

Abstract
Background
Burkitt lymphoma is a fast‐growing mature B cell malignancy, whose genetic hallmark is translocation and activation of the c‐myc gene. Prompt multiagent immunochemotherapy regimens can have favorable outcomes, but prognosis is poor in refractory or relapsed disease. We previously identified a novel family of near‐infrared heptamethine carbocyanine fluorescent dyes (HMCD or DZ) with tumor‐homing properties via organic anion‐transporting peptides. These membrane carriers have uptake in tumor cells but not normal cells in cell culture, mouse and dog tumor models, patient‐derived xenografts, and perfused kidney cancers in human patients.
Methods
Here we report the cytotoxic effects of a synthesized conjugate of DZ with cisplatin (CIS) on B cell lymphoma CA46, Daudi, Namalwa, Raji, and Ramos cell lines in cell culture and in xenograft tumor formation. Impaired mitochondrial membrane permeability was examined as the mechanism of DZ‐CIS–induced lymphoma cell death.
Results
The new conjugate, DZ‐CIS, is cytotoxic against Burkitt lymphoma cell lines and tumor models. DZ‐CIS retains tumor‐homing properties to mitochondrial and lysosomal compartments, does not accumulate in normal cells and tissues, and has no nephrotoxicity in mice. DZ‐CIS accumulated in Burkitt lymphoma cells and tumors induces apoptosis and retards tumor cell growth in culture and xenograft tumor growth in mice.
Conclusion
DZ‐CIS downregulated c‐myc and overcame CIS resistance in myc‐driven TP53‐mutated aggressive B cell Burkitt lymphoma. We propose that DZ‐CIS could be used to treat relapsed/refractory aggressive Burkitt lymphomas.
Keywords: Burkitt lymphoma, heptamethine carbocyanine, cisplatin, conjugate, cell death
Short abstract
The cytotoxic effect of a synthesized conjugate between a tumor cell–specific heptamethine carbocyanine and cisplatin on Burkitt lymphoma cells is evaluated. The conjugate inhibits c‐myc expression, reduces mitochondrial membrane permeability, and induces lymphoma cell death in culture and in xenograft tumors in athymic mice.
Introduction
Burkitt lymphoma, a highly aggressive B lymphocyte non‐Hodgkin lymphoma, presents in extranodal and nodal sites1 and also manifests as B cell acute lymphoblastic leukemia.2 A characteristic c‐myc oncogene translocation between the long arm of chromosome 8 and the immunoglobulin heavy chain gene on chromosome 14 is seen in 80% of Burkitt lymphoma cases or between the long arm of chromosome 8 and the kappa light chain gene on chromosome 2 or the lambda light chain gene on chromosome 22 in 20% cases.3 These translocations induce active c‐myc protein expression in tumor cells, unlike the tight c‐myc regulation in normal B cells.4
Mortality is high in Burkitt lymphoma due to rapid‐growth advanced disease at diagnosis.5 Short intensive chemotherapy regimens have favorable outcomes,6 but prognosis in refractory or relapsed disease is poor. The median overall survival is 2.7 months.7 Salvage high‐dose cisplatin (CIS) and carboplatin chemotherapy and autologous hematopoietic stem cell transplantation have modest survival benefits in eligible patients.8
Drug resistance is a major obstacle in cancer therapy.9 Monoclonal antibodies and small molecule tyrosine‐kinase or serine/threonine kinase inhibitors have not achieved improvements in survival in refractory tumors,10, 11 although antibody–drug conjugates12 and small molecule ligand and drug conjugates using delivery strategies such as σ2‐opioid receptor,13 folate receptor,14 carbonic anhydrase IX inhibitor,15 PSMA,16 or HSP9017 ligands and cytotoxic drugs have shown promise.
We identified a family of near‐infrared (NIR) heptamethine carbocyanine dyes (DZs) with tumor imaging and tumor‐homing properties.18 Cancer cells express a group of cell membrane organic anion‐transporting polypeptides (OATPs)19 at higher levels than normal cells and thus have preferential uptake in tumors.20, 21, 22, 23, 24 OATPs facilitate Na+‐independent uniport or exchange membrane transport of amphiphilic anions and neutral or cationic substrates.25 We previously developed DZs or their analogs conjugated docetaxel,21 gemcitabine,22 and clorgyline26 for tumor‐targeted delivery. In the present study, DZ‐CIS conjugate was designed to home in on the mitochondrial and lysosomal compartments and safely kill CIS‐resistant aggressive Burkitt lymphoma cells in vitro and in vivo without renal or liver toxicity.
Materials and Methods
Synthesis of DZ‐CIS Conjugate
All chemicals and reagents were purchased from Sigma‐Aldrich (St. Louis, Missouri) or VWR International (Radnor, Pennsylvania). Deionized water (18.2 Ω) for solutions was obtained from the Milli‐Q Direct Ultrapure Water System (Merck Millipore, Billerica, Massachusetts). All intermediates were characterized by proton nuclear magnetic resonance (1H‐NMR) and mass spectrometry, and the purity of compounds was analyzed by high‐performance liquid chromatography (HPLC). 1H‐NMR data were collected on Bruker 400‐MHz spectrometers (Bruker, Billerica, Massachusetts) using standard parameters; chemical shifts are reported in ppm (δ) in residual nondeuterated solvent. Electrospray ionization mass spectrometry analysis of new compounds was performed at the Mass Spectrometry and Biomarker Discovery Core of Cedars‐Sinai Medical Center using a Thermo Fisher LTQ Orbitrap Elite system (Thermo Fisher Scientific, Waltham, Massachusetts).
CIS (Selleck Chemicals, Houston, Texas) was oxidized with hydrogen peroxide to form cis, cis, trans‐diaminedichloro‐dihydroxyplatinum (IV) (oxoplatin) 2.27 The asymmetric sulfonic acid analog of heptamethine carbocyanine, DZ 1 was synthesized as reported.22 DZ (500 mg, 0.71 mmol) was added to oxoplatin compound (350 mg, 1.05 mmol) in dimethyl sulfoxide (DMSO, 20 mL) followed by 1‐ethyl‐3‐(3‐dimethyllaminopropyl) N’‐ethylcarbodiimide hydrochloride (136 mg, 0.071 mmol) and dimethylaminopyridine (80 mg, 0.65 mmol) and stirred for 20 hours at room temperature. The solution was filtered and DMSO was removed by lyophilization, purified by C18‐RP silica chromatography, and eluted with methanol‐water to produce DZ‐CIS 3 (275 mg; yield, 38%).
Cell Culture
The human Burkitt lymphoma cell lines Namalwa, Raji, Daudi, Ramos, and CA46 from the American Type Culture Collection (Manassas, Virginia) were obtained from Professor H. Phillip Koeffler’s laboratory (Cedars‐Sinai Medical Center, Los Angeles, California).28 Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Life Technologies, Carlsbad, California) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, Georgia), 100 IU/mL penicillin, and 100 µg/mL streptomycin (Thermo Fisher Scientific) at 37°C in a humidified incubator with 5% CO2.
Cell Proliferation Assay
Cells (1 × 104/well) of quadruplet wells in 96‐well plates (USA Scientific, Irvine, California) were exposed to exponentially increasing concentrations (0.25 μM to 64 μM) of CIS or DZ‐CIS for 24 hours. The final concentration of solvent DMSO (Sigma‐Aldrich) never exceeded 1%. Cells were treated with 10% 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide (Sigma‐Aldrich) for 4 hours and decolorized with 100 μL 2‐propanol (Sigma‐Aldrich). Supernatant extinction was read at 595 nm by microplate reader (Bio‐Rad Laboratories, Hercules, California).
Stromal Cell Coculture
CCD16Lu normal human lung stromal cells obtained from American Type Culture Collection were established by previously described protocols.29 For green fluorescent protein (GFP) tagging, lentiviral particles of MISSION pLKO.1‐puro‐CMV‐TurboGFP (Sigma‐Aldrich) were used to infect stromal cells, subjected to puromycin (2 μg/mL) for 2 weeks to remove noninfected cells. GFP‐tagged cells were maintained in RPMI 1640 medium containing 10% fetal bovine serum without puromycin. Burkitt lymphoma cells were cocultured on a monolayer of stromal cells at a 5:1 ratio in 6‐well plates and maintained for 2 days before treatment.
Apoptosis Assay
Cell death rates were determined by counting viable cells in trypan blue‐stained samples on a TC20 automatic cell counter (Bio‐Rad Laboratories). Caspase 3/7 enzymatic activities were measured in cells treated with increasing concentrations (16‐64 μM) of CIS or DZ‐CIS for 12 hours by the Caspase‐Glo 3/7 Assay System (Promega, Madison, Wisconsin). Luminescence intensity was measured by LUMIstar Omega microplate luminometer (BMG Labtech, BioTek, Winooski, Vermont). Western blot analysis of caspase activation in whole cell lysates from treated lymphoma cells used antibodies to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), caspase 9, caspase 3, and poly(adenosine diphosphate‐ribose) polymerase (PARP) from Cell Signaling Technology (Danvers, Massachusetts) and horseradish peroxidase (HRP)‐conjugated secondary antibodies (Santa Cruz Biotechnology, Dallas, Texas).
Flow Cytometry
Lymphoma cells were treated with 4 μM of DZ‐CIS for 15 minutes, washed 3 times with phosphate‐buffered saline (PBS) containing 1% bovine serum albumin (Sigma‐Aldrich), and analyzed fir intracellular DZ‐CIS accumulation on a BD Accuri C6 flow cytometer (Becton Dickinson Biosciences, San Jose, California) using FlowJo software (FlowJo LLC, Ashland, Oregon). Data were expressed as a mean percentage from 3 independent experiments. To assess mitochondrial membrane permeability, cells were treated with 5,5’,6,6’‐Tetrachloro‐1,1’,3,3’‐tetraethylbenzimidazolylcarbocyanine chloride (JC‐1, GeneCopoeia, Rockville, Maryland) for 1 hour with the JC‐1 Mitochondrial Membrane Potential Detection Kit. After washing off free dyes, cells were treated with DZ‐CIS (8 µM) for 6 hours and subjected to flow cytometric analysis.
Western Blotting
Cell lysates were prepared using 50 mM Tris, pH 8, 150 mM NaCl, 0.02% NaN3, 0.1% SDS, 1% NP‐40 and 0.5% sodium deoxycholate lysis buffer containing 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Roche Applied Science, Indianapolis, Indiana). Protein concentration was determined by Bradford assay using Coomassie Plus Protein Reagent (Thermo Fisher Scientific). Proteins (50 μg) were resolved on a 4%‐12% Bis‐Tris gradient SDS‐PAGE (Bio‐Rad Laboratories) under reducing conditions and transferred to nitrocellulose membrane (Bio‐Rad Laboratories). Primary antibodies to p‐PI3K, ALDH1A1, nuclear factor κB (NF‐κB), p‐NF‐κB, SRC, p‐SRC, max, p27, p21, p‐c‐JUN, p53, p‐p53, Ku70, DNMT1, DNMT3a, DNMT3b, HDAC2, and β‐actin were purchased from Santa Cruz Biotechnology. All other antibodies used in this study were from Cell Signaling Technology. HRP‐conjugated secondary antibodies were from Santa Cruz Biotechnology. Protein bands were detected by SuperSignal West Dura Supstrate Western Blot Detection Reagent (Thermo Fisher Scientific).
Fluorescence Microscopy
Namalwa cells on chamber slides (Nalge Nunc International, Rochester, New York) were treated with 4 μM CIS, DZ‐CIS or vehicle for 24 hours and then centrifuged at 200 g for 5 minutes, counterstained with 4’,6‐diamidino‐2‐phenyindole dihydrochloride (Thermo Fisher Scientific), and fixed for 10 minutes in 4% paraformaldehyde (Sigma‐Aldrich) with glass coverslips in aqueous mounting medium (Sigma‐Aldrich) for NIR fluorescence dye uptake analyzed by Nikon Eclipse Ti‐E confocal microscope (Nikon Corporation, Tokyo), quantitated by ImageJ (NIH, Bethesda, Maryland).
Tumor Xenografts
Animal studies were approved by the Institutional Animal Care and Use Committee. Male 4‐ to 6‐week‐old athymic NCr nude (Foxn1Nu/Foxn1Nu) mice (Taconic Biosciences, Hudson, New York) and NOD Scid (NOD.CB17‐ Prkdcscid/J) mice (The Jackson Laboratory, Bar Harbor, Maine) underwent xenograft tumor formation by injecting 107 lymphoma cells in 100 μL PBS subcutaneously (n = 15 mice/group) with 2 and 4 tumors per mouse to the flank, respectively. When tumors reached 100 mm3, mice were randomized into 3 groups and treated intraperitoneally twice weekly with either 5 mg/kg (NOD Scid) or 10 mg/kg (NCr nude) of CIS, DZ‐CIS, or vehicle (n = 5 per treatment) and dissolved in PBS with 5% DMSO and 15% PEG 40 (Sigma‐Aldrich). Tumor size was measured by caliper 3 times weekly. Tumor volume was calculated as: tumor volume = (length × width2)/2. At the endpoint tumors, livers and kidneys were excised, fixed in formalin, and paraffin‐embedded for histological and immunocytochemical analysis.
Tumor Cell Imaging
Animals underwent NIR fluorescence imaging 48 hours after the last DZ‐CIS intraperitoneally injection with whole body imaging using an IVIS Lumina XR Imaging System (PerkinElmer, Waltham, Massachusetts) with fluorescent filter sets (excitation/emission, 783/840 nm), with automatic subtraction of background fluorescence. After euthanasia, tumors and organs were retrieved for biodistribution studies using the same imaging system.
Subcellular Localization of DZ‐CIS by Fluorescence Microscopy
To determine where DZ‐CIS accumulates, we treated Namalwa cells with DZ‐CIS (10 μM) for 1 hour followed by costaining with MitoTracker Green (100 nM) or LysoTracker Green (100 nM) (Thermo Fisher Scientific) for 30 minutes and counterstaining with Hoechst 33342 (200 ng/mL) (Sigma‐Aldrich) for 10 minutes. After fixation in 4% (vol/vol) paraformaldehyde solution for 15 minutes, fluorescence microscopy using a BZ‐X710 microscope (Keyence America, Elmwood Park, New Jersey) with built‐in software was used to for colocalization of DZ‐CIS, MitoTracker and LysoTracker signals.
Immunohistochemistry
Immunohistochemistry of tumor xenografts and organs included antibodies against Ki‐67, survivin (Santa Cruz Biotechnology), cleaved caspase 3, c‐myc (Cell Signaling Technology) and M30 soluble caspase‐cleaved keratin 18 (Sigma‐Aldrich). Formalin‐fixed paraffin‐embedded sections (4 μm) were stained as described previously,30 treated for 30 minutes with EnVision+ Dual Link System‐HRP (Dako, Carpinteria, California) and developed with 3’3‐diaminobenzidine (Dako) with image acquisition (×400 magnification) by digital camera and software (Nikon Corporation, Minato, Tokyo).
Statistical Analysis
All results are presented as the mean ± standard error of the mean for at least 3 independent tests. Normality of distribution was assessed by Kolmogorov‐Smirnov test. Parametric data were compared using a Student t test; nonparametric data were compared using a Mann‐Whitney U test and SPSS software version 15 (IBM Corporation, Armonk, New York). Half maximal inhibitory concentration (IC50) values were calculated using a nonlinear regression method after normalization by GraphPad Prism version 5 Software (GraphPad Software, La Jolla, California).
Results
DZ‐CIS Conjugate Inhibits Burkitt Lymphoma Cell Viability and Proliferation
DZ is tumor cell–specific, efficiently accumulating in xenograft tumors 24 hours after administration.31, 32, 33 The synthesis of DZ‐CIS conjugate used a succinic ester linker (Fig. 1). We assess whether DZ‐CIS could overcome CIS insensitivity or resistance in Burkitt lymphoma cells because these cells are inherently refractory to CIS treatment (Fig. 2A).34 DZ‐CIS effectively killed Burkitt lymphoma cells (Fig. 2A), as confirmed by trypan blue staining (Fig. 2B). DZ‐CIS treatment dose‐dependently killed all 5 Burkitt lymphoma cell lines with IC50 values ranging from 0.72 μM for Namalwa cells to 2.53 μM for Daudi cells (Fig. 2A, Table 1). Using 10 μM DZ‐CIS, lymphoma cell death was direct and efficient, with morphological changes starting at 6 hours and cell death at 12 hours (Fig. 2C). DZ‐CIS was comparatively more effective than doxorubicin, docetaxel, targeted tyrosine kinase inhibitors, and mammalian target of rapamycin inhibitors, whose effects take days or weeks.35
Figure 1.
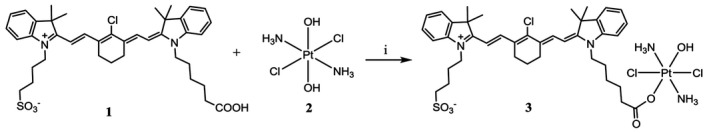
Synthesis of the heptamethine carbocyanine fluorescent dye–cisplatin conjugate (DZ‐CIS). DZ‐CIS conjugate was synthesized through a succinic ester linker. DZ‐1 cis‐ and trans‐diaminedichloro‐dihydroxyplatinum (IV) (oxoplatin) 2 were mixed and reacted as described in Materials and Methods by adding N’‐ethylcarbodiimide hydrochloride and dimethylaminopyridine in dimethyl sulfoxide solution.
Figure 2.
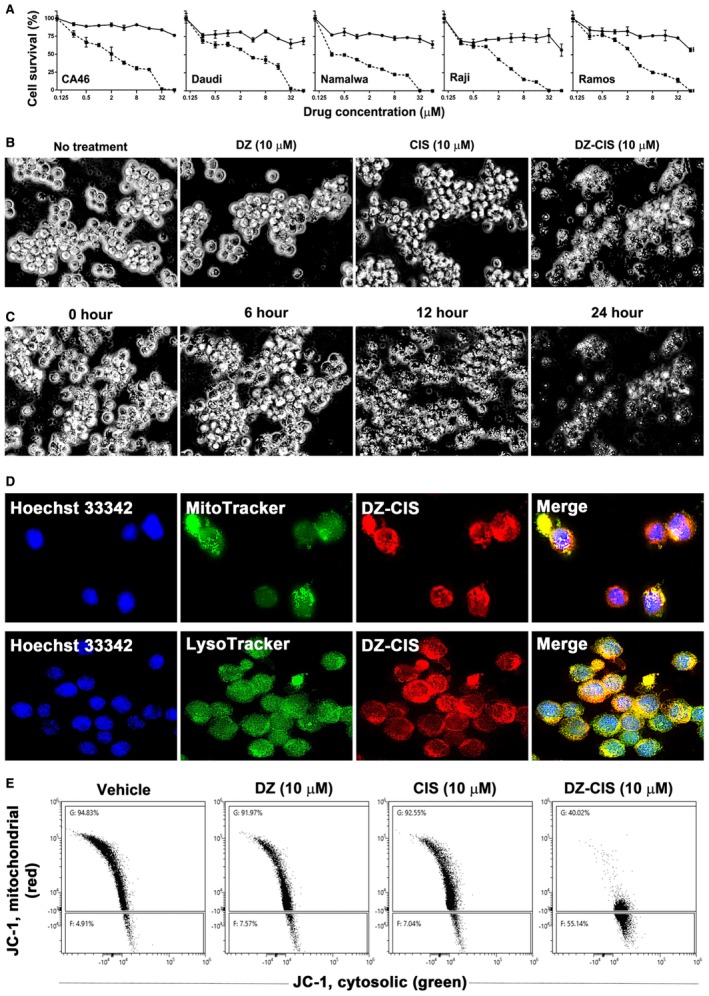
Heptamethine carbocyanine fluorescent dye–cisplatin conjugate (DZ‐CIS) cytotoxicity. (A) Dose‐effect curve. All Burkitt lymphoma cell lines were insensitive to CIS (circles on solid line) but dose‐dependently sensitive to DZ‐ CIS (squares on dotted line). (B) DZ‐CIS‐induced death of Namalwa cells 24 hours after treatment. Black and white images of trypan blue–stained cells (magnification ×100). (C) DZ‐CIS (10 μM) caused marked lymphoma cell death with morphological changes starting 6 hours into the treatment. Cell death became conspicuous around 12 hours (trypan blue stain, magnification ×100). (D) DZ‐CIS preferentially accumulates to mitochondria and lysosomes, demonstrated by colocalization (magnification ×600) of the NIR signal with MitoTracker (upper row) and LysoTracker (lower row). (E) Flow cytometry indicates that 6‐hour treatment with DZ‐CIS disrupts mitochondrial membrane integrity. JC‐1 in the cytosol lost its red fluorescence as monomeric JC‐1 (which emits green fluorescence).
Table 1.
Cytotoxicity (IC50, µM) of the DZ‐CIS Conjugate
| Cell line | CIS | 95% CI | DZ‐CIS | 95% CI |
|---|---|---|---|---|
| Daudi | >64 | ND | 2.53 | 1.833‐3.489 |
| Raji | >64 | ND | 1.23 | 1.004‐1.502 |
| Namalwa | >64 | ND | 0.72 | 0.511‐1.024 |
| Ramos | >64 | ND | 2.40 | 2.029‐2.841 |
| CA‐46 | >64 | ND | 2.00 | 1.604‐2.512 |
Abbreviations: CI, confidence interval; CIS, cisplatin; DZ‐CIS, heptamethine carbocyanine fluorescent dye–cisplatin; ND, not determined.
Results were calculated with quadruplet 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide assay data points after 24 hours of treatment.
DZ accumulates in subcellular organelles including mitochondria and lysosomes18, 23, 32 and DZ‐CIS colocalized by MitoTracker and LysoTracker in Namalwa cells (Fig. 2D), markedly altered mitochondrial membrane potential, with leakage of JC‐1 dimer from the intramitochondrial compartment into cytosol at 6 hours (Fig. 2E).
DZ‐CIS Accumulates in Burkitt Lymphoma Cells and Tumors
Two separate experiments assessed DZ‐CIS targeting specificity by established protocols,21, 22, 26 evaluating intracellular accumulation microscopically and by flow cytometry. After a 15‐minute treatment with 4 μM DZ‐CIS, 99.4% of lymphoma cells stained positively by NIR fluorescence (Fig. 3A and B). We observed that the uptake of DZ‐CIS into cancer cells cannot be effectively blocked by bromsulphthalein, a competitive OATP inhibitor, but can be blocked by telmisartan and rifampicin, nonspecific OATP inhibitors (data not shown), suggesting that unlike heptamethine carbocyanine fluorescent dyes, DZ‐CIS is no longer transported into cancer cells by the classical OATPs, possibly due to modification of chemical structure of DZ to become a DZ‐CIS conjugate.
Figure 3.
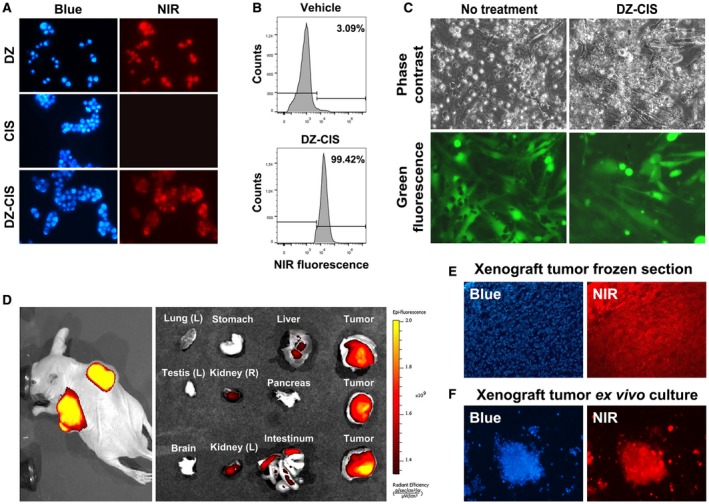
The heptamethine carbocyanine fluorescent dye–cisplatin conjugate (DZ‐CIS) acts specifically on Burkitt lymphoma cells. (A) Near‐infrared (NIR) microscopy of Namalwa cells after treatment with 4 μM DZ‐CIS for 15 minutes. DZ‐CIS accumulated in all lymphoma cells. Blue fluorescence of the nuclei was from Hoechst 33342 co‐stain (200×). (B) Namalwa cells treated with 4 μM DZ‐CIS for 15 minutes were assayed for rapid uptake of the fluorescent dye–drug conjugate in live cells using flow cytometry. (C) Images of lymphoma and stromal cell coculture show that DZ‐CIS preferentially kills lymphoma but not stromal cells. In coculture with GFP‐tagged CCD16Lu normal human lung mesenchymal stromal cells, 24‐hour DZ‐CIS treatment preferentially killed Burkitt lymphoma cells (Namalwa), while stromal cells survived and displayed healthy morphology (magnification ×200). DZ‐CIS–induced death of lymphoma cells in coculture was confirmed with trypan blue stain. (D) Tumor‐specific uptake and retention of DZ‐CIS in vivo were evaluated with NCrnu/nu mice bearing Namalwa xenograft tumors. After treatment with DZ‐CIS for 2 weeks, mice were subjected to whole body NIR imaging (left). Necropsy tumor samples together with host organs were subjected to ex vivo NIR imaging (right). (E) Frozen sections of xenograft tumors were stained with 4’,6‐diamidino‐2‐phenyindole dihydrochloride (DAPI) and examined for retention of DZ‐CIS by NIR microscopy (magnification ×100). (F) Xenograft tumors diced in ex vivo culture were stained with DAPI (magnification ×100). Most tumor cells still carried DZ‐CIS signals after 3 days in the culture.
DZ exhibits tumor cell specificity, accumulates preferentially in tumors in vivo and clears efficiently from normal tissues and organs.18, 31 DZ‐CIS has similar tumor cell targeting specificity and killed Burkitt lymphoma cells preferentially. Mesenchymal stromal cells tagged with GFP survived treatment (Fig. 3C).
DZ‐CIS targeting specificity was further assessed in a mouse subcutaneous tumor xenograft model36 to image tumor growth. Mice bearing Namalwa tumors received DZ‐CIS (5 mg/kg) 3 times a week for 2 weeks. DZ‐CIS accumulated specifically in the xenografts (Fig. 3D). DZ‐CIS NIR signals were detected at least 7 days after a single administration, like our prototype IR783 and MHI‐148 analogs.18, 21, 22, 23, 31 DZ‐CIS is prevented from diffusing out from cancer cells by interaction with cellular nucleic acids and proteins.37, 38
After whole body NIR imaging, mice were euthanized at 24 hours and necropsy specimens were again imaged by NIR. Higher NIR signals were detected in xenograft tumors than host tissues (Fig. 3D). Most tumor cells exhibited NIR fluorescence (Fig. 3E). In ex vivo tumor tissue culture, most cells carried NIR signals for 3 days (Fig. 3F). These results validated the targeting specificity of DZ‐CIS.
DZ‐CIS Treatment Induces Caspase Activity in Burkitt Lymphoma Cells
A Caspase‐Glo 3/7 assay detected dose‐dependent caspase activation by increased enzymatic activity of caspases 3 and 7 in lymphoma cells after 12‐hour treatment with 8 to 32 μM DZ‐CIS (Fig. 4A). Western blotting revealed enhanced cleavage of caspase 3, caspase 9, and PARP apoptotic cascade proteins in Namalwa cells treated with DZ‐CIS, but not CIS (Fig. 4B). The pan‐caspase inhibitor Z‐VAD (10 μM) displayed transient protection between 6 and 12 hours of treatment but could not prevent complete cell death at 24 hours (data not shown). This suggests DZ‐CIS‐induced lymphoma cell death is not mediated solely by caspase activation.
Figure 4.
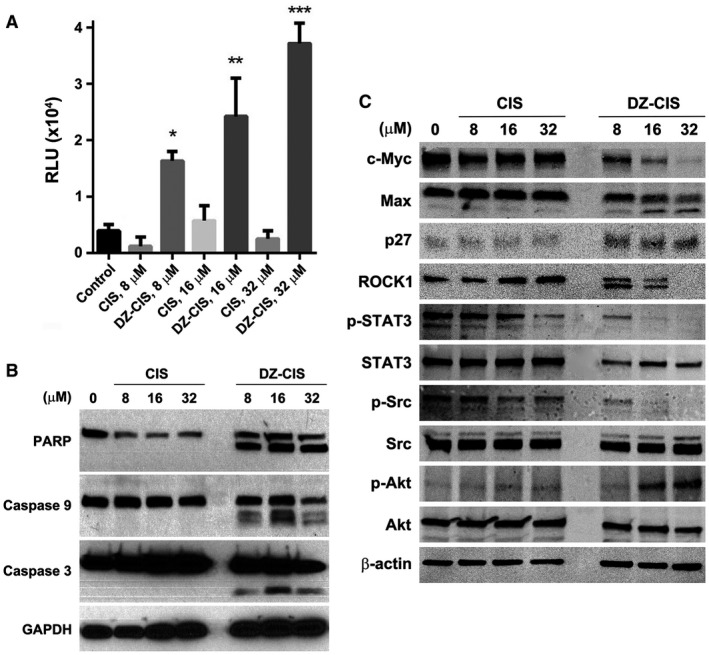
Heptamethine carbocyanine fluorescent dye–cisplatin conjugate (DZ‐CIS)–induced caspase cascade activation. (A) Namalwa cells treated with DZ‐CIS were assayed for the involvement of caspases with a CaspaseGlo 3/7 luminescent assay 24 hours after treatment. Results are presented with the mean ± standard deviation of quadruple measurements (* P < .05, ** P < .01, *** P < .0001). (B) Whole cell lysates of the treated cells were subjected to western blotting to detect activation of the caspase cascade. (C) DZ‐CIS–treated Namalwa cells were examined for protein level and protein modification changes by western blotting. DZ‐CIS treatment showed decreased expression of c‐myc and induced Max protein cleavage and increased p27 protein expression, suggesting growth arrest of treated cells. DZ‐CIS treatment caused cleavage of the ROCK1 protein. c‐Jun, STAT3, and Src protein phosphorylation was suppressed. Paradoxically, DZ‐CIS caused increased phosphorylation of AKT, suggesting its activation.
DZ‐CIS c‐myc Signaling Network in Burkitt Lymphoma Cells
To investigate whether lymphoma cell death was due to disturbed intracellular signaling transduction, key signaling network proteins were examined in Burkitt lymphoma Namalwa cells after 24‐hour exposure to DZ‐CIS by western blotting. CIS displayed little influence. DZ‐CIS markedly altered cellular protein expression, suppressing c‐myc levels and inducing Max protein cleavage and upregulation of p27 protein expression (Fig. 4C). DZ‐CIS treatment caused ROCK1 protein cleavage, one of the downstream targets of caspase 3 cleavage, resulting in cell death.39
Reduced c‐myc protein levels were accompanied by changes in posttranslational protein phosphorylation, suppressing phosphorylation of c‐Jun, STAT3 and Src, 3 important proto‐oncogene proteins (Fig. 4C). We also explored the possible action of DZ‐CIS in other signaling pathways. The results show that DZ‐CIS induced elevated Akt protein phosphorylation and changes in expression levels of p‐c‐JUN, SAPK/JNK, p‐SAPK/JNK, Mdr1, SMAD2, p‐SMAD2, p‐CREB, Ku70, and p‐p53 but had little influence on the expression or phosphorylation of ALDH1A1, NF‐κB, p53, PI3K, Mek1/2, Erk, and p38 MAPK signaling proteins. In addition, DZ‐CIS treatment altered the expression of epigenetic regulatory proteins DNMT1, DNMT3a, DNMT3b, and HDAC2 (data not shown).
DZ‐CIS Inhibited Xenograft Tumor Growth Without Renal or Liver Toxicity
NCr nude and NOD Scid mice bearing subcutaneous human Burkitt lymphoma Namalwa tumors were treated intraperitoneally with either 5 (NOD Scid) or 10 (NCr nude) mg/kg twice a week. Tumor volume significantly decreased in both groups with DZ‐CIS, but not CIS or vehicle‐treatment groups (P < .05) (Fig. 5A).
Figure 5.
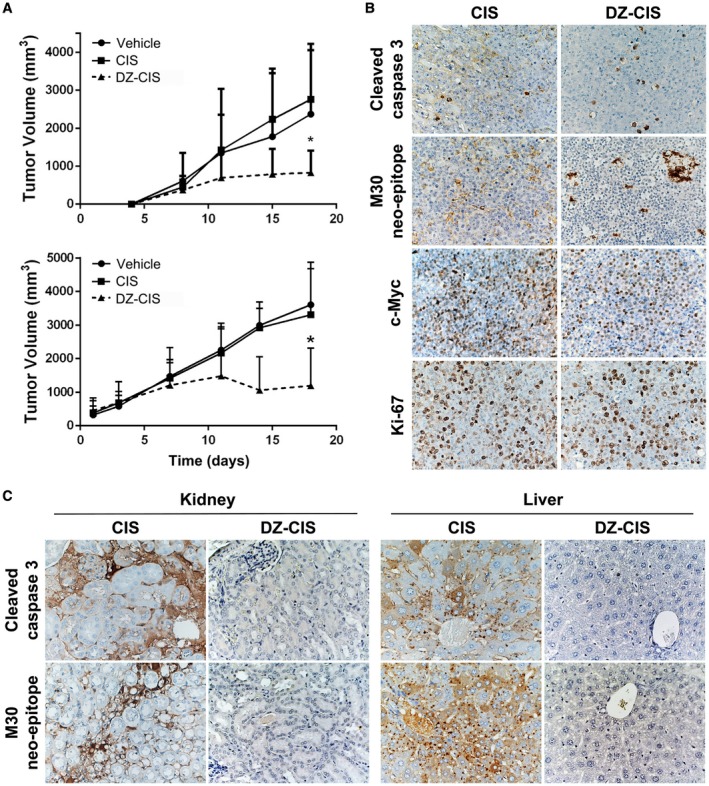
The heptamethine carbocyanine fluorescent dye–cisplatin conjugate (DZ‐CIS) inhibited xenograft tumor growth. (A) NCrnu/nu mice (top, n = 5 nude mice bearing 8‐10 Namalwa tumors per treatment group) and NOD Scid mice (bottom, n = 5 Scid mice bearing 16 Namalwa tumors per treatment group) were treated intraperitoneally with either 5 (NOD Scid) or 10 (NCr nude) mg/kg DZ‐CIS twice a week, from day 4 and day 1, respectively, resulting in significantly decreased tumor volume compared with CIS or vehicle treatment groups (P < .05). (B) CIS and DZ‐CIS–treated xenograft tumors were analyzed by immunohistochemistry for cell proliferation, survival, and death biomarker protein expression. (C) Mice were evaluated for renal and hepatic injuries with immunohistochemistry for cell death markers.
When mice were examined for cytotoxic effects, apoptosis markers of cleaved caspase 3 and the M30 neo‐epitope increased in the DZ‐CIS treated group, with an increase in intensity of cleaved caspase 3 and M30 compared with the CIS‐treated group (Fig. 5B). Decreased Ki‐67 staining in the DZ‐CIS group implying reduced tumor cell proliferation was accompanied by c‐myc loss in treated Namalwa cells in vitro (Fig. 4C). The expression of c‐myc decreased dramatically in DZ‐CIS–treated tumors (Fig. 5B). A major side effect of CIS chemotherapy is renal toxicity,40, 41 and CIS‐treated mice had renal tubular dilatation and increased cleaved caspase 3, together with caspase‐cleaved and formalin‐resistant cytokeratin 18 neo‐epitope (M30) in kidney glomeruli and proximal tubules and diffusely in liver hepatocytes (Fig. 5C). DZ‐CIS treatment showed no evidence of cleaved caspase 3 and M30 staining in mouse kidney and liver. Histological and immunohistochemical data suggest that DZ‐CIS conjugate was nontoxic to normal mouse kidney and liver tissues.
Discussion
Most studies agree that CIS cytotoxic effects are initiated by interaction with purine bases resulting in DNA–DNA intrastrand crosslink, simultaneously activating both prosurvival and proapoptotic signaling pathways. Activation of DNA damage recognition proteins and pathways involving p53 and p38 MAPK induce growth arrest and cell death.42 CIS, as a platinum‐containing compound, is also toxic to enucleated cells.43 Nonetheless, some cancer cells adapt to CIS treatment, and resistance eventually develops.44 Tumors resistant to CIS often show cross‐resistance to other unrelated cytotoxic drugs, suggesting a common drug‐resistance mechanism.45 Even in CIS‐sensitive tumor cells, CIS accumulation and cytotoxicity are not correlated with the dose.46 CIS cytotoxicity may relate to the proportion of cells in the S phase of the cell cycle, or to the ability to repair genomic damage, since normal tissues with slower proliferation rates and higher DNA damage–repair activities are less affected than tumors.46, 47, 48 Tumor cell–specific delivery and longer CIS retention could thus be exploited to overcome resistance and increase CIS antitumor efficacy.
We synthesized and characterized DZ‐CIS to deliver CIS directly to CIS‐insensitive Burkitt lymphoma cells, in which a modified heptamethine carbocyanine moiety facilitates tumor‐specific uptake. DZ‐CIS was potently cytotoxic against Burkitt lymphoma cells (Fig. 2) and tumors (Fig. 5) in a concentration‐ and time‐dependent manner, without harmful side effects in normal tissues, such as kidney and liver (Figs. 3 and 5). These results agree with previous reports of a linear correlation between total DNA‐bound platinum and the magnitude of platinum cytotoxicity in cancer cells.49
In this study, Burkitt lymphoma cells and the mouse tumor model both accumulated DZ and DZ‐CIS, confirming our earlier observations that DZ can deliver CIS specifically to tumor cells but not normal cells.18 FACS analysis detected rapid DZ‐CIS accumulation in 96.9% of Burkitt lymphoma Namalwa cells after 15 minutes (Fig. 3). NIR fluorescence microscopy showed the tumor specificity of DZ‐CIS uptake (Fig. 3), probably due to induction of OATP carrier expression by DZ‐CIS.19 Thus, DZ‐CIS showed preferential accumulation in tumors compared with all other normal organs (P < .0001) (Fig. 3). These data revealed an unexpected fact: the chemical conjugation of DZ and CIS created a new cytocidal antitumor agent with pharmacological features unseen in either the precursor DZ targeting ligand or CIS. How DZ‐CIS kills Burkitt lymphoma cells and tumors remains undetermined. We propose that DZ‐CIS transported into cancer cells by nonspecific OATP membrane carriers kills cancer cells by interrupting mitochondrial membrane integrity and lysosomal functions to trigger apoptosis. We obtained evidence that DZ‐CIS inhibits mitochondrial functions such as oxidative phosphorylation and mitophagy, and also perturbs lysosomal functions by accumulating unfolded proteins and disturbing autophagy to trigger apoptotic cell death.50 This could explain why the pan‐caspase inhibitor Z‐VAD had limited antagonistic effects on DZ‐CIS‐mediated lymphoma cell death (Fig. 4) and why caspase activation occurred after (~24 hours) rather than before the marked loss of mitochondrial membrane potential (~6 hours) resulting in cytochrome c release (Fig. 2D).
Alternatively, DZ‐CIS disturbs intracellular signaling and alters gene expression, leading to Burkitt lymphoma cell death. DZ‐CIS treatment caused differential protein expression relative to untreated cells or CIS‐treated groups (Fig. 4). DZ‐CIS treatment, for instance, resulted in cleavage of the Max protein, a partner protein for c‐myc function, and its downstream target ROCK1 protein, an important promoter of apoptosis. DZ‐CIS also decreased expression of c‐myc and survivin and inhibited phosphorylation of c‐Jun, STAT3 and Src proteins, which may suppress the antiapoptotic properties of these regulatory proteins. DZ‐CIS may kill lymphoma cells by interfering with signaling transduction. However, the exact mechanism of DZ‐CIS–mediated lymphoma cell killing appears complex, because treatment induced a counterintuitive upregulation of Akt phosphorylation, commonly considered as a prosurvival signaling event.
Because DZ‐CIS as a new antitumor drug showed unique pharmacologic properties quite different from those of CIS, it is not surprising that DZ‐CIS did not cause kidney and liver toxicity in treated mice (Fig. 5). DZ‐CIS also inhibited the expression of cell proliferation (Ki‐67) and cell survival marker (c‐myc and survivin) expression in Burkitt Namalwa tumor xenografts. The absence of side effects will facilitate future translational trials of DZ‐CIS on human Burkitt lymphoma in a clinical setting.
Funding Support
This work was supported by the Cedars‐Sinai Endowed Cancer Research Chair (to Leland W. K. Chung) and a Phileoever Foundation grant (to Yi Zhang, Gina Chia‐Yi Chu, and Leland W. K. Chung).
Conflict of Interest Disclosures
DZ‐CIS is licensed to DaZen Theranostics, Inc., where Leland W. K. Chung is currently serving as the Chairman of the Board and Chief Scientist for the development of one of the licensed lead compounds from laboratory to the clinic and for which Stefan Mrdenovic, Yi Zhang, Ruoxiang Wang, and Gina Chia‐Yi Chu are shareholders. Leland W. K. Chung also has a patent pending (US 2016/0310604 A1).
Author Contributions
Stefan Mrdenovic: Experimental data collection, animal model analysis, and writing (original draft). Yi Zhang: Chemical synthesis and purification. Ruoxiang Wang: Quality control, data validation, and writing (review and editing). Lijuan Yin: Experimental data collection. Gina Chia‐Yi Chu: Animal model analysis and data collection and validation. Liyuan Yin: Experimental data collection. Michael Lewis: Data collection and validation. Marija Heffer: Supervising and data interpretation. Haiyen E. Zhau: Data validation and interpretation. Leland W. K. Chung: Conceptualization, resources, project administration, supervision, and writing (review and editing).
We thank H Phillip Koeffler for insightful assistance.
References
- 1. Molyneux EM, Rochford R, Griffin B, et al. Burkitt’s lymphoma. Lancet. 2012;379:1234‐1244. [DOI] [PubMed] [Google Scholar]
- 2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumors of haematopoietic and lymphoid tissues, Revised Fourth Edition. Geneva, Switzerland: World Health Organization; 2017. [Google Scholar]
- 3. Brown JR, Freedman AS, Aster JC. Pathobiology of Burkitt lymphoma. UpToDate. https://www.uptodate.com/contents/pathobiology-of-burkitt-lymphoma. Accessed July 1, 2018.
- 4. ar‐Rushdi A, Nishikura K, Erikson J, Watt R, Rovera G, Croce CM. Differential expression of the translocated and the untranslocated c‐myc oncogene in Burkitt lymphoma. Science. 1983;222:390‐393. [DOI] [PubMed] [Google Scholar]
- 5. Yang F, Du J, Zhang H, et al. Serum metabolomics of Burkitt lymphoma mouse models. PLoS One. 2017;12:e0170896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoelzer D, Walewski J, Dohner H, et al. Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood. 2014;124:3870‐3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Short NJ, Jabbour E, Sasaki K, et al. Impact of complete molecular response on survival in patients with Philadelphia chromosome‐positive acute lymphoblastic leukemia. Blood. 2016;128:504‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. National Comprehensive Cancer Network . Recent updates to NCCN clinical practice guidelines in oncology, B‐cell lymphoma. Version 2, 2018. https://www.nccn.org/professionals/physician_gls/recently_updated.aspx. Accessed May 15, 2018.
- 9. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714‐726. [DOI] [PubMed] [Google Scholar]
- 10. Davis C, Naci H, Gurpinar E, Poplavska E, Pinto A, Aggarwal A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009‐13. BMJ. 2017;359:j4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeVita VT Jr, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643‐8653. [DOI] [PubMed] [Google Scholar]
- 12. Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand‐targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14:203‐219. [DOI] [PubMed] [Google Scholar]
- 13. Hashim YM, Spitzer D, Vangveravong S, et al. Targeted pancreatic cancer therapy with the small molecule drug conjugate SW IV‐134. Mol Oncol. 2014;8:956‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reddy JA, Dorton R, Westrick E, et al. Preclinical evaluation of EC145, a folate‐vinca alkaloid conjugate. Cancer Res. 2007;67:4434‐4442. [DOI] [PubMed] [Google Scholar]
- 15. Krall N, Pretto F, Decurtins W, Bernardes GJ, Supuran CT, Neri D. A small‐molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl. 2014;53:4231‐4235. [DOI] [PubMed] [Google Scholar]
- 16. Kumar A, Mastren T, Wang B, Hsieh JT, Hao G, Sun X. Design of a small‐molecule drug conjugate for prostate cancer targeted theranostics. Bioconjug Chem. 2016;27:1681‐1689. [DOI] [PubMed] [Google Scholar]
- 17. Heske CM, Mendoza A, Edessa LD, et al. STA‐8666, a novel HSP90 inhibitor/SN‐38 drug conjugate, causes complete tumor regression in preclinical mouse models of pediatric sarcoma. Oncotarget. 2016;7:65540‐65552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang X, Shi C, Tong R, et al. Near IR heptamethine cyanine dye‐mediated cancer imaging. Clin Cancer Res. 2010;16:2833‐2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu JB, Shao C, Li X, et al. Near‐infrared fluorescence imaging of cancer mediated by tumor hypoxia and HIF1alpha/OATPs signaling axis. Biomaterials. 2014;35:8175‐8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo S, Zhang E, Su Y, Cheng T, Shi C. A review of NIR dyes in cancer targeting and imaging. Biomaterials. 2011;32:7127‐7138. [DOI] [PubMed] [Google Scholar]
- 21. Wu J, Pan D, Chung LW. Near‐infrared fluorescence and nuclear imaging and targeting of prostate cancer. Transl Androl Urol. 2013;2:254‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu JB, Shi C, Chu GC, et al. Near‐infrared fluorescence heptamethine carbocyanine dyes mediate imaging and targeted drug delivery for human brain tumor. Biomaterials. 2015;67:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang X, Shao C, Wang R, et al. Optical imaging of kidney cancer with novel near infrared heptamethine carbocyanine fluorescent dyes. J Urol. 2013;189:702‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuan J, Yi X, Yan F, et al. Nearinfrared fluorescence imaging of prostate cancer using heptamethine carbocyanine dyes. Mol Med Rep. 2015;11:821‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hagenbuch B, Stieger B. The SLCO (former SLC21) superfamily of transporters. Mol Aspects Med. 2013;34:396‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu JB, Lin TP, Gallagher JD, et al. Monoamine oxidase A inhibitor‐near‐infrared dye conjugate reduces prostate tumor growth. J Am Chem Soc. 2015;137:2366‐2374. [DOI] [PubMed] [Google Scholar]
- 27. Shi Y, Liu SA, Kerwood DJ, Goodisman J, Dabrowiak JC. Pt(IV) complexes as prodrugs for cisplatin. J Inorg Biochem. 2012;107:6‐14. [DOI] [PubMed] [Google Scholar]
- 28. Kawano S, Miller CW, Gombart AF, et al. Loss of p73 gene expression in leukemias/lymphomas due to hypermethylation. Blood. 1999;94:1113‐1120. [PubMed] [Google Scholar]
- 29. Sun X, He H, Xie Z, et al. Matched pairs of human prostate stromal cells display differential tropic effects on LNCaP prostate cancer cells. In Vitro Cell Dev Biol Anim. 2010;46:538‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu P, Chu GC, Zhu G, et al. Multiplexed quantum dot labeling of activated c‐Met signaling in castration‐resistant human prostate cancer. PLoS One. 2011;6:e28670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. An J, Zhao N, Zhang C, et al. Heptamethine carbocyanine DZ‐1 dye for near‐infrared fluorescence imaging of hepatocellular carcinoma. Oncotarget. 2017;8:56880‐56892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi C, Wu JB, Pan D. Review on near‐infrared heptamethine cyanine dyes as theranostic agents for tumor imaging, targeting, and photodynamic therapy. J Biomed Opt. 2016;21:50901. [DOI] [PubMed] [Google Scholar]
- 33. Zhang C, Zhao Y, Zhang H, et al. The application of heptamethine cyanine dye DZ‐1 and indocyanine green for imaging and targeting in xenograft models of hepatocellular carcinoma. Int J Mol Sci. 2017;18:E1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gougelet A, Mansuy A, Blay JY, Alberti L, Vermot‐Desroches C. Lymphoma and myeloma cell resistance to cytotoxic agents and ionizing radiations is not affected by exposure to anti‐IL‐6 antibody. PLoS One. 2009;4:e8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Millar AW, Lynch KP. Rethinking clinical trials for cytostatic drugs. Nat Rev Cancer. 2003;3:540‐545. [DOI] [PubMed] [Google Scholar]
- 36. Donnou S, Galand C, Touitou V, Sautes‐Fridman C, Fabry Z, Fisson S. Murine models of B‐cell lymphomas: promising tools for designing cancer therapies. Adv Hematol. 2012;2012:701704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Silva GL, Ediz V, Yaron D, Armitage BA. Experimental and computational investigation of unsymmetrical cyanine dyes: understanding torsionally responsive fluorogenic dyes. J Am Chem Soc. 2007;129:5710‐5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stadler AL, Renikuntla BR, Yaron D, Fang AS, Armitage BA. Substituent effects on the assembly of helical cyanine dye aggregates in the minor groove of a DNA template. Langmuir. 2011;27:1472‐1479. [DOI] [PubMed] [Google Scholar]
- 39. Shi J, Wei L. Rho kinase in the regulation of cell death and survival. Arch Immunol Ther Exp (Warsz). 2007;55:61‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Manohar S, Leung N. Cisplatin nephrotoxicity: a review of the literature. J Nephrol. 2018;31:15‐25. [DOI] [PubMed] [Google Scholar]
- 41. Palipoch S, Punsawad C. Biochemical and histological study of rat liver and kidney injury induced by Cisplatin. J Toxicol Pathol. 2013;26:293‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Basu A, Krishnamurthy S. Cellular responses to Cisplatin‐induced DNA damage. J Nucleic Acids. 2010;2010:201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265‐7279. [DOI] [PubMed] [Google Scholar]
- 46. Stewart DJ, Benjamin RS, Luna M, et al. Human tissue distribution of platinum after cis‐diamminedichloroplatinum. Cancer Chemother Pharmacol. 1982;10:51‐54. [DOI] [PubMed] [Google Scholar]
- 47. Fichtinger‐Schepman AM, Vendrik CP, van Dijk‐Knijnenburg WC, et al. Platinum concentrations and DNA adduct levels in tumors and organs of cisplatin‐treated LOU/M rats inoculated with cisplatin‐sensitive or ‐resistant immunoglobulin M immunocytoma. Cancer Res. 1989;49:2862‐2867. [PubMed] [Google Scholar]
- 48. Pujol JL, Cupissol D, Gestin‐Boyer C, Bres J, Serrou B, Michel FB. Tumor‐tissue and plasma concentrations of platinum during chemotherapy of non‐small‐cell lung cancer patients. Cancer Chemother Pharmacol. 1990;27:72‐75. [DOI] [PubMed] [Google Scholar]
- 49. Martens‐de Kemp SR, Dalm SU, Wijnolts FM, et al. DNA‐bound platinum is the major determinant of cisplatin sensitivity in head and neck squamous carcinoma cells. PLoS One. 2013;8:e61555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Piao S, Amaravadi RK. Targeting the lysosome in cancer. Ann N Y Acad Sci. 2016;1371:45‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]