

Abstract
In the nematode Caenorhabditis elegans, the heat shock response (HSR) is regulated at the organismal level by a network of thermosensory neurons that senses elevated temperatures and activates the HSR in remote tissues. Which neuronal receptors are required for this signaling mechanism and in which neurons they function are largely unanswered questions. Here we used worms that were engineered to exhibit RNA interference hypersensitivity in neurons to screen for neuronal receptors that are required for the activation of the HSR and identified a putative G-protein coupled receptor (GPCR) as a novel key component of this mechanism. This gene, which we termed GPCR thermal receptor 1 (gtr-1), is expressed in chemosensory neurons and has no role in heat sensing but is critically required for the induction of genes that encode heat shock proteins in non-neural tissues upon exposure to heat. Surprisingly, the knock-down of gtr-1 by RNA interference protected worms expressing the Alzheimer's-disease-linked aggregative peptide Aβ3–42 from proteotoxicity but had no effect on lifespan. This study provides several novel insights: (1) it shows that chemosensory neurons play important roles in the nematode's HSR-regulating mechanism, (2) it shows that lifespan and heat stress resistance are separable, and (3) it strengthens the emerging notion that the ability to respond to heat comes at the expense of protein homeostasis (proteostasis).
Introduction
Upon exposure to elevated temperature, organisms activate a protective mechanism that enables the restoration of functional homeostasis that is known as the “heat shock response” (HSR; Morimoto, 1998). When heat is sensed, the heat shock factor 1 (HSF-1) trimerizes, enters the nucleus, and activates the expression of its target genes, including genes that encode the “heat shock proteins” (Hsp's; Sarge et al., 1993). An additional key transcription factor that promotes heat stress resistance is DAF-16/FOXO (Lithgow et al., 1995). The target gene network of DAF-16/FOXO is different yet partially overlaps with the genes downstream of HSF-1 (McElwee et al., 2003; Murphy et al., 2003). In the nematode Caenorhabditis elegans, HSF-1 and DAF-16 are also required for protection from other environmental insults, including UV radiation (Murakami and Johnson, 1996), oxidative stress (Honda and Honda, 1999), and exposure to pathogenic bacteria (innate immunity; Singh and Aballay, 2006).
In the worm, the activities of DAF-16 and of HSF-1 are negatively regulated by the insulin/IGF signaling (IIS) pathway. Upon activation, the worm's sole insulin/IGF receptor, DAF-2, initiates a signaling cascade that promotes phosphorylation events that retain DAF-16 (Henderson and Johnson, 2001; Lee et al., 2001) and HSF-1 (Chiang et al., 2012) in the cytosol, preventing them from regulating their target gene networks. Therefore, IIS reduction elevates stress resistance by hyperactivating DAF-16 and HSF-1 (Lithgow et al., 1995; Honda and Honda, 1999). This manipulation also extends lifespan (Kenyon et al., 1993) and protects from proteotoxicity (Hsu et al., 2003; Morley and Morimoto, 2004; Cohen et al., 2006; Teixeira-Castro et al., 2011; Zhang et al., 2011), raising the question of whether these functions are mechanistically linked.
When exposed to a temperature gradient, worms migrate from areas of elevated or decreased temperatures toward their cultivation temperature (for review, see Ma and Shen, 2012). This navigational behavior, which was termed “thermotaxis,” depends on the activity of several neurons that compose the thermosensory circuit: the AFD thermosensory neurons and their postsynaptic partners, the AIY interneurons (Mori, 1999), the AIZ interneurons (Mori and Ohshima, 1995), the AWC odor-sensory neurons (Biron et al., 2008), and the ASI chemosensory neurons (Beverly et al., 2011).
The key roles of the neural thermosensory circuit in heat sensation raised the prospect that this neural network also regulates the HSR. Recently, a functional link between the thermosensory circuit and HSR activation has been demonstrated. Worms that carry mutated gcy-8, a gene that encodes an AFD-specific receptor-type guanylyl cyclase, or mutated ttx-3, a gene that encodes an AIY-specific, LIM homeodomain protein, exhibit mitigated HSR activation when exposed to heat (Prahlad et al., 2008). What neural components are required for the activation of the HSR in remote tissues is largely unknown.
In the present study, we used worms that were engineered to be hypersensitive for RNA interference (RNAi) in neurons to search for neuronal receptors that are required for HSR activation and identified a putative GPCR as a critical component of this mechanism. The knock-down of this gene, gtr-1, largely abolished the induction of DAF-16- and HSF-1-regulated Hsp's and significantly reduced the survival rates of worms that were exposed to heat. Interestingly, the knock-down of gtr-1 had no effect on lifespan but provided partial protection from proteotoxicity.
Materials and Methods
Worm and RNAi strains.
N2 (wild-type, Bristol), daf-2(e1370) mutant worm, TU3335 (unc-119p::YFP + unc-119p::sid-1 + mec-6p::mec-6), CL2006 (unc-54p::human Aβ3–42), PY1322 (gcy-8p::GFP), OH103 (lin-11p::GFP), OH99 (ttx-3p::GFP), and CL2070 (hsp-16.2p::GFP) worms were obtained from the Caenorhabditis Genetics Center. The worms were grown at 20°C. CF512 (fer-15(b26)II; fem-1(hc17)IV) animals are heat-sensitive and sterile and were routinely grown at 15°C. To avoid progeny, CF512 worms were hatched at 20°C and L1 larvae transferred to 25°C for 48 h and back to 20°C until harvested. To reduce gene expression, bacterial strains expressing dsRNA: empty vector (pAD12), daf-2 (pAD48), and daf-16 (pAD43), were used. hsf-1, gtr-1, and gcy-8 dsRNA-expressing bacteria were from a genomic RNAi library (J. Ahringer). octr-1 dsRNA-expressing bacteria were from a cDNA library (M. Vidal). Each RNAi bacteria colony was grown at 37°C in LB with 100 μg/ml ampicillin, and then seeded onto Nematode Growth media (NG)-ampicillin plates supplemented with 100 mm isopropyl β-d-1-thiogalactopyranoside (IPTG; ∼4 mm final concentration).
Creation of worm strains and RNAi constructs.
The following primers were used to amplify the promoter region of gtr-1 (including restriction sites): forward-5′ CAGAAG CTTCCCCACTCTCTACCCAACG 3′; reverse-TGACCCGGGTGAAAATGTGTTCTGAAAAAAAAAACGAATTCGATA. The PCR product was cloned between BamH1 and XmaI restriction sites upstream of tdTomato. The final construct was microinjected into N2 worms. The promoter region of hsp-12.6 was amplified using the following primer set (including restriction sites): forward-CAAGTCGACAATTTGTTTATGTAAATGCGTTTTAGTGTG; reverse-ACGGGATCCATCATCTTGGCAAAAGTTTTTGGG. The PCR product was cloned upstream of tdTomato and the obtained plasmid was injected into N2 worms. Selection was performed using fluorescence microscopy.
RNAi construct toward the gtr-1 3′UTR was constructed using gBlock synthesis (Integrated DNA Technologies):
AGCGAGCTATTTACCAACTTTTCCCCTTTTTTGCTGTGTTTAATTTTATCACTTTTGTAAATCTAAAGATCTCATTTTCATTCAATTCACGGTTATTTTAATAAATATTCTGTTCGGCTAGCTCG. The gBlock construct was digested with NheI and SacI and cloned into pL4440 plasmid.
Heat and innate immunity stress assays.
Synchronous eggs were placed on NG plates seeded with the RNAi bacterial strain (as indicated) and supplemented with 100 mm IPTG (∼4 mm final). The worms were developed and maintained at 20°C. For heat stress assays, 120 day 1 adult animals were transferred onto fresh plates (12 animals per plate) that were spotted with RNAi bacteria, and exposed to 35°C. Survival rates were recorded in 3 h intervals or after 10, 12, 15, or 20 h, as indicated. To evaluate resistance to toxic bacteria (innate immunity), eggs of daf-2(e1370) worms were placed on plates seeded with the indicated RNAi bacteria and grown to day 1 of adulthood. The worms were then transferred onto plates seeded with Pseudomonas aeruginosa and survival rates were scored daily.
Microscopy and signal quantification.
Synchronous worms were washed twice with PBS and snap frozen in liquid nitrogen. Fluorescent images were obtained using a Nikon AZ100 microscope and NIS elements software. Quantitative fluorescence analysis was made using ImageJ software. Neural expression patterns were tested using confocal microscopy. The worms were washed twice with M9, anesthetized using 18 mm sodium azide (#S-2002; Sigma) and placed on an agar pad for visualization. Images were obtained using a Zeiss LSM 710 laser scanning microscope.
Thermotaxis assay.
Synchronized, 1-d-old, daf-2(e1370) mutant worms that were developed on the indicated RNAi bacteria were placed on fresh NG plates and exposed to the temperature gradient. The temperatures of each zone of the plate were monitored using a laser-guided, infrared thermometer. Photographs were taken before exposing the worms to the temperature gradient and 12 min thereafter.
RNA isolation and quantitative real-time PCR.
Total RNA was isolated from synchronized worm populations using QIAzol reagent (catalog #79306; Qiagen) and the RNeasy lipid tissue kit (catalog #74804; Qiagen). cDNA was synthesized using the iScript cDNA synthesis kit (catalog #170–8891; Bio-Rad). Quantitative real-time PCRs (qPCRs) were performed with EvaGreen SuperMix (catalog #172–5204; Bio-Rad). Quantities were normalized to levels of act-1 cDNA. hsp-12.6 primer set: forward-TTCCAGTGATGGCTGACG, reverse: GGCTTCTAGGCCTACTTCG. hsp-70 primer set: forward: GGTTGGGGGATCAACTCG, reverse: CACCAAAGGCTACTGCTTCG; hsf-1 primer set: forward: TTGACGACGACAAGCTTCCAGT, reverse: AAAGCTTGCACCAGAATCATCCC; daf-16 primer set: forward: CTTCAAGCCAATGCCACTACC, reverse: GGAGATGAGTTGGATGTTGATAGC; act-1 primer set: forward: GAGCACGGTATCGTCACCAA, reverse: TGTGATGCCAGATCTTCTCCAT.
SDS-PAGE and Western blot analysis.
Twelve thousand CL2070 worms were treated with RNAi bacteria as indicated and homogenized using a dounce homogenizer. The worm homogenates were centrifuged for 3 min at 850 × g (3000 rpm in a desktop centrifuge) to sediment debris. The postdebris supernatants were collected, protein amounts were measured (catalog #500–0006; Bio-Rad), supplemented with loading buffer (10% glycerol, 125 mm Tris base, 1% SDS), boiled for 10 min, and 10 μg of total protein was loaded into each well. Proteins were separated on polyacrylamide gels, transferred onto PVDF membranes (Pierce), and probed with either GFP antibody (catalog #2956; Cell Signaling Technology) or anti γ-tubulin antibody clone GTU-88 (catalog #T-6557; Sigma). HRP-conjugated secondary antibody, a chemiluminescence system, and a luminescent image analyzer (Las-3000; Fujifilm) were used to detect protein signals.
Lifespan assay.
Synchronized worm eggs were placed on master NG-ampicillin plates seeded with the indicated RNAi bacterial strain and supplemented with 100 mm IPTG (∼4 mm final). The worms were developed to adulthood at 20°C and 120 animals per treatment were transferred onto small NG-ampicillin plates (12 animals per plate). Worms that failed to move their tips when tapped twice with a platinum wire were scored as dead. Survival rates were scored daily.
Egg-laying assay.
To follow the reproductive profiles of daf-2(e1370) animals, synchronized eggs were placed on plates seeded with empty vector (EV), daf-16, or gtr-1 RNAi bacteria. At the L4 larval stage, 15 animals were transferred onto new plates seeded with the same RNAi strain (one worm per plate). The worms were transferred onto new plates in 24 h intervals and the number of progeny in each plate was counted 72 h thereafter.
Dauer formation assay.
Eggs of daf-2(e1370) animals were transferred onto plates seeded with RNAi bacteria as indicated and were incubated at either 20°C or at 25°C. Dauer larvae and adult worms were counted 3 d thereafter.
Paralysis assay.
Synchronous CL2006 worm populations were grown on NG plates containing 100 μg/ml ampicillin (∼4 mm final) spotted with E. coli cultures expressing dsRNA as indicated. On the first day of adulthood, 100 worms were placed on 10 plates (10 animals per plate). The plates were divided randomly to five sets (two plates, 20 worms per set). The worms were tested daily for paralysis by tapping their noses with a platinum wire. Worms that moved their noses but failed to move their bodies were scored as “paralyzed” and were removed from the plates. To avoid scoring old animals as paralyzed, paralysis assays were terminated at day 12 of adulthood.
Statistical analyses.
Statistical significance of the results was performed using the Student's t test using one-tailed distribution and two-sample equal variance. The analyses were done using at least three independent biological repeats of each experiment as indicated. Statistical information of lifespan experiments is presented in Table 1.
Table 1.
Lifespans of CF512 and e1370 worms
| Strain | RNAi treatment | Mean lifespan ± SEM (days) | Number of animals/initial number | Mean lifespan (% of control) | p (against control) |
|---|---|---|---|---|---|
| CF512 | EV | 17.02 ± 0.42 | 114/124 | ||
| gtr-1 | 16.19 ± 0.38 | 118/122 | 95% | 0.128 | |
| daf-16 | 14.41 ± 0.33 | 115/125 | 85% | 1.94E-06 | |
| e1370 | EV | 42.3 ± 1.80 | 64/120 | ||
| gtr-1 | 39.74 ± 1.84 | 86/125 | 94% | 0.321 | |
| daf-16 | 26.24 ± 0.66 | 61/121 | 62% | 2.47E-16 |
Results
gtr-1 is required for the survival of worms at elevated temperatures
The limited penetrance of double-stranded RNA (dsRNA) into neurons of C. elegans (Timmons et al., 2001) has notably restricted the efficiency of feeding RNAi bacteria as a screening technique for the identification of genes, which encode products that are required for neuronal function. Recently, this technical obstacle has been resolved by the creation of a worm strain that expresses the transmembrane protein SID-1, which is crucial for the uptake of dsRNA by the nematode's cells (Winston et al., 2002) under the regulation of the pan-neural unc-119 promoter. This expression not only enables an efficient gene-specific knock-down within neurons, but also reduces the RNAi-mediated gene knock-down in other tissues (Calixto et al., 2010). We used the neuronal-expressing sid-1 worms (strain TU3335, unc-119p::sid-1) to search for receptors that are required for survival in elevated temperature and found that worms that were fed bacteria expressing dsRNA toward gtr-1 (encoded by F25E2.1) exhibited significantly reduced viability over time when exposed to 35°C (Fig. 1A). Four independent experiments indicated that whereas the average survival rates of control worm populations (fed with bacteria that harbor the empty RNAi vector, EV) was ∼42%, only 15% of the gtr-1 RNAi-treated worms were alive after 10 h of exposure to heat (Fig. 1B). This survival rate was comparable to that of worms that were fed bacteria expressing dsRNA toward gcy-8, which was shown to be required for HSR activation (Prahlad et al., 2008).
Figure 1.

Knock-down of gtr-1 sensitizes worms to heat stress. A–B, Worms expressing sid-1 under the regulation of the pan-neuronal unc-119 promoter (strain TU3335) were treated with RNAi toward either gcy-8 or gtr-1 or left untreated (EV), exposed to 35°C, and their survival rates were recorded in 3 h intervals (A). Similarly, the survival rates of RNAi-treated TU3335 worms (as in A) exposed to 35°C for 10 h were recorded (B). gtr-1 RNAi-treated animals exhibited reduced survival rates compared with their untreated counterparts. The survival rates of gtr-1 and gcy-8 RNAi-treated animals were comparable. C–F, gtr-1 RNAi-treated wild-type worms (strain N2) (C,D) and CF512 animals (E,F) exhibited significantly reduced survival rates in 35°C compared with their untreated counterparts. Three independent single-time-point survival assays with each strain (12 and 15 h of exposure to 35°C for N2 and CF512 worms, respectively) showed that daf-16 and gtr-1 RNAi-treated animals exhibited similarly reduced lifespan after exposure to heat compared with untreated animals (D,F). G, Similarly to daf-16, the knock-down of gtr-1 by RNAi abolished the elevated stress resistance of daf-2(e1370) mutant worms that were exposed to heat (35°C) for 20 h. In B, D, F, and G, bars represent mean survival ± SEM, p < 0.01. H, CF512 worms that were treated with RNAi toward the 3′UTR region of gtr-1 and exposed to 35°C exhibited significantly reduced survival rates compared with the control group (EV).
We then tested whether gtr-1 is also required for the survival of wild-type worms (strain N2) in elevated temperature. N2 worms that were treated with either gtr-1 or daf-16 RNAi and exposed to heat as described above exhibited similarly reduced survival rates over time compared with their untreated counterparts (Fig. 1C). These results were confirmed by three independent experiments in which an average of 19% of the gtr-1 RNAi-treated N2 worms survived after 12 h of exposure to heat compared with 38% of the untreated (EV) and 9% of daf-16 RNAi-treated animals (Fig. 1D). Similarly, temperature-sensitive sterile worms (strain CF512) that were treated with either gtr-1 or daf-16 RNAi exhibited reduced heat stress resistance compared with untreated (EV) animals of the same strain over time (Fig. 1E), as well as in three independent experiments in which CF512 worms were treated with RNAi as described above and exposed to heat for 15 h (Fig. 1F). CF512 worms were exposed to 25°C during development to prevent them from having progeny; however, this mild treatment does not activate their HSR (Volovik et al., 2012). These results show that the knock-down of gtr-1 results in reduced survival of three different worm strains upon heat shock.
The elevated heat stress resistance of daf-2 mutants (Lithgow et al., 1995) led us to investigate whether this phenotype is dependent on the activity of gtr-1. We used daf-2(e1370) mutant worms that harbor a weak daf-2 allele. The worms were either fed control bacteria (EV) or treated with RNAi toward either daf-16 or gtr-1 throughout development, exposed to 35°C at day 1 of adulthood, and survival rates were tested after 20 h of exposure to heat. Three independent experiments revealed that whereas ∼85% of the untreated daf-2(e1370) mutant animals were alive, only ∼10% of the gtr-1 and of daf-16 RNAi-treated worms survived after the heat insult (Fig. 1G). This indicates that gtr-1 is also critically required for the increased resistance of daf-2 mutant animals to thermal stress.
To further establish our observations and to verify the specificity of the RNAi-mediated knock-down of gtr-1, we created an RNAi construct that is directed toward the 3′UTR of gtr-1. Using CF512 worms, we found that knocking down gtr-1 by the new RNAi construct led to an identical effect of significantly (p < 0.01) reduced survival in 35°C observed when gtr-1 was knocked down by the library's RNAi strain or by daf-16 RNAi (Fig. 1H).
The observations that gtr-1 RNAi-treated daf-2(e1370) mutant, N2, and CF512 worms show reduced heat resistance were puzzling given that in these animals the penetrance of RNAi to neurons is limited. Two hypotheses can explain these observations: (1) gtr-1 is tightly regulated and the relatively low efficiency of RNAi-facilitated gtr-1 knock-down in neurons is sufficient to sensitize the worms to heat, or (2) that gtr-1 executes its heat resistance functions in a non-neural tissue.
To distinguish between these two possibilities and to analyze the spatial and temporal expression patterns of gtr-1, we created worms that express the fluorescent protein tdTomato (Shaner et al., 2004) under the regulation of the gtr-1 promoter (3 kb upstream of the gtr-1 open reading frame, strain EHC101).
gtr-1 is expressed in chemosensory neurons
To determine in which worm tissues gtr-1 is expressed, we visualized EHC101 worms using fluorescent microscopy and found that the expression of tdTomato was confined to neurons (Fig. 2A). Prominent expression was observed in the head and tail neurons and along the ventral cord. Some of the head neural cell bodies appeared to be located in close proximity to the pharynx (Figs. 2A). The key role of gtr-1 in the worm's resistance to heat and its expression in neurons that are located near the pharynx raised the prospect that among other neuronal cells, gtr-1 is expressed in the AFD thermosensory neurons. To examine this hypothesis, we crossed EHC101 worms with animals that express the green fluorescent protein (GFP) under the regulation of the gcy-8 promoter (strain PY1322) to obtain animals that express GFP in AFD neurons and tdTomato in gtr-1-expressing cells. Confocal microscopy clearly indicated that gtr-1 is not expressed in AFD neurons, because GFP and tdTomato exhibited distinct expression patterns (Fig. 2B). Similarly, we crossed EHC101 with animals that express GFP under the regulation of the ttx-3 promoter (strain OH99) and found that gtr-1 is not expressed in the AIY interneurons (Fig. 2C).
Figure 2.

gtr-1 is expressed in chemosensory neurons. A, Fluorescent visualization of worms that express tdTomato under the regulation of gtr-1 promoter (strain EHC101) revealed that gtr-1 is expressed in neurons of the head ganglia, the ventral cord, and the tail. Scale bar, 50 μm. B–C, gtr-1 is expressed in neither the AFD thermosensory neurons (labeled by GFP driven by the AFD-specific, gcy-8 promoter; B) nor in AIY interneurons (labeled with GFP driven by the ttx-3 promoter; C). D, Visualization of worms that express GFP under the regulation of the chemosensory specific lin-11 promoter (green channel) and tdTomato driven by the gtr-1 promoter (red channel) shows colocalization. In B and D, scale bars, 10 μm.
We then examined whether gtr-1 is expressed in other neurons of the thermosensory circuit (Ma and Shen, 2012). EHC101 worms were crossed with animals that express GFP under the regulation of the lin-11 promoter (strain OH103). lin-11 encodes a LIM homeodomain protein that is expressed in several head and tail neurons and is crucially required for the activity of the thermoregulatory neural network (Hobert et al., 1998). Accordingly, OH103 animals express GFP in the head chemosensory neurons ADF and ADL as well as in the AIZ, AVG, and RIC interneurons (Hobert et al., 1998). Confocal visualization of the obtained worms revealed identical expression patterns of GFP and tdTomato, which is indicative of coexpression of lin-11 and gtr-1 in the same neuronal cells (Fig. 2D).
The coexpression of gtr-1 and lin-11 in chemosensory neurons led us to investigate whether these two genes are also temporally coexpressed. GFP driven by the lin-11 promoter can be first detected in the early comma stage of embryonic development (Hobert et al., 1998). Therefore, we sought to determine whether gtr-1 is also expressed during embryogenesis. Fertile EHC101 worms were placed for 40 min (the “laying period”) on plates seeded with EV bacteria to obtain highly synchronized eggs. The worms were then removed and the eggs were visualized by fluorescent microscopy in 30 min intervals after the laying period. A faint tdTomato signal could be detected in eggs for the first time 90 min after the laying period in a single location. This time point, which corresponds to the 1.5-fold stage, is estimated to be 400 min after fertilization (Fig. 3A, 1.5-fold, arrow; Baugh et al., 2003). At the 3-fold stage, tdTomato signal was observed in two distinct areas of the developing embryo (Fig. 3A, arrows). At late morphogenesis, clear signals were observed in the head and tail and along the developing embryo's body (Fig. 3A, late morphogenesis). These results indicate that gtr-1 is first expressed at the early stage of embryogenesis and is widely expressed in multiple sites during later stages. This temporal insight suggests that although the expression of gtr-1 may lag slightly behind the expression of lin-11, both genes are expressed in similar temporal patterns (Hobert et al., 1998), and also raises the prospect that the products of these genes might be functionally interrelated.
Figure 3.

Temporal analysis of gtr-1 expression. A, Eggs of EHC101 worms were visualized in 30 min intervals by a fluorescent microscope. tdTomato signal was first observed in a single location at the 1.5-fold embryonic stage (arrow). At the 3-fold stage, clear signals were detected in two areas corresponding to the head and tail regions of the developing embryo (arrows), whereas at late morphogenesis, tdTomato signals were seen in the head and tail and along the developing embryo's body. B, tdTomato signals were observed in the head, tail, and ventral cord of L1 larvae and in ADL and AIZ neurons of L2 and L3 larvae.
Clear fluorescent signals were also observed throughout larval development in the head, tail, and ventral cord neurons (Fig. 3B). At the L2 and L3 larval stages, the tdTomato signal, which is indicative of gtr-1 expression, was observed in the ADL neurons and AIZ interneurons.
Knock-down of gtr-1 does not affect thermosensation
Neurons of the thermoregulatory circuit exhibit two major functions upon exposure to elevated temperature: heat sensation and the initiation of signaling that activates the HSR in other tissues (Prahlad et al., 2008). We sought to determine in which of these neural functions gtr-1 plays its roles. First we investigated whether gtr-1 is essential for heat sensation by performing a worm migration assay on a temperature gradient. In this set of experiments, we used daf-2(e1370) mutant worms, a strain that exhibited the most prominent reduction in heat resistance upon RNAi-mediated gtr-1 knock-down (Fig. 1G). The worms were either grown throughout development on EV bacteria or fed with gtr-1 RNAi bacteria and placed on temperature gradient plates (6–38°C). The plates were photographed immediately after placing the worms and 12 min thereafter. Both untreated worms (EV) (Fig. 4A, top) and gtr-1 RNAi-treated animals (Fig. 4A, bottom) rapidly migrated to the central region of the plate where the temperature was similar to their cultivation temperatures (18–20°C; Fig. 4A, rectangles). To quantify the worms' migration behavior, we divided each plate to six equal zones along the temperature gradient and counted the worms that were present in each zone at the beginning of the experiment and at the 12 min time point in four independent experiments. Our results (Fig. 4B) confirmed the significance of the indistinguishable thermotactic behaviors of the worm groups. The migration of gtr-1 RNAi-treated worms away from the hot and cold regions indicates that this gene product is dispensable for heat sensation and suggests that it functions in the signaling mechanism that activates the HSR.
Figure 4.

Knock-down of gtr-1 by RNAi has no effect on heat sensing. A, daf-2(e1370) mutant worms were placed on temperature gradient plates. The plates were photographed right after placing the worms and 12 min thereafter. Both untreated (EV; top) and gtr-1 RNAi-treated (bottom) animals migrated away from regions of high (∼38°C) and low (∼6°C) temperatures to populate the central region of the plates where the temperatures were similar to their cultivation temperature (18–20°C, rectangles). B, Four independent experiments as in A confirmed that untreated and gtr-1 RNAi-treated worms exhibited indistinguishable thermotactic behavior.
gtr-1 is required for the induction of the HSR mechanisms downstream of both HSF-1 and DAF-16
To examine the possibility that gtr-1 is required for the HSR-activating neuronal signaling mechanism, we investigated whether the knock-down of this gene impaired the induction of heat shock proteins in remote tissues upon exposure to high temperature. First, we used worms that express GFP under the regulation of the hsp-16.2 promoter (strain CL2070). hsp-16.2 is a well established HSF-1 target, encoding a small heat shock protein that is predominantly expressed in the worm's intestine (Link et al., 1999). CL2070 worms were left untreated (EV) or treated with RNAi toward either hsf-1 or gtr-1 from hatching to the first day of adulthood. The worms were exposed to heat shock (33°C, 3 h) and GFP was visualized by fluorescent microscopy. Although a clear GFP signal was observed in the intestine of untreated animals, gtr-1 RNAi treatment largely abolished the induction of GFP in the worms (Fig. 5A). Signal quantification (Fig. 5B) indicated that although gtr-1 RNAi exhibited a less prominent effect on the expression of GFP compared with RNAi toward hsf-1, the effect of gtr-1 RNAi was significant (p < 0.01). To further establish and quantify this phenomenon, we treated CL2070 animals with RNAi and exposed them to heat as described above. The worms were homogenized and subjected to Western blot analysis using a GFP antibody. Our results (Fig. 5C) showed that the knock-down of gtr-1 by RNAi (Fig. 5C, lane 3) resulted in a remarkably reduced GFP level compared with the level seen in untreated worms (Fig. 5C, lane 1). The effect of gtr-1 RNAi treatment on the activity of the hsp-16.2 promoter was comparable to that of hsf-1 RNAi (Fig. 5C, lane 2) but was not as prominent. It is plausible that the prominence of hsf-1 RNAi compared with gtr-1 RNAi treatment in this set of experiments emanates from the lower efficiency of RNAi-mediated gene knock-down in neurons (Timmons et al., 2001).
Figure 5.
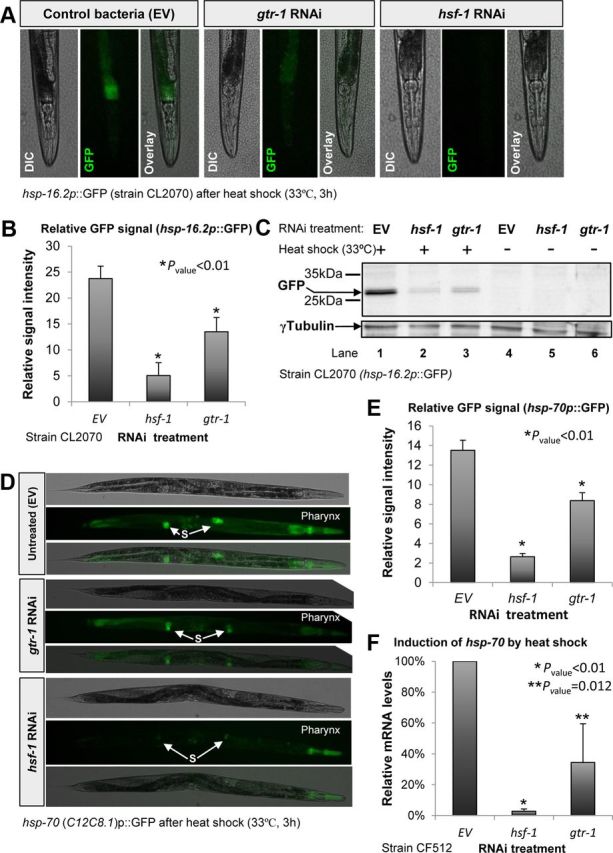

Knock-down of gtr-1 prevents the induction of the heat shock response downstream of both DAF-16 and HSF-1. A, Worms expressing GFP under the control of the hsp-16.2 promoter (strain CL2070) that were treated with gtr-1 RNAi and exposed to heat shock exhibited reduced GFP signal compared with their untreated (EV) counterparts. B–C, Measurement of GFP signal intensities (B) and Western blot analysis using GFP antibody (C) indicated that knock-down of gtr-1 resulted in remarkable reduction in the induction of hsp-16.2 by heat. This effect was significant (p < 0.01) but less prominent than the effect of hsf-1 RNAi. D–E, Knock-down of gtr-1 by RNAi reduces the signal intensity of worms expressing GFP under the regulation of hsp-70 (C12C8.1) promoter as visualized by fluorescent microscope. This effect was most prominent in the pharynx (D) and less in the spermatheca (“S”). Signal quantification (>20 worms per group; E) confirmed the significance of this observation (p < 0.01). F, Knock-down of gtr-1 by RNAi reduced the induction of hsp-70 (C12C8.1) by heat shock as measured by qPCR in CF512 worms (p = 0.012). G–H, Induction level of hsp-12.6 was significantly (p < 0.01) reduced by the knock-down of gtr-1 by RNAi as visualized (G) and quantified (H) in EHC102 worms that express tdTomato under the regulation of the hsp-12.6 promoter. This effect was most prominent in the vulva (insets). I, qPCR analysis confirmed the necessity of gtr-1 for the induction of hsp-12.6 in heat-shocked daf-2(e1370) worms (error bars represent ± SEM).
To further investigate the role of gtr-1 in HSR activation, we examined whether it is required for the induction of the inducible hsp-70 (encoded by the gene C12C8.1), a pivotal and well characterized HSR-induced, HSF-1 target gene in C. elegans (Snutch et al., 1988). To visualize directly the activity of the hsp-70 promoter, we used worms that express GFP under its regulation (Morley and Morimoto, 2004). The worms were treated with RNAi, exposed to heat shock as described above, and visualized by fluorescent microscopy. Much like the hsp-16.2 promoter, the activity of the hsp-70 promoter was largely reduced consequently to the knock-down of gtr-1 (Fig. 5D). The significance of this phenomenon, which was most notable in the pharynx, was confirmed by the measurement of signal intensities (Fig. 5E, p < 0.01). qPCR analysis of the hsp-70 mRNA levels in CF512 worms that were exposed to heat shock (33°C, 3 h) confirmed the key role of gtr-1 in the induction of this gene (Fig. 5F, p = 0.012).
We then investigated whether gtr-1 is also required for the activation of the other arm of the HSR mechanism, downstream of DAF-16, upon exposure to heat shock. We focused on hsp-12.6, a small heat shock protein that is a member of the α-crystallin family (Leroux et al., 1997) that is coregulated by DAF-16 (Murphy et al., 2003) and by HSF-1 (Hsu et al., 2003). To follow the expression of hsp-12.6 visually, we created worms that express the fluorescent protein tdTomato under the regulation or the hsp-12.6 promoter (2697 bp upstream of the hsp-12.6 open reading frame, strain EHC102). EHC102 worms were grown from hatching to day 1 of adulthood on bacteria expressing either gtr-1 or daf-16 RNAi or on EV bacteria and exposed to heat shock (33°C, 3 h). Much like daf-16 RNAi treatment, the knock-down of gtr-1 reduced the tdTomato signal (Fig. 5G) in the vulva (insets) and intestine of the worms, indicative of reduced induction of the hsp-12.6 promoter by heat shock. Signal quantification (Fig. 5H) and qPCR analysis (Fig. 5I) revealed that whereas the knock-down of daf-16 reduced the induction of hsp-12.6 by 80–90%, the knock-down of gtr-1 by RNAi had a less prominent but significant (p < 0.01) effect of ∼60% reduction compared with control worms.
No role for gtr-1 in the regulation of lifespan, resistance to pathogenic bacteria, or IIS downstream developmental functions
Lifespan extension is a hallmark of IIS reduction (Kenyon et al., 1993). Therefore, we sought to determine whether gtr-1 plays any role in the determination of lifespan. First, we examined whether the knock-down of gtr-1 has an effect on the lifespan of CF512 worms. The worms were fed control bacteria (EV) or bacteria expressing dsRNA toward either gtr-1 or daf-16 throughout development and adulthood and their lifespans were recorded. Unlike daf-16 RNAi treatment, the knock-down of gtr-1 had no significant effect on the worms' lifespans (Fig. 6A, Table 1). To assess whether gtr-1 is required for the longevity phenotype of daf-2(e1370) mutant worms, we treated the animals with daf-16 or gtr-1 RNAi as described above and followed their lifespan. Although the knock-down of daf-16 reduced the lifespan of the animals remarkably, gtr-1 RNAi treatment had no significant effect on the lifespan of these long-lived worms (Fig. 6B, Table 1).
Figure 6.

gtr-1 is dispensable for the determination of lifespan, for innate immunity, and for the developmental functions of the IIS. A–B, gtr-1 RNAi-treated and untreated (EV) CF512 worms had indistinguishable lifespans (A). Similarly, the knock-down of gtr-1 by RNAi had no significant effect on the lifespans of daf-2(e1370) mutant worms (B). C–D, Knock-down of gtr-1 affected neither the egg-laying pattern of daf-2(e1370) mutant worms (C) nor the percentage of dauer larvae in a worm population that was hatched and grown at 25°C (D). E, In contrast to hsf-1 RNAi, gtr-1 RNAi treatment during larval development had no effect on the survival rates of daf-2(e1370) mutant worms that were fed with the pathogenic bacteria P. aeruginosa during adulthood (p = 0.63). F, Knock-down of octr-1 by RNAi had no effect on the heat stress resistance of daf-2(e1370) mutant worms.
We then investigated whether gtr-1 is involved in the developmental functions of the IIS and found that knocking down this gene affected neither the modified egg-laying pattern of daf-2(e1370) worms (Dillin et al., 2002) nor their rates of dauer larvae formation when grown at 25°C (Fig. 6C,D, respectively).
The recent finding that the putative neuronal GPCR OCTR-1 regulates the innate immune response of C. elegans (Sun et al., 2011) and the critical necessity of HSF-1 (Singh and Aballay, 2006) and DAF-16 (Garsin et al., 2003) for this stress response prompted us to investigate whether gtr-1 is also required for the survival of worms that were exposed to pathogenic bacteria. daf-2(e1370) mutant worms were allowed to hatch and develop on EV bacteria or were treated with hsf-1 or gtr-1 RNAi, transferred onto plates seeded with P. aeruginosa, and their survival rates were recorded daily. Although animals that were treated with hsf-1 RNAi exhibited reduced survival, worms that were developed on gtr-1 RNAi bacteria and control animals (EV) had similar lifespans when grown on the pathogenic bacteria (Fig. 6E), indicating that gtr-1 has no role in the innate immune response.
To further scrutinize the possible relations between heat stress resistance and the innate immune response of the nematode, we investigated whether OCTR-1 (Sun et al., 2011) is also required for heat stress resistance. daf-2(e1370) mutant animals were developed on EV bacteria or on bacteria expressing dsRNA toward either daf-16 or octr-1. At day 1 of adulthood, the worms were exposed to heat shock (35°C) and rates of survival were followed in 4 h intervals. Our results (Fig. 6F) showed no role for octr-1 in the neuronal regulation of heat stress resistance, supporting the notion that gtr-1 and octr-1 function in distinct neuronal stress response mechanisms.
The ability to respond to heat comes at the expense of the capability to cope with proteotoxicity
In addition to their key roles in stress response, DAF-16, HSF-1, and heat shock proteins are also instrumental for the maintenance of proteostasis (for review, see Cohen and Dillin, 2008). Although these roles of heat shock proteins suggest that abolishing the worm's ability to induce the HSR will result in impaired proteostasis, it was reported recently that worms that carry mutated gcy-8 or ttx-3 are partially protected from proteotoxicity (Prahlad and Morimoto, 2011). Therefore, we investigated whether the knock-down of gtr-1 counters proteotoxicity or if it worsens the toxic phenotype associated with protein aggregation. To clarify this, we used worms that were engineered to express the Alzheimer's-disease-associated human Aβ3–42 peptide in their body wall muscles (Aβ worms, strain CL2006; Link, 1995). The expression of Aβ3–42 (McColl et al., 2009) results in progressive paralysis of the worm population. This paralysis phenotype can be alleviated by the knock-down of daf-2 by RNAi in a DAF-16- and HSF-1-dependent manner (Cohen et al., 2006).
To determine whether the knock-down of gtr-1 affects Aβ-associated toxicity, we treated Aβ worms with RNAi toward daf-2, hsf-1, or gtr-1 or left them untreated (EV), followed the rates of paralysis in the worm groups, and found that the knock-down of gtr-1 ameliorated Aβ toxicity (Fig. 7A). Three independent paralysis assays confirmed the significance (p = 0.045) of this observation (Fig. 7B) and supported the idea that abolishing the worm's ability to activate the HSR mitigates proteotoxicity.
Figure 7.

Knock-down of gtr-1 partially protects from Aβ proteotoxicity. A, CL2006 worms (expressing Aβ3–42 in their body wall muscles) were either left untreated (EV) or were treated with daf-2, hsf-1, or gtr-1 RNAi and the rates of paralysis within the worm populations were recorded daily. The rate of paralysis within the gtr-1 RNAi-treated population was lower than that of the control group (EV), but higher than that of daf-2 RNAi-treated worms. B, The counter-proteotoxic effect of gtr-1 RNAi treatment was confirmed by three independent paralysis assays. Bars represent the relative slopes of the paralysis graphs as in A (p = 0.045).
Discussion
gtr-1 is expressed in amphid, chemosensory neurons
In this study, we identified gtr-1 as a new critical component of the neuronal signaling mechanism that is required for HSR activation in non-neural tissues. gtr-1 (F25E2.1) is located on chromosome X and holds eight exons that encode a protein of 329 aa. GTR-1 is predicted to be a seven-transmembrane GPCR and has close homologs in other nematodes. However, the role of gtr-1 as a GPCR has not been established and the possibility that it possesses other function cannot be excluded. gtr-1 is coexpressed with lin-11 in chemosensory neurons (Hobert et al., 1998), but has no apparent role in thermotaxis. This finding indicates that gtr-1 is not required for heat sensing and suggests that this putative GPCR is solely needed for the induction of the neural signaling that activates the HSR in remote tissues upon exposure to heat. Interestingly, the similar effects of RNAi toward gtr-1 and gcy-8 on the worms' survival after heat shock (Fig. 1A,B) suggest that the thermosensory and chemosensory neurons are equally important for HSR induction. Although it is unclear how this signaling mechanism acts, it was reported recently that the worm's thermotactic behavior is dependent upon the activity of HSF-1 and the estrogen signaling pathway (Sugi et al., 2011). In the light of the results of this study, it will be interesting to determine whether GTR-1 is also functionally interrelated with the estrogen signaling pathway. It is also important to characterize the neuronal secretion mechanism that is influenced by GTR-1: does it affect dense core vesicle secretion mechanism or is it perhaps involved in mediating neurotransmitter release?
It was reported previously that the AFD neurons are pivotal for the activation of the HSR in remote tissues (Prahlad et al., 2008). In the present study, we show that the activation of the heat shock response is not exclusively controlled by this pair of neurons, but rather is also dependent upon the activity of additional components of the thermosensory circuit, the chemosensory neurons. This finding raises the key questions of how neurons of this circuit communicate to integrate environmental cues and how the decision to activate the HSR is made at the cellular and interneuronal levels. It will be also important to determine whether the chemosensory neurons are involved in sending the HSR-activating signal or if they play their roles exclusively in the decision making process.
The similar temporal and spatial expression patterns of gtr-1 and lin-11 and their key roles in the activity of the thermosensory circuit strongly suggest that these genes possess their functions in the same neuronal mechanism. Accordingly, it is tempting to speculate that these gene products interact functionally to promote HSR-activating signaling. It is also possible that LIN-11 and GTR-1 interact physically and perhaps form a functional protein complex with additional proteins. In addition, it would be interesting to study the possible functional links between GTR-1 and other neuronal LIM homeobox proteins that are required for thermosensation, such as CEH-14 (Cassata et al., 2000). Biochemical experimental techniques will be needed to address these questions.
Stress resistance, longevity, and counter-proteotoxic mechanisms are separable
The GPCR OCTR-1 acts in two pairs of sensory neurons, ASH and ASI, as a regulator of the worm's innate immune response in non-neural tissues (Sun et al., 2011). The lack of effect of gtr-1 knock-down on the survival rates of worms that were grown on P. aeruginosa and the dispensability of octr-1 for heat stress resistance (Fig. 6E,F, respectively) indicate that different G-protein-coupled receptors have specific roles as activators of distinct stress-signaling pathways. The distinction between mechanisms that respond to dissimilar insults was demonstrated by Chatzigeorgiou et al. (2010), who discovered that in C. elegans, PVD neurons use different molecular components to respond to low temperature and to harsh mechanical stimuli. Our results and the role of OCTR-1 in the regulation of innate immunity (Sun et al., 2011) indicate that chemosensory neurons are also capable of responding differentially to distinct environmental insults.
Because the IIS downstream transcription factors DAF-16 and HSF-1 are lifespan determinants (Kenyon, 2010), we also investigated whether the knock-down of gtr-1 shortens the lifespan of worms with a natural aging program and that of long-lived, daf-2 mutant animals and found no shortening effect in either case. This observation is consistent with reports showing that the knock-down of genes that encode superoxide dismutases, which are critical for oxidative stress resistance, either have no effect on lifespan (Van Raamsdonk and Hekimi, 2012) or extend the lifespan of C. elegans in one case (Van Raamsdonk and Hekimi, 2009). Our present study supports the idea that stress resistance and lifespan are separable and shows that, similarly to the mechanism that allows oxidative stress resistance, the ability to cope with heat stress has little or no role in the regulation of lifespan.
The intriguing finding that the knock-down of either gcy-8 or ttx-3 leads to a mild activation of the HSR and to the mitigation of proteotoxicity (Prahlad and Morimoto, 2011) led us to assess the effect of gtr-1 knock-down on Aβ-associated toxicity. Using worms expressing Aβ in the body wall muscle and the well established paralysis assay, we confirmed the previous conclusion that the abolishment of the worm's ability to respond to heat protects it from proteotoxicity, and also extended these findings to the toxic effects of Aβ aggregation. Together with the previous study, our present results suggest that an impairment of HSR-activating neural signaling pathways can reduce the tightness of the HSR negative regulatory mechanisms in the signal-accepting cells. This could provide protection from toxic protein aggregation, reduce the rate of neural loss, slow the progression of neurodegenerative disorders, and alleviate their symptoms once manifested. Therefore, selective knock-down of neuronal signaling components may be a novel intervention strategy to combat neurodegenerative disorders.
Footnotes
This work was supported by a Marie Curie Reintegration grant (Grant #279134 to E.C.), the Israel Science Foundation (Grant #671/11 to E.C.), and the U.S. Agency for International Development's American Schools and Hospitals Abroad program (for the procurement of the confocal microscope LSM710). We thank Dr. Tziona Ben-Gedalya and Dr. Lorna Moll for critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Baugh LR, Hill AA, Slonim DK, Brown EL, Hunter CP. Composition and dynamics of the Caenorhabditis elegans early embryonic transcriptome. Development. 2003;130:889–900. doi: 10.1242/dev.00302. [DOI] [PubMed] [Google Scholar]
- Beverly M, Anbil S, Sengupta P. Degeneracy and neuromodulation among thermosensory neurons contribute to robust thermosensory behaviors in Caenorhabditis elegans. J Neurosci. 2011;31:11718–11727. doi: 10.1523/JNEUROSCI.1098-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron D, Wasserman S, Thomas JH, Samuel AD, Sengupta P. An olfactory neuron responds stochastically to temperature and modulates Caenorhabditis elegans thermotactic behavior. Proc Natl Acad Sci U S A. 2008;105:11002–11007. doi: 10.1073/pnas.0805004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calixto A, Chelur D, Topalidou I, Chen X, Chalfie M. Enhanced neuronal RNAi in C. elegans using SID-1. Nat Methods. 2010;7:554–559. doi: 10.1038/nmeth.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassata G, Kagoshima H, Andachi Y, Kohara Y, Dürrenberger MB, Hall DH, Bürglin TR. The LIM homeobox gene ceh-14 confers thermosensory function to the AFD neurons in Caenorhabditis elegans. Neuron. 2000;25:587–597. doi: 10.1016/s0896-6273(00)81062-4. [DOI] [PubMed] [Google Scholar]
- Chatzigeorgiou M, Yoo S, Watson JD, Lee WH, Spencer WC, Kindt KS, Hwang SW, Miller DM, 3rd, Treinin M, Driscoll M, Schafer WR. Specific roles for DEG/ENaC and TRP channels in touch and thermosensation in C. elegans nociceptors. Nat Neurosci. 2010;13:861–868. doi: 10.1038/nn.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Ching TT, Lee HC, Mousigian C, Hsu AL. HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell. 2012;148:322–334. doi: 10.1016/j.cell.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci. 2008;9:759–767. doi: 10.1038/nrn2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Dillin A, Crawford DK, Kenyon C. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science. 2002;298:830–834. doi: 10.1126/science.1074240. [DOI] [PubMed] [Google Scholar]
- Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM. Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science. 2003;300:1921. doi: 10.1126/science.1080147. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Hobert O, D'Alberti T, Liu Y, Ruvkun G. Control of neural development and function in a thermoregulatory network by the LIM homeobox gene lin-11. J Neurosci. 1998;18:2084–2096. doi: 10.1523/JNEUROSCI.18-06-02084.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13:1385–1393. [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Lee RY, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950–1957. doi: 10.1016/s0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- Leroux MR, Ma BJ, Batelier G, Melki R, Candido EP. Unique structural features of a novel class of small heat shock proteins. J Biol Chem. 1997;272:12847–12853. doi: 10.1074/jbc.272.19.12847. [DOI] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD, Cypser JR, Johnson CJ, Johnson TE. Direct observation of stress response in Caenorhabditis elegans using a reporter transgene. Cell Stress Chaperones. 1999;4:235–242. doi: 10.1379/1466-1268(1999)004<0235:doosri>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lithgow GJ, White TM, Melov S, Johnson TE. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci U S A. 1995;92:7540–7544. doi: 10.1073/pnas.92.16.7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Shen Y. Structural basis for degeneracy among thermosensory neurons in Caenorhabditis elegans. J Neurosci. 2012;32:1–3. doi: 10.1523/JNEUROSCI.5112-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl G, Roberts BR, Gunn AP, Perez KA, Tew DJ, Masters CL, Barnham KJ, Cherny RA, Bush AI. The Caenorhabditis elegans A beta 1–42 model of Alzheimer disease predominantly expresses A beta 3–42. J Biol Chem. 2009;284:22697–22702. doi: 10.1074/jbc.C109.028514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwee J, Bubb K, Thomas JH. Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell. 2003;2:111–121. doi: 10.1046/j.1474-9728.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- Mori I. Genetics of chemotaxis and thermotaxis in the nematode Caenorhabditis elegans. Annu Rev Genet. 1999;33:399–422. doi: 10.1146/annurev.genet.33.1.399. [DOI] [PubMed] [Google Scholar]
- Mori I, Ohshima Y. Neural regulation of thermotaxis in Caenorhabditis elegans. Nature. 1995;376:344–348. doi: 10.1038/376344a0. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Johnson TE. A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditis elegans. Genetics. 1996;143:1207–1218. doi: 10.1093/genetics/143.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Prahlad V, Morimoto RI. Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci U S A. 2011;108:14204–14209. doi: 10.1073/pnas.1106557108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, Cornelius T, Morimoto RI. Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science. 2008;320:811–814. doi: 10.1126/science.1156093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol. 1993;13:1392–1407. doi: 10.1128/mcb.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Singh V, Aballay A. Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc Natl Acad Sci U S A. 2006;103:13092–13097. doi: 10.1073/pnas.0604050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snutch TP, Heschl MF, Baillie DL. The Caenorhabditis elegans hsp70 gene family: a molecular genetic characterization. Gene. 1988;64:241–255. doi: 10.1016/0378-1119(88)90339-3. [DOI] [PubMed] [Google Scholar]
- Sugi T, Nishida Y, Mori I. Regulation of behavioral plasticity by systemic temperature signaling in Caenorhabditis elegans. Nat Neurosci. 2011;14:984–992. doi: 10.1038/nn.2854. [DOI] [PubMed] [Google Scholar]
- Sun J, Singh V, Kajino-Sakamoto R, Aballay A. Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science. 2011;332:729–732. doi: 10.1126/science.1203411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira-Castro A, Ailion M, Jalles A, Brignull HR, Vilaça JL, Dias N, Rodrigues P, Oliveira JF, Neves-Carvalho A, Morimoto RI, Maciel P. Neuron-specific proteotoxicity of mutant ataxin-3 in C. elegans: rescue by the DAF-16 and HSF-1 pathways. Hum Mol Genet. 2011;20:2996–3009. doi: 10.1093/hmg/ddr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A. 2012;109:5785–5790. doi: 10.1073/pnas.1116158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volovik Y, Maman M, Dubnikov T, Bejerano-Sagie M, Joyce D, Kapernick EA, Cohen E, Dillin A. Temporal requirements of heat shock factor-1 for longevity assurance. Aging Cell. 2012;11:491–499. doi: 10.1111/j.1474-9726.2012.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston WM, Molodowitch C, Hunter CP. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 2002;295:2456–2459. doi: 10.1126/science.1068836. [DOI] [PubMed] [Google Scholar]
- Zhang T, Mullane PC, Periz G, Wang J. TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum Mol Genet. 2011;20:1952–1965. doi: 10.1093/hmg/ddr076. [DOI] [PMC free article] [PubMed] [Google Scholar]