

Abstract
G-protein-coupled receptors (GPCRs) may form heteromeric complexes and cooperatively mediate cellular responses. Although heteromeric GPCR complexes are suggested to occur in many neurons, their contribution to neuronal function remains unclear. We address this question using two GPCRs expressed in cerebellar Purkinje cells: adenosine A1 receptor (A1R), which regulates neurotransmitter release and neuronal excitability in central neurons, and type-1 metabotropic glutamate receptor (mGluR1), which mediates cerebellar long-term depression, a form of synaptic plasticity crucial for cerebellar motor learning. We examined interaction between these GPCRs by immunocytochemical, biochemical, and Förster resonance energy transfer analyses in cultured mouse Purkinje cells and heterologous expression cells. These analyses revealed that the GPCRs closely colocalized and formed heteromeric complexes on the cell surfaces. Furthermore, our electrophysiological analysis showed that CSF levels (40–400 nm) of adenosine or synthetic A1R agonists with comparable potencies blocked mGluR1-mediated long-term depression of the postsynaptic glutamate-responsiveness (glu-LTD) of cultured Purkinje cells. A similar dose of the A1R agonist decreased the ligand affinity of mGluR1 and did not affect depolarization-induced Ca2+ influx, which is an essential factor in inducing glu-LTD. The A1R agonist did not affect glu-LTD mimicked by direct activation of protein kinase C. These results suggest that A1R blocked glu-LTD by decreasing the ligand sensitivity of mGluR1, but not the coupling efficacy from mGluR1 to the intracellular signaling cascades. These findings provide a new insight into neuronal GPCR signaling and demonstrate a novel regulatory mechanism of synaptic plasticity.
Introduction
G-protein-coupled receptors (GPCRs) are a large protein family the genes of which comprise ∼5% of the total human genes. GPCRs respond to various ligands and mediate diverse modalities of cellular responses (Franco et al., 1997; Pierce et al., 2002; Boyden et al., 2004). The modality is further broadened by heteromeric complex formation between GPCRs. There is increasing evidence that such complexes mediate responses that neither of the constituent GPCRs can mediate by itself (Selbie and Hill, 1998; Rios et al., 2001). Many central neurons express multiple types of GPCRs and some neuronal GPCRs are shown to form heteromeric complexes (Rios et al., 2001; Fribourg et al., 2011; Liu et al., 2011). However, it remains unclear how GPCR complexes play functional roles in central neurons.
We address this question using type-1 metabotropic glutamate receptor (mGluR1, a Gq/11-protein-coupled receptor) and adenosine A1 receptor (A1R, a Gi/o-protein-coupled receptor) in cerebellar Purkinje cells. Purkinje cells integrate motor commands and sensory information carried by the synaptic inputs from the parallel fibers (PFs). mGluR1 on the postsynaptic membrane of the PF-Purkinje cell synapses mediates long-term depression (LTD) at these synapses (cerebellar LTD), a form of synaptic plasticity crucial for cerebellar motor learning (Ito, 2002; Kano et al., 2008). Among various forms of synaptic plasticity in the cerebellum, cerebellar LTD has been studied most intensively (Boyden et al., 2004) and is one of the pivotal paradigms for investigating the mechanism of synaptic plasticity in the CNS. A1R is also suggested to function at the postsynaptic sites of Purkinje cells (Tabata et al., 2007; Akiyama et al., 2009). Adenosine or its precursor is released from neurons and astrocytes as a neuromodulator, co-neurotransmitter, or gliotransmitter, and adenosine accumulates to a submicromolar level in the CSF (Ballarin et al., 1991; Higgins et al., 1994; Rossi et al., 2007). Adenosine in the CSF can indeed activate A1R (Haas and Selbach, 2000) and this leads to suppression of neurotransmitter release from the presynaptic terminals. This suppression is believed to contribute to the control of arousal level, the prevention of excitotoxic neural degeneration, and pathophysiological reactions against hypoxia and ischemia (Phillis and Wu, 1981). In contrast, the postsynaptic function of A1R has not been explored intensively. This study is the first step in elucidating this issue by focusing on the A1R-mediated mGluR1 signaling modulation occurring at the postsynaptic sites. We found that A1R colocalized with mGluR1 on the postsynaptic structures of Purkinje cells. Our coimmunoprecipitation and Förster resonance energy transfer (FRET) analyses in a heterologous expression cell system clearly showed complex formation between A1R and mGluR1. We further determined the domain of the receptor proteins required for complex formation. Moreover, activation of A1R with submicromolar levels of agonists led to the suppression of mGluR1-mediated long-term depression of cultured Purkinje cell's glutamate -responsiveness, which is the cellular basis for cerebellar LTD. These findings provide a new insight into neuronal GPCR signaling and demonstrate a novel regulatory mechanism of synaptic plasticity.
Materials and Methods
Cell culture.
The experiments in this study were approved by the committees on animal experiments of Juntendo University, University of Tokyo, and University of Toyama. Cerebellar cell cultures were prepared as described previously (Tabata et al., 2000). Briefly, cerebellar cells were obtained from perinatal C57BL/6 mouse embryos, dissociated with trypsin, and plated on plastic dishes or low-fluorescence plastic films (Sumilon, MS-92132; Sumitomo). The cells were cultured in a low-serum medium for 10–22 d. Purkinje cells were identified by their large somata (18 μm or greater) and thick primary dendrites. Neuro2a and HEK-293 cells were passaged in 10% FBS/DMEM. Expression vectors were transfected into cells with Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's protocol. These cells were used 18–24 h after transfection to avoid overexpression and aggregation (Sakurai et al., 2008). A stable cell line that expresses A1R and mGluR1 in response to doxycycline application was created using the Flp-In and Jump-In T-Rex HEK293 cell system (Invitrogen) according to the manufacturer's protocol. Flp-In/Jump-In T-Rex HEK293 cells were transfected with pcDNA5/FRT/TO-mGluR1 and pcDNA3/TO-A1R-phiC31 and a stable double transfectant was selected using blasticidin, hygromycin, and G418.
Antibodies.
We used the following commercially available primary antibodies: mouse anti-mGluR1 (BD Biosciences); rabbit anti-A1R (Abcam); mouse anti-GFP (Invitrogen), and highly cross-adsorbed secondary antibodies to mouse or rabbit IgG conjugated with Alexa Fluor 488 or 594 (Invitrogen). Antibodies were raised in rabbits against synthetic peptides corresponding to the intracellular domain (aa 1116–1130) of rat mGluR1a, the intracellular domain (aa 309–326) of rat A1R (Ciruela et al., 2001), recombinant mCherry, and GFP, and were affinity purified with antigens (Sakurai et al., 2008).
Coimmunoprecipitation.
mGluR1 and A1R were immunoprecipitated as described previously (Tabata et al., 2004) Briefly, young adult mouse cerebella were homogenized with ice-cold buffer containing 0.32 m sucrose and 10 mm Tris-HCl, pH 7.5, with a protease inhibitor mixture (cOmplete Protease Inhibitor Cocktail; Roche Diagnostics) and centrifuged at 1000 × g at 4°C for 10 min. The crude membrane fraction was obtained by centrifugation of the supernatant at 17,000 × g for 30 min and solubilized in RIPA buffer containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 25 mm Tris-HCl, pH 7.5, 137 mm NaCl, 3 mm KCl with cOmplete Protease Inhibitor Cocktail at 4°C for 60 min. After centrifugation at 20,000 × g for 60 min, the supernatant was incubated with a specific antibody (5–12 μg) against mGluR1 or A1R bound to protein A magnetic beads (Invitrogen) at 4°C for ≥ 3 h. The immunoprecipitates were washed 4 times with cold RIPA buffer and dissolved with 2× lithium dodecylsulfate sample buffer (Invitrogen) with dithiothreitol at room temperature for 1 h. The bound samples were electrophoresed and immunoblotted as described previously (Sakurai et al., 2008). Neuro2a cells were lysed in RIPA buffer directly and lysates were immunoprecipitated with a specific antibody against GFP (0.3 μg/ml) or mCherry (0.2 μg/ml) as described above.
Immunofluorescence staining.
Cerebellar neurons on films were fixed sequentially with 0.1 m sodium phosphate buffer containing 4% paraformaldehyde at 4°C for 30 min and with 100% methanol at room temperature for 10 min. The fixed preparations were rinsed with PBS and then treated with PBS containing 0.1% Triton X-100 and 5% FBS or horse serum at room temperature for 30 min (Kamikubo et al., 2006). The treated cells were incubated with a primary antibody against mGluR1 or A1R (5 μg/ml) at 4°C overnight and then treated with a secondary antibody conjugated with Alexa Fluor 488 or 594 (10 μg/ml) at room temperature for 2.5 h. The cells were examined, using a confocal laser microscope (LSM510; Zeiss or FV1000; Olympus).
Plasmids.
For rat A1R and mGluR1, full-length cDNA and fragments were cloned into pENTR/D-TOPO vector (Invitrogen). For construction of expression vectors, pcDNA3.1/Hygro(+) was converted to a Gateway vector by inserting the Gateway cassette into a EcoRV site. GFP variant (cyan fluorescent protein [CFP], modified yellow fluorescent protein [Venus]) or mCherry cDNA was amplified by PCR and inserted into pcDNA3.1-GW digested with NheI and HindIII (for N-terminal fluorescent tag) or with XhoI and ApaI (for C-terminal fluorescent tag). The Gateway conversion cassette was similarly introduced into the EcoRV site of pSecTag2/Hygro A vector. Fluorescent protein cDNA was amplified by PCR and inserted into pSecTag2-GW digested with AscI and HindIII. For A1R expression, A1R cDNA was transferred from the entry vector to pcDNA3.1-GW vector by Gateway LR reaction. For the expression of mGluR1 with N-terminal fluorescent tag, mGluR1 cDNA without signal peptide sequence was transferred to pSecTag2-GW by recombination. For the expression of mGluR1 with C-terminal tag, full-length mGluR1 cDNA was transferred to pcDNA3.1-GW vector. For introduction of the mutations, we used a PCR-based method. All constructs were verified by DNA sequencing (Applied Biosystems).
Förster resonance energy transfer.
Using a confocal laser scanning microscope (SP5/TCS; Leica Microsystems), CFP was excited with a 458 nm argon laser and the emission was collected through a 465–505 nm spectrophotometer prism and sliders. Venus (Nagai et al., 2002) was excited with a 514 nm argon laser and the emission was collected through a 525–600 nm spectrophotometer prism and sliders. The acceptor (Venus) was bleached with 20–50 scan cycles with the 514 nm laser line at maximum intensity. Using the Leica Application Suite, FRET efficiency was calculated from the difference of fluorescence intensity of the donor (CFP) before and after photobleaching (Kenworthy and Edidin, 1998; Lippincott-Schwartz et al., 2001). FRET efficiency (FRETeff) was calculated as follows:
where F458post and F458pre are the fluorescence intensities of the CFP after and before photobleaching, respectively.
Live-cell multicolor total internal reflection fluorescence microscopy.
For multicolor total internal reflection fluorescence microscopy (TIRFM), we used a Leica AM TIRF MC custom-equipped from the manufacturer with a 100×, 1.46 numerical aperture (NA) oil-immersion objective (Leica Microsystems); 405, 488, 561, and 635 nm lasers; and the EM-CCD camera system (ImagEM; Hamamatsu Photonics). HEK293 or Neuro2a cell were plated on a collagen-coated handmade glass-bottom dish.
Electrophysiology.
Somatic whole-cell voltage-clamp recordings were made from cultured Purkinje cells in the dishes using a perforated-patch technique (holding potential after the correction of a liquid junction potential between the pipette and bath solutions, −70 mV). The recording pipette contained the following (in mm): 95 Cs2SO4, 15 CsCl, 0.4 CsOH, 8 MgCl2, 10 HEPES, and 500 μg/ml amphotericin B, pH 7.35. The bath was perfused at a rate of 0.6–2 ml/min with saline consisting of the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 0.8 MgCl2, 10 HEPES, pH 7.3, at 25°C. For evaluating glu-LTD, saline was supplemented with 0.3–0.5 μm tetrodotoxin (TTX) and 10 μm (-)-bicuculline methochloride (Bic). For evaluating the dose–response relationship in mGluR1-mediated responses, the examined cell was perfused with saline supplemented with 0.3 μm TTX and 10 μm 6-nitro-7-sulfamoylbenzo (f) quinoxaline-2,3-dione (NBQX), 10 μm (3-[(R)-2-carboxypiperazin-4 yl]propyl-1-phosphonic acid, and 10 μm Bic during test trials. Signals were low-pass filtered at 0.1–2 kHz and sampled at 0.25–10 kHz using an amplifier (EPC-8 or EPC-9/2; HEKA) controlled by PULSE software (HEKA). The data were obtained after the series resistance became ≤100 MΩ (for the analysis of the dose response of the R,S-3,5-dihydroxyphenylglycine (DHPG)-evoked current, typically ≤35 MΩ).
Ca2+ imaging.
Cerebellar neurons on a film were loaded with fura-2 acetoxymethyl ester (5 μm) at 37°C for 15 min and the film was placed on a glass-based recording chamber and perfused at a rate of 0.6 ml/min with the above-mentioned saline. During test trials, the perfusate was switched to the saline supplemented with 0.3 μm TTX, 10 μm NBQX, and 10 μm Bic. The intracellular free Ca2+ concentration ([Ca2+]i)-dependent fluorescence signals were captured at 2–5 Hz using an imaging system (Polychrome II; TILL) attached to an inverted microscope (IX70 with a 20× objective lens of NA 0.75; Olympus). The amplitude of a [Ca2+]i rise was expressed as a change in the ratio of somatic fluorescence signals alternatively excited at 340 and 380 nm (exposure duration, 40 and 20 ms, F340/F380, respectively).
LANCE cAMP assay.
The LANCE cAMP competitive immunoassay (PerkinElmer) was performed as described by the manufacturer. Assay plates were prepared containing 10 μl of test compound in buffer containing 0.1 μm (final concentration) forskolin. The HEK293 cells stably expressing mGluR1 and A1R were treated with pertussis toxin (PTX) or a drug-free medium (untreated cells) for >12 h and then added in buffer containing 100 nm (final concentration) [R]-N6-(1-methyl-2-phenylethyl) adenosine (R-PIA) and LANCE Alexa Fluor anti-cAMP antibody. After incubation for 30 min at room temperature, the cell-containing buffer was incubated with LANCE detection buffer (20 μl) for 60 min at room temperature. The plates were then quantitated using an EnVision multilabel plate reader (PerkinElmer).
Drug application.
Glutamate was applied iontophoretically (30 ms, 0.05 Hz) through a glass pipette filled with 50 mm l-glutamate and 50 mm HEPES (pH adjusted to 7.1 with sodium hydroxide) and located ∼20 μm from the branching point of a primary dendrite of the examined cell. The level of ejection current was adjusted (40–800 nA, 30 ms) so that the peak amplitude of basal glutamate-evoked currents ranged from 100 to 320 pA. Local application of DHPG, R-PIA, and 75 mm K+ saline was done by delivering the drug-containing saline through a wide-tipped pipette located near the examined cell under the control of gravity. Bath application of other reagents was done by perfusing the recording chamber at rate of 1–2 ml/min with the drug-containing saline.
Data analysis.
The peak amplitude of a response was measured as a difference from the prestimulus level to the maximal deflection throughout the record (glutamate-evoked current) or during a 10 s (DHPG-evoked [Ca2+]i rises) or 1 s (K+-evoked [Ca2+]i rises) agonist application. Groups of numerical data are presented as mean ± SEM throughout the text and figures. Statistical differences were examined by a two-sided Mann–Whitney U test unless otherwise stated.
Results
Complex formation and colocalization of A1R and mGluR1
We assessed the possibility of complex formation and colocalization of A1R and mGluR1 in central neurons using newly raised antibodies that recognize the GPCR's C termini. We performed coimmunoprecipitation experiments on the lysates of the crude synaptosome fraction derived from mouse cerebella. In the immunoblot of the fractions precipitated with the anti-A1R antibody, immunoreactivities for mGluR1 monomer and dimer were detected at the corresponding molecular weights (∼140 and ∼300 kDa, respectively; Fig. 1A). Furthermore, in the immunoprecipitates obtained with the anti-mGluR1 antibody, an immunoreactivity for A1R was found (Fig. 1B). This result suggests complex formation between A1R and mGluR1. We then performed double immunostaining with these antibodies in cultured Purkinje cells. Intense immunofluorescent signals of both antigens were observed in the dendritic spines and branches and soma, suggesting the colocalization of A1R and mGluR1 at the postsynaptic structures (Fig. 1C).
Figure 1.
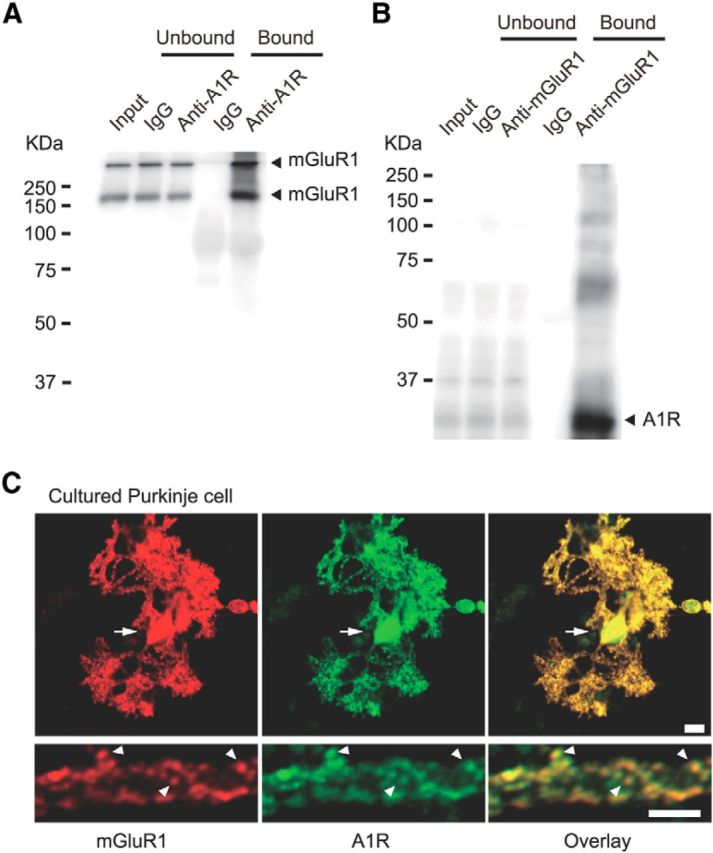
Endogenous interaction between A1R and mGluR1 in cerebella and colocalization of A1R and mGluR1 in cultured Purkinje cells. A, Coimmunoprecipitation of mGluR1 with A1R in the lysate of the crude synaptosome fractions derived from mouse cerebella. Monomeric and dimeric bands were detected by immunoblotting with anti-mGluR1 antibody in the anti-A1R immunoprecipitate. B, A1R was detected by immunoblotting with the anti-A1R antibody in the anti-mGluR1 immunoprecipitate. C, Double-immunofluorescence staining of a cultured Purkinje cell (16-d-old in vitro) with anti-mGluR1 (red) and anti-A1R (green) antibodies. Arrows and arrowheads, soma and dendritic spines with colocalized signals, respectively. Scale bars, 10 μm.
We explored the molecular basis for the interaction between A1R and mGluR1 in a heterologous expression system. We performed coimmunoprecipitation studies using Neuro2a cells transfected with full-length A1R fused with Venus at the N terminus (Venus-A1R) and full-length mGluR1 fused with mCherry at the N terminus (mCh-mGluR1). In the fraction precipitated with a Venus-reactive anti-GFP antibody, an immunoreactivity for mCh-mGluR1 was detected (Fig. 2A). In a previous study, the membrane-proximal C-terminal peptide (C-tail) of A1R (i.e., the fourth cytoplasmic domain [A1RC4], 34 aa, positions 293–326) was assumed to be folded into an α-helical structure and this domain was shown to be important for signal transduction and trafficking (Pankevych et al., 2003). To examine the involvement of this domain in interaction with mGluR1, we performed coimmunoprecipitation on the extracts of cells expressing mCh-mGluR1 and GFP variant (CFP or Venus)-fused whole wild-type A1RC4 (A1RC4Wt), its fragment (Fig. 2F,G), or the third cytoplasmic domain of A1R (A1RC3). Bands corresponding to mGluR1 monomer and dimer were found only in the immunoprecipitates from cells expressing Venus-fused A1RC4Wt and CFP-fused membrane-proximal fragment of A1R C-tail (A1RC4a, 16 aa, positions 293–308; Fig. 2A). Conversely, in the immunoprecipitates from cells coexpressing Venus-fused mGluR1, an ∼37 kDa band corresponding to mCherry-fused A1RCWt and an ∼30 kDaband corresponding to mCherry-fused A1RC4a were detected (Fig. 2B). In contrast, coimmunoprecipitation was not observed between mCherry- or Venus-fused mGluR1 and CFP- or mCherry-fused membrane-distal fragment of A1R C-tail (A1RC4b, 18 aa, positions 309–326) or Venus-fused A1RC3 (Fig. 2A,B). In addition, coimmunoprecipitation was observed between A1RC4a and mGluR5, which has a structural similarity to mGluR1 (Fig. 2C). These results suggest that the first half of A1RC4 is responsible for interaction with mGluR1. Moreover, replacement of F295, F299, W303, and F307 of A1RC4 with alanines (A1RC4Mt) abolished coimmunoprecipitation of Venus-fused full-length mGluR1 (Venus-mGluR1) (Fig. 2D,E). This result suggests that the aromatic amino acid residues of the A1RC4 are important for A1R-mGluR1 interaction.
Figure 2.
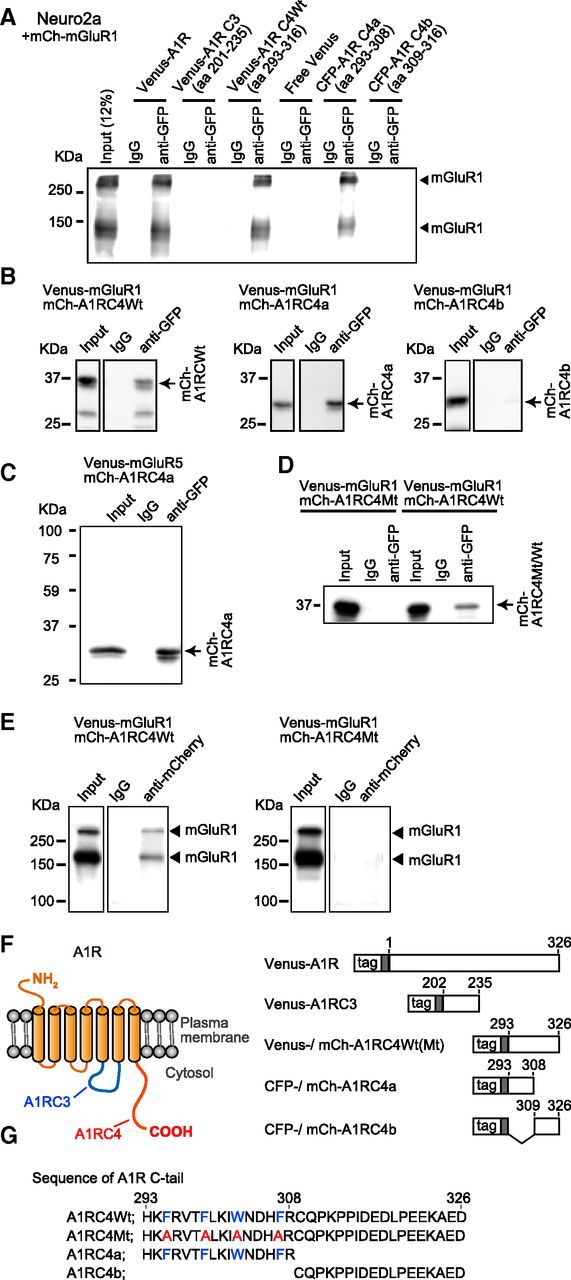
Complex formation of A1R and mGluR1 in Neuro2a cells. A, Coimmunoprecipitation of mCherry-mGluR1 with Venus- or CFP-fused full-length A1R or A1R C-terminal tail. Lysates from Neuro2a cells expressing Venus- or CFP-fused full-length or cytoplasmic domains of A1R and mCherry-mGluR1 were immunoprecipitated with the rabbit anti-GFP antibody. The precipitated fractions were immunoblotted with the mouse anti-mGluR1 antibody. B, Coimmunoprecipitation of mCherry-fused full-length A1R and A1R C-terminal tail with Venus-mGluR1. Neuro2a cells were transfected with Venus-mGluR1 and either mCherry-fused A1R C-tail (mCh-A1RC4Wt, aa 293–316), mCherry-fused A1R C-tail membrane-proximal region (mCh-A1RC4a, aa 293–308), or mCherry-fused the A1R C-tail membrane-distal region (mCh-A1RC4b, aa 309–316). Crude cell lysate (Input) and the immunoprecipitates with the anti-GFP or control IgGs were immunoblotted with the anti-mCherry antibody. C, Immunoprecipitation with the anti-GFP antibody from the extracts of Neuro2a cells expressing Venus-mGluR1 and either mCh-A1RC4Wt or a mCherry-fused mutant A1R C-tail (mCh-A1RC4Mt). In mCh-A1RC4Mt, F295, F299, W303, and F307 were replaced with alanines. Crude cell lysate (Input) and the immunoprecipitates with the anti-GFP or control IgGs were immunoblotted with the anti-mCherry antibody. Arrow, mCherry-fused A1RC4Wt, A1RC4a, or A1RC4b. D, Immunoprecipitation with the anti-mCherry antibody from the extracts of Neuro2a cells expressing Venus-mGluR1 and either mCh-A1RC4Wt or mCh-A1RC4Mt. Crude cell lysate (Input) and the immunoprecipitates with the anti-mCherry or control IgGs were immunoblotted with the anti-GFP antibody. Filled triangles, mGluR1 monomer and dimer. E, Schematic structure of A1R and its cytoplasmic domains used for the coimmunoprecipitation studies. F, Amino acid sequence of the A1R C-terminal tail and the alanine mutant.
Physical interaction between A1R and mGluR1
We scrutinized the physical interaction between A1R with mGluR1 in living cells, measuring FRET efficiency by the acceptor photobleaching method (Miyawaki and Tsien, 2000). We used Venus-A1R- and mCh-mGluR1-transfected Neuro2a cells in which colocalization of A1R and mGluR1 occurred (Fig. 3A).
Figure 3.
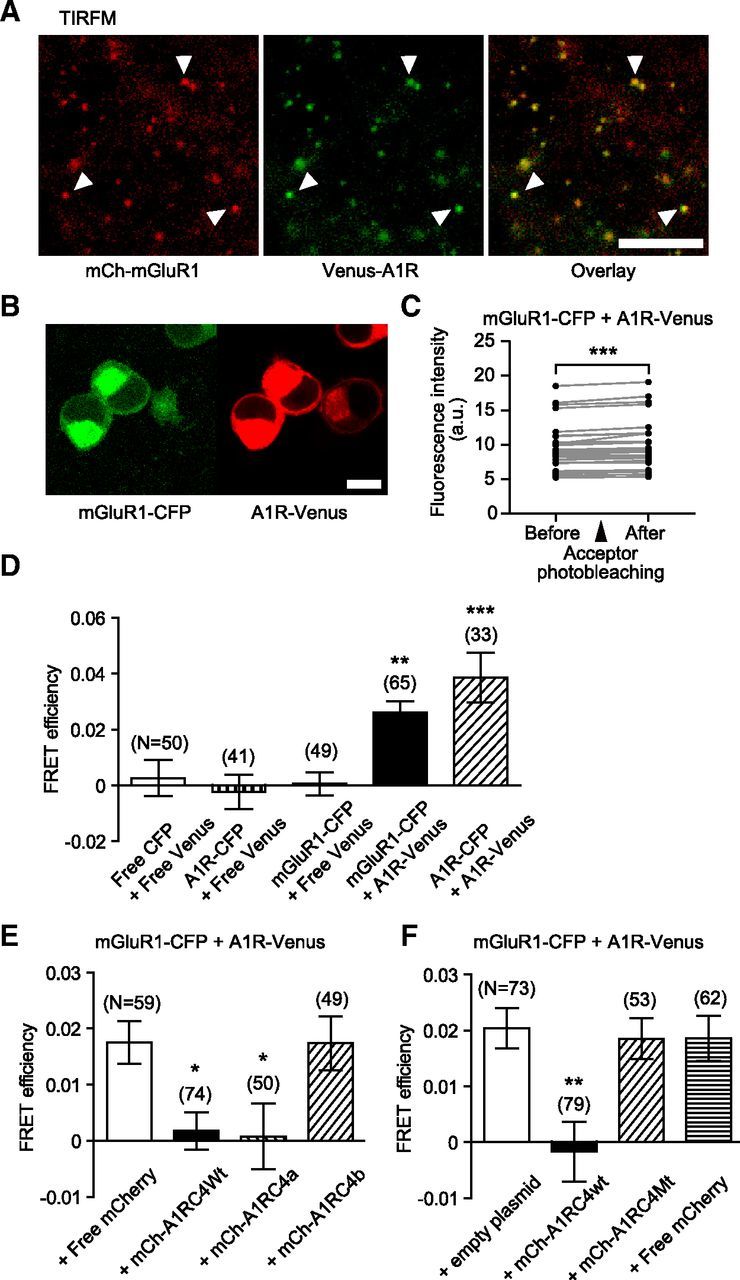
Physical interaction of A1R and mGluR1 in living cells. A, TIRF microscopic images of Neuro2a cells transiently expressing mCherry-fused mGluR1 (mCh-mGluR1, red) and Venus-fused A1R (Venus-A1R, green). Arrowheads, colocalized mCh-mGluR1 and Venus-A1R, respectively. Scale bars, 10 μm. B, Confocal laser microscopic images of Neuro2a cells transiently expressing full-length mGluR1 with the C-terminal CFP-tag (mGluR1-CFP, green) and full-length A1R with the C-terminal Venus-tag (A1R-Venus, red). Scale bars, 10 μm. C, Intensities of mGluR1-CFP fluorescence before and after photobleaching of A1R-Venus. CFP was excited at 458 nm and its emission detected around 480 nm. ***p < 0.001, paired t test. D, FRET efficiency between CFP and Venus in living Neuro2a cells. FRET efficiency was evaluated by acceptor photobleaching. A1R homodimer and mGluR1-CFP + A1R-Venus pair displayed significant FRET signals. Free CFP + free Venus pair, A1R-CFP + free Venus pair, and mGluR1-CFP + free Venus pair did not show a significant FRET signal. E, F, Reduction of FRET efficiency between mGluR1-CFP and A1R-Venus by A1R C-terminal tail. FRET efficiency between mGluR1-CFP and A1R-Venus in living Neuro2a cells was reduced by the coexpression of mCh-A1RC4Wt or mCh-A1RC4a, but not by free mCherry, mCh-A1RC4b, or A1RC4Mt. The overall difference was analyzed using ANOVA and then differences among every pair of the data groups were analyzed using Tukey-Kramer multiple-comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars, ±SEM.
We transfected Neuro2a cells with genes encoding CFP fused to the C terminus of the full-length mGluR1 (mGluR1-CFP, FRET donor) and Venus fused to the C terminus of the full-length A1R (A1R-Venus, FRET acceptor) (Fig. 3B). We verified that the fluorescence intensity of mGluR1-CFP increased after photobleaching of A1R-Venus (Fig. 3C). In these preparations, FRET signals from mGluR1-CFP + A1R-Venus pair were as strong as positive control (A1R-CFP + A1R-Venus pair; Fig. 3D). In contrast, FRET signals from mGluR1-CFP + A1R-Venus pair were significantly stronger than their negative controls (free CFP + free Venus, A1R-CFP + free Venus, and mGluR1-CFP + free Venus pairs; Fig. 3D). These results clearly indicate close physical interaction of A1R and mGluR1 in living cells. We further examined the involvement of the A1R C-tail in the A1R-mGluR1 interaction. Overexpression of mCherry-fused A1RC4Wt or A1RC4a decreased the FRET signal from the mGluR1-CFP + A1R-Venus pair (Fig. 3E,F). In contrast, overexpression of mCherry-fused A1RC4b or A1RC4Mt did not decrease the FRET signal (Fig. 3E,F). These results indicate that the aromatic amino acid residues of A1R C-tail are required for the A1R-mGluR1 interaction.
A1R activation blocks glu-LTD
We assessed whether and how A1R acts on mGluR1-mediated glu-LTD in cultured Purkinje cells, a cellular basis for cerebellar LTD (see Introduction). A1R is expressed in the presynaptic terminals of PFs and the postsynaptic structures of Purkinje cells (Dittman and Regehr, 1996; Ciruela et al., 2001). Therefore, in the cerebellar slices, pharmacological manipulation of A1R should affect not only the postsynaptic, but also the presynaptic A1R. Therefore, we used the cellular preparations rather than cerebellar slices to focus on the action of A1R agonists at the postsynaptic site of Purkinje cells.
In cultured Purkinje cells, we monitored the postsynaptic glutamate responsiveness by measuring an inward current evoked by iontophoretically applied glutamate (30 ms, 0.05 Hz) to a dendrite (Iglu) in a perforated-patch whole-cell mode. Long-term depression of this inward current (i.e., glu-LTD) was induced by 20 sets of conjunctive somatic depolarization and glutamate iontophoresis. In the normal saline, the peak amplitude of Iglu was reduced to 71.4 ± 8.8% (n = 7) of the basal level at 50–52 min after the conjunctive stimuli (Fig. 4A, control). In contrast, in the continuous presence of the A1R-selective agonist R-PIA (100 nm), the peak amplitude was not reduced throughout the recording sessions (110.5 ± 7.3%, n = 7 at 50–52 min; Fig. 4A, R-PIA). Similarly, the continuous presence of 2-chloro-N6-cyclopentyladenosine (CCPA, 500 nm), another A1R-selective agonist, blocked glu-LTD (102.8 ± 4.6%, n = 5, at 40–52 min; Fig. 4A, CCPA). The relative peak amplitudes of Iglu at 50–52 min were significantly larger with these A1R agonists than that of the control (Fig. 5D). The above experiments were performed in saline containing a relatively high concentration (5 mm) of K+ and at a relatively low temperature (25°C). The effect of the A1R agonist on glu-LTD was also observed under a more physiological condition with 2.5 mm K+ and 30°C (Fig. 5E).
Figure 4.
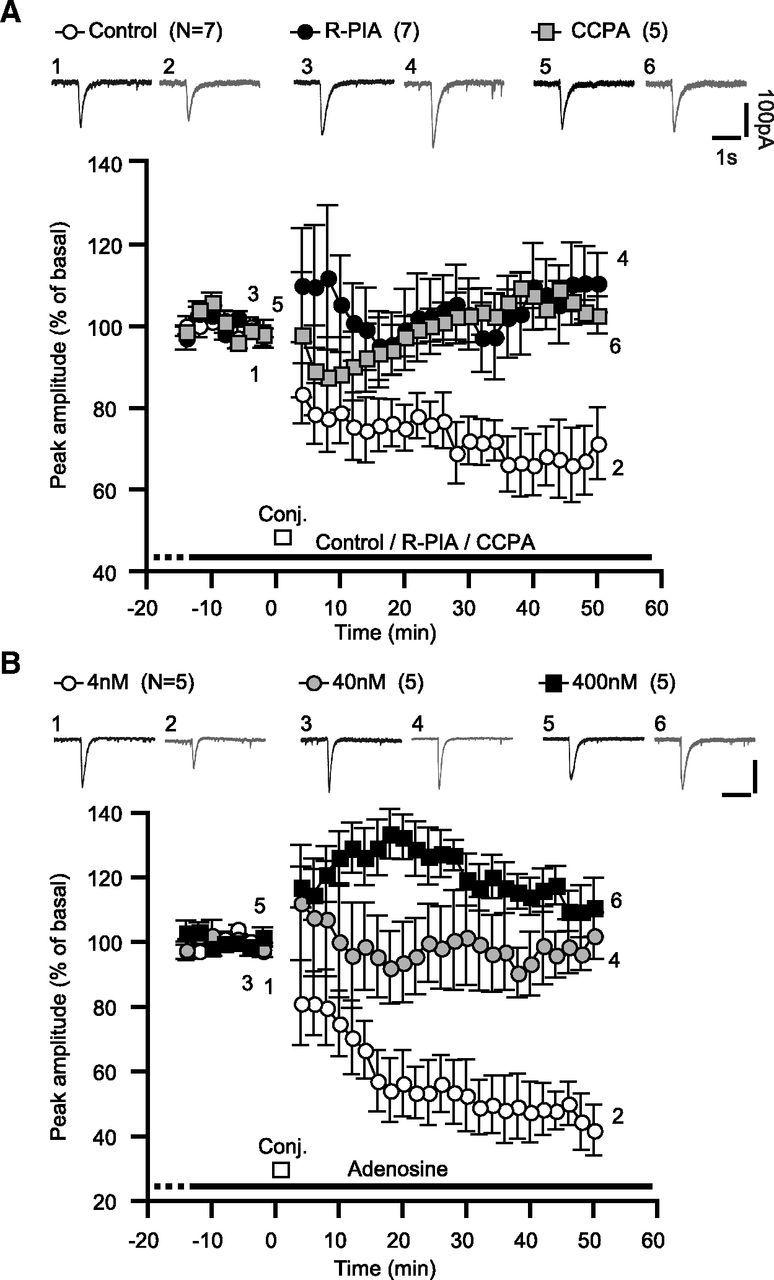
A1R activation blocks glu-LTD in cerebellar Purkinje cells. A, A1R-selective agonists blocks glu-LTD in cultured Purkinje cells. Each pair of traces indicates sample glutamate-evoked currents of a cell before and after the conjunctive depolarization and glutamate iontophoresis in the absence (Control) or continuous presence of the A1R-selective agonist R-PIA (100 nm) or CCPA (500 nm). Iglu was monitored under voltage clamp at −70 mV. The conjunctive stimuli were 12 sets of simultaneous depolarizing voltage jumps (0 mV, 3 s) and glutamate iontophoresis. Each plot indicates the time course of the mean peak amplitude of Iglu. B, The endogenous A1R agonist adenosine blocked glu-LTD in a concentration-dependent fashion. Each pair of traces indicates sample Iglu of a cell before and after the conjunctive stimuli in the continuous presence of the labeled dose of adenosine. Each plot indicates the time course of the mean peak amplitude of Iglu. Scale bars, 1 s and 100 pA. White bars, timing of the conjunctive stimuli; dots and error bars, mean ± SEM of the data for every 2 min period.
Figure 5.
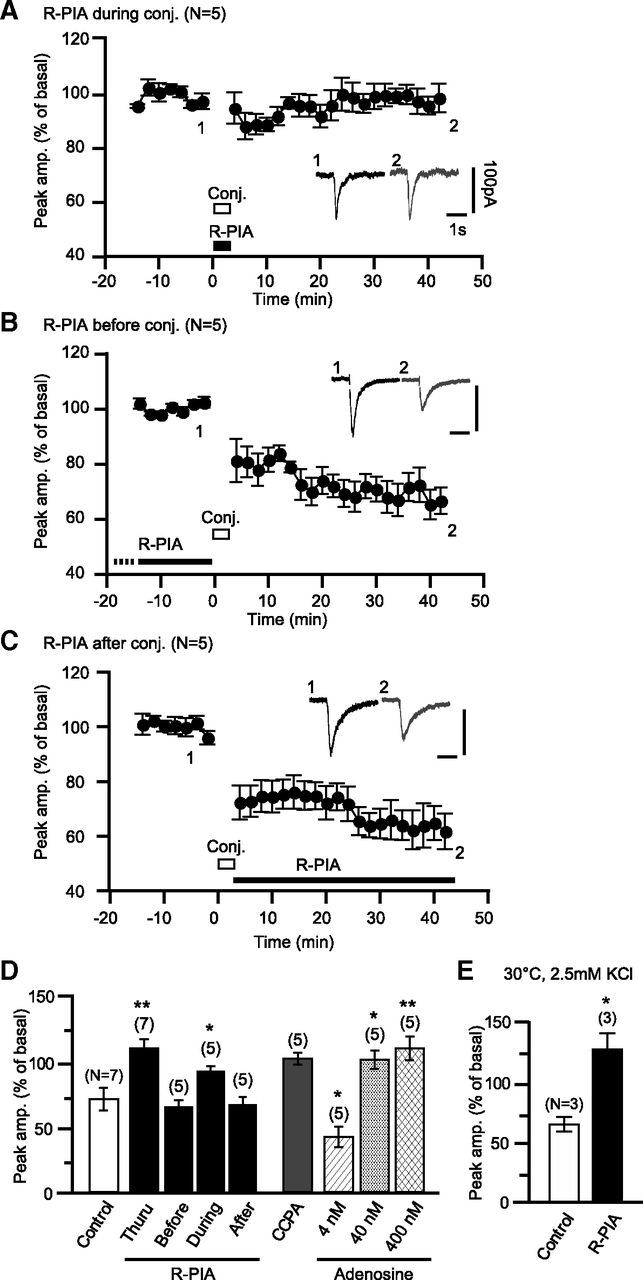
A1R activation during the induction phase of glu-LTD is sufficient to block glu-LTD. A, R-PIA (100 nm) application restricted to the period of the conjunctive depolarization/glutamate stimuli blocks glu-LTD. The conjunctive stimuli were 12 sets of simultaneous depolarizing voltage jumps (0 mV, 3 s) and glutamate iontophoresis. Traces indicate sample Iglu of a cell before and after the conjunctive stimuli. Each plot indicates the time course of the mean peak amplitude of Iglu. B, C, R-PIA (100 nm) application before (B) or after (C) the conjunctive stimuli did not affect glu-LTD. D, Summary of the effects of the labeled manipulations on glu-LTD. Columns and error bars, mean ± SEM of the peak amplitudes of Iglu at 40–50 min of the onset of the conjunctive stimuli. E, The effect of R-PIA on glu-LTD was seen in saline containing 2.5 mm KCl and at 30°C. Columns and error bars, mean ± SEM of the peak amplitudes of Iglu at 35 min of the onset of the conjunctive stimuli. Scale bars, 1 s and 100 pA. White bars, timing of the conjunctive stimuli; dots and error bars, mean ± SEM of the data for every 2 min period. *p < 0.05; **p < 0.01.
Next, we examined the effect of the endogenous A1R agonist on glu-LTD. Previous studies using pharmacological techniques and microdialysis estimate that the basal level of adenosine in the extracellular fluid in some brain regions ranges from 20 to 400 nm (Ballarin et al., 1991; Dunwiddie and Diao, 1994). A1R has a ligand affinity of ∼70 nm and could possibly respond to submicromolar levels of adenosine(Dunwiddie and Masino, 2001). We found that the continuous presence of these levels of adenosine blocked glu-LTD (with 40 nm, 102.1 ± 7.2%, n = 5 at 50–52 min; with 400 nm, 110.8 ± 9.1%, n = 5; Figs. 4B, 5D). In contrast, in the continuous presence of 4 nm adenosine, glu-LTD with a similar extent (50.2 ± 6.6%, n = 5) to the control (Figs. 4B, 5D) was observed. These results demonstrate that the physiologically possible levels of adenosine can block glu-LTD.
A1R agonist-induced glu-LTD blockade is attributable to the desensitization of mGluR1
We explored how A1R activation influences glu-LTD induction. Even when R-PIA application was restricted to the period of the conjunctive stimuli, glu-LTD was blocked (97.7 ± 4.6%, n = 5, at 40–42 min; Fig. 5A,D). In contrast, when applied only before (63.0 ± 5.7%, n = 5, at 40–42 min) or after (64.9 ± 6.2%, n = 5, at 40–42 min) the conjunctive stimuli, R-PIA failed to block glu-LTD (Fig 5B–D). These results suggest that the A1R-mediated glu-LTD blockade is due to the modulation of the induction process but not the maintenance process of glu-LTD.
In cultured cerebellar Purkinje cells, simultaneous glutamate-evoked mGluR1 signaling and depolarization-evoked Ca2+ influx are known to be the sufficient factors to induce glu-LTD (Narasimhan and Linden, 1996; Kamikubo et al., 2007). Therefore, A1R activation could possibly block glu-LTD by reducing either or both of these factors. As to the former possibility, we examined the effect of R-PIA on mGluR1 signaling by monitoring the dose–response relationship of an inward current evoked by whole-cell application of DHPG, a group I mGluR agonist. R-PIA (50 nm) shifted the Kd from 36.9 ± 4.3 μm (n = 10) to 68.0 ± 9.2 μm (n = 10; Fig. 6A–C). R-PIA did not change the absolute amplitude of inward currents with the saturating dose of DHPG (from 161.5 ± 34.0 to 128.2 ± 19.5 pA; Fig. 6D). These results suggest that A1R activation may attenuate the glutamate analog responsiveness of Purkinje cells by decreasing mGluR1's ligand sensitivity, but not by decreasing the coupling efficacy of mGluR1 to its subsequent cascade. To further examine the influence of A1R on the downstream mGluR1 signaling cascade, we measured the effect of R-PIA on inositol trisphosphate receptor (IP3R)-mediated Ca2+ release from the intracellular store (Masu et al., 1991) using fura-2 AM fluorometry. This reaction is a major step of the mGluR1 signaling cascade essential for glu-LTD induction (Kamikubo et al., 2007). A1R-coupled Gi/o-protein may facilitate IP3R-mediated Ca2+ release (Basheer et al., 2002). If such facilitation increased the persistent Ca2+ release in Purkinje cells, this could possibly cause partial depletion of stored Ca2+ and a decrease in Ca2+ release after mGluR1 activation. However, R-PIA (50 nm) had little effect on Ca2+ release from the intracellular store (Fig. 7A–D), which does not support these possibilities. Moreover, our fura-2 AM fluorometry showed that R-PIA (50 nm) had little effect on both depolarization-evoked [Ca2+]i rises and the resting [Ca2+]i (Fig. 7E–H). This result suggests that blockade of glu-LTD is not due to a reduction of the depolarization-evoked Ca2+ influx.
Figure 6.
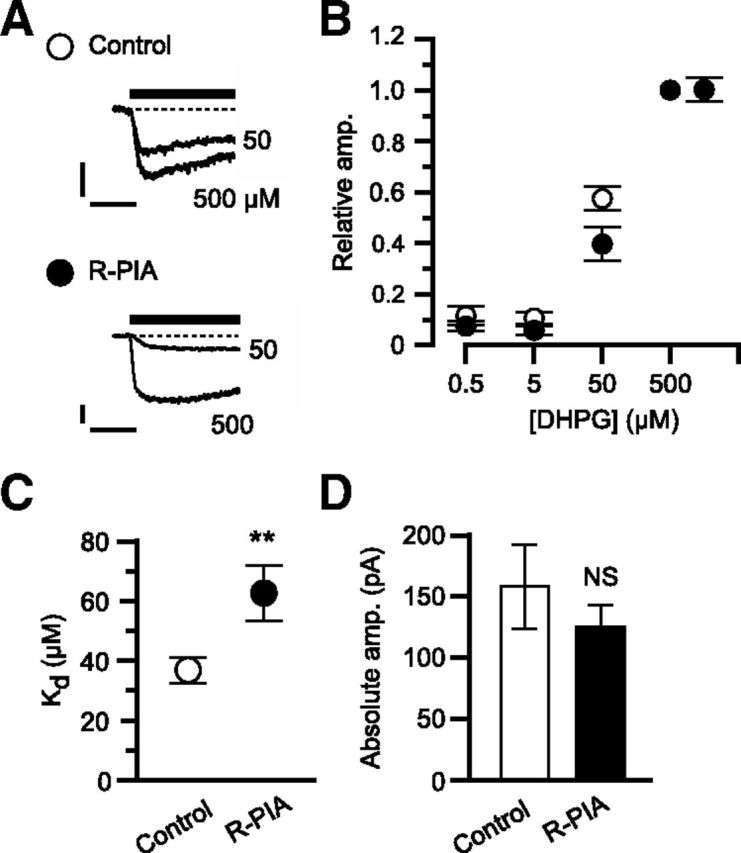
A1R activation decreases mGluR1's ligand sensitivity. A–D, DHPG-induced inward currents in the absence (Control, n = 10) or presence of R-PIA (50 nm, R-PIA, n = 10). A, Each set of superimposed traces indicates sample inward current evoked by saturating (500 μm) and unsaturating (50 μm) doses of DHPG in a Purkinje cell. Holding potential, −70 mV. Scale bars, 10 s and 50 pA. B, Mean relative peak amplitude of the inward currents as a function of DHPG dose. The amplitude was normalized to the value with 500 μm DHPG for each cell. C, Mean apparent Kd. D, Mean absolute peak amplitudes of inward currents evoked by the saturating dose of DHPG. **p < 0.01; NS, p > 0.05, unpaired t test. Error bars, ±SEM.
Figure 7.
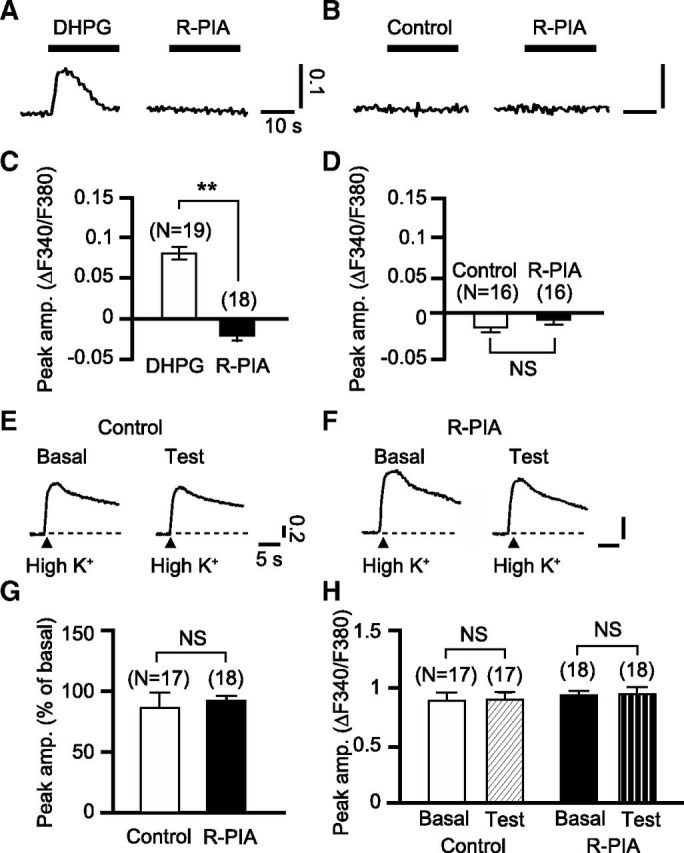
A1R activation does not induce an elevation of [Ca2+]i in Purkinje cells. A, [Ca2+]i responses to R-PIA (50 nm, 30 s) were measured in the cells that displayed Ca2+ release from the intracellular stores in response to DHPG (50 μm, 20 s). Thick bars, timing of drug application; vertical scale bars, changes in F340/F380. B, [Ca2+]i responses to R-PIA were measured in the cells that had been exposed only to the vehicle, not to DHPG. C, D, Mean peak amplitudes of the [Ca2+]i responses to the labeled drugs measured as shown in A and B. R-PIA did not increase the cytoplasmic Ca2+ level, indicating that A1R activation with the submicromolar level of the agonist does not facilitate constitutive Ca2+ release. E, F, Each set of traces indicates sample depolarization-evoked [Ca2+]i rises obtained from a cell before (Basal) and after (Test) a 12 min local application of the normal (Control) or R-PIA (50 nm)-containing saline (R-PIA). To depolarize the cells, high-K+ (75 mm)-containing saline was applied locally for 1 s (arrowheads). Vertical scale bars, changes in F340/F380. G, Mean peak amplitudes of the [Ca2+]i rises after an application of the normal saline or R-PIA measured as shown in E and F. Amplitude is expressed as the percentage of the basal level for each cell. H, Mean resting [Ca2+]i before and after an application of the normal saline or R-PIA. **p < 0.01; NS, p > 0.05, paired t test. Error bars, ±SEM.
mGluR1 signaling and voltage-gated Ca2+ influx together activate protein kinase C (PKC), which in turn initiates AMPA receptor internalization, the final step of LTD (Narasimhan and Linden, 1996; Ito, 2002). We investigated whether A1R activation blocks glu-LTD by inhibiting PKC or the subsequent signaling cascades (Fig. 8A). Previous studies (Linden and Connor, 1991; Matsuda et al., 2000) showed that PKC agonists can induce glu-LTD by directly activating PKC-coupled AMPAR internalization. Accordingly, we observed that 12-O-tetradecanoylphorbol-13-acetate (TPA, 200 nm, 10 min), a PKC activator, reduced Iglu. The relative peak amplitude at 60 min from the onset of TPA application was 64.8 ± 9.7% (n = 6) of the basal level. The continuous presence of R-PIA in the saline did not hamper TPA-induced reduction (60.4 ± 8.0%, n = 7). This result suggests that glu-LTD blockade is not due to inhibition of PKC or its subsequent signaling cascades.
Figure 8.
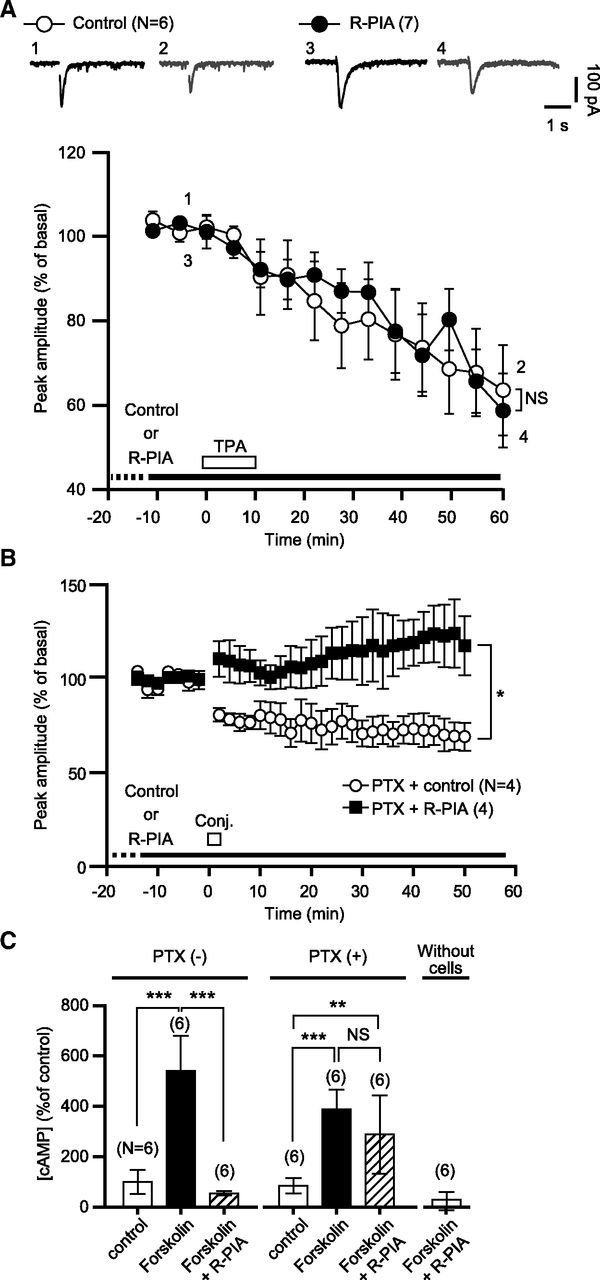
A1R-mediated blockade of glu-LTD is not due to modulation of the PKC or PKA cascade. A, Each pair of traces indicates sample Iglu of a cell before and after a bath application of TPA (200 nm, 10 min) in the absence (Control) or continuous presence of R-PIA (50 nm). Each plot indicates the time course of the mean peak amplitude of Iglu. White bar, timing of TPA application; dots and error bars, mean ± SEM of the data of every 5 min period. B, PTX, a Gi/o-protein inhibitor, does not abolish R-PIA-induced glu-LTD blockade. Each plot indicates the time course of the mean peak amplitude of Iglu of a PTX (500 ng/ml, over 16 h)-pretreated cell before and after the conjunctive depolarization/glutamate stimuli in the absence (Control) or continuous presence (R-PIA) of R-PIA (100 nm). Dots and error bars, mean ± SEM of the data for every 2 min period. C, Comparison of the effects of the labeled test agents on the cAMP production of the HEK293 cells stably expressing A1R and mGluR1 by LANCE cAMP competitive immunoassay. The assay was performed after a 30 min test agent application. PTX(-), cells-without a PTX pretreatment; PTX(+), PTX (500 ng/ml, > 12 h)-pretreated cells. Error bars, ±SD. Without cells, mock-up assay with cell-free buffers.
To determine the involvement of Gi/o-protein, the primary messenger coupled to A1R, we assessed glu-LTD in Purkinje cells pretreated with PTX, a Gi/o-protein inhibitor (500 ng/ml, over 16 h). In the normal saline, the conjunctive stimuli induced glu-LTD in the PTX-pretreated cells, although the magnitude of glu-LTD was relatively small (69.2 ± 7.4%, n = 4, at 50 min; Fig. 8B). Addition of R-PIA (100 nm) to the saline blocked glu-LTD in the PTX-treated cells (117.5 ± 15.8%, n = 4). The LANCE cAMP assay showed that PTX used in the pretreatment was indeed effective (Fig. 8C). Direct stimulation of adenylyl cyclase with folskolin increased cAMP production in both the PTX-pretreated and untreated cells. Activation of A1R by R-PIA inhibited the increase of cAMP production in the untreated cells, indicating that R-PIA application resulted in Gi/o-protein activation. However, the R-PIA-induced inhibition of cAMP production was not seen in the PTX-pretreated cells, suggesting that the pretreatment prevented Gi/o-protein activation. These results suggest that the A1R-mediated glu-LTD blockade was independent of Gi/o-protein.
Discussion
Here we have demonstrated clearly for the first time the colocalization of native A1R and mGluR1 in central neurons (Fig. 1A). Our immunocytochemical analysis showed that the A1R-mGluR1 complex may occur at the postsynaptic structures of cultured Purkinje cells (Fig. 1B). Colocalization might be due to an intrinsic property of the GPCRs, because it was observed in multiple cell types (i.e., Purkinje cells and Neuro2a cells; Fig. 3A). Colocalization may enable local functional interplay between the GPCRs through direct or indirect physical interactions (Lu et al., 2004a; Lu et al., 2004b; Tateyama and Kubo, 2007).
Our coimmunoprecipitation and FRET analyses (Figs. 2, 3) showed that A1R and mGluR1 have a high propensity to form complexes, as was suggested previously in a different type of heterologous expression system and rat cerebellum (Ciruela et al., 2001). The analyses also showed that the membrane-proximal region of A1R C-terminal tail is important for A1R-mGluR1 interaction. The previous study suggested that the membrane-proximal region is folded into an α-helical structure and contains four aromatic amino acids arrayed on one side (Pankevych et al., 2003). Mutation of these aromatic amino acids to alanines disrupted interaction with mGluR1 (Figs. 2, 3). This result indicates that π-π stacking conferred by the aromatic amino acid residues might be important for A1R-mGluR1 interaction (Nugent and Jones, 2012).
We found that A1R activation blocked glu-LTD induced by the conjunctive glutamate/depolarization stimuli in cultured Purkinje cells (Fig. 4). The blockade became evident immediately after induction of glu-LTD (Fig. 4). An application restricted to the period of the conjunctive stimuli was enough for the A1R agonist to exert blockade, whereas A1R applied only before or after the conjunctive stimuli did not block glu-LTD (Fig. 5). These results suggest that A1R activation influences the induction process but not the maintenance process of glu-LTD. In cultured Purkinje cells, mGluR1 signaling and depolarization-induced Ca2+ influx are known as the sufficient factors to trigger glu-LTD (Narasimhan and Linden, 1996; Kamikubo et al., 2007). Our previous (Tabata et al., 2007) and present (Fig. 6) studies show that A1R activation attenuates mGluR1 signaling. Conversely, at the dose at which such an attenuation was observed, R-PIA did not reduce Ca2+ influx through voltage-gated channels (Fig. 7). These results suggest that A1R-mediated glu-LTD blockade is due to a reduction of mGluR1 signaling. A submicromolar concentration of R-PIA reduced the relative amplitude of mGluR1-mediated inward currents evoked by the unsaturating dose of DHPG without changing the absolute amplitude with the saturating dose of DHPG (Fig. 6). This result suggests that A1R blocked glu-LTD mainly by decreasing the ligand sensitivity of mGluR1. Our measurement shown in Figure 6 could underestimate the extent of A1R-mediated reduction of mGluR1's ligand responsiveness. A recent study showed that the expression level of A1R mRNA is only 1/50 of that of mGluR1a mRNA in mouse cerebellar Purkinje cells (Akiyama et al., 2009), indicating that only a small fraction of the mGluR1 population can interact with A1R. Therefore, whole-cell DHPG application activates not only A1R-interacting mGluR1, but also noninteracting mGluR1. The extent of A1R-mediated reduction estimated with whole-cell DHPG application could be lowered by the influence of the noninteracting mGluR1 lacking the reduction.
Several observations suggest that modulation of the downstream cascade of mGluR1 signaling is less important for A1R-mediated blockade of glu-LTD. A previous study reported that the βγ-subunit complex of Gi/o-protein facilitates PLC/IP3R-mediated Ca2+ release from the intracellular stores (Park et al., 1993). If such facilitation occurred in Purkinje cells, the continuous activation of A1R could increase constitutive Ca2+ release and the resultant partial depletion of stored Ca2+ could reduce the amplitude of mGluR1-mediated Ca2+ release. However, a prolonged application of R-PIA did not produce an increase in the cytoplasmic Ca2+ level, which would reflect facilitated constitutive release (Fig. 7). Moreover, R-PIA did not block the depression of Iglu induced by the PKC activator (Fig. 8), suggesting that A1R-mediated blockade of glu-LTD is not due to modulation of PKC and the subsequent signaling cascades, which are the common targets of mGluR1 signaling and Ca2+ entering through voltage-gated channels (Narasimhan and Linden, 1996).
The precise concentration of adenosine at the postsynaptic membrane of Purkinje cells is unknown. A previous study showed that the extracellular fluid in some regions of normally functioning rat brain was estimated to contain 40–400 nm adenosine (Ballarin et al., 1991; Dunwiddie and Diao, 1994). Neuronal A1R is thought to be usually exposed to a concentration considerably lower than these values for several reasons. The above study (Ballarin et al., 1991) also showed that adenosine was continuously taken up by the neural tissue. It is known that A1R of many cell types docks adenosine deaminase, an extracellular adenosine-degrading enzyme (Franco et al., 1997). However, the concentration of adenosine might sometimes reach a level that is enough for A1R to induce glu-LTD (40–400 nm; Fig. 4B) in an activity-dependent manner. When excited, many neurons, including cerebellar climbing fibers, are reported to release adenosine and/or its precursors (Do et al., 1991) A biochemical study (Balaban et al., 1984) showed that the 5′-nucleotidase's activity which catalyzes the precursors into adenosine increases around the PF–Purkinje cell synapses after stimulation of the climbing fibers. PFs may also release adenosine in an activity-dependent manner (Wall and Dale, 2007). Moreover, an arrest of the uptake mechanisms may also raise the concentration of adenosine in the CSF (Ballarin et al., 1991).
In the cerebellar cortex, activation of A1R on the presynaptic membrane may inhibit neurotransmitter release via Gi/o-protein and this may also result in a decrease in mGluR1 signaling. Therefore, to gauge the relative contributions of presynaptic and postsynaptic A1R to cerebellar LTD in situ awaits the innovation of a new technique to selectively manipulate presynaptic or postsynaptic A1R.
The inhibition of Gi/o-protein with pertussis toxin did not inhibit A1R-mediated glu-LTD blockade (Fig. 8B). This result is consistent with the previous study (Tabata et al., 2007) showing that Gi/o protein inhibition with pertussis toxin does not block A1R-mediated attenuation of mGluR1 signaling in cultured Purkinje cells. Interestingly, activation of another type of Gi/o-protein-coupled GPCR (B-type GABA receptor, GABABR) augments mGluR1 signaling and glu-LTD (Kamikubo et al., 2007). The fact that A1R and GABABR, GPCRs coupled to the same class of G-protein mediate the opposing effects on mGluR1 strongly suggests the existence of Gi/o-protein-independent linkage between A1R and mGluR1.
We do not exclude the possibility of the involvement of mechanisms other than those studied here in glu-LTD blockade. However, the present results suggest that an A1R-mediated decrease in the ligand responsiveness of mGluR1 is at least one of the major causes of glu-LTD blockade. These findings demonstrate that distinct neuronal GPCRs, each of which plays a different role by itself, may interplay and thereby cooperatively regulate the induction of synaptic plasticity.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research (KAKENHI 21700359 to Y.K.; 19045019, 21026011, 20500284, 20022025, and 23500384 to T.T.; 21220006 and 25000015 to M.K.) and the Strategic Research Program for Brain Sciences (Development of biomarker candidates for social behavior) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT); a Grant-in-Aid for Scientific Research on Innovative Areas (Foundation of Synapse and Neurocircuit Pathology) from MEXT; the MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2011–2015; the Uehara Memorial Foundation and the Narishige Neuroscience Research Foundation (to Y.K.); and the University of Toyama Presidential Grant (to T.T.). We thank T. Murayama for helpful discussion.
References
- Akiyama K, Nakanishi S, Naito T. Gene expression profiling of mouse cerebellar Purkinje cell. Neurosci Res. 2009;65:S77. doi: 10.1016/j.neures.2009.09.282. [DOI] [Google Scholar]
- Balaban CD, Wurpel JN, Severs WB. A specific harmaline-evoked increase in cerebellar 5′-nucleotidase activity. Neurosci Lett. 1984;50:111–116. doi: 10.1016/0304-3940(84)90471-3. [DOI] [PubMed] [Google Scholar]
- Ballarín M, Fredholm BB, Ambrosio S, Mahy N. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand. 1991;142:97–103. doi: 10.1111/j.1748-1716.1991.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Basheer R, Arrigoni E, Thatte HS, Greene RW, Ambudkar IS, McCarley RW. Adenosine induces inositol 1,4,5-trisphosphate receptor-mediated mobilization of intracellular calcium stores in basal forebrain cholinergic neurons. J Neurosci. 2002;22:7680–7686. doi: 10.1523/JNEUROSCI.22-17-07680.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Katoh A, Raymond JL. Cerebellum-dependent learning: the role of multiple plasticity mechanisms. Annu Rev Neurosci. 2004;27:581–609. doi: 10.1146/annurev.neuro.27.070203.144238. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Escriche M, Burgueño J, Angulo E, Casadó V, Soloviev MM, Canela EI, Mallol J, Chan WY, Lluis C, McIlhinney RA, Franco R. Metabotropic glutamate 1α and adenosine A1 receptors assemble into functionally interacting complexes. J Biol Chem. 2001;276:18345–18351. doi: 10.1074/jbc.M006960200. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do KQ, Vollenweider FX, Zollinger M, Cuénod M. Effect of climbing fibre deprivation on the K+-evoked release of endogenous adenosine from rat cerebellar slices. Eur J Neurosci. 1991;3:201–208. doi: 10.1111/j.1460-9568.1991.tb00080.x. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J Pharmacol Exp Ther. 1994;268:537–545. [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Ciruela F, Saura C, Mallol J, Canela EI, Lluis C. Cell surface adenosine deaminase: much more than an ectoenzyme. Prog Neurobiol. 1997;52:283–294. doi: 10.1016/S0301-0082(97)00013-0. [DOI] [PubMed] [Google Scholar]
- Fribourg M, Moreno JL, Holloway T, Provasi D, Baki L, Mahajan R, Park G, Adney SK, Hatcher C, Eltit JM, Ruta JD, Albizu L, Li Z, Umali A, Shim J, Fabiato A, MacKerell AD, Jr, Brezina V, Sealfon SC, Filizola M, et al. Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell. 2011;147:1011–1023. doi: 10.1016/j.cell.2011.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Selbach O. Functions of neuronal adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:375–381. doi: 10.1007/s002100000314. [DOI] [PubMed] [Google Scholar]
- Higgins MJ, Hosseinzadeh H, MacGregor DG, Ogilvy H, Stone TW. Release and actions of adenosine in the central nervous system. Pharm World Sci. 1994;16:62–68. doi: 10.1007/BF01880657. [DOI] [PubMed] [Google Scholar]
- Ito M. The molecular organization of cerebellar long-term depression. Nat Rev Neurosci. 2002;3:896–902. doi: 10.1038/nrn962. [DOI] [PubMed] [Google Scholar]
- Kamikubo Y, Egashira Y, Tanaka T, Shinoda Y, Tominaga-Yoshino K, Ogura A. Long-lasting synaptic loss after repeated induction of LTD: independence to the means of LTD induction. Eur J Neurosci. 2006;24:1606–1616. doi: 10.1111/j.1460-9568.2006.05032.x. [DOI] [PubMed] [Google Scholar]
- Kamikubo Y, Tabata T, Kakizawa S, Kawakami D, Watanabe M, Ogura A, Iino M, Kano M. Postsynaptic GABAB receptor signalling enhances LTD in mouse cerebellar Purkinje cells. J Physiol. 2007;585:549–563. doi: 10.1113/jphysiol.2007.141010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Tabata T. Type-1 metabotropic glutamate receptor in cerebellar Purkinje cells: a key molecule responsible for long-term depression, endocannabinoid signalling and synapse elimination. Philos Trans R Soc Lond B Biol Sci. 2008;363:2173–2186. doi: 10.1098/rstb.2008.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy AK, Edidin M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 Å using imaging fluorescence resonance energy transfer. J Cell Biol. 1998;142:69–84. doi: 10.1083/jcb.142.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Snapp E, Kenworthy A. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol. 2001;2:444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- Liu XY, Liu ZC, Sun YG, Ross M, Kim S, Tsai FF, Li QF, Jeffry J, Kim JY, Loh HH, Chen ZF. Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell. 2011;147:447–458. doi: 10.1016/j.cell.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Yan H, Othman T, Rivkees SA. Cytoskeletal protein 4.1G is a binding partner of the metabotropic glutamate receptor subtype 1 α. J Neurosci Res. 2004a;78:49–55. doi: 10.1002/jnr.20230. [DOI] [PubMed] [Google Scholar]
- Lu D, Yan H, Othman T, Turner CP, Woolf T, Rivkees SA. Cytoskeletal protein 4.1G binds to the third intracellular loop of the A1 adenosine receptor and inhibits receptor action. Biochem J. 2004b;377:51–59. doi: 10.1042/BJ20030952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Launey T, Mikawa S, Hirai H. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 2000;19:2765–2774. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000;327:472–500. doi: 10.1016/S0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Narasimhan K, Linden DJ. Defining a minimal computational unit for cerebellar long-term depression. Neuron. 1996;17:333–341. doi: 10.1016/S0896-6273(00)80164-6. [DOI] [PubMed] [Google Scholar]
- Nugent T, Jones DT. Membrane protein structural bioinformatics. J Struct Biol. 2012;179:327–337. doi: 10.1016/j.jsb.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Pankevych H, Korkhov V, Freissmuth M, Nanoff C. Truncation of the A1 adenosine receptor reveals distinct roles of the membrane-proximal carboxyl terminus in receptor folding and G protein coupling. J Biol Chem. 2003;278:30283–30293. doi: 10.1074/jbc.M212918200. [DOI] [PubMed] [Google Scholar]
- Park D, Jhon DY, Lee CW, Lee KH, Rhee SG. Activation of phospholipase C isozymes by G protein βγ subunits. J Biol Chem. 1993;268:4573–4576. [PubMed] [Google Scholar]
- Phillis JW, Wu PH. The role of adenosine and its nucleotides in central synaptic transmission. Prog Neurobiol. 1981;16:187–239. doi: 10.1016/0301-0082(81)90014-9. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Rios CD, Jordan BA, Gomes I, Devi LA. G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther. 2001;92:71–87. doi: 10.1016/S0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci. 2007;10:1377–1386. doi: 10.1038/nn2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Kaneko K, Okuno M, Wada K, Kashiyama T, Shimizu H, Akagi T, Hashikawa T, Nukina N. Membrane microdomain switching: a regulatory mechanism of amyloid precursor protein processing. J Cell Biol. 2008;183:339–352. doi: 10.1083/jcb.200804075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbie LA, Hill SJ. G protein-coupled-receptor cross-talk: the fine-tuning of multiple receptor-signalling pathways. Trends Pharmacol Sci. 1998;19:87–93. doi: 10.1016/S0165-6147(97)01166-8. [DOI] [PubMed] [Google Scholar]
- Tabata T, Sawada S, Araki K, Bono Y, Furuya S, Kano M. A reliable method for culture of dissociated mouse cerebellar cells enriched for Purkinje neurons. J Neurosci Methods. 2000;104:45–53. doi: 10.1016/S0165-0270(00)00323-X. [DOI] [PubMed] [Google Scholar]
- Tabata T, Araishi K, Hashimoto K, Hashimotodani Y, van der Putten H, Bettler B, Kano M. Ca2+ activity at GABAB receptors constitutively promotes metabotropic glutamate signaling in the absence of GABA. Proc Natl Acad Sci U S A. 2004;101:16952–16957. doi: 10.1073/pnas.0405387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, Kawakami D, Hashimoto K, Kassai H, Yoshida T, Hashimotodani Y, Fredholm BB, Sekino Y, Aiba A, Kano M. G protein-independent neuromodulatory action of adenosine on metabotropic glutamate signalling in mouse cerebellar Purkinje cells. J Physiol. 2007;581:693–708. doi: 10.1113/jphysiol.2007.129866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateyama M, Kubo Y. Coupling profile of the metabotropic glutamate receptor 1α is regulated by the C-terminal domain. Mol Cell Neurosci. 2007;34:445–452. doi: 10.1016/j.mcn.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Wall MJ, Dale N. Auto-inhibition of rat parallel fibre-Purkinje cell synapses by activity-dependent adenosine release. J Physiol. 2007;581:553–565. doi: 10.1113/jphysiol.2006.126417. [DOI] [PMC free article] [PubMed] [Google Scholar]