

Abstract
BACE1 is the rate-limiting enzyme that cleaves amyloid precursor protein (APP) to produce the amyloid β peptides that accumulate in Alzheimer's disease (AD). BACE1, which is elevated in AD patients and APP transgenic mice, also cleaves the β2-subunit of voltage-gated sodium channels (Navβ2). Although increased BACE1 levels are associated with Navβ2 cleavage in AD patients, whether Navβ2 cleavage occurs in APP mice had not yet been examined. Such a finding would be of interest because of its potential impact on neuronal activity: previous studies demonstrated that BACE1-overexpressing mice exhibit excessive cleavage of Navβ2 and reduced sodium current density, but the phenotype associated with loss of function mutations in either Navβ-subunits or pore-forming α-subunits is epilepsy. Because mounting evidence suggests that epileptiform activity may play an important role in the development of AD-related cognitive deficits, we examined whether enhanced cleavage of Navβ2 occurs in APP transgenic mice, and whether it is associated with aberrant neuronal activity and cognitive deficits. We found increased levels of BACE1 expression and Navβ2 cleavage fragments in cortical lysates from APP transgenic mice, as well as associated alterations in Nav1.1α expression and localization. Both pyramidal neurons and inhibitory interneurons exhibited evidence of increased Navβ2 cleavage. Moreover, the magnitude of alterations in sodium channel subunits was associated with aberrant EEG activity and impairments in the Morris water maze. Together, these results suggest that altered processing of voltage-gated sodium channels may contribute to aberrant neuronal activity and cognitive deficits in AD.
Introduction
Alzheimer's disease (AD) is characterized by progressive decline in memory and cognition (Blennow et al., 2006). Although the amyloid precursor protein (APP) and amyloid β (Aβ) peptides cleaved from it play a major role in AD pathophysiology (Bertram et al., 2010; Mucke and Selkoe, 2012), the precise mechanisms leading to cognitive deficits are poorly understood.
AD is associated with a 5- to 10-fold increase in seizure incidence (Amatniek et al., 2006; Lozsadi and Larner, 2006), and transgenic mouse models of AD exhibit brain-wide aberrant neuronal and epileptiform activity (Hsiao et al., 1995; LaFerla et al., 1995; Moechars et al., 1999; Lalonde et al., 2005; Palop et al., 2007; Minkeviciene et al., 2009; Vogt et al., 2009; Harris et al., 2010; Roberson et al., 2011; Sanchez et al., 2012). Aberrant activity induces hippocampal dysfunction and memory deficits; modulating aberrant activity and seizure susceptibility modulate cognitive deficits in APP mice (Palop et al., 2007; Roberson et al., 2007, 2011; Sanchez et al., 2012; Verret et al., 2012) and patients with amnestic mild cognitive impairment, a condition that confers risk for AD (Bakker et al., 2012). Although seizures were previously thought to be secondary to disease progression, aberrant activity and/or seizures may directly contribute to cognitive deficits early in disease progression.
An observation of relevance is that BACE1, the rate-limiting enzyme in Aβ production, also cleaves the β2-subunit of voltage-gated sodium channels (Navβ2) in cortical regions (Kim et al., 2007). Similar to APP, Navβ2 is a single transmembrane domain protein that is cleaved by BACE1 to produce a C-terminal fragment (CTF) subsequently cleaved by γ-secretase to release an intracellular domain (ICD) (Wong et al., 2005; Kim et al., 2007). Navβ2-ICD translocates to the nucleus and triggers expression of the pore-forming 1.1α subunit (Nav1.1α). BACE1 overexpression in transgenic mice or cells results in excessive cleavage of Navβ2 and surplus expression of Nav1.1α that is intracellularly retained, leading to reduced surface levels (Kim et al., 2007). Such alterations decrease action potential propagation and neuronal activity (Chen et al., 2002; Lopez-Santiago et al., 2006; Kim et al., 2007). The in vivo phenotype associated with loss-of-function mutations in Navβ-subunits or α-subunits is epilepsy (Catterall, 2010). Disinhibition from loss of GABAergic activity could contribute, but loss of neuronal activity in other populations may also cause imbalances between excitation and inhibition.
BACE1 is elevated in AD patients and APP mice (Fukumoto et al., 2002; Holsinger et al., 2002; Sun et al., 2002; Yang et al., 2003; Li et al., 2004; Tesco et al., 2007; Zhao et al., 2007; Hebert et al., 2008), and increased BACE1 levels/activity are associated with Navβ2 cleavage in AD patients (Kim et al., 2007). However, whether Navβ2 cleavage occurs in APP mice has not been examined. Such a finding would be of interest because of implications for neuronal activity. Moreover, because APP mice have no experimental manipulations of BACE1 levels/activity, such a finding would demonstrate that increased levels of Aβ can drive increases in BACE1 levels that are sufficient to exert physiological consequences.
Materials and Methods
Transgenic mice.
Tg2576 mice express human APPSwe (Hsiao et al., 1996). PSAPP mice express human APPSwe and human presenilinM146L (Holcomb et al., 1998). Mice were singly housed with ad libitum access to food and water. For brain harvesting, mice were flush-perfused transcardially with PBS. Hemibrains were frozen on dry ice for biochemistry or postfixed in 4% paraformaldehyde for immunohistochemistry. All procedures were conducted in accordance with the Wyeth Research Institutional Animal Care and Use Committee and the Thomas Jefferson University Institutional Animal Care and Use Committee.
Western blot analysis.
The cortex from one hemibrain of each mouse was isolated and homogenized with a Polytron tissue homogenizer in ice-cold RIPA buffer containing (in mm): 50 Tris-HCl, pH 7.6, 150 NaCl, 2 EDTA, 0.1% SDS, 0.01% NP-40, Protease Inhibitor Cocktail (Roche). Lysates were centrifuged at 5000 × g for 10 min. Equal amounts of protein were resolved by SDS-PAGE on 4–12% gels and transferred to nitrocellulose, and probed with antibodies against Navβ2 (Alomone), Nav1.1α (Alomone), BACE1 (Cell Signaling Technology), or actin (Millipore). For testing of anti-Navβ2 specificity, antibody was incubated with control peptide antigen (Alomone) at ratio of 1:5 (antibody/peptide) or PBS for 1 h at room temperature, before incubation with the blot. IR-dye-conjugated secondary antibodies were used for detection and quantification using a LI-COR Odyssey infrared imaging system.
Immunohistochemistry.
Brain sections (30 μm) were stained with avidin-biotin/immunoperoxidase using rabbit-anti-calbindin (Swant), rabbit-anti-NPY (ImmunoStar), and rabbit-anti-Navβ2 (Alomone), and signal detected using biotinylated goat-anti-rabbit antibody (Vector Laboratories). Diaminobenzidine was used as a chromagen. For double-labeling experiments, brain sections were stained with rabbit-anti-Navβ2 and mouse anti-GAD67 (Millipore), and signal was detected using FITC-conjugated goat-anti-rabbit and rhodamine-conjugated goat-anti-mouse secondary antibodies (Jackson ImmunoResearch). Immunofluorescence was visualized with confocal microscopy (Olympus FluoView 1000). Nontransgenic (NTG) and APP mice were processed simultaneously, and stained sections were analyzed as described previously (Chin et al., 2005) using AxioVision and NIH ImageJ, or MetaMorph, to acquire and quantify images.
Cell surface biotinylation assay.
Mice were cervically dislocated and brains rapidly placed into ice-cold Kreb's solution containing (in mm): 125 NaCl, 4 KCl, 1.25 KH2PO4, 10 glucose, 1.5 MgSO4, 26 NaHCO3, 1.5 CaCl2. A Vibratome was used to cut 350 μm brain slices. Slices were equilibrated in Kreb's solution bubbled with 95% oxygen and 5% carbon dioxide in a 30°C water bath before biotinylation in 1 mg/ml biotin solution (Pierce) at 4°C. Cortical/hippocampal regions were microdissected and homogenized as above, and equal amounts of protein were incubated with NeutrAvidin-agarose beads (Pierce) to pull down biotinylated proteins.
Morris water maze.
Male Tg2576 and NTG mice (5 months old, 9–10 per genotype) were tested in a white circular pool (1.5 m diameter) filled with 22°C water made opaque with nontoxic white paint. The platform (15 cm diameter) used for hidden platform training sessions was submerged 1 cm below the water surface. The platform (30 cm diameter) used during cued platform sessions was at water surface level and had a flowerpot in the center. During cued platform training, a curtain encircled the pool preventing visual access to extra-mazes cues. Performance was video-tracked and analyzed using EthoVision (Noldus Information Technology).
Hidden platform training involved 8 trials/d over 7 d. Sixty-second trials were separated by a 10 min intertrial interval. During acquisition trials, platform location remained constant and mice entered the pool from randomized locations. Latency and distance traveled to the platform were analyzed. After the last training trial, the platform was removed and mice were given a 60 s probe trial test. Mice were then tested in cued platform sessions. Latency to reach the platform was measured for four trials, each starting from different locations, to assess visual acuity and motor performance. The experimenter was blinded to genotypes during testing.
EEG surgery and recordings.
Mice (5–7 months) were implanted with an indwelling electrode (Plastics One) consisting of three polyimide-coated wires with bare tips, implanted at a depth of 1 mm from the dorsal surface of the brain over the hippocampus (2.5 mm posterior, 2.0 mm lateral from bregma) and in frontal cortices (1 and 2 mm rostral from hippocampal wire). This implant provided a differential signal recorded at 1 kHz, amplified 1000×, and bandpass filtered from 0.1 to 500 Hz with a 60 Hz notch filter in line, using a CED Micro1401 data acquisition system and Spike2 (Cambridge Electronic Design) software. EEG signals were interpolated to 128 Hz and bandpass filtered from 1 to 64 Hz. Power spectral densities (PSDs) were calculated with fast Fourier transform to convert the waveform data into a power spectrum with 0.5 Hz resolution. For each frequency bin (0.5 Hz), the value of power in that frequency was divided by the sum of all power in all frequencies up to 64 Hz. To calculate the amount of time spent at >6 Hz (time in high frequency), PSDs were generated every 30 s for each recording. The signal processing ensured integer values representing the dominant frequency (DF) in hertz for each 30 s epoch. Average DF was calculated for each mouse from each DF in each 30 s epoch (3600/30 s = 120 epochs) in its recording. The time in high frequency was calculated by summing the number of 30 s epochs that had a DF ≥6 Hz and dividing it by the total number of 30 s epochs (total time of the recording) for each mouse.
Results
Alterations in BACE1 expression, cortical Navβ2 cleavage, and Nav1.1α expression in APP mice
Previous studies demonstrated that BACE1 overexpression results in increased cleavage of Navβ2, and altered expression and surface localization of Nav1.1α as illustrated in Figure 1A (Kim et al., 2007). Because BACE1 levels are increased in AD patients and other lines of APP mice (Fukumoto et al., 2002; Holsinger et al., 2002; Sun et al., 2002; Yang et al., 2003; Li et al., 2004; Tesco et al., 2007; Zhao et al., 2007; Hebert et al., 2008), we examined whether APP mice exhibit similar alterations in voltage-gated sodium channels. Using Western blot analysis of cortical lysates, we confirmed that the 5- to 6-month-old, pre-plaque APP mice used in this study also exhibit increased BACE1 expression (Fig. 1B,D). We also found increased levels of Navβ2 CTFs produced by BACE1 cleavage, as well as increased expression of Nav1.1α (Fig. 1B,E,F). Incubation of the anti-Navβ2 antibody with control peptide antigen before exposure to the membrane demonstrated that the bands ∼45–50 kDa and 13 kDa are specific Navβ2 bands (Fig. 1C) similar to those observed in previous studies (Messner and Catterall, 1985; Kim et al., 2007; Gersbacher et al., 2010). Surface biotinylation assays of hippocampal slices demonstrated that the increased expression of Nav1.1α was retained intracellularly, and was not detected on neuronal cell surfaces (Fig. 1G,H). Thus, the aberrantly expressed Nav1.1α subunits likely remain nonfunctional, as previously described (Kim et al., 2007). These alterations in sodium channel levels appear to have functional consequences, because we found an inverse relationship between surface (functional) expression of Nav1.1α and performance in the hidden platform portion of the Morris water maze, a hippocampal-dependent memory task (Fig. 1I). We did not find sodium channel alterations in hippocampal lysates from APP mice (data not shown), suggesting that enhanced cortical Navβ2 cleavage influences hippocampal function. Total levels of Nav1.1α and Navβ2 CTFs also appeared to be increased in cortical lysates from brains of AD patients relative to controls (Fig. 1J), similar to that previously demonstrated (Kim et al., 2007).
Figure 1.

Voltage-gated sodium channel alterations in APP mice and AD patients. A, Cleavage of Navβ2 by BACE1 and γ-secretase and alterations in Nav1.1α. Modified from Kim et al. (2007). B, Increased BACE1 expression and Navβ2-CTFs in cortical lysates from 5- to 6-month-old APP compared with NTG mice (middle and bottom). Navβ2-CTFs are further cleaved by γ-secretase to produce an ICD that translocates to the nucleus to increase Nav1.1α expression, which is also increased in APP mice (top). C, Incubation of the anti-Navβ2 antibody with blocking peptide eliminated the bands around 50 and 13 kDa, indicating that these bands are Navβ2-specific bands. D–F, Quantification of blots in B, n = 8–10/genotype. G, Reduced surface levels of Nav1.1α in APP mice. Cortical slices were biotinylated and immunoprecipated for cell-surface proteins. APP mice had less surface Nav1.1α despite increased total levels, quantified in H, n = 6/genotype. I, Relationship between surface levels of Nav1.1α and performance in the hidden platform portion of the water maze, a hippocampal-dependent memory task. J, Cortical lysates from AD patients also exhibit increased Navβ2-CTFs and total Nav1.1α levels, similar to previously published (Kim et al., 2007). *p < 0.05, **p < 0.01.
Nuclear accumulation of Navβ2 cleavage fragments in GABAergic and non-GABAergic neurons
To identify neuronal populations susceptible to Navβ2 cleavage, we looked for brain regions with enhanced nuclear localization of Navβ2-ICD fragments produced after cleavage by BACE1 and γ-secretase. The antibody used in this study recognizes a 19 aa sequence at the most C-terminal portion of Navβ2, a region contained in the ICD fragment that translocates to the nucleus (Wong et al., 2005; Kim et al., 2007). We found an increase in Navβ2-positive nuclei in the cortex of APP mice (Fig. 2A,B). The increase was not specific to a certain region of cortex, although it was qualitatively more obvious in somatosensory and entorhinal cortical areas. This result suggests that cortical neurons are subject to increased Navβ2 cleavage. We also found increased numbers of Navβ2-positive nuclei in the cortex of transgenic mice that express both mutant human APP and mutant human presenilin (PSAPP mice), another well characterized transgenic mouse model of AD, demonstrating that these findings are not unique to only one transgenic model of AD (Fig. 2C). To evaluate whether the neurons that exhibited increased nuclear levels of Navβ2 are GABAergic and/or glutamatergic, we performed double-labeling for Navβ2 and GAD67 (Fig. 2D,E), a marker of GABAergic interneurons that is expressed by nearly 100% of interneurons in the neocortex (Rudy et al., 2011). Confocal imaging indicated that both GABAergic and non-GABAergic neurons exhibited nuclear accumulation of Navβ2.
Figure 2.
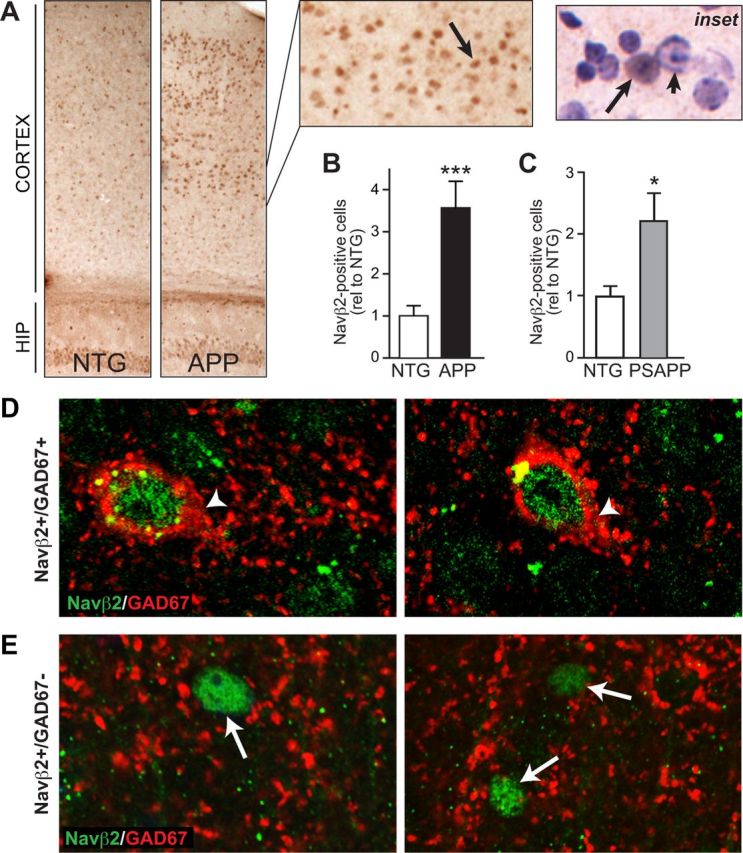
Increased numbers of Navβ2-positive nuclei in cortex of APP mice. Navβ2-positive nuclei were assessed in brain sections from APP mice after immunohistochemical staining with an antibody that recognizes Navβ2-ICD. A, APP mice exhibit increased numbers of Navβ2-positive nuclei in cortical regions. Arrow points to a Navβ2-positive nucleus. Inset, Hematoxylin counterstain demonstrates nuclear Navβ2 localization (arrow). Arrowhead, Nucleus with no Navβ2. B, Quantification of nuclear Navβ2 staining in APP and NTG mice, n = 7–9/genotype. C, Quantification of Navβ2 staining in brain sections from PSAPP mice reveals a significant increase in Navβ2-positive nuclei also in this mouse model, n = 5–7/genotype. D, E, Double-labeling of Navβ2 and GAD67 in brain sections from APP mice. Confocal images of Navβ2 and GAD67 immunostaining demonstrate that some Navβ2-positive nuclei are found in cells that also express GAD67 (D, arrowheads), but others do not express GAD67 (E, arrows). *p < 0.05, ***p < 0.001.
Aberrant neuronal activity is associated with sodium channel alterations in APP mice
Epileptiform activity and/or seizures have been previously reported in some lines of APP mice with high levels of Aβ; however, the data for Tg2576 mice are variable (Hsiao et al., 1995; LaFerla et al., 1995; Moechars et al., 1999; Lalonde et al., 2005; Palop et al., 2007; Oberheim et al., 2008; Minkeviciene et al., 2009; Roberson et al., 2011; Sanchez et al., 2012; Verret et al., 2012). Therefore, to determine whether the sodium channel alterations and memory deficits we observed might be related to aberrant neuronal activity, we performed video-EEG recordings to verify that these mice exhibit aberrant neuronal activity. We found that 5- to 7-month-old APP mice exhibited spike-wave discharges and abnormal EEG patterns (Fig. 3). APP mice exhibited longer durations of higher frequency brain activity, suggesting increased synchrony (Fig. 3A–C), although they did not exhibit obvious spikes or seizures. We confirmed that APP mice with epileptiform activity exhibited increased Navβ2 cleavage and increased total levels of Nav1.1α (Fig. 3D,E), although there was no direct one-to-one correlation between time spent in high-frequency and Navβ2 CTFs (p = 0.27, n = 7) or Nav1.1α (p = 0.78, n = 7) for individual APP mice (data not shown). The lack of one-to-one correlation between Nav1.1α/Navβ2 and time spent in high-frequency may reflect the fact that lysate from the entire cortex was used to measure Nav1.1α/Navβ2, rather than specific regions that might contribute more directly to epileptiform activity. APP mice exhibited decreased calbindin expression in the dentate gyrus and ectopic NPY expression in mossy fibers of the hippocampus, both of which are well documented hallmarks of chronic excitotoxicity that serve neuroprotective roles but that impair neuronal function (Molinari et al., 1996; Vezzani et al., 1999; Nägerl et al., 2000; Palop et al., 2007) (Fig. 3F,G). Calbindin expression correlated inversely, and NPY expression correlated positively, with the EEG measure of time spent in high-frequency (Fig. 3H,I). Decreased calbindin expression in the dentate gyrus is also observed in AD patients, and the magnitude of the decrease corresponds with the degree of dementia (Palop et al., 2003). In APP mice, decreases in calbindin in the dentate gyrus also correlate tightly with hippocampal-dependent learning/memory deficits (Palop et al., 2003).
Figure 3.

Aberrant neuronal activity in APP mice. A, EEG traces demonstrate increased amplitude and frequency of brain activity in two representative NTG and two APP mice at 5–7 months of age. B, Radar plots show time (2 h) as a continuous parameter, and frequency from 0 to 10 Hz in concentric circles. C–E, APP mice exhibit longer durations of higher frequency activity than NTG mice (C), which is associated with increased total Nav1.1α levels (D) and increased Navβ2 cleavage (E) (n = 7–9/genotype). F, G, APP mice exhibit decreased calbindin expression in dentate gyrus (F) and ectopic NPY expression in mossy fibers (G). H, I, Calbindin expression in the dentate gyrus correlated inversely (H) and NPY expression in mossy fibers correlated positively (I) with the EEG parameter of time spent in high-frequency. *p < 0.05.
Together, these results suggest that aberrant sodium channel cleavage may contribute to aberrant neuronal activity and cognitive deficits in APP mice. BACE1 inhibitors may provide therapeutic benefit by both reducing Aβ production, and normalizing sodium channel function and aberrant neuronal activity.
Discussion
A role for network dysfunction in AD-related cognitive deficits was recently highlighted in a number of studies, underscoring the need to identify cellular mechanisms that contribute to altered neuronal activity (Palop et al., 2007; Minkeviciene et al., 2009; Roberson et al., 2011; Bakker et al., 2012; Sanchez et al., 2012; Verret et al., 2012). Here we report increased Navβ2 processing and reduction of Nav1.1α surface expression in cortex of APP mice. Intracellular retention of Nav1.1α may result from impairment of trafficking due to intracellular accumulation of Nav1.1α subunits; from elevated BACE1 activity in APP mice, which impairs trafficking; or from a relative loss of Navβ2, which is necessary for surface expression of Nav1.1α (Kovacs et al., 2010). These alterations decrease sodium channel density and action potential propagation in other systems (Chen et al., 2002; Lopez-Santiago et al., 2006; Kim et al., 2007), suggesting that activity in cortical neurons may be impaired in APP mice. Because Navβ2 is expressed in inhibitory and excitatory neurons (Lai and Jan, 2006), the network consequence of increased Navβ2 cleavage could be very different depending on the cell type in which it occurs. Our studies suggest that both glutamatergic and GABAergic neurons are susceptible to enhanced Navβ2 processing. Moreover, although normally Nav1.1α is more highly, but not exclusively, expressed in inhibitory interneurons (Gong et al., 1999; Ogiwara et al., 2007), its expression could also be altered in excitatory neurons after increased processing of Navβ2.
A recent study in another line of APP mice demonstrated decreased levels of Nav1.1α only in parvalbumin-positive interneurons, which led to impaired interneuron function and aberrant neuronal activity that could be normalized by overexpressing Nav1.1α in interneurons (Verret et al., 2012). The mechanisms by which Nav1.1α alterations arise in that model remain to be defined. Although our studies demonstrate increased rather than decreased total levels of Nav1.1α, we found that surface Nav1.1α levels are reduced (Fig. 1G,H). Thus, both studies demonstrate decreased levels of functional Nav1.1α in cortex of APP mice. These findings indicate that sodium channels can be regulated by more than one mechanism, and present an important target by which neuronal activity might be modulated in APP mice.
Coordinated cortical activity and oscillations strongly influence function of the hippocampus, which receives information about spatial contexts from a number of cortical areas including entorhinal, perirhinal, retrosplenial, posterior parietal, and prefrontal cortex (Buzsaki, 2002; Gordon, 2011; Vann and Albasser, 2011). Encoding of spatial information depends on the integration of information from these dynamically interconnected regions, and is modulated by attention (Muzzio et al., 2009; Derdikman and Moser, 2010). Indeed, we found that alterations in cortical Nav1.1α levels were associated with aberrant EEG activity, and correlated inversely with performance in the water maze (Figs. 1I, 3).
The APP mice used here have no experimental manipulations of BACE1 levels/activity. Thus, overexpression of APP/Aβ is sufficient to initiate a pathway leading to sodium channel cleavage, possibly through regulation of BACE1 levels (Bourne et al., 2007; Shimmyo et al., 2008). Mice that overexpress BACE1 exhibit not only sodium channel alterations but also impairments in learning and memory (Rockenstein et al., 2005; Kim et al., 2007). However, seizures and epileptiform activity have not yet been described in BACE1-transgenic mice. Ablation of BACE1 leads to opposite changes in sodium channel levels as well as epileptiform activity and cognitive deficits, indicating that a balance of BACE1 activity must be maintained to preserve normal neuronal activity and network function (Ohno et al., 2004; Kobayashi et al., 2008; Hitt et al., 2010; Hu et al., 2010; Kim et al., 2011).
Recent studies have suggested several cellular mechanisms for aberrant neuronal activity in AD and related mouse models, including depolarized resting membrane potential and increased excitability of cortical pyramidal neurons (Minkeviciene et al., 2009), impaired inhibition (Busche et al., 2008), and decreased activity of parvalbumin-positive interneurons due to loss of voltage-gated sodium channel subunits (Verret et al., 2012). Together, these and other mechanistic studies are beginning to identify the range of cellular mechanisms by which neuronal activity and network function may become dysregulated in AD. Further studies are necessary to understand how and when these cellular alterations occur during disease progression and how they fit together in the manifestation of aberrant neuronal activity in AD and related models.
Footnotes
We thank A. Silverio for technical assistance and Dr. E. Masliah for human brain tissue from the Alzheimer's Disease Research Center at the University of California San Diego. The JC lab is grateful for the support of the Alzheimer's Association and the Margaret Q. Landenberger Research Foundation.
References
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Bourne KZ, Ferrari DC, Lange-Dohna C, Rossner S, Wood TG, Perez-Polo JR. Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J Neurosci Res. 2007;85:1194–1204. doi: 10.1002/jnr.21252. [DOI] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/S0896-6273(02)00586-X. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Bharucha V, Chen Y, Westenbroek RE, Brown A, Malhotra JD, Jones D, Avery C, Gillespie PJ, 3rd, Kazen-Gillespie KA, Kazarinova-Noyes K, Shrager P, Saunders TL, Macdonald RL, Ransom BR, Scheuer T, Catterall WA, Isom LL. Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci U S A. 2002;99:17072–17077. doi: 10.1073/pnas.212638099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdikman D, Moser EI. A manifold of spatial maps in the brain. Trends Cogn Sci. 2010;14:561–569. doi: 10.1016/j.tics.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Gersbacher MT, Kim DY, Bhattacharyya R, Kovacs DM. Identification of BACE1 cleavage sites in human voltage-gated sodium channel beta 2 subunit. Mol Neurodegener. 2010;5:61. doi: 10.1186/1750-1326-5-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Rhodes KJ, Bekele-Arcuri Z, Trimmer JS. Type I and type II Na(+) channel alpha-subunit polypeptides exhibit distinct spatial and temporal patterning, and association with auxiliary subunits in rat brain. J Comp Neurol. 1999;412:342–352. doi: 10.1002/(SICI)1096-9861(19990920)412:2<342::AID-CNE11>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Gordon JA. Oscillations and hippocampal-prefrontal synchrony. Curr Opin Neurobiol. 2011;21:486–491. doi: 10.1016/j.conb.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu GQ, Palop JJ, Masliah E, Mucke L. Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010;68:428–441. doi: 10.1016/j.neuron.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitt BD, Jaramillo TC, Chetkovich DM, Vassar R. BACE1−/− mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol Neurodegener. 2010;5:31. doi: 10.1186/1750-1326-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor β-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, Wilson CG, Yan R. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci. 2010;30:8819–8829. doi: 10.1523/JNEUROSCI.1334-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Gersbacher MT, Inquimbert P, Kovacs DM. Reduced sodium channel Nav1.1 levels in BACE1-null mice. J Biol Chem. 2011;286:8106–8116. doi: 10.1074/jbc.M110.134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ, Kovacs DM. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol. 2007;9:755–764. doi: 10.1038/ncb1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi D, Zeller M, Cole T, Buttini M, McConlogue L, Sinha S, Freedman S, Morris RG, Chen KS. BACE1 gene deletion: impact on behavioral function in a model of Alzheimer's disease. Neurobiol Aging. 2008;29:861–873. doi: 10.1016/j.neurobiolaging.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Kovacs DM, Gersbacher MT, Kim DY. Alzheimer's secretases regulate voltage-gated sodium channels. Neurosci Lett. 2010;486:68–72. doi: 10.1016/j.neulet.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer's Abeta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Strazielle C. Neurobehavioral characterization of APP23 transgenic mice with the SHIRPA primary screen. Behav Brain Res. 2005;157:91–98. doi: 10.1016/j.bbr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, Wong P, Price D, Shen Y. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci U S A. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Pertin M, Morisod X, Chen C, Hong S, Wiley J, Decosterd I, Isom LL. Sodium channel beta2 subunits regulate tetrodotoxin-sensitive sodium channels in small dorsal root ganglion neurons and modulate the response to pain. J Neurosci. 2006;26:7984–7994. doi: 10.1523/JNEUROSCI.2211-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozsadi DA, Larner AJ. Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer's disease. Dement Geriatr Cogn Disord. 2006;22:121–124. doi: 10.1159/000093664. [DOI] [PubMed] [Google Scholar]
- Messner DJ, Catterall WA. The sodium channel from rat brain: separation and characterization of subunits. J Biol Chem. 1985;260:10597–10604. [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moechars D, Lorent K, Van Leuven F. Premature death in transgenic mice that overexpress a mutant amyloid precursor protein is preceded by severe neurodegeneration and apoptosis. Neuroscience. 1999;91:819–830. doi: 10.1016/S0306-4522(98)00599-5. [DOI] [PubMed] [Google Scholar]
- Molinari S, Battini R, Ferrari S, Pozzi L, Killcross AS, Robbins TW, Jouvenceau A, Billard JM, Dutar P, Lamour Y, Baker WA, Cox H, Emson P. Deficits in memory and hippocampal long-term potentiation in mice with reduced calbindin D28K expression. Proc Natl Acad Sci U S A. 1996;93:8028–8033. doi: 10.1073/pnas.93.15.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzzio IA, Kentros C, Kandel E. What is remembered? Role of attention on the encoding and retrieval of hippocampal representations. J Physiol. 2009;587:2837–2854. doi: 10.1113/jphysiol.2009.172445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Mody I, Jeub M, Lie AA, Elger CE, Beck H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca2+ influx because of a loss of calbindin-D28K in temporal lobe epilepsy. J Neurosci. 2000;20:1831–1836. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M. Loss of astrocytic domain organization in the epileptic brain. J Neurosci. 2008;28:3264–3276. doi: 10.1523/JNEUROSCI.4980-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/S0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mante M, Alford M, Adame A, Crews L, Hashimoto M, Esposito L, Mucke L, Masliah E. High β-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-β levels: implications for the treatment of Alzheimer's disease. J Biol Chem. 2005;280:32957–32967. doi: 10.1074/jbc.M507016200. [DOI] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2011;71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimmyo Y, Kihara T, Akaike A, Niidome T, Sugimoto H. Epigallocatechin-3-gallate and curcumin suppress amyloid beta-induced beta-site APP cleaving enzyme-1 upregulation. Neuroreport. 2008;19:1329–1333. doi: 10.1097/WNR.0b013e32830b8ae1. [DOI] [PubMed] [Google Scholar]
- Sun A, Koelsch G, Tang J, Bing G. Localization of beta-secretase memapsin 2 in the brain of Alzheimer's patients and normal aged controls. Exp Neurol. 2002;175:10–22. doi: 10.1006/exnr.2002.7875. [DOI] [PubMed] [Google Scholar]
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vann SD, Albasser MM. Hippocampus and neocortex: recognition and spatial memory. Curr Opin Neurobiol. 2011;21:440–445. doi: 10.1016/j.conb.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Sperk G, Colmers WF. Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci. 1999;22:25–30. doi: 10.1016/S0166-2236(98)01284-3. [DOI] [PubMed] [Google Scholar]
- Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging. 2011;32:1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, De Strooper B, Saftig P, Nukina β Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem. 2005;280:23009–23017. doi: 10.1074/jbc.M414648200. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]