

Abstract
Dysregulated hippocampal neurogenesis has been associated with neurodegenerative disorders, including Alzheimer's disease (AD), in which it may potentially represent an auto-reparatory mechanism that could counteract neuronal loss and cognitive impairment. We evaluated hippocampal neurogenesis in TgCRND8 mice and reported that, at 32 weeks of age, corresponding to an advanced AD-like neuropathology stage, increased numbers of proliferating cells, doublecortin-expressing progenitors/neuroblasts, and early postmitotic calretinin-expressing neurons were present compared with wild-type (WT) littermates. When hippocampal neural progenitor cells (NPCs) were isolated from TgCRND8 mice, we demonstrated that (1) their neurogenic potential was higher compared with WT NPCs; (2) medium conditioned by TgCRND8 NPC promoted neuronal differentiation of WT NPCs; and (3) the proneurogenic effect of TgCRND8-conditioned medium was counteracted by blockade of the receptor for advanced glycation end products (RAGE)/nuclear factor-κB (NF-κB) axis. Furthermore, we showed that β-amyloid 1–42 (Aβ1–42) oligomers, but not monomers and fibrils, and the alarmin high-mobility group box-1 protein (HMGB-1) could promote neuronal differentiation of NPCs via activation of the RAGE/NF-κB axis. Altogether, these data suggest that, in AD brain, an endogenous proneurogenic response could be potentially triggered and involve signals (Aβ1–42 oligomers and HMGB-1) and pathways (RAGE/NF-κB activation) that also contribute to neuroinflammation/neurotoxicity. A more detailed analysis confirmed no significant increase of new mature neurons in hippocampi of TgCRND8 compared with WT mice, suggesting reduced survival and/or integration of newborn neurons. Therapeutic strategies in AD should ideally combine the ability of sustaining hippocampal neurogenesis as well as of counteracting an hostile brain microenvironment so to promote survival of vulnerable cell populations, including adult generated neurons.
Introduction
Receptor for advanced glycation end products (RAGE) is a membrane-bound receptor activated by multiple ligands, including advanced glycation end products, β-amyloid (Aβ) peptides and oligomers, the alarmin high-mobility group box-1 protein (HMGB-1), and calcium-binding proteins S100/calgranulins. In the adult CNS, RAGE is expressed by several cell types, and, during ligand binding, it produces a sustained cellular activation, commonly regarded as deleterious because it participates in inflammation and cytotoxicity in both acute and chronic conditions (Yan et al., 1996; Schmitt et al., 2006; Muhammad et al., 2008; Han et al., 2011). Recently, a vast array of information has raised attention on the potential contribution of RAGE to Alzheimer's disease (AD) pathophysiology. The expression levels of the receptor and its ligands are indeed increased in AD brain (Sasaki et al., 2001; Shuvaev et al., 2001; Takata et al., 2003; Lüth et al., 2005; Shepherd et al., 2006; Gruden et al., 2007; Cruz-Sánchez et al., 2010). Moreover, the expression of soluble RAGE isoforms, which may act as decoy receptors, is reduced in brain and plasma of AD patients with early cognitive deterioration (Nozaki et al., 2007). According to the current knowledge, RAGE has been suggested to play manifold roles in AD pathophysiology by increasing Aβ influx across the blood–brain barrier (Deane et al., 2004; Silverberg et al., 2010; Maślińska et al., 2011), neuroinflammation (Du Yan et al., 1997; Lue et al., 2001; Fang et al., 2010), Aβ-induced oxidative stress (Takuma et al., 2009), and tau phosphorylation (Esposito et al., 2008; Li et al., 2012). In a mouse familial AD (FAD) model, RAGE overexpression was shown to accelerate disease progression (Arancio et al., 2004).
Dysregulated adult hippocampal neurogenesis has also been associated with several neurodegenerative disorders, including AD (Bondolfi et al., 2002; Jin et al., 2004a,b; López-Toledano and Shelanski, 2004; Ziabreva et al., 2006; Heo et al., 2007; Chen et al., 2008; López-Toledano et al., 2010; Rodríguez and Verkhratsky, 2011). Although at present the contribution of hippocampal neurogenesis to disease pathophysiology is uncertain (Chuang, 2010; Crews et al., 2010), the formation of new neurons in hippocampus (HP) may potentially represent an endogenous auto-reparatory mechanism that could attenuate neuronal loss and cognitive impairment associated with AD (Schaeffer et al., 2009; DeCarolis and Eisch, 2010). Interestingly, RAGE appeared to be expressed by bromodeoxyuridine (BrdU)-labeled cells in the adult rat subgranular zone (SGZ) after chronic fluoxetine administration, a treatment promoting hippocampal neurogenesis (Malberg et al., 2000; Manev et al., 2003). Moreover, intracerebroventricular infusion of the RAGE ligand S100B enhanced hippocampal neurogenesis and, in parallel, ameliorated cognitive performance after traumatic brain injury (Kleindienst et al., 2005). Altogether, these findings suggest that RAGE may potentially regulate a proneurogenic response to brain injury in the hippocampal area. This hypothesis is corroborated by previous data demonstrating that activation of the RAGE/nuclear factor-κB (NF-κB) axis promotes both proliferation and neuronal differentiation of adult SVZ neural progenitor cells (NPCs) in vitro (Meneghini et al., 2010).
Based on these observations, we decided to investigate the putative role of the RAGE/NF-κB axis in the modulation of adult hippocampal neurogenesis and its potential relevance in AD pathophysiology.
Materials and Methods
Animals.
For in vivo studies, transgenic (Tg) (n = 32) and wild-type (WT) (n = 30) mice of the TgCRND8 line (Chishti et al., 2001; Hyde et al., 2005) were used at different age points, ranging from 8 to 32 weeks. For in vitro studies, 4- to 6-month-old male CD1 mice, purchased from Charles River Laboratories, 6-to 8-month-old male Tg and WT TgCRND8 mice and 4-to 6-month-old NF-κB p50−/− (B6; 129P2-Nfκb1 tm1 Bal/J) and WT (B6; 129PF2) mice, obtained from The Jackson Laboratory, were used. Mice, kept three to four per cage with access to water and food ad libitum, were maintained in high-efficiency particulate air-filtered Thoren units (Thoren Caging System) at the University of Eastern Piedmont animal facility. Animal care and treatments were performed in accordance with European Community Directive 86/609/EEC guidelines and were reviewed and approved by the local Institutional Animal Care and Use Committee.
Neuropathological assessment of TgCRND8 mice.
Eight-, 15-, 25-, and 32-week-old TgCRND8 mice (three to six per group) and their age-matched non-Tg littermates were used. Mice were transcardially perfused with saline first and then with 4% paraformaldehyde (PFA). After removal, brains were postfixed and then washed with 0.01 m PBS, 50% ethanol, and 70% ethanol, and finally stored in 70% ethanol at 4°C until embedding processing. Brains were embedded in paraffin, and serial sections were taken from the anterior pole to cerebellum with a microtome (Leica Lasertechnik) at 8 μm thickness. Sections were incubated overnight at 4°C with primary antibodies. To identify Aβ plaques and astrogliosis, sections were incubated overnight at 4°C with a mouse anti-Aβ protein monoclonal antibody (1:100; Merck Millipore) and with a mouse monoclonal antibody to glial fibrillary acidic protein (GFAP) (1:50; Boehringer Mannheim), respectively. After washing in PBS, slides were incubated with biotinylated horse anti-mouse antibody (1:200; Vector Laboratories), followed by ABC reagent (Vector Laboratories) for 30 min at room temperature and developed using DAB peroxidase (Sigma-Aldrich). Three slices per mouse at the level of the HP, each separated by ∼320 μm, were used at each age for quantification. Immunostained sections were acquired through a CCD video camera connected to the microscope (10× objective for amyloid plaques and 20× for GFAP), and images were processed with a computerized image analysis system (Image Pro Plus software; Media Cybernetics), as described previously (Hyde et al., 2005).
Immunohistochemistry analyses.
Mice (n = 5–6 per group) were injected with BrdU (150 mg/kg body weight, i.p.) and killed 24 h later. Alternatively, mice were injected twice a day with BrdU (100 mg/kg body weight, i.p.) for 5 consecutive days and killed 21 d after the last injection. The following primary antibodies were used: rabbit polyclonal anti-Sox-2 (SRY-related HMG-box gene 2) (1:800; Merck Millipore), goat polyclonal anti-RAGE (1:800; R&D Systems), rat monoclonal anti-BrdU (1:200; Novus Biologicals), rabbit polyclonal anti-doublecortin (DCX) (1:200; Cell Signaling Technology), rabbit polyclonal anti-calretinin (CR) (1:4000; Swant), and mouse monoclonal anti-NeuN antibody (1:100; Merck Millipore). For quantification and phenotypic characterization of newborn cells, a modified unbiased stereology protocol was used, as described previously (Denis-Donini et al., 2008).
Isolation and culture of adult hippocampal neurospheres.
For hippocampal neurosphere (NS) preparation from adult mice, the detailed procedure was as described previously (Denis-Donini et al., 2008). Primary [passage 1 (P1)] NS were dissociated after 7 d in vitro, whereas P2–P30 NS were dissociated every 5 d in vitro. At each passage, cells were grown as described previously (Denis-Donini et al., 2008). P3–P30 NS were used in this study.
Preparation of Aβ1–42 forms.
Synthetic Aβ1-42 peptide (Sigma-Aldrich) was initially dissolved to 1 mm in hexafluoroisopropanol (Sigma-Aldrich) and aliquoted in sterile microcentrifuge tubes. Hexafluoroisopropanol was removed under vacuum, and the peptide film was stored dessicated at −20°C. For the aggregation protocol, the peptide was dissolved in dry dimethylsulfoxide (Sigma-Aldrich) at a concentration of 5 mm. For preparation of Aβ oligomers and fibrils, the protocol developed by Dahlgren et al. (2002) was adopted. Peptide preparations were characterized by using Tris–tricine separation gels. For detection, proteins were stained with Proteosilver Silver Stain kit (Sigma-Aldrich).
ELISA assay.
Human Aβ1-42 levels were quantified in conditioned medium derived from WT and TgCRND8 NPC cultures by using the Aβ42 Human ELISA kit (Invitrogen), in accordance with the instructions of the manufacturer.
NPC differentiation.
For NPC differentiation, the detailed procedure was described previously (Meneghini et al., 2010). NPCs were treated in the presence of 0.01–10 ng/ml HMGB-1 (Sigma-Aldrich), 100–500 nm Aβ1–42 forms, or vehicle for 24 h. For RAGE blockade, 60 min before RAGE ligand or vehicle addition, 20 μg/ml neutralizing polyclonal anti-RAGE antibody (R&D System) or preimmune IgG was added to NPC culture medium. For NF-κB inhibition, 10 μg/ml SN-50 peptide, 10 μg/ml SN-50M inactive control, 3 μm JSH-23 [4-methyl-N1-(3-phenylpropyl)benzene-1,2-diamine], and 3 μm SC-514 (3-amino-5-thiophen-3-ylthiophene-2-carboxamide) (Calbiochem-Merck) were added to NPC cultures 60 min before RAGE ligand or vehicle, as described previously (Valente et al., 2012). After 24 h, cells were washed in PBS and fixed by ice-cold 4% PFA for 20 min at room temperature for subsequent immunofluorescent analysis. In each experiment, five fields per well were counted using a fluorescent microscope with a 60× objective. For experiments based on media switch between differentiated WT or TgCRND8 NPCs, conditioned media were collected and centrifuged at 235 × g for 10 min, and supernatants were stored at 4°C before use. All experiments were run in triplicates by using different cell preparations and repeated at least three times.
Assessment of cell viability.
Necrosis evaluation in NPC cultures was performed as described previously (Meneghini et al., 2010). For apoptosis quantification, an in situ terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) assay was performed by the In Situ Cell Death Detection kit (Roche Diagnostics), in accordance with the instructions of the manufacturer. All experiments were run in triplicates in different cell preparations and repeated at least three times.
Immunocytochemistry.
After fixation, undifferentiated or differentiated NPCs were washed three times in PBS and permeabilized in PBS containing 0.48% (v/v) Triton X-100. Permeabilization was omitted for RAGE immunostaining. The detailed procedure was described previously (Meneghini et al., 2010). The following primary antibodies were used: rabbit monoclonal anti-MAP-2 (1:600; Merck Millipore), mouse monoclonal anti-GFAP (1:400; Merck Millipore), goat polyclonal anti-RAGE (1:600; Merck Millipore), chicken polyclonal anti-nestin (1:4000; Neuromics), rabbit polyclonal anti-Sox-2 (1:500; Merck Millipore), rabbit polyclonal anti-CR (1:500; Swant), rabbit polyclonal anti-p65 (1:500; Santa Cruz Biotechnology), and rabbit monoclonal NG-2 (1:500; Merck Millipore). Nuclei were counterstained with 0.8 ng/ml Hoechst diluted in PBS. Fluorescent microscope or laser-scanning confocal microscope were used for analysis. Adobe Photoshop CS (Adobe Systems) was used for digital processing of the images. Only light intensity, brightness, and contrast adjustments were applied to improve information.
Protein isolation and Western blot analyses.
For protein isolation, tissues and NS from adult CD1 mice were disrupted in lysis buffer, homogenates were incubated on ice for 60 min and centrifuged at 15700 × g for 30 min at 4°C, and the supernatant was recovered. Western blot analysis was performed as described previously (Meneghini et al., 2010).
Statistical analyses.
For in vivo neurogenesis evaluation, statistical analysis was performed using the unpaired, two-sided t test comparison (Student's t test). For amyloid burden and astrogliosis quantification in TgCRND8 mice, comparison among ages was performed by one-way ANOVA, and data were expressed as mean ± SEM values. For in vitro experiments, data are reported as mean ± SEM, with at least three replicates per group and analyzed by two-sided t test comparison (Student's t test). Statistical significance level was set for p values <0.05.
Results
RAGE is expressed by nestin+ and Sox-2+ NPCs of adult mouse HP
By Western blot analysis, different RAGE isoforms can be detected using two antibodies recognizing N- or C-terminal epitopes (Fig. 1A). In lung protein extracts, used as positive control, both antibodies identified three bands with apparent molecular weight (MW) of ∼43, 54, and 57 kDa (Gefter et al., 2009). Additionally, the N-terminus antibody revealed bands corresponding to 48 and 70 kDa. Based on previous reports (Harashima et al., 2006; Hudson et al., 2008; Gefter et al., 2009; Kalea et al., 2009), the 54 kDa species is likely to correspond to full-length RAGE (FL-RAGE), whereas the 48 and 70 kDa bands may represent endogenous secretory RAGE (esRAGE) proteins obtained by C-terminal cleavage, because they are not detected by the C-terminal antibody. The 57 kDa band presumably corresponds to xRAGE, a less characterized membrane-bound RAGE isoform (Gefter et al., 2009). Based on the MW of human homologs of RAGE isoforms, we can suppose that the band around 43 kDa presumably represents the mouse RAGE_v18, homolog of the human RAGE_v4 (Hudson et al., 2008; Kalea et al., 2009). In protein extracts of HP and undifferentiated neural progenitors growing in NS, we detected a band pattern mostly similar to that observed in lung extracts and including the most characterized FL-RAGE and esRAGE. In addition, the antibody recognizing the C-terminal epitope revealed a band around 45 kDa only in HP and NS protein extracts, which may potentially represent the murine homolog of the human membrane-bound N-truncated isoform, the MW of which is 42 kDa (Yonekura et al., 2003), but was not detected by the antibody against N-terminal epitope. Subsequent immunocytochemical analysis of hippocampal NS demonstrated the presence of RAGE immunoreactivity in undifferentiated progenitors (Fig. 1B,E), which are characterized by the expression of the intermediate neurofilament nestin (Fig. 1C) and the transcription factor Sox-2 (Fig. 1F). Double-immunolabeling experiments confirmed colocalization of RAGE with nestin (Fig. 1D) and Sox-2 (Fig. 1G) in adult hippocampal neural progenitors. Finally, immunohistochemical analysis of the adult HP of CD1 mice showed that a subpopulation of Sox-2-expressing (Sox-2+) neural stem/NPCs in the dentate gyrus (DG) also coexpresses RAGE. In particular, RAGE immunoreactivity mainly decorated the process that those cells project toward the external part of the granular cell layer (GCL) (Fig. 1H).
Figure 1.
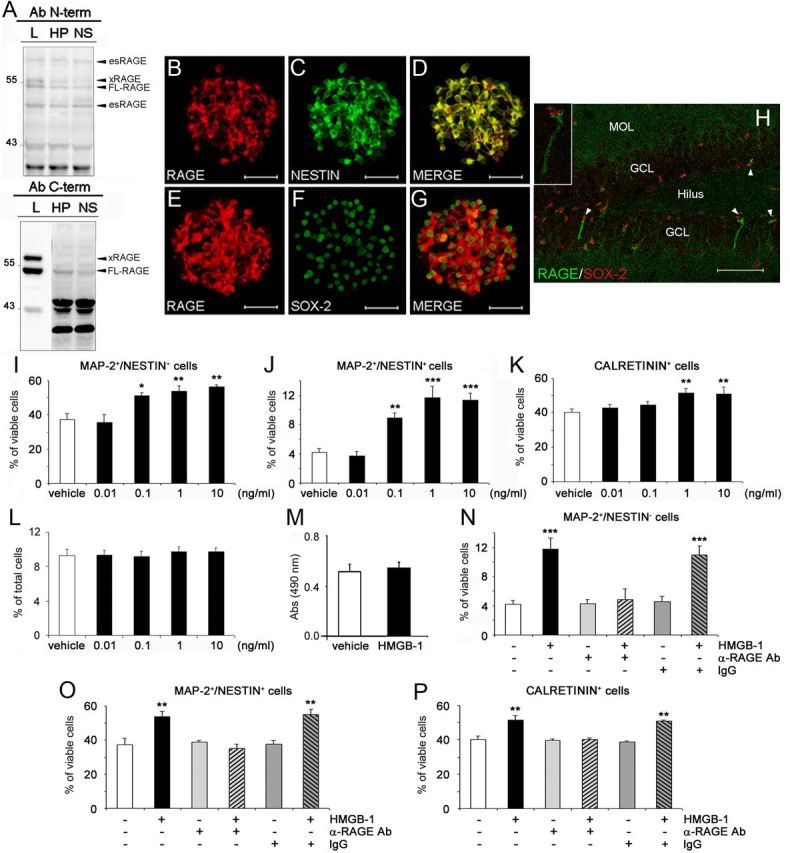
RAGE, expressed by adult hippocampal NPCs, mediates proneurogenic effects elicited by HMGB-1. A, Representative immunoblots confirming the presence of different RAGE isoforms in extracts from undifferentiated neural progenitors grown in NS, HP, or lung (L) by using two antibodies (Ab) recognizing different N- and C-terminal epitopes. Bands corresponding to RAGE-specific isoforms are marked. B–G, Representative fluorescent microscopy images of NS immunolabeled with antibodies against RAGE (B, E, red), nestin (C, green), and Sox-2 (F, green). The merged images showed that RAGE immunoreactivity colocalized with nestin (D) and Sox-2 (G). Magnification, 600×. Scale bar, 56.2 μm. H, Representative confocal microscopy image of the adult mouse DG immunostained with antibodies against RAGE (green) and Sox-2 (red). Some Sox-2+ NPCs (indicated by arrowheads) were also positive for RAGE. MOL, Molecular layer. Magnification, 400×. Scale bar, 70 μm. Inset, Higher magnification of a Sox-2+/RAGE+ cell. I–K, Under differentiating conditions, 24 h treatment of adult hippocampal NPCs with 0.1–10 ng/ml HMGB-1 significantly increased, in a concentration-dependent manner, the percentage of MAP-2+/nestin+ (I), MAP-2+/nestin− (J), and CR+ (K) cells compared with vehicle-treated cells. L, M, No significant difference was observed between vehicle- or HMGB-1-treated cultures in the percentage of apoptotic nuclei (L) and in LDH activity in media (M) of differentiated cell cultures. N–P, A neutralizing anti-RAGE antibody (α-RAGE, 20 μg/ml), but not preimmune IgG serum (IgG), abolished the proneurogenic effects of 1 ng/ml HMGB-1 on MAP-2+/nestin− (N), MAP-2+/nestin+ (O), and CR+ (P) cells. I–P, Data represent the mean ± SEM of n = 3 experiments, run in triplicates. *p < 0.05, **p < 0.01, ***p < 0.001 versus vehicle (Student's t test).
RAGE activation promotes differentiation of adult hippocampal NPCs toward the neuronal lineage
RAGE activation by different ligands, including HMGB-1, promotes neuronal differentiation of adult SVZ-derived NPCs (Meneghini et al., 2010). Therefore, we decided to evaluate whether RAGE had a similar functional role in adult hippocampal NPCs.
After removal of growth factors from the culture medium, adult NPCs stop dividing and differentiate onto laminin-coated dishes. Double immunolabeling for markers of neurons (MAP-2) and undifferentiated progenitor cells (nestin) was performed to dissect in vitro the different stages of neuronal differentiation of NPCs derived from the HP of adult CD1 mice (Valente et al., 2012) in response to HMGB-1, a RAGE ligand. After 24 h, a concentration-dependent increase in the percentage of MAP-2+/nestin+ immature neurons was observed in cells treated with 0.01–10 ng/ml HMGB-1 with a maximal effect elicited at 1 ng/ml (percentage of increase over vehicle-treated cells, +44.1 ± 11.6%, p < 0.01; Fig. 1I). In parallel, we observed a concentration-dependent increase in the percentage of MAP-2+/nestin− mature neurons in HMGB-1-treated cells, with a maximal effect elicited at the same concentration (percentage of increase over vehicle-treated cells, +176.3 ± 32.7%, p < 0.001; Fig. 1J). Similarly, HMGB-1 treatment increased the percentage of CR+ postmitotic neurons compared with vehicle-treated cells (p < 0.01 at 1–10 ng/ml; Fig. 1K), further confirming the proneurogenic effect of HMGB-1 on adult hippocampal NPCs. HMGB-1 effects on glial differentiation were also evaluated by antibodies directed against GFAP and NG-2, markers of astrocytes and oligodendrocyte precursors, respectively. In our experimental conditions, 1 ng/ml HMGB-1 had no effect on GFAP+ (vehicle, 6.64 ± 1.05%; 1 ng/ml HMGB-1, 6.44 ± 1.97%) and NG-2+ cell populations (vehicle, 14.68 ± 3.60%; 1 ng/ml HMGB-1, 17.47 ± 2.69%). To investigate whether HMGB-1 could promote cell survival in addition to neuronal differentiation, we analyzed both the number of apoptotic cells and the amount of lactate dehydrogenase (LDH) released by necrotic cells in the presence of HMGB-1 or vehicle. By TUNEL assay, no difference was observed in the apoptotic rate in vehicle- versus HMGB1-treated cells (Fig. 1L). Similarly, no difference in LDH activity was detected at 1 ng/ml HMGB-1 (Fig. 1M), as well as other HMGB-1 concentrations (data not shown). Finally, we demonstrated that HMGB-1 effect on neuronal differentiation of adult NPCs was RAGE mediated because the HMGB-1-induced increase in the percentage of MAP-2+/nestin− mature neurons (Fig. 1N), MAP-2+/nestin+ immature neurons (Fig. 1O), and CR+ postmitotic neurons (Fig. 1P) could be prevented in the presence of a neutralizing anti-RAGE antibody but not by preimmune IgG (α-RAGE + HMGB-1 vs HMGB-1: p < 0.001, p < 0.01, and p < 0.01 for MAP-2+/nestin−, MAP-2+/nestin+, and CR+ cells, respectively). When applied alone, α-RAGE had no significant effect on NPC differentiation (Fig. 1N–P). Overall, these data confirmed that, in vitro, HMGB-1 promoted neuronal differentiation of adult hippocampal NPCs via RAGE activation.
NF-κB signaling is involved in HMGB-1-mediated neuronal differentiation of adult hippocampal NPCs
NF-κB signaling lies downstream of RAGE activation in several cell types, including SVZ-derived adult NPCs (Li and Schmidt, 1997; Bierhaus et al., 2001; Meneghini et al., 2010). Additionally, NF-κB proteins are involved in the regulation of adult hippocampal neurogenesis (Denis-Donini et al., 2008; Koo et al., 2010). For these reasons, we decided to investigate the involvement of NF-κB activation in RAGE-mediated effects on adult hippocampal NPCs.
NF-κB p65 immunoreactivity can be detected in the cytoplasm of the majority of vehicle-treated hippocampal NPCs (Fig. 2A). Within 30 min after cell exposure to 1 ng/ml HMGB-1, NF-κB p65 nuclear translocation could be observed (Fig. 2B). Furthermore, 3 μm JSH-23, a cell-permeable selective blocker of NF-κB p65 nuclear translocation (IC50 = 7 μm), completely prevented HMGB-1-mediated increase of both MAP-2+/nestin+ immature neurons (JSH-23 + HMGB-1 vs HMGB-1: p < 0.01; Fig. 2C) and MAP-2+/nestin− mature neurons (JSH-23 + HMGB-1 vs HMGB-1: p < 0.001; Fig. 2D). Similar results were obtained in the presence of 10 μg/ml SN-50, a cell-permeable peptide that inhibits NF-κB p50 nuclear translocation (Lin et al., 1995) [SN-50 + HMGB-1 vs HMGB-1: p < 0.01 and p < 0.001 for MAP-2+/nestin+ (Fig. 2C) and MAP-2+/nestin− (Fig. 2D) cells, respectively]. Interestingly, when applied alone, SN-50 peptide produced a statistically significant decrease in the percentage of MAP-2+/nestin− mature neurons when compared with vehicle-treated cells (p < 0.05; Fig. 2D). As a negative control, 10 μg/ml SN-50M inactive peptide (Lin et al., 1995) had no effect alone or in the presence of HMGB-1 (Fig. 2C,D). Finally, SC-514, a selective and reversible inhibitor of IκB kinase 2 (IKK2) (IC50 = 3–12 μm), completely prevented HMGB-1-mediated effect on MAP-2+/nestin+ immature neurons (SC-514 + HMGB-1 vs HMGB-1: p < 0.01; Fig. 2C) and on MAP-2+/nestin− mature neurons (SC-514 + HMGB-1 vs HMGB-1: p < 0.001; Fig. 2D) at a concentration of 3 μm. At all tested concentrations, JSH-23, SN-50, and SC-514 had no effect on the survival of NPCs or their progeny when added alone or in the presence of HMGB-1 (data not shown). To further confirm the involvement of NF-κB signaling, we isolated and expanded in vitro hippocampal NPCs derived from adult p50−/− mice and their WT littermates. As expected, in WT NPCs, 24 h exposure to 1 ng/ml HMGB-1 significantly increased the number of both MAP-2+/nestin+ immature neurons (p < 0.01; Fig. 2E) and MAP-2+/nestin− mature neurons (p < 0.001; Fig. 2F) compared with vehicle-treated cells. Conversely, HMGB-1 had no effect in p50−/− NPCs (Fig. 2E,F). Interestingly, in vehicle-treated conditions, we observed a reduced number of MAP-2+/nestin− cells in p50−/− compared with WT cultures (5.5 ± 0.5 and 3.1 ± 0.3% in WT and p50−/− cultures, respectively; p < 0.05; Fig. 2F). Overall, these data demonstrated the involvement of NF-κB signaling in the proneurogenic effects of HMGB-1 on adult hippocampal NPCs.
Figure 2.

NF-κB signaling is involved in HMGB1-mediated neuronal differentiation of hippocampal NPCs. A, B, HMGB-1 induced nuclear translocation of NF-κB p65 in adult hippocampal NPCs under differentiating conditions. Confocal microscopy images showing cytoplasmatic or nuclear (indicated by arrows) NF-κB p65 immunostaining. NPCs were exposed for 30 min to vehicle (A) or 1 ng/ml HMGB-1 (B). Magnification, 800×. Scale bar, 37.5 μm. C, D, The inhibition of NF-κB signaling activation by SN-50 peptide (10 μg/ml), JSH-23 (3 μm), and SC-514 (3 μm) completely abolished the increase of MAP-2+/nestin+ (C) and MAP-2+/nestin− (D) cells induced by 1 ng/ml HMGB-1. When applied alone, SN-50 peptide decreased the percentage of MAP-2+/nestin− mature neurons compared with vehicle-treated cells. SN-50M (negative control peptide) did not counteract HMGB-1-mediated effect on NPC neuronal differentiation. Data represent the mean ± SEM of n = 3 experiments, run in triplicates. *p < 0.05, **p < 0.01, ***p < 0.001 versus vehicle (Student's t test). E, F, Twenty-four hours of treatment of WT and NF-κB p50−/− hippocampal NPCs with 1 ng/ml HMGB-1 or vehicle. Compared with vehicle, HMGB-1 significantly increased the percentage of MAP-2+/nestin+ (E) and MAP-2+/nestin− (F) neurons in WT but not in NF-κB p50−/− cultures. E, F, Data represent the mean ± SEM of n = 3 experiments, run in triplicates. *p < 0.05, **p < 0.01, ***p < 0.001 versus vehicle-treated WT cells (Student's t test).
Increased cell proliferation, generation of newborn neuroblasts, and postmitotic neurons in hippocampi of 32-week-old TgCRND8 mice
TgCRND8 mice represent a well-established disease model characterized by an early onset and rapidly progressing AD-like pathology (Chishti et al., 2001; Hyde et al., 2005). To confirm that progression, heterozygous TgCRND8 mice and their WT littermates were examined at four different ages, namely 8, 15, 25, and 32 weeks. The time course of amyloid burden in cortex and hippocampi of TgCRND8 mice was quantified by computer-assisted image analysis. An age-dependent increase in the percentage of area occupied by plaques was observed, ranging from 0.01 ± 0.00% at 8 weeks to 3.75 ± 0.14% at 32 weeks of age (Fig. 3A). A statistically significant difference was reported in 25-week-old TgCRND8 mice versus both 15-week-old (p < 0.05) and 8-week-old (p < 0.01) animals and in 32-week-old versus all other ages (p < 0.001). In addition to Aβ deposition, an inflammatory process reminiscent of AD also occurs in TgCRND8 mice (Chishti et al., 2001; Hyde et al., 2005). Immunostaining with a GFAP-specific antibody indicated the presence of astrocytosis that progressively increased with age in cortex and HP of Tg mice (data not shown). GFAP-positive cells, showing the typical morphology of reactive astrocytes with large cell bodies and thick processes, started to be detected at 25 weeks of age, and patches of immunolabeled cells could be seen in the cortex, whereas at 32 weeks, astrocytic reaction was spread to the whole cortex and hippocampi. Image analysis allowed to quantify the extension and age progression of astrogliosis in TgCRND8 versus age-matched WT controls (Fig. 3B). The percentage of area occupied by GFAP-positive cells increased from 0.71 ± 0.28 at 8 weeks, to 2.34 ± 0.78 at 15 weeks, 9.76 ± 2.01 at 25 weeks, and 36.19 ± 2.47 at 32 weeks. A statistically significant difference was reported in 25-week-old TgCRND8 mice versus both 15-week-old (p < 0.05) and 8-week-old (p < 0.01) animals and in 32-week-old versus all other ages (p < 0.001). In brain of WT animals, there was no age-dependent difference in the percentage of area occupied by GFAP-positive cells (Fig. 3B), being on average ∼0.48 ± 0.13%.
Figure 3.

Increased cell proliferation in the SGZ of TgCRND8 mice at an advanced stage of AD-like neuropathology (32 weeks of age). A, Quantification of amyloid burden in cortex/hippocampi of TgCRND8 mice at 8, 15, 25, and 32 weeks showed an age-dependent increase in the area percentage occupied by plaques. A statistically significant difference was reported in 25-week-old TgCRND8 mice versus both 15-week-old (p < 0.05) and 8-week-old (p < 0.01) animals and in 32-week-old versus all other ages (p < 0.001). B, Quantification of the area percentage occupied by GFAP-positive cells demonstrated an age-dependent progression of astrogliosis in cortex of TgCRND8 versus age-matched WT controls. A statistically significant difference was reported in 25-week-old TgCRND8 mice versus both 15-week-old (p < 0.05) and 8-week-old (p < 0.01) animals and in 32-week-old versus all other ages (p < 0.001). In WT animals, there was no age-dependent difference in the percentage of area occupied by GFAP-positive cells. A, B, Comparison among ages was performed by one-way ANOVA, and data were expressed as mean values ± SEM. **p < 0.01, ***p < 0.001 versus 8-week-old TgCRND8 mice; #p < 0.05, ###p < 0.001 versus 15-week-old TgCRND8 mice; §§§p < 0.001 versus 25-week-old TgCRND8 mice. C–H, Representative images of BrdU immunostaining in DG of WT (C, E, G) and TgCRND8 (D, F, H) mice at 8 (C, D), 20 (E, F), and 32 (G, H) weeks of age, killed 24 h after thymidine analog administration. Arrowheads indicated BrdU+ proliferating cells in the SGZ. Asterisks (H) indicated Aβ plaques. Magnification, 200×. Scale bar, 50 μm. I–K, Quantification of BrdU-IR cells in the SGZ, GCL, and total DG of 8-week-old (I), 20-week-old (J), and 32-week-old (K) mice revealed a statistically significant increase in the number of SGZ BrdU+ cells in 32-week-old TgCRND8 mice compared with age-matched WT littermates (K). Data represent the mean ± SD. n = 5 mice per group. *p < 0.05 versus WT mice (Student's t test).
Based on these neuropathology findings, we decided to focus subsequent analysis on the following age points: (1) 8 weeks, which we consider a pre-plaque stage; and (2) 20 and 32 weeks, which we refer to as initial and advanced stage of amyloid pathology and neuroinflammation, respectively. In TgCRND8 mice and their WT littermates, we initially evaluated hippocampal cell proliferation at different ages. Eight-, 20-, and 32-week-old TgCRND8 and WT mice were administered BrdU (150 mg/kg, i.p.) and killed 24 h later. We then quantified BrdU-immunoreactive (IR) cells in the SGZ and GCL of both WT and TgCRND8 mice. Regardless of age, as expected, BrdU-IR cells were distributed throughout the neurogenic area, with the large majority of them being mainly located in the SGZ in both WT (66–76%) and Tg (65–70%) mice. As shown in Figure 3, C, D, and I, BrdU-IR cell counts were comparable in the SGZ and GCL in 8-week-old Tg and WT mice. No significant change could be observed when the total number of BrdU+ cells in the DG was evaluated. Similar results were obtained in 20-week-old mice of both genotypes (Fig. 3E,F,J). Conversely, 32-week-old TgCRND8 mice had significantly more BrdU-IR cells in the SGZ compared with age-matched WT mice (mean ± SD number, 132 ± 67 and 261 ± 116 in WT and TgCRND8 mice, respectively; p < 0.05; +198% vs WT; Fig. 3G,H,K). No significant difference was observed between 32-week-old WT and TgCRND8 mice in the GCL region (mean ± SD number, 61 ± 68 and 133 ± 92 in WT and Tg mice, respectively; p = 0.152; Fig. 3K). When cell counts in the entire DG were analyzed, an increased, although not statistically significant, number of BrdU-IR cells was observed in TgCRND8 mice compared with WT littermates (mean ± SD number, 193 ± 126 and 395 ± 185 in WT and Tg mice, respectively; p = 0.052; Fig. 3K). Moreover, analysis of BrdU-IR cells in the DG of 8-, 20-, and 32-week-old mice revealed an age-dependent reduction in the number of SGZ BrdU-positive cells in both genotypes (Fig. 3I–K), an observation consistent with previous data showing a precocious age-related decline in murine hippocampal cell proliferation (Seki and Arai, 1995). Overall, these results suggested the occurrence of increased cell proliferation in the SGZ, a region in which neural stem/NPCs lie and divide (Rietze et al., 2000; Kempermann et al., 2003), of TgCRND8 mice at an advanced stage of amyloid pathology.
To understand whether increased cell proliferation in the SGZ of 32-week-old TgCRND8 mice compared with WT littermates had an impact on the generation of adult-born neuroblasts and neurons in that area, we then quantified the total number of hippocampal cells expressing the neuroblast marker DCX in mutant and WT mice of that age. A significantly increased number of DCX+ cells was found in TgCRND8 GCL compared with age-matched WT mice (mean ± SD number, 266 ± 70 and 616 ± 344 in WT and Tg mice GCL, respectively, p < 0.01; 417 ± 139 and 532 ± 282 in WT and Tg mice SGZ, respectively, p = 0.30; 683 ± 188 and 1148 ± 610 in WT and Tg mice DG, respectively, p < 0.05; Fig. 4A). DCX is expressed by proliferating progenitor cells and newly generated neuroblasts (Brown et al., 2003). For these reasons, we determined the number of BrdU+/DCX+ cells in 32-week-old animals that were killed 24 h after administration of the thymidine analog. An increased number of DCX+/BrdU+ cells was reported in the SGZ and not in the GCL of TgCRND8 mice compared with age-matched WT controls (in the SGZ, mean ± SD, 64 ± 17 and 174 ± 82 in WT and Tg mice, respectively, p < 0.05; in the GCL, mean ± SD, 30 ± 36 and 68 ± 37 in WT and Tg mice, respectively, p = 0.19; in the DG, mean ± SD, 94 ± 44 and 242 ± 101 in WT and Tg mice, respectively; p < 0.05; Fig. 4B). Altogether, these data also suggested an increased proliferation rate in DCX+ NPCs/neuroblasts of TgCRND8 mice compared with their WT counterpart. To evaluate whether survival and/or neuronal commitment of newly generated cells was indeed increased in hippocampi of 32-week-old TgCRND8 mice, an additional group of animals of both genotypes (n = 5–6) was administered BrdU and killed 21 d later. When BrdU immunoreactivity was analyzed, as expected, IR cells appeared to be distributed in the DG, with the large majority of them being located in the GCL in both WT (73%) and Tg (64%) mice. No significant difference in the absolute number of surviving BrdU-IR cells could be observed between WT and TgCRND8 mice (mean ± SD number, 190.4 ± 79.3 and 379 ± 269 in WT and Tg mice GCL, respectively; p = 0.170). Additionally, we evaluated the number of DCX+/BrdU+ cells in the hippocampi of 32-week-old TgCRND8 and WT mice. A significantly increased number of DCX+/BrdU+ cells was present in the GCL of TgCRND8 versus WT mice (mean ± SD, 35 ± 23 and 86 ± 43 in WT and Tg mice, respectively; p = 0.036; Fig. 4C). Overall, these data suggested that an increased number of BrdU-labeled cells had indeed acquired a DCX+ phenotype in TgCRND8 mice compared with WT controls. CR is a calcium-binding protein transiently expressed at an early postmitotic stage by adult generated DG granule cells (Brandt et al., 2003). Counting of CR+ cells also revealed a striking difference in the DG of 32-week-old TgCRND8 mice compared with age-matched WT littermates. Significantly more CR+ cells were indeed present in the SGZ, GCL, and DG of TgCRND8 compared with age-matched WT mice (mean ± SD number, 734 ± 236 and 1578 ± 476 in WT and Tg mice SGZ, respectively, p < 0.05, +115% vs WT control; 454 ± 111 and 1386 ± 600 in WT and Tg mice GCL, respectively, p < 0.05, +205% vs WT control; 1188 ± 317 and 2964 ± 618 in WT and Tg mice DG, respectively, p < 0.01; +149% vs WT control; Fig. 4D).
Figure 4.

TgCRND8 neural progenitors generate an increased number of DCX+ neuroblasts and CR+ postmitotic neurons in vivo. A, Quantification of DCX-IR cells in SGZ, GCL, and DG of 32-week-old WT and TgCRND8 mice revealed a significantly increased number in the GCL and DG of TgCRND8 mice compared with their WT littermates. B, C, Quantification of DCX+/BrdU+ cells in 32-week-old WT and TgCRND8 mice killed 24 h (B) and 21 d (C) after BrdU injection. D, Quantitative analysis of CR-IR cells in the SGZ, GCL, and total DG of 32-week-old mice revealed a significantly increased number in all hippocampal subregions TgCRND8 mice compared with WT littermates. A–D, Data represent the mean number ± SD. *p < 0.05, **p < 0.01 versus WT mice (Student's t test).
Overall, these data suggested that, at an advanced stage of AD-like neuropathology, TgCRND8 mice displayed an increased number of hippocampal proliferating cells, DCX+ proliferating progenitors/neuroblasts, and CR+ postmitotic neurons compared with their age-matched WT littermates.
Increased in vitro neurogenic potential of adult hippocampal NPCs derived from TgCRND8 mice compared with their WT-derived counterpart: role of Aβ1–42 oligomers and RAGE/NF-κB axis activation
We then decided to isolate and culture hippocampal NPCs derived from 32-week-old WT and TgCRND8 mice. Under differentiating conditions, we observed that, similarly to what reported in vivo, TgCRND8 NPCs gave rise to a significantly higher percentage of CR+ postmitotic neurons compared with WT NPCs (44.1 ± 2.5 and 56.6 ± 1.9% in WT and TgCRND8 cultures, respectively; +28% vs WT NPCs; p < 0.001; Fig. 5A–C). Under the same experimental conditions, no significant difference was observed in cell death rate of WT versus TgCRND8-derived NPCs and their progeny, as evaluated by TUNEL assay (Fig. 5D) or by measuring LDH activity in the culture medium (data not shown). Interestingly, in the presence of 20 μg/ml α-RAGE neutralizing antibody (Fig. 5E) and 10 μg/ml SN-50 peptide (Fig. 5F), the in vitro neurogenic potential of TgCRND8 NPCs was reduced and similar to that of WT NPC. Overall, these data confirmed that activation of the RAGE/NF-κB axis was involved in the increased neurogenic potential of TgCRND8-derived hippocampal NPCs compared with their WT counterpart.
Figure 5.
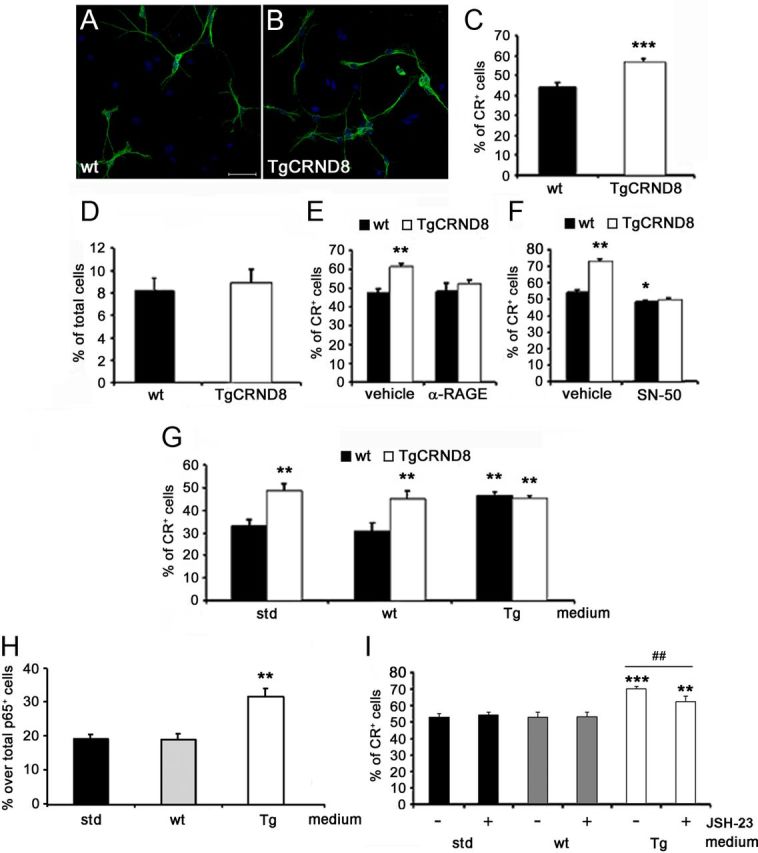
TgCRND8 neural progenitors generate an increased number of CR+ postmitotic neurons in vitro via RAGE/NF-κB axis. A, B, Confocal microscopy images of CR+ neurons (green) generated in vitro from adult hippocampal NPCs isolated from WT (A) and TgCRND8 (B) mice. Nuclei were counterstained with Hoechst (blue). Magnification, 600×. Scale bar, 56.25 μm. C, D, TgCRND8 NPCs gave rise to a higher percentage of CR+ neurons over total viable cells compared with WT NPCs (C) in the absence of differences in the apoptotic rate (D). Data represent the mean ± SEM of n = 3 experiments, run in triplicates. ***p < 0.001 versus WT cultures (Student's t test). E, F, In the presence of a neutralizing anti-RAGE antibody (E; α-RAGE, 20 μg/ml) or SN-50 peptide (F; 10 μg/ml) the percentage of CR+ neurons derived from TgCRND8 NPCs was similar to that of WT cultures. Treatment with SN-50 peptide also decreased the percentage of CR+ neurons in WT cultures compared with vehicle (F). Data represent the mean ± SEM of n = 3 experiments, run in triplicates. *p < 0.05, **p < 0.01 versus vehicle-treated WT cells (Student's t test). G, WT NPCs exposed to TgCRND8 NPC-conditioned medium (Tg) gave rise to a significantly higher percentage of CR+ cells compared with WT cells differentiated in the presence of standard (std) or WT NPC-conditioned (wt) media. Media switch experiments had no effect on neuronal differentiation of TgCRND8 NPC cultures. Data represent the mean ± SEM of n = 3 experiments, run in triplicates. **p < 0.01 versus WT cells differentiated in standard medium (Student's t test). H, Quantification of p65+ nuclei in WT NPCs after 2 h exposure to WT- or TgCRND8-conditioned media. Data are expressed as percentage of p65+ nuclei over total p65 IR cells. **p < 0.01 versus WT cells differentiated in standard medium (Student's t test). I, Percentage of CR+ cells in WT NPCs after 24 h exposure to WT- or TgCRND8-conditioned media in the presence or absence of JSH-23 (3 μm). I, Data represent the mean ± SEM of n = 3 experiments, run in triplicates. **p < 0.01, ***p < 0.001 versus WT-conditioned medium; ##p < 0.01 versus vehicle-treated TgCRND8 medium (Student's t test).
We then collected conditioned media from TgCRND8 and WT NPC cultures. When WT NPCs were exposed for 24 h to TgCRND8-conditioned medium, a significantly higher number of CR+ cells was counted compared with WT NPCs differentiated in standard medium or WT NPC-conditioned medium (p < 0.01; Fig. 5G). Conversely, when TgCRND8 NPCs were differentiated in the presence of conditioned medium from WT cells, no difference was observed in the number of CR+ neurons compared with TgCRND8 cells differentiated in standard medium or in TgCRND8 NPC-conditioned medium (Fig. 5G). These data suggested that soluble factors potentially released by TgCRND8-derived NPCs could contribute to increased neuronal differentiation in vitro. Finally, we evaluated, by immunofluorescence, p65 nuclear translocation in WT NPCs exposed for 2 h to standard, TgCRND8-conditioned or WT-conditioned medium. Under these experimental conditions, the percentage of cells with p65+ nuclei over the total number of p65-labeled cells significantly increased in WT NPCs treated with TgCRND8-conditioned medium compared with cultures exposed to WT-conditioned or standard medium (31.63 ± 2.42, 19.13 ± 1.64, and 19.12 ± 1.23% in TgCRND8-, WT-conditioned, and standard media, respectively; p < 0.01; Fig. 5H). To further confirm the role of NF-κB signaling pathway activation, we exposed WT NPCs to Tg-conditioned or standard media in the presence of 3 μm JSH-23 or vehicle. Inhibition of p65 nuclear translocation significantly reduced the effect of Tg-conditioned medium on the number of CR+ cells [vehicle (TgCRND8-conditioned medium) vs JSH23 (TgCRND8-conditioned medium): p < 0.01; Figure 5I]. As shown previously, JSH-23 alone had no effect on WT NPC cultures (Fig. 5I). We then evaluated, by ELISA, the presence of Aβ1–42 species in the conditioned media from TgCRND8 and WT-derived NPC cultures. Interestingly, Aβ1–42 could be detected in medium that was conditioned by TgCRND8-derived NPCs under differentiating conditions (60.30 ± 11.28 pg/ml), whereas, as expected, no detectable amounts were present in WT-conditioned media.
Based on these results, we treated WT NPC cultures for 24 h with different concentrations (100–500 nm) of Aβ1–42 monomers, oligomers, or fibrils, whose nature was confirmed by running onto Tris–tricine gels (Fig. 6A). Interestingly, Aβ1–42 oligomers significantly increased, in a concentration-dependent manner, the percentage of CR+ postmitotic neurons (Fig. 6B), MAP-2+/nestin+ immature neurons (Fig. 6C), and MAP-2+/nestin− mature neurons (Fig. 6D) generated by WT NPCs when compared with vehicle. Maximal effect by Aβ1–42 oligomers was elicited at 200 nm (percentage of increase over vehicle-treated cells: +53.5 ± 5.2% CR+ cells, p < 0.01; +33.1 ± 8.8% MAP-2+/nestin+ cells, p < 0.01; +166.8 ± 29.6% MAP-2+/nestin− cells, p < 0.001). Conversely, neither Aβ1–42 monomers nor Aβ1–42 fibrils had positive effects on neuronal differentiation of WT NPC (Fig. 6B–D). Interestingly, 500 nm Aβ1–42 fibrils significantly decreased the percentage of MAP-2+/nestin− mature neurons compared with vehicle-treated cells (Fig. 6D, p < 0.05), whereas it had no effect on CR+ and MAP-2+/nestin+ cells (Fig. 6B,C). Under the same experimental conditions, no difference was observed between vehicle- and Aβ1–42-treated cultures in the apoptotic (Fig. 6E) or necrotic (Fig. 6F) rate. Again, in WT NPCs, 20 μg/ml α-RAGE antibody, 3 μm JSH-23, and 10 μg/ml SN-50, but not 10 μg/ml SN-50M inactive peptide, abolished the effect of Aβ1–42 oligomers on CR+ postmitotic neurons (Aβ1–42 oligomers vs α-RAGE + Aβ1–42 oligomers: p < 0.01; Aβ1–42 oligomers vs JSH-23 + Aβ1–42 oligomers: p < 0.001; Aβ1–42 oligomers vs SN-50 + Aβ1–42 oligomers: p < 0.001; Fig. 6G), MAP-2+/nestin+ immature neurons (Aβ1–42 oligomers vs α-RAGE + Aβ1–42 oligomers: p < 0.01; Aβ1–42 oligomers vs JSH-23 + Aβ1–42 oligomers: p < 0.05; Aβ1–42 oligomers vs SN-50 + Aβ1–42 oligomers: p < 0.01; Fig. 6H), and MAP-2+/nestin− mature neurons (Aβ1–42 oligomers vs α-RAGE + Aβ1–42 oligomers: p < 0.01; Aβ1–42 oligomers vs JSH-23 + Aβ1–42 oligomers: p < 0.01; Aβ1–42 oligomers vs SN-50 + Aβ1–42 oligomers: p < 0.001; Fig. 6I). No effect was elicited by the neutralizing antibody or by JSH-23 alone (Fig. 6G–I), whereas, as reported previously, SN-50 treatment reduced the percentage of CR+ and MAP-2+/nestin− cells in WT NPC cultures (Fig. 6G,I). To further confirm the role of NF-κB signaling pathway activation, hippocampal NPCs derived from adult p50−/− mice and their WT littermates were differentiated in the presence of 200 nm Aβ1–42 oligomers. As expected, in WT NPCs, Aβ1–42 oligomers increased the percentage of CR+ cells (percentage of increase over vehicle-treated cells: +60.4 ± 8.13%; p < 0.01), MAP-2+/nestin+ immature neurons (percentage of increase over vehicle-treated cells: +34.7 ± 1.34%; p < 0.01), and MAP-2+/nestin− mature neurons (percentage of increase over vehicle-treated cells: +91.1 ± 9.65%; p < 0.001). Conversely, Aβ1–42 oligomers had no effect in p50−/− NPCs (percentage over vehicle-treated cells: −2.6 ± 3.72, +10.5 ± 6.32, and −1.3 ± 1.27% for CR+, MAP-2+/nestin+, and MAP-2+/nestin− cells, respectively). Overall, these data demonstrated that activation of the RAGE/NF-κB axis mediated the proneurogenic effects elicited by Aβ1–42 oligomers on adult hippocampal NPCs.
Figure 6.

Aβ1–42 oligomers promote neuronal differentiation of adult hippocampal NPCs via activation of the RAGE/NF-κB axis. A, Representative image of monomeric (mon), oligomeric (olig), and fibrillary (fib) Aβ1-42 preparations separated onto Tris–tricine gel and revealed by silver staining. B–D, One hundred to 500 nm Aβ1–42 oligomers significantly increased, compared with vehicle, the percentage of CR+ (B), MAP-2+/nestin+ (C), and MAP-2+/nestin− (D) neurons generated in WT NPC cultures. Aβ1–42 monomers and fibrils had no positive effect on neuronal differentiation of WT NPCs, with 500 nm Aβ1–42 fibrils significantly reducing the number of MAP-2+/nestin− mature neurons in cultures. E, No difference in the number of apoptotic cells in vehicle-treated and monomeric, oligomeric, and fibrillary Aβ1–42-treated WT NPC cultures. F, No difference in LDH activity released in media was observed between vehicle-treated and 200 nm monomeric, oligomeric, and fibrillary Aβ1–42-treated WT NPC cultures. G–I, In WT NPCs, the neutralizing anti-RAGE antibody (α-RAGE, 20 μg/ml), SN-50 peptide (10 μg/ml), and JSH-23 (3 μm) prevented the effect of Aβ1–42 oligomers (200 nm) on CR+ (G), MAP-2+/nestin+ (H), and MAP-2+/nestin− (I) neurons. SN-50 alone reduced the percentage of CR+ (G) and MAP-2+/nestin− (I) cells in WT NPC cultures, whereas SN-50M had no effect alone or in the presence of Aβ1–42 oligomers. B–I, Data represent the mean ± SEM of n = 3 experiments, run in triplicates. *p < 0.05, **p < 0.01, ***p < 0.001 versus vehicle-treated NPC cultures (Student's t test).
The absolute number of newly generated mature neurons is not significantly different in 32-week-old WT and TgCRND8 mice hippocampi
Based on the observation that 32-week-old TgCRND8 mice have an increased number of CR+ postmitotic neurons, we administered BrdU (100 mg/kg, i.p., twice a day for 5 consecutive days) to WT and TgCRND8 (n = 5 per group) of corresponding age and collected their brains 3 weeks after the last BrdU injection. We then performed a double immunostaining for BrdU and NeuN (a marker of mature neurons) on brain sections. BrdU-IR cells from a set of 1 in 8 sections throughout the hippocampal DG extension of each animal were analyzed by confocal microscopy and 3D reconstruction to establish colocalization with NeuN. As shown in Figure 7, A, B, and E, as expected, BrdU+/NeuN− cells were significantly increased in the GCL of TgCRND8 compared with age-matched WT mice (96 ± 40 and 418 ± 290 in WT and Tg mice GCL, respectively; p < 0.05). Surprisingly, despite the presence of a significantly increased number of DCX+ progenitors/neuroblasts and CR+ postmitotic neurons, we documented no significant change in the number of mature newly generated neurons identified as BrdU+/NeuN+ cells in the GCL of the two mouse groups (mean ± SD number: 146 ± 20 and 154 ± 79 in WT and Tg mice, respectively; Fig. 7C–E).
Figure 7.

Similar numbers of BrdU+/NeuN+ neurons in the DG of 32-week-old TgCRND8 and WT mice. A,B and C,D, Representative confocal microscopy images of BrdU (blue) and NeuN (red) double immunostaining in DG of 32-week-old WT (A, C) and TgCRND8 (B, D) mice. Arrowheads indicate BrdU+/NeuN− (A, B) and BrdU+/NeuN+ (C, D) cells. Scale bar, 47.7 μm. E, Confocal microscopy and 3D reconstruction analysis revealed a significant increase in the number of BrdU+/NeuN− cells but no difference in the number of mature BrdU+/NeuN+ neurons in the GCL of 32-week-old TgCRND8 mice compared with age-matched WT littermates. Data represent the mean ± SD. *p < 0.05 versus WT mice (Student's t test).
Discussion
In a recent study, we proposed a novel role for the RAGE/NF-κB axis, namely promotion of neuronal differentiation in SVZ-derived adult NPCs (Meneghini et al., 2010). Here we further extend our observations providing evidence for RAGE expression in Sox-2+ hippocampal neural progenitors in vivo and in vitro. In addition, we demonstrated that the alarmin HMGB-1 promoted neuronal differentiation of adult hippocampal neural progenitors via RAGE activation. Interestingly, under the same experimental conditions, HMGB-1 had no effect on astrogliogenesis, oligodendrogenesis, or cell survival. Activation of the NF-κB signaling pathway appeared to be involved in the proneurogenic effect elicited by HMGB-1, because it was counteracted by inhibitors of p50 and p65 nuclear translocation and IKK2. Additionally, HMGB-1 proneurogenic effect was absent in NPC cultures derived from p50−/− mice. These data are in line with previous reports suggesting the contribution of the NF-κB signaling to the positive regulation of adult neurogenesis (Denis-Donini et al., 2005, 2008; Rolls et al., 2007; Meneghini et al., 2010; Grilli and Meneghini, 2012). Conversely, NF-κB signaling, and in particular p65 activation, has also been involved in the antineurogenic effects of stress (Koo et al., 2010). The fact that both induction and inhibition of adult neurogenesis may rely on activation of NF-κB signaling is likely to reflect high-level complexity within the pathway. Indeed, NF-κB proteins represent a composite family of transcription factors (p50, p65, p52, c-rel, relB) whose members can combine to form heterodimers and homodimers of different composition, which in turn can be differentially activated in a given cell type and exert different, even opposite, functions through different sets of gene target activation (Grilli and Memo, 1997; Grilli et al., 2003; Bonini et al., 2011; Grilli and Meneghini, 2012).
There are increasing reports showing that deregulated adult neurogenesis may be associated with AD (Chuang, 2010; Crews et al., 2010). Hippocampal neurogenesis and/or proliferation have been suggested to be increased in AD patients (Jin et al., 2004b). Data in FAD animal models are mostly contradictory, with both increased and decreased neurogenesis being described (Chuang, 2010; Crews et al., 2010; Marlatt and Lucassen, 2010). Despite the paucity of information, relevant therapeutic implications for modulation of adult hippocampal neurogenesis in AD can be envisioned because of the functional impact of this process on synaptic plasticity, learning, and memory (Schaeffer et al., 2009; Gonzalez-Castaneda et al., 2011; Iqbal and Grundke-Iqbal, 2011). Therefore, research efforts are required for a better understanding of the receptors and signals that may regulate hippocampal neurogenesis in AD.
We evaluated hippocampal neurogenesis in TgCRND8 mice, a FAD animal model characterized by age-related accumulation of amyloid deposits in cortex, HP, and white matter accompanied by extensive neuroinflammation and severe cognitive impairment within the first year of life (Chishti et al., 2001; Hyde et al., 2005). By analyzing the number of BrdU-incorporating cells in the SGZ and GCL of adult TgCND8 and WT mice 24 h after administration of the thymidine analog, we observed no significant difference in the percentage of IR cells between the two genotypes at 8 weeks (pre-plaque stage) and 20 weeks (initial stage of amyloid pathology) of age. Conversely, 32-week-old TgCRND8 mice had significantly more BrdU-IR cells in the SGZ, but not in GCL, compared with WT animals, suggesting an increased cell proliferation in the region in which neural stem/NPCs lie and divide. In the 24 h BrdU paradigm, we could also observe an increased number of DCX+/BrdU+ cells in the SGZ of 32-week-old TgCRND8 mice compared with WT littermates, suggesting an increased proliferation rate of DCX+ cells in the AD mouse model. These changes were accompanied by a significant increase in the number of DCX+/BrdU+ neuroblasts in the GCL of 32-week-old TgCRND8 compared with WT littermates, as measured 21 d after BrdU labeling. We propose that these changes were not merely attributable to increased survival of BrdU-labeled cells in mutant mice because, in parallel, no significant difference could be observed in the absolute number of BrdU+ cells in hippocampi of TgCRND8 and WT mice at 21 d after BrdU injection. Finally, we demonstrated that the absolute number of DCX+ cells as well as of CR+ postmitotic adult-born neurons were significantly increased in the hippocampi of TgCRND8 mice compared with WT animals. Overall, these results are in line with data obtained in other APP Tg mouse models, in which an increased cell proliferation and/or an enhanced formation of newborn neuroblasts and postmitotic neurons was observed only at a post-plaque deposition stage (Jin et al., 2004a; Ermini et al., 2008; Kolecki et al., 2008; Yu et al., 2009).
By using TgCRND8- and WT-derived hippocampal NPCs, we further investigated the molecular mechanisms potentially involved in the increased number of progenitors, neuroblasts, and postmitotic neurons in TgCRND8 hippocampi. We demonstrated that, also in vitro, TgCRND8-derived NPCs had a higher neurogenic potential compared with WT-derived progenitors and that this increased effect could be abolished by blocking activation of the RAGE/NF-κB axis. Additionally, conditioned media from TgCRND8 NPC cultures also increased the number of neurons generated in vitro by adult WT NPC cultures, and again, this effect correlated with NF-κB activation and in particular with p65 nuclear translocation. In an attempt to identify the soluble factors that were responsible for that effect, we could prove the presence of human Aβ1–42 in TgCRND8-conditioned media and that only Aβ1–42 oligomers but not monomers or fibrils could act as proneurogenic signal when added to cultures of adult hippocampal NPCs. These data are in agreement with previous studies suggesting that oligomeric Aβ can increase the number of newborn neurons derived from neonatal hippocampal and adult SVZ neural progenitors (López-Toledano and Shelanski, 2004; Heo et al., 2007). Herein we provide novel evidence that the proneurogenic effect of Aβ1–42 oligomers is RAGE mediated and requires nuclear translocation of NF-κB p50/p65 dimers. As shown for HMGB-1, the proneurogenic effect of oligomeric Aβ was absent in NPC cultures derived from p50−/− mice. Although not conclusive, these data potentially suggest that the activation of RAGE/NF-κB axis by Aβ oligomers (or by HMGB-1) in adult NPCs may potentially contribute to the increased number of DCX+ and CR+ cells in hippocampi of TgCRND8 mice. Furthermore, these data corroborate the idea that not all effects elicited by Aβ oligomers in brain are invariably deleterious.
Over the years, extensive research work has contributed to the concept of RAGE involvement in AD-associated inflammation and neurodegeneration (Yan et al., 1997; Lue et al., 2001; Arancio et al., 2004; Deane et al., 2004; Esposito et al., 2008; Takuma et al., 2009; Fang et al., 2010; Perrone et al., 2012), events that are usually correlated with the toxic effects of Aβ oligomers rather than fibrils (Walsh and Selkoe, 2004; Glabe, 2005; Glabe and Kayed 2006). Indeed, several groups have proposed blockade of RAGE as a strategy for therapeutic intervention in AD (Aderinwale et al., 2010; Sabbagh et al., 2011; Deane et al., 2012; Perrone et al., 2012). The novel proneurogenic role of RAGE/NF-κB axis activation adds complexity to that scenario, opening the possibility that RAGE engagement by HMGB-1, whose expression levels are increased in AD (Takata et al., 2003), or by Aβ oligomers could also promote the differentiation of adult hippocampal NPCs toward the neuronal lineage in vivo. In a murine model of traumatic brain injury, Kleindienst et al. (2005) demonstrated that the proneurogenic effects induced by intracerebroventricular administration of the RAGE ligand S100B correlated with improved cognitive recovery. Conversely, despite the increased number of DCX+ neuroblasts and CR+ postmitotic neurons, TgCRND8 mice are cognitively impaired (Chishti et al., 2001; Hyde et al., 2005). Indeed, when we extended our analysis to establish the final destiny of adult-born neurons in 32-week-old TgCRND8 mice, no significant difference could be observed in the absolute number of mature BrdU+/NeuN+ cells in TgCRND8 and WT mice hippocampi. These findings potentially suggest that, unlike acute damage, in a chronic neurodegenerative disease, the RAGE-mediated hippocampal proneurogenic response is quantitatively and/or temporally inadequate to counteract age-progressing pathophysiological events such as amyloid deposition, neurodegeneration, and/or neuroinflammation. It could also be envisioned that, in AD pathology, final integration and/or full maturation of adult generated neurons may be hampered by a hostile microenvironment (Li et al., 2008; Schaeffer et al., 2009). Surprisingly, in such respects, Aβ oligomers are not deleterious but may actually promote differentiation of adult NPCs toward the neuronal lineage.
Altogether, our data propose a novel molecular mechanism that may regulate hippocampal neurogenesis in AD brain and rely on Aβ oligomers and HMGB-1-induced activation of RAGE/NF-κB axis in adult NPCs. Although this potentially reparative attempt is not sufficient to attenuate damage and cognitive impairment, we believe that these findings would reinforce the need for multitargeted drug strategies in AD. Ideally, therapeutic interventions should combine the ability of promoting and sustaining hippocampal neurogenesis as well as counteracting the hostile brain microenvironment, so to allow survival and complete maturation/integration of adult newborn neurons.
Footnotes
Acknowledgments: This work was supported by grants from Ministero della Istruzione Università Ricerca (MIUR), under the Progetti di Interesse Nazionale (PRIN 2007), from Ministero della Salute (Ricerca Finalizzata), and from Fondazione Cariplo to M.G.
The authors declare no competing financial interests.
References
- Aderinwale OG, Ernst HW, Mousa SA. Current therapies and new strategies for the management of Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2010;25:414–424. doi: 10.1177/1533317510372372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, Stern DM, Du Yan SS. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004;23:4096–4105. doi: 10.1038/sj.emboj.7600415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Klöting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Häring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini SA, Ferrari-Toninelli G, Uberti D, Montinaro M, Buizza L, Lanni C, Grilli M, Memo M. Nuclear factor κB-dependent neurite remodeling is mediated by Notch pathway. J Neurosci. 2011;31:11697–11705. doi: 10.1523/JNEUROSCI.1113-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt MD, Jessberger S, Steiner B, Kronenberg G, Reuter K, Bick-Sander A, von der Behrens W, Kempermann G. Transient calretinin expression defines early postmitotic step of neuronal differentiation in adult hippocampal neurogenesis of mice. Mol Cell Neurosci. 2003;24:603–613. doi: 10.1016/s1044-7431(03)00207-0. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Després S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Chen Q, Nakajima A, Choi SH, Xiong X, Sisodia SS, Tang YP. Adult neurogenesis is functionally associated with AD-like neurodegeneration. Neurobiol Dis. 2008;29:316–326. doi: 10.1016/j.nbd.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Chuang TT. Neurogenesis in mouse models of Alzheimer's disease. Biochim Biophys Acta. 2010;1802:872–880. doi: 10.1016/j.bbadis.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Crews L, Rockenstein E, Masliah E. APP transgenic modeling of Alzheimer's disease: mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct Funct. 2010;214:111–126. doi: 10.1007/s00429-009-0232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Sánchez FF, Gironès X, Ortega A, Alameda F, Lafuente JV. Oxidative stress in Alzheimer's disease hippocampus: a topographical study. J Neurol Sci. 2010;299:163–167. doi: 10.1016/j.jns.2010.08.029. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, Thiyagarajan M, Zarcone T, Fritz G, Friedman AE, Miller BL, Zlokovic BV. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarolis NA, Eisch AJ. Hippocampal neurogenesis as a target for the treatment of mental illness: a critical evaluation. Neuropharmacology. 2010;58:884–893. doi: 10.1016/j.neuropharm.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis-Donini S, Caprini A, Frassoni C, Grilli M. Members of the NF-kappaB family espresse in zones of active neurogenesis in the postnatal and adult mouse brain. Brain Res Dev Brain Res. 2005;154:81–89. doi: 10.1016/j.devbrainres.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Denis-Donini S, Dellarole A, Crociara P, Francese MT, Bortolotto V, Quadrato G, Canonico PL, Orsetti M, Ghi P, Memo M, Bonini SA, Ferrari-Toninelli G, Grilli M. Impaired adult neurogenesis associated with short-term memory defects in NF-kappaB p50-deficient mice. J Neurosci. 2008;28:3911–3919. doi: 10.1523/JNEUROSCI.0148-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stern D, Schmidt AM. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:5296–5301. doi: 10.1073/pnas.94.10.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermini FV, Grathwohl S, Radde R, Yamaguchi M, Staufenbiel M, Palmer TD, Jucker M. Neurogenesis and alterations of neural stem cells in mouse models of cerebral amyloidosis. Am J Pathol. 2008;172:1520–1528. doi: 10.2353/ajpath.2008.060520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Scuderi C, Lu J, Savani C, De Filippis D, Iuvone T, Steardo L, Jr, Sheen V, Steardo L. S100B induces tau protein hyperphosphorylation via Dickopff-1 up-regulation and disrupts the Wnt pathway in human neural stem cells. J Cell Mol Med. 2008;12:914–927. doi: 10.1111/j.1582-4934.2008.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Yan S, Schmidt AM, Chen JX, Yan SS. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease. FASEB J. 2010;24:1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gefter JV, Shaufl AL, Fink MP, Delude RL. Comparison of distinct protein isoforms of the receptor for advanced glycation end-products expressed in murine tissues and cell lines. Cell Tissue Res. 2009;337:79–89. doi: 10.1007/s00441-009-0791-0. [DOI] [PubMed] [Google Scholar]
- Glabe CC. Amyloid accumulation and pathogenesis of Alzheimer's disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem. 2005;38:167–177. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66(2 Suppl 1):S74–S78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Castaneda RE, Galvez-Contreras AY, Luquín S, Gonzalez-Perez O. Neurogenesis in Alzheimer's disease: a realistic alternative to neuronal degeneration? Curr Signal Transduct Ther. 2011;6:314–319. doi: 10.2174/157436211797483949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilli M, Memo M. Transcriptional pharmacology of neurodegenerative disorders: novel venue towards neuroprotection against excitotoxicity? Mol Psychiatry. 1997;2:192–194. doi: 10.1038/sj.mp.4000252. [DOI] [PubMed] [Google Scholar]
- Grilli M, Meneghini V. NF-κB proteins in adult neurogenesis: relevance for learning and memory in physiology and pathology. In: Albensi BC, editor. Transcription factors CREB and NF-κB: involvement in synaptic plasticity and memory formation. Oak Park, IL: Bentham Sciences Publishers; 2012. pp. 199–250. [Google Scholar]
- Grilli M, Ferrari Toninelli G, Uberti D, Spano P, Memo M. Alzheimer's disease linking neurodegeneration with neurodevelopment. Funct Neurol. 2003;18:145–148. [PubMed] [Google Scholar]
- Gruden MA, Davidova TB, Malisauskas M, Sewell RD, Voskresenskaya NI, Wilhelm K, Elistratova EI, Sherstnev VV, Morozova-Roche LA. Differential neuroimmune markers to the onset of Alzheimer's disease neurodegeneration and dementia: autoantibodies to Abeta(25–35) oligomers, S100b and neurotransmitters. J Neuroimmunol. 2007;186:181–192. doi: 10.1016/j.jneuroim.2007.03.023. [DOI] [PubMed] [Google Scholar]
- Han SH, Kim YH, Mook-Jung I. RAGE: the beneficial and deleterious effects by diverse mechanisms of actions. Mol Cells. 2011;31:91–97. doi: 10.1007/s10059-011-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harashima A, Yamamoto Y, Cheng C, Tsuneyama K, Myint KM, Takeuchi A, Yoshimura K, Li H, Watanabe T, Takasawa S, Okamoto H, Yonekura H, Yamamoto H. Identification of mouse orthologue of endogenous secretory receptor for advanced glycation end-products: structure, function and expression. Biochem J. 2006;396:109–115. doi: 10.1042/BJ20051573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo C, Chang KA, Choi HS, Kim HS, Kim S, Liew H, Kim JA, Yu E, Ma J, Suh YH. Effects of the monomeric, oligomeric, and fibrillar Abeta42 peptides on the proliferation and differentiation of adult neural stem cells from subventricular zone. J Neurochem. 2007;102:493–500. doi: 10.1111/j.1471-4159.2007.04499.x. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, Grant PJ, Schmidt AM. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008;22:1572–1580. doi: 10.1096/fj.07-9909com. [DOI] [PubMed] [Google Scholar]
- Hyde LA, Kazdoba TM, Grilli M, Lozza G, Brusa R, Zhang Q, Wong GT, McCool MF, Zhang L, Parker EM, Higgins GA. Age-progressing cognitive impairments and neuropathology in transgenic CRND8 mice. Behav Brain Res. 2005;160:344–355. doi: 10.1016/j.bbr.2004.12.017. [Erratum (2005) 163:277; Brussa, Rosella (corrected to Brusa, Rossella)] [DOI] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I. Opportunities and challenges in developing Alzheimer disease therapeutics. Acta Neuropathol. 2011;122:543–549. doi: 10.1007/s00401-011-0878-z. [DOI] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A. 2004a;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004b;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalea AZ, Reiniger N, Yang H, Arriero M, Schmidt AM, Hudson BI. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009;23:1766–1774. doi: 10.1096/fj.08-117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development. 2003;130:391–399. doi: 10.1242/dev.00203. [DOI] [PubMed] [Google Scholar]
- Kleindienst A, McGinn MJ, Harvey HB, Colello RJ, Hamm RJ, Bullock MR. Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotrauma. 2005;22:645–655. doi: 10.1089/neu.2005.22.645. [DOI] [PubMed] [Google Scholar]
- Kolecki R, Lafauci G, Rubenstein R, Mazur-Kolecka B, Kaczmarski W, Frackowiak J. The effect of amyloidosis-beta and ageing on proliferation of neuronal progenitor cells in APP-transgenic mouse hippocampus and in culture. Acta Neuropathol. 2008;116:419–424. doi: 10.1007/s00401-008-0380-4. [DOI] [PubMed] [Google Scholar]
- Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107:2669–2674. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Yamamori H, Tatebayashi Y, Shafit-Zagardo B, Tanimukai H, Chen S, Iqbal K, Grundke-Iqbal I. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J Neuropathol Exp Neurol. 2008;67:78–84. doi: 10.1097/nen.0b013e318160c5db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem. 1997;272:16498–16506. doi: 10.1074/jbc.272.26.16498. [DOI] [PubMed] [Google Scholar]
- Li XH, Lv BL, Xie JZ, Liu J, Zhou XW, Wang JZ. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol Aging. 2012;33:1400–1410. doi: 10.1016/j.neurobiolaging.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- López-Toledano MA, Shelanski ML. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J Neurosci. 2004;24:5439–5444. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Toledano MA, Ali Faghihi M, Patel NS, Wahlestedt C. Adult neurogenesis: a potential tool for early diagnosis in Alzheimer's disease? J Alzheimers Dis. 2010;20:395–408. doi: 10.3233/JAD-2010-1388. [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- Lüth HJ, Ogunlade V, Kuhla B, Kientsch-Engel R, Stahl P, Webster J, Arendt T, Münch G. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer's disease brains. Cereb Cortex. 2005;15:211–220. doi: 10.1093/cercor/bhh123. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manev H, Uz T, Manev R. Glia as a putative target for antidepressant treatments. J Affect Disord. 2003;75:59–64. doi: 10.1016/s0165-0327(02)00044-7. [DOI] [PubMed] [Google Scholar]
- Marlatt MW, Lucassen PJ. Neurogenesis and Alzheimer's disease: biology and pathophysiology in mice and men. Curr Alzheimer Res. 2010;7:113–125. doi: 10.2174/156720510790691362. [DOI] [PubMed] [Google Scholar]
- Maślińska D, Laure-Kamionowska M, Taraszewska A, Deręgowski K, Maślińska S. Immunodistribution of amyloid beta protein (Aβ) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol. 2011;49:295–300. [PubMed] [Google Scholar]
- Meneghini V, Francese MT, Carraro L, Grilli M. A novel role for the receptor for advanced glycation end-products in neural progenitor cells derived from adult subventricular zone. Mol Cell Neurosci. 2010;45:139–150. doi: 10.1016/j.mcn.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki I, Watanabe T, Kawaguchi M, Akatsu H, Tsuneyama K, Yamamoto Y, Ohe K, Yonekura H, Yamada M, Yamamoto H. Reduced expression of endogenous secretory receptor for advanced glycation endproducts in hippocampal neurons of Alzheimer's disease brains. Arch Histol Cytol. 2007;70:279–290. doi: 10.1679/aohc.70.279. [DOI] [PubMed] [Google Scholar]
- Perrone L, Sbai O, Nawroth PP, Bierhaus A. The complexity of sporadic Alzheimer's disease pathogenesis: the role of RAGE as therapeutic target to promote neuroprotection by inhibiting neurovascular dysfunction. Int J Alzheimers Dis. 2012;2012:734956. doi: 10.1155/2012/734956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietze R, Poulin P, Weiss S. Mitotically active cells that generate neurons and astrocytes are present in multiple regions of the adult mouse hippocampus. J Comp Neurol. 2000;424:397–408. [PubMed] [Google Scholar]
- Rodríguez JJ, Verkhratsky A. Neurogenesis in Alzheimer's disease. J Anat. 2011;219:78–89. doi: 10.1111/j.1469-7580.2011.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25:206–212. doi: 10.1097/WAD.0b013e318204b550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N, Toki S, Chowei H, Saito T, Nakano N, Hayashi Y, Takeuchi M, Makita Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer's disease. Brain Res. 2001;888:256–262. doi: 10.1016/s0006-8993(00)03075-4. [DOI] [PubMed] [Google Scholar]
- Schaeffer EL, Novaes BA, da Silva ER, Skaf HD, Mendes-Neto AG. Strategies to promote differentiation of newborn neurons into mature functional cells in Alzheimer brain. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1087–1102. doi: 10.1016/j.pnpbp.2009.06.024. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Bigl K, Meiners I, Schmitt J. Induction of reactive oxygen species and cell survival in the presence of advanced glycation end products and similar structures. Biochem Biophys Acta. 2006;1763:927–936. doi: 10.1016/j.bbamcr.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Seki T, Arai Y. Age-related production of new granule cells in the adult dentate gyrus. Neuroreport. 1995;6:2479–2482. doi: 10.1097/00001756-199512150-00010. [DOI] [PubMed] [Google Scholar]
- Shepherd CE, Goyette J, Utter V, Rahimi F, Yang Z, Geczy CL, Halliday GM. Inflammatory S100A9 and S100A12 proteins in Alzheimer's disease. Neurobiol Aging. 2006;27:1554–1563. doi: 10.1016/j.neurobiolaging.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Shuvaev VV, Laffont I, Serot JM, Fujii J, Taniguchi N, Siest G. Increased protein glycation in cerebrospinal fluid of Alzheimer's disease. Neurobiol Aging. 2001;22:397–402. doi: 10.1016/s0197-4580(00)00253-0. [DOI] [PubMed] [Google Scholar]
- Silverberg GD, Miller MC, Messier AA, Majmudar S, Machan JT, Donahue JE, Stopa EG, Johanson CE. Amyloid deposition and influx transporter expression at the blood-brain barrier increase in normal aging. J Neuropathol Exp Neurol. 2010;69:98–108. doi: 10.1097/NEN.0b013e3181c8ad2f. [DOI] [PubMed] [Google Scholar]
- Takata K, Kitamura Y, Kakimura J, Shibagaki K, Tsuchiya D, Taniguchi T, Smith MA, Perry G, Shimohama S. Role of high mobility group protein-1 (HMG1) in amyloid-beta homeostasis. Biochem Biophys Res Commun. 2003;301:699–703. doi: 10.1016/s0006-291x(03)00024-x. [DOI] [PubMed] [Google Scholar]
- Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, Sosunov A, McKhann G, Funatsu Y, Nakamichi N, Nagai T, Mizoguchi H, Ibi D, Hori O, Ogawa S, Stern DM, Yamada K, Yan SS. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci U S A. 2009;106:20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente MM, Bortolotto V, Cuccurazzu B, Ubezio F, Meneghini V, Francese MT, Canonico PL, Grilli M. α2δ ligands act as positive modulators of adult hippocampal neurogenesis and prevent depression-like behavior induced by chronic restraint stress. Mol Pharmacol. 2012;82:271–280. doi: 10.1124/mol.112.077636. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Yan SD, Stern D, Schmidt AM. What's the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest. 1997;27:179–181. doi: 10.1046/j.1365-2362.1996.00072.x. [DOI] [PubMed] [Google Scholar]
- Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, Okamoto H, Watanabe T, Yamamoto H. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003;370:1097–1109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, He J, Zhang Y, Luo H, Zhu S, Yang Y, Zhao T, Wu J, Huang Y, Kong J, Tan Q, Li XM. Increased hippocampal neurogenesis in the progressive stage of Alzheimer's disease phenotype in an APP/PS1 double transgenic mouse model. Hippocampus. 2009;19:1247–1253. doi: 10.1002/hipo.20587. [DOI] [PubMed] [Google Scholar]
- Ziabreva I, Perry E, Perry R, Minger SL, Ekonomou A, Przyborski S, Ballard C. Altered neurogenesis in Alzheimer's disease. J Psychosom Res. 2006;61:311–316. doi: 10.1016/j.jpsychores.2006.07.017. [DOI] [PubMed] [Google Scholar]