor 1 (Tgfbr1) is conditionally knocked out in the trigeminal ganglia and DRG, resulted in reduced Cdk5 activity and attenuated thermal hyperalgesia after peripheral inflammation, suggesting a positive role for TGF-β1 in inflammation-induced hyperalgesia (Utreras et al., 2012). In support of this finding, we observed in the present study that an intraplantar injection of TGF-β1 induced immediate thermal hyperalgesia. Thus, TGF-β1 may positively modulate sensitivity of nociceptive primary afferent neurons in peripheral inflammation and bone cancer pain.
TGF-β1 is involved in bone cancer-induced thermal hyperalgesia by sensitizing TRPV1
Previous studies revealed that, after cancer infiltration, TRPV1 expression increased in the DRG and contributed to thermal hyperalgesia (Niiyama et al., 2007; Pan et al., 2010; Han et al., 2012). Selective blockade of TRPV1 resulted in a significant attenuation of nociceptive behavior in various models of cancer pain (Ghilardi et al., 2005; Niiyama et al., 2007). Consistently, in the present study, we observed a significant upregulation in TRPV1 expression in the DRG on PTD 14 in bone cancer rats. In addition to an increase in TRPV1 expression, enhanced TRPV1 currents and a decreased desensitization rate to repetitive capsaicin application were also observed in DRG neurons from PTD 14 bone cancer rats. These results suggest that both transcriptional changes in TRPV1 expression and post-translational changes in the sensitivity of the TRPV1 channel may be involved in bone cancer-induced thermal hyperalgesia.
TRPV1 is present on sensory fibers in mineralized bone and bone marrow (Ghilardi et al., 2005). There are at least two mechanisms in bone cancer that may contribute to the sensitization of TRPV1. One such mechanism is involved in the sensitivity of TRPV1 to acid. Osteoclast-mediated acidosis and lysis of acidic tumor cells result in an acidic environment (Griffiths, 1991; Boyle et al., 2003), which can, at normal body temperatures, activate TRPV1 on the primary afferent fibers that innervate the affected bone (Tominaga et al., 1998). Another possible mechanism is sensitization of TRPV1 by chemical mediators, including bradykinin, SP, ATP, NGF, and activin (Woolf and Salter, 2000; Zhang et al., 2007; Zhu et al., 2007). We have now shown that TGF-β1 can also sensitize TRPV1 within 20 min of its application, consistent with its abilities to increase the action potential firing frequencies of small DRG neurons and induce thermal hyperalgesia after intraplantar injection. Furthermore, TGF-β1-induced thermal hyperalgesia can be abolished by the TRPV1 antagonist capsazepine; in addition, TGF-β1-induced thermal hyperalgesia only occurred in wide-type, but not in TRPV1−/− mice, suggesting that the sensitization of TRPV1 is necessary for TGF-β1-induced thermal hyperalgesia. In addition to increasing the sensitivity of TRPV1, TGF-β1 was also shown to upregulate TRPV1 expression and antagonize TRPV1 desensitization in DRG neurons. It is plausible that excessive levels of TGF-β1 in bone tumors contribute to bone cancer-induced thermal hyperalgesia via increasing the sensitivity and expression density of TRPV1.
TGF-β1 sensitization of TRPV1 by TGF-βR-mediated PKC and TAK/p38 signaling pathways
Bioactive TGF-β binds with high affinity and selectivity to TGFβRII, which then recruits and activates TGFβRI, enabling the subsequent phosphorylation of downstream substrates (de Caestecker, 2004; Krieglstein et al., 2011). The TGF-β1 receptor subunits TGFβRI and TGFβRII are present in DRG neurons, and both types of receptors are coexpressed with TRPV1. After the development of bone cancer pain, TGFβRI and TGFβRII levels were significantly increased in the affected DRG on PTD 14. The TGF-β1-induced sensitization of TRPV1 and bone cancer-induced thermal hyperalgesia were blocked by the TGFβRI kinase inhibitor SD-208, suggesting that this receptor kinase is required for the TGF-β1-induced sensitization of TRPV1 in DRG neurons and thermal hyperalgesia in bone cancer rats (Fig. 8).
Figure 8.

Putative model for elucidating the relationship between TGF-β1 and bone cancer pain. With the development of bone cancer, cancer cells secrete factors that result in osteoclastogenesis. TGF-β1 is released by osteoclast bone resorption and binds with high affinity and selectivity to the membrane-spanning TGFβRII on nociceptors. Activated TGFβRII recruits and activates TGFβRI, which enables the subsequent phosphorylation of downstream intracellular signals, such as PKC and TAK1-p38, leading to the sensitization of TRPV1.
Activation of TGFβRI leads to the downstream propagation through Smad-dependent and Smad-independent pathways (Derynck and Zhang, 2003; Krieglstein et al., 2011). In our study, the TGFβ1-induced enhancement of TRPV1 sensitivity occurred within 20 min and was not inhibited by the Smad3 inhibitor SIS3, indicating that the Smad-dependent signaling pathway is not involved in this rapid response. Recently, several Smad-independent pathways, including various branches of the MAP kinase (ERK, JNK, and p38) and PKC pathways, have been reported in different cell types (Zhang, 2009; Nagaraj and Datta, 2010). PKC, in particular the PKCε isoform, has been implicated in the sensitization of TRPV1 by NGF (Zhu and Oxford, 2007), SP (Zhang et al., 2007), lysophosphatidic acid (Pan et al., 2010), and the TGF-β family member activin A (Zhu et al., 2007). In the present study, we further demonstrated that PKCε is required for the TGF-β1-induced sensitization of TRPV1. In addition to PKC-mediated TRPV1 sensitization, we also found that the inhibition of TAK1, a member of the MAPKKK family (upstream of MAPK), completely blocked TGFβ1-induced TRPV1 sensitization and thermal hyperalgesia, suggesting an important role for the MAPK pathway in the cross talk between TGFβ1 and TRPV1. Utreras et al. (2012) found that TGFβ1 induced an increase in phosphor-ERK1/ERK2 in rat B104 neuroblastoma cells. In contrast, activin A did not promote phosphorylation of ERK, and inhibition of ERK failed to block the sensitization of TRPV1 by activin in cultured DRG neurons (Zhu et al., 2007). In the present study, our data strongly implicated the activation of the TAK1-p38/MAPK pathway, but not the ERK and JNK pathways, as being critical for the TGFβ1-induced sensitization of TRPV1.
In conclusion, we have demonstrated a pivotal role for TGF-β1-mediated intracellular signaling in thermal hyperalgesia in advanced bone cancer. The upregulation of TRPV1 expression level and sensitization of TRPV1 in nociceptors by TGF-β might be an important peripheral mechanism of bone cancer pain. The present study highlights the possibility of a new clinical strategy to alleviate bone cancer pain in advanced cancer.
Footnotes
This work was supported by the National Basic Research Program of China (2013CB531900) and the National Natural Science Foundation of China (31121061, 31271183, and 31070973).
The authors declare no competing financial interests.
References
- Adami S. Bisphosphonates in prostate carcinoma. Cancer. 1997;80(Suppl 8):1674–1679. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1674::aid-cncr18>3.3.co;2-d. [DOI] [PubMed] [Google Scholar]
- Ai X, Cappuzzello J, Hall AK. Activin and bone morphogenetic proteins induce calcitonin gene-related peptide in embryonic sensory neurons in vitro. Mol Cell Neurosci. 1999;14:506–518. doi: 10.1006/mcne.1999.0798. [DOI] [PubMed] [Google Scholar]
- Ananth S, Knebelmann B, Grüning W, Dhanabal M, Walz G, Stillman IE, Sukhatme VP. Transforming growth factor beta1 is a target for the von Hippel-Lindau tumor suppressor and a critical growth factor for clear cell renal carcinoma. Cancer Res. 1999;59:2210–2216. [PubMed] [Google Scholar]
- Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM, Cornell JE, Pollock BH, Mundy GR, Sun LZ. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006;66:6714–6721. doi: 10.1158/0008-5472.CAN-05-3565. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci U S A. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/S0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Dallas SL, Rosser JL, Mundy GR, Bonewald LF. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts: a cellular mechanism for release of TGF-beta from bone matrix. J Biol Chem. 2002;277:21352–21360. doi: 10.1074/jbc.M111663200. [DOI] [PubMed] [Google Scholar]
- de Caestecker M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15:1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Duan KZ, Xu Q, Zhang XM, Zhao ZQ, Mei YA, Zhang YQ. Targeting A-type K(+) channels in primary sensory neurons for bone cancer pain in a rat model. Pain. 2012;153:562–574. doi: 10.1016/j.pain.2011.11.020. [DOI] [PubMed] [Google Scholar]
- Echeverry S, Shi XQ, Haw A, Liu H, Zhang ZW, Zhang J. Transforming growth factor-beta1 impairs neuropathic pain through pleiotropic effects. Mol Pain. 2009;5:16. doi: 10.1186/1744-8069-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghilardi JR, Röhrich H, Lindsay TH, Sevcik MA, Schwei MJ, Kubota K, Halvorson KG, Poblete J, Chaplan SR, Dubin AE, Carruthers NI, Swanson D, Kuskowski M, Flores CM, Julius D, Mantyh PW. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci. 2005;25:3126–3131. doi: 10.1523/JNEUROSCI.3815-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths JR. Are cancer cells acidic? Br J Cancer. 1991;64:425–427. doi: 10.1038/bjc.1991.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto DT, Khasabov SG, Cain DM, Simone DA. Tumor-evoked sensitization of C nociceptors: a role for endothelin. J Neurophysiol. 2008;100:2300–2311. doi: 10.1152/jn.01337.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Li Y, Xiao X, Liu J, Meng XL, Liu FY, Xing GG, Wan Y. Formaldehyde up-regulates TRPV1 through MAPK and PI3K signaling pathways in a rat model of bone cancer pain. Neurosci Bull. 2012;28:165–172. doi: 10.1007/s12264-012-1211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering S, Isken E, Knabbe C, Janott J, Jost C, Pommer A, Muhr G, Schatz H, Pfeiffer AF. TGFbeta1 and TGFbeta2 mRNA and protein expression in human bone samples. Exp Clin Endocrinol Diabetes. 2001;109:217–226. doi: 10.1055/s-2001-15109. [DOI] [PubMed] [Google Scholar]
- Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-beta1 to the bone. Endocr Rev. 2005;26:743–774. doi: 10.1210/er.2004-0001. [DOI] [PubMed] [Google Scholar]
- Juárez P, Guise TA. TGF-beta in cancer and bone: implications for treatment of bone metastases. Bone. 2011;48:23–29. doi: 10.1016/j.bone.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Kawamata T, Niiyama Y, Yamamoto J, Furuse S. Reduction of bone cancer pain by CB1 activation and TRPV1 inhibition. J Anesth. 2010;24:328–332. doi: 10.1007/s00540-010-0919-0. [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Zheng F, Unsicker K, Alzheimer C. More than being protective: functional roles for TGF-beta/activin signaling pathways at central synapses. Trends Neurosci. 2011;34:421–429. doi: 10.1016/j.tins.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Lantero A, Tramullas M, Díaz A, Hurlé MA. Transforming growth factor-beta in normal nociceptive processing and pathological pain models. Mol Neurobiol. 2012;45:76–86. doi: 10.1007/s12035-011-8221-1. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH, Duong V, Dunn LK, Mauviel A, Guise TA. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011;71:175–184. doi: 10.1158/0008-5472.CAN-10-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Gudey SK, Landström M. Non-Smad signaling pathways. Cell Tissue Res. 2012;347:11–20. doi: 10.1007/s00441-011-1201-y. [DOI] [PubMed] [Google Scholar]
- Mulsow JJ, Watson RW, Fitzpatrick JM, O'Connell PR. Transforming growth factor-beta promotes pro-fibrotic behavior by serosal fibroblasts via PKC and ERK1/2 mitogen activated protein kinase cell signaling. Ann Surg. 2005;242:880–887. doi: 10.1097/01.sla.0000189606.58343.cd. discussion 887–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals PJ, Koteliansky V, Arteaga CL. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109:1551–1559. doi: 10.1172/JCI15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj NS, Datta PK. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs. 2010;19:77–91. doi: 10.1517/13543780903382609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niiyama Y, Kawamata T, Yamamoto J, Omote K, Namiki A. Bone cancer increases transient receptor potential vanilloid subfamily 1 expression within distinct subpopulations of dorsal root ganglion neurons. Neuroscience. 2007;148:560–572. doi: 10.1016/j.neuroscience.2007.05.049. [DOI] [PubMed] [Google Scholar]
- Niiyama Y, Kawamata T, Yamamoto J, Furuse S, Namiki A. SB366791, a TRPV1 antagonist, potentiates analgesic effects of systemic morphine in a murine model of bone cancer pain. Br J Anaesth. 2009;102:251–258. doi: 10.1093/bja/aen347. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues. J Biol Chem. 2002;277:13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- Pan HL, Zhang YQ, Zhao ZQ. Involvement of lysophosphatidic acid in bone cancer pain by potentiation of TRPV1 via PKCepsilon pathway in dorsal root ganglion neurons. Mol Pain. 2010;6:85. doi: 10.1186/1744-8069-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland-Goldsmith MA, Maruyama H, Matsuda K, Idezawa T, Ralli M, Ralli S, Korc M. Soluble type II transforming growth factor-beta receptor attenuates expression of metastasis-associated genes and suppresses pancreatic cancer cell metastasis. Mol Cancer Ther. 2002;1:161–167. [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/S0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Tramullas M, Lantero A, Díaz A, Morchón N, Merino D, Villar A, Buscher D, Merino R, Hurlé JM, Izpisúa-Belmonte JC, Hurlé MA. BAMBI (bone morphogenetic protein and activin membrane-bound inhibitor) reveals the involvement of the transforming growth factor-beta family in pain modulation. J Neurosci. 2010;30:1502–1511. doi: 10.1523/JNEUROSCI.2584-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utreras E, Keller J, Terse A, Prochazkova M, Iadarola MJ, Kulkarni AB. Transforming growth factor-β1 regulates Cdk5 activity in primary sensory neurons. J Biol Chem. 2012;287:16917–16929. doi: 10.1074/jbc.M111.329979. inelevel0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulchanova L, Schuster DJ, Belur LR, Riedl MS, Podetz-Pedersen KM, Kitto KF, Wilcox GL, McIvor RS, Fairbanks CA. Differential adeno-associated virus mediated gene transfer to sensory neurons following intrathecal delivery by direct lumbar puncture. Mol Pain. 2010;6:31. doi: 10.1186/1744-8069-6-31. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Xu P, Van Slambrouck C, Berti-Mattera L, Hall AK. Activin induces tactile allodynia and increases calcitonin gene-related peptide after peripheral inflammation. J Neurosci. 2005;25:9227–9235. doi: 10.1523/JNEUROSCI.3051-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cang CL, Kawasaki Y, Liang LL, Zhang YQ, Ji RR, Zhao ZQ. Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: a novel pathway for heat hyperalgesia. J Neurosci. 2007;27:12067–12077. doi: 10.1523/JNEUROSCI.0496-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Li GD, Zhao ZQ. State-dependent phosphorylation of epsilon-isozyme of protein kinase C in adult rat dorsal root ganglia after inflammation and nerve injury. J Neurochem. 2003;85:571–580. doi: 10.1046/j.1471-4159.2003.01675.x. [DOI] [PubMed] [Google Scholar]
- Zhu W, Oxford GS. Phosphoinositide-3-kinase and mitogen activated protein kinase signaling pathways mediate acute NGF sensitization of TRPV1. Mol Cell Neurosci. 2007;34:689–700. doi: 10.1016/j.mcn.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Xu P, Cuascut FX, Hall AK, Oxford GS. Activin acutely sensitizes dorsal root ganglion neurons and induces hyperalgesia via PKC-mediated potentiation of transient receptor potential vanilloid I. J Neurosci. 2007;27:13770–13780. doi: 10.1523/JNEUROSCI.3822-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]