

Abstract
Pain is a complex experience composed of sensory and affective components. Although the neural systems of the sensory component of pain have been studied extensively, those of its affective component remain to be determined. In the present study, we examined the effects of corticotropin-releasing factor (CRF) and neuropeptide Y (NPY) injected into the dorsolateral bed nucleus of the stria terminalis (dlBNST) on pain-induced aversion and nociceptive behaviors in rats to examine the roles of these peptides in affective and sensory components of pain, respectively. In vivo microdialysis showed that formalin-evoked pain enhanced the release of CRF in this brain region. Using a conditioned place aversion (CPA) test, we found that intra-dlBNST injection of a CRF1 or CRF2 receptor antagonist suppressed pain-induced aversion. Intra-dlBNST CRF injection induced CPA even in the absence of pain stimulation. On the other hand, intra-dlBNST NPY injection suppressed pain-induced aversion. Coadministration of NPY inhibited CRF-induced CPA. This inhibitory effect of NPY was blocked by coadministration of a Y1 or Y5 receptor antagonist. Furthermore, whole-cell patch-clamp electrophysiology in dlBNST slices revealed that CRF increased neuronal excitability specifically in type II dlBNST neurons, whereas NPY decreased it in these neurons. Excitatory effects of CRF on type II dlBNST neurons were suppressed by NPY. These results have uncovered some of the neuronal mechanisms underlying the affective component of pain by showing opposing roles of intra-dlBNST CRF and NPY in pain-induced aversion and opposing actions of these peptides on neuronal excitability converging on the same target, type II neurons, within the dlBNST.
Introduction
Pain is a complex experience consisting of sensory and affective components. Although the neural systems of the sensory component of pain have been studied extensively, those of the negative affective component are only beginning to be understood. Recently, some behavioral studies using a conditioned place paradigm have revealed neural mechanisms underlying the negative affective component of pain. Johansen et al. (2001) reported the crucial role of the anterior cingulate cortex in conditioned place aversion (CPA) induced by the intraplantar injection of formalin. We reported that the central amygdaloid nucleus and basolateral amygdaloid nucleus (BLA) were differently involved in intraplantar formalin-induced and intraperitoneal acetic acid-induced CPA (Tanimoto et al., 2003). In addition to these brain areas, we found that the excitotoxic lesions of the bed nucleus of the stria terminalis (BNST) reduced pain-induced aversion without reducing nociceptive behaviors (Deyama et al., 2007). Moreover, we recently demonstrated that noradrenergic neurotransmission within the ventral part of BNST mediated the negative affective component of pain (Deyama et al., 2008, 2009, 2011).
The dorsolateral part of the BNST (dlBNST) is densely innervated with corticotropin-releasing factor (CRF)-containing fibers (Sakanaka et al., 1986; Morin et al., 1999) and expresses CRF receptors (Van Pett et al., 2000). Intra-BNST CRF has been implicated in negative affective states, such as anxiety, fear, and aversion. Intra-BNST infusion of CRF has been shown to elicit anxiety-associated behaviors in the elevated plus maze test (Sahuque et al., 2006) and to enhance startle responses (Lee and Davis, 1997). Furthermore, it has been reported that intra-BNST administration of CRF produced CPA (Sahuque et al., 2006). However, the role of CRF-mediated neurotransmission within the dlBNST in the negative affective component of pain remains unclear.
Neuropeptide Y (NPY)-containing fibers are also observed in the dlBNST (Walter et al., 1991), and its receptors, Y1, Y2, Y4, and Y5 subtypes, are expressed in this brain region (Parker and Herzog, 1999). Although a large body of literature exists on the anxiolytic and anti-aversive effects of NPY (Heilig, 1995; Kask et al., 1997; Nakajima et al., 1998; Gutman et al., 2008), the role of this peptide in pain-induced aversion remains to be determined.
In this study, we examined the effects of intra-dlBNST injection of CRF and NPY on pain-induced aversion and nociceptive behaviors to examine the roles of these peptides in affective and sensory components of pain, respectively, and revealed their opposing roles in the negative affective component of pain. Furthermore, whole-cell patch-clamp electrophysiology in dlBNST slices showed opposing actions of these peptides on neuronal excitability specifically in type II dlBNST neurons.
Materials and Methods
Animals.
Male Sprague Dawley rats (Japan SLC) (180–280 g or 20 to 50 d old) were used for the behavioral or electrophysiological experiments, respectively. The rats were maintained at a constant ambient temperature (22 ± 1°C) under a 12 h light/dark cycle with food and water available ad libitum. All experiments were performed with the approval of the Institutional Animal Care and Use Committee at Hokkaido University.
Drugs.
NBI27914 (a selective CRF1 receptor antagonist), antisauvagine-30 (AS-30; a selective CRF2 receptor antagonist), and L-152,804 (a selective NPY Y5 receptor antagonist) were purchased from Tocris Bioscience. CRF was purchased from Peptide Institute or Bachem AG. NPY and BIBP3226 (a selective NPY Y1 antagonist) were from Abgent and Bachem AG, respectively. Tetrodotoxin (TTX) was from Wako Pure Chemical Industries and SR95531, ZD7288, and kynurenic acid were from Sigma.
For the behavioral experiments, AS-30 was dissolved in PBS, pH 7.4, containing 0.1% bovine serum albumin (BSA; Sigma). NBI27914 was dissolved in dimethylsulfoxide (DMSO), then diluted with PBS containing 0.1% BSA; the solution contained DMSO at a final concentration of 8.4%. The final concentration of these antagonists was 0.3 nmol/0.5 μl or 1.0 nmol/0.5 μl. NPY was dissolved in PBS containing 0.1% BSA at a concentration of 0.1 nmol/0.5 μl or 0.3 nmol/0.5 μl. CRF was dissolved in saline containing 0.1% BSA and 0.03% acetic acid, then mixed with PBS containing 0.1% BSA at a ratio of 2:1. The final concentration of CRF was 0.1 nmol/0.6 μl or 0.3 nmol/0.6 μl. When the effects of NPY in the presence or absence of NPY antagonists on CRF-induced CPA were examined, CRF solution was mixed with these drugs dissolved in PBS containing 0.1% BSA at a ratio of 2:1. L-152,804 was dissolved in DMSO, then diluted with PBS containing 0.1% BSA; the solution contained DMSO at a final concentration of 16.7%.
For the electrophysiological experiments, the stock solutions for CRF and NPY were prepared at concentrations of 1 mm in H2O containing 0.1% BSA. The stock solutions for NBI27914 and AS-30 were prepared at a concentration of 300 μm in DMSO and in H2O containing 0.1% BSA, respectively. The stock solutions for BIBP3226 and L-152,804 were prepared at a concentration of 1 mm in H2O and in DMSO, respectively. The stock solution for ZD7288 was prepared at a concentration of 10 mm in H2O. These stock solutions were stored at –30°C until use. Before bath application, these stock solutions were diluted to final concentrations with normal Ringer's solution containing 0.01% BSA. We confirmed that bath application of DMSO alone at the final concentration (0.1%) did not affect either membrane potential or input resistance (data not shown).
In vivo microdialysis.
In vivo microdialysis was performed using a peptide microdialysis system (AtmosLM; Eicom). Under sodium pentobarbital anesthesia (50 mg/kg, i.p.), each rat was implanted unilaterally with a microdialysis guide cannula (outer diameter, o.d.; 0.72 mm, PEG-6; Eicom) 1.0 mm above the dlBNST (–0.3 mm rostral, 1.7 mm lateral, 5.5 mm ventral to bregma) (Paxinos and Watson, 1998). After surgery, rats were housed individually in cages. One day after the implantation of the guide cannula, microdialysis experiments were performed in unanesthetized and freely moving rats. Microdialysis probes (dialysis membrane: 1000 kDa molecular weight cutoff polyethylene membrane, length 1.0 mm; o.d., 0.44 mm; PEP-6–01; Eicom) were inserted through the guide cannula and continuously perfused with Ringer's solution (Na+ 147 mm, K+ 4 mm, Ca2+ 2.3 mm, and Cl− 155.6 mm) containing 0.15% BSA at a flow rate of 10 μl /min. The rats were then placed in a Plexiglas chamber (30 × 30 × 35 cm: width × length × height) for the 2 h preconditioning period. After the preconditioning period, the flow rate was changed to 1 μl/min. After an additional stabilization period of 1 h, 11 15 min dialysates were collected in polypropylene tubes at 4°C. The first three samples were taken as baseline samples. Immediately after collection of the last baseline sample, each rat was administered an intraplantar injection (right hindpaw) of 100 μl of 2% formalin. Dialysate samples were stored at –20°C. CRF concentrations in the samples were measured using a competitive enzyme immunoassay kit (Phoenix Pharmaceuticals).
Surgery and microinjection.
Under sodium pentobarbital anesthesia (50 mg/kg, i.p.), rats were implanted bilaterally with 25 gauge stainless steel guide cannulae (o.d., 0.5 mm; inner diameter, i.d., 0.22 mm) 1.5 mm above the dlBNST (–0.3 mm rostral, 1.7 mm lateral, 5.0 mm ventral to bregma) or lateral ventricle (–0.3 mm rostral, 1.4 mm lateral, 2.7 mm ventral to bregma) (Paxinos and Watson, 1998). After surgery, rats were housed individually in plastic cages with woodchip bedding, allowed to recover for at least 5 d, and handled for 1–2 min each day for 3 consecutive days before behavioral experiments. For microinjection, 33 gauge stainless steel injection cannulae (o.d., 0.2 mm; i.d., 0.08 mm) were inserted bilaterally into the guide cannulae. The injection cannulae protruded 1.5 mm from the tip of the guide cannulae to reach the dlBNST or lateral ventricle. Injection cannulae were attached to a microinjection pump (CMA) via PE 8 polyethylene tubing. Drug or vehicle was administrated bilaterally in a volume of 0.5–0.6 μl/side at a rate of 0.5 μl/min, and the injection cannulae were left in place for an additional 1 min after microinjection to prevent backflow.
Conditioned CPA.
CPA tests were conducted as described previously (Deyama et al., 2007, 2008, 2009). A shuttle box composed of two equal-sized compartments (30 × 30 × 30 cm) with distinct visual and tactile cues (one compartment was black with a smooth floor, and the other was white with a textured floor) under dim illumination (25 ± 5 lux at the center of the box) was used for a 4 consecutive day experimental procedure. On day 1 (habituation session) and day 2 (preconditioning session), rats explored the two compartments ad libitum for 900 s; the time spent in each compartment during the exploring period was measured automatically (KN-80; Natsume Seisakusho). Rats that spent >80% (>720 s) of the total time (900 s) in one side on day 2 or showed a difference of >200 s in the time spent in one side between days 1 and 2 were eliminated from subsequent procedures. Additionally, after behavioral tests, histological analyses were performed, and data from rats with misplacement of both or either of the bilateral microinjection cannulae were eliminated from statistical analyses. Both before and after such eliminations, no significant difference (p > 0.05 (n = 353) and p > 0.05 (n = 197), respectively) was observed between the time spent in the black (442.6 ± 6.3 s and 445.8 ± 7.1 s, respectively) and white (457.4 ± 6.3 s and 454.2 ± 7.1 s, respectively) compartments, indicating the absence of any significant bias in compartment preference before conditioning.
Formalin-induced CPA was used to evaluate the affective component of pain (Johansen et al., 2001; Tanimoto et al., 2003; Johansen and Fields, 2004; Deyama et al., 2008, 2011). In this CPA test, we used a bias-like protocol (Tzschentke, 1998). Specifically, we designated the compartment in which each rat spent more time (>450 s) on day 2 (preconditioning session) as each animal's pain-paired compartment. This type of protocol was successfully used to examine the CPA induced by opioid withdrawal (Kosten, 1994; Rafieian-Kopaei et al., 1995; Nakagawa et al., 2005) and by visceral and somatic pain (Tanimoto et al., 2003; Deyama et al., 2007, 2008, 2009). On day 3 (conditioning session), place conditioning was performed as follows. In the vehicle control session (conducted between 08:00 and 12:00), each rat was given an intraplantar injection of saline (100 μl) into the left hindpaw and then immediately confined in the nonpain-paired compartment for 1 h. After at least 4 h, in the pain-conditioning session (conducted between 14:00 and 18:00), each rat was injected with NBI27914 (0.3 nmol/side or 1 nmol/side), AS-30 (0.3 nmol/side or 1 nmol/side), NPY (0.1 nmol/side or 0.3 nmol/side), or vehicle into the bilateral dlBNST or lateral ventricle. At 10 min after the intra-dlBNST injection, the rats were given an intraplantar injection of 2% formalin (100 μl) into the right hindpaw and then confined in the pain-paired compartment for 1 h. On day 4 (test session), each rat was allowed to explore the two compartments ad libitum, and the time spent in each compartment during the exploring period (900 s) was recorded automatically. CPA scores were calculated by subtracting the time spent in the pain-paired compartment during the test session from the time spent in this compartment during the preconditioning session.
In the experiments investigating intra-dlBNST CRF-induced aversion (a 6 consecutive day experimental protocol), we designated the compartment in which the rat spent more time (>450 s) on day 2 (preconditioning session) as the drug-paired compartment for each animal. On days 3–5, place conditioning was performed over 3 consecutive days as follows. The rats were divided into two groups (Groups 1 and 2). In the morning session (conducted between 08:00 and 12:00), rats in Group 1 were given an intra-dlBNST injection of drugs or vehicle and were immediately confined in the drug-paired compartment for 30 min. On the other hand, rats in Group 2 were confined in the nondrug-paired compartment for 30 min without being given an intra-dlBNST injection. In the afternoon session (conducted between 13:00 and 18:00), rats in Group 1 were confined in the nondrug-paired compartment for 30 min without being given an intra-dlBNST injection, and rats in Group 2 were given an intra-dlBNST injection of drugs or vehicle and were immediately confined in the drug-paired compartment for 30 min. On day 6, in the test session, each rat was allowed to explore the two compartments freely, and the time spent in each compartment during the exploring period (900 s) was recorded. CPA scores were calculated as in the CPA test for pain-induced aversion.
Formalin test.
Formalin-induced nociceptive behaviors were examined to evaluate the sensory component of pain (Johansen et al., 2001; Tanimoto et al., 2003; Johansen and Fields, 2004; Deyama et al., 2008, 2011). Each rat was placed in a Plexiglas cylinder (30 cm diameter; 30 cm height) for 30 min to acclimatize it to the experimental environment. Drugs or vehicle were injected bilaterally into the dlBNST of each rat, and the animals were returned to the cylinder. At 10 min after the intra-dlBNST injection, the rats were given an intraplantar injection of 2% formalin (100 μl) into the right hindpaw and immediately returned to the cylinder. The amount of time the rat spent lifting, licking, shaking, or biting the injected paw was measured for each 5 min period over 60 min. Measurement of nociceptive behaviors was done live by an observer blind to the treatment conditions. Nociception was quantified using a rating scale method by assigning weights to the following categories of nociceptive behaviors: category 0 = weight is evenly distributed among all paws; category 1 = injected paw is lifted; and category 2 = injected paw is licked, shaken, or bitten. The nociceptive score was calculated for each 5 min (300 s) period using the following formula: nociceptive score = [{time (s) spent with lifting the injected paw} × 1 + {time (s) spent with licking, shaking, or biting the injected paw} × 2]/300 (s).
Histology.
After the in vivo microdialysis experiments and behavioral tests, histological analyses were performed. Rats were decapitated, and brains were removed rapidly and frozen in powdered dry ice. Coronal sections (50 μm) including the BNST were prepared with a cryostat, thaw-mounted onto slides, stained with thionine, and examined under a microscope (×40). Data from the rats with extensive tissue damage, misplacement of the microdialysis probes, or misplacement of both or either of the bilateral injection cannulae were excluded from statistical analyses.
Slice preparation for electrophysiology.
Rats were decapitated under isoflurane anesthesia, and the brains were quickly removed and submerged in ice-cold artificial CSF containing the following (in mm): 130 NaCl, 3.5 KCl, 1.1 KH2PO4, 1.0 CaCl2, 6.0 MgCl2, 30 NaHCO3, 10 glucose, and 2 kynurenic acid, saturated with 95% O2/5% CO2. Coronal slices (250 μm thick) containing the BNST region were prepared using a microslicer (VT1200S; Leica). Slices were incubated in a chamber containing normal Ringer's solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, 26 NaHCO3, and 25 glucose, saturated with 95% O2/5% CO2 at 33–35°C for 0.5 h, and then placed at room temperature for >0.5 h before recording.
Recording procedures for electrophysiology.
Slices were placed in a recording chamber on an upright microscope (BX-51WI; Olympus) and continuously superfused with normal Ringer's solution (34°C) saturated with 95% O2/5% CO2 at a flow rate of 1–1.5 ml/min. Pipettes were pulled from thin-walled borosilicate glass capillaries with a micropipette puller (Model P-1000; Sutter Instrument). Tip resistance was 5–8 MΩ when pipettes were filled with an internal solution containing the following (in mm): 150 KOH, 2 MgCl2, 10 KCl, 0.2 EGTA, 2 Na2-ATP, 0.3 Na2-GTP, 10 HEPES, and 0.1 spermine. The pH was adjusted to 7.3–7.4 with gluconic acid. Recordings were obtained from dlBNST neurons visualized with infrared video microscopy (model IR-1000; Dage-MTI). Because dlBNST neurons have been categorized into three distinct types (Hammack et al., 2007), we first identified cell types by assessing the membrane potential responses to hyperpolarizing and depolarizing current injections. To monitor hyperpolarizing responses, we set the initial membrane potential at –60 mV and incremental currents (400 ms) ranging from 0 to −360 pA were injected. To monitor depolarizing and firing responses, we set the initial membrane potential at –80 mV, and incremental currents (400 ms) ranging from 0 to 160 pA were injected. Neurons that exhibited a depolarizing sag in response to hyperpolarizing current injection and a regular firing pattern in response to depolarizing current injection were classified as type I. Neurons that exhibited a depolarizing sag in response to hyperpolarizing current injection and burst firings in response to both the termination of the hyperpolarizing current injections and depolarizing current injections were classified as type II. Neurons that exhibited no depolarizing sag in response to hyperpolarizing current injection and a regular firing pattern in response to depolarizing current injection were classified as type III. Neurons were excluded from analyses if the amplitude of the action potential, which was determined as the difference between threshold and the peak, was <30 mV. Data were acquired with a Multiclamp 700B amplifier and the pClamp10 software (Molecular Devices).
Measurement of membrane potentials and input resistance.
The initial membrane potentials were set to approximately –70 mV (–69.32 ± 0.23 mV, n = 50) by injecting negative or positive currents. To inhibit action potential-induced synaptic transmission, 500 nm TTX was added to superfused Ringer's solution. To monitor the changes in membrane potentials, a window with a 1 s duration was placed every 5 s, and the average membrane potential within the window was determined. To monitor changes in input resistance, input resistance was calculated by measuring the change in membrane potential evoked by a negative current (–80 pA, 200 ms) every 5 s. Then, the membrane potentials and input resistance during a 1 min period were calculated by averaging 12 of these values. After confirming stable membrane potentials and input resistance for >3 min, 1 μm CRF or 1 μm NPY was perfused through the recording chamber for 2 min except for the experiment shown in Figure 8, where NPY was perfused until the end of the experiment. In experiments assessing the effects of antagonists for CRF or NPY receptors on CRF- or NPY-induced change in membrane potentials, the antagonists were perfused for >10 min before bath application of CRF or NPY to the end of the experiments. Membrane potentials and input resistance were measured in the periods of 0–3 min before and 13–16 min after drug application. The latter period was chosen because the effects of CRF and NPY emerged gradually and became maximal in this period in most of the neurons tested. The effects of drugs were evaluated by comparing the average values observed during these two periods.
Figure 8.

Inhibitory effects of NPY on CRF-induced depolarization in type II dlBNST neurons. A, Representative trace from current-clamp recording in a type II dlBNST neuron. The effects of the peptides were evaluated in the periods indicated by shading (Control, 0–3 min before NPY application; NPY, 0–3 min before CRF application; NPY + CRF, 12–15 min after CRF application). B, C, Effects of CRF on membrane potential (B) and input resistance (C) in the presence of NPY. Gray symbols and lines show data obtained from individual neurons (n = 5), and black symbols and lines show averaged data obtained from five neurons. *p < 0.05 compared with control (Newman–Keuls post hoc test).
Analyses of steady-state membrane current–voltage relationships.
Steady-state membrane current–voltage (I–V) relationships were examined in type II dlBNST neurons. Cells were voltage clamped at –70 mV in the presence of 1 μm TTX. To assess the I–V relationship, we used a voltage ramp protocol described in a previous study (Hara and Nakaya, 1997) with some modifications. Briefly, the membrane potential was first depolarized to 0 mV and then hyperpolarized to –120 mV at a rate of 0.1 mV/ms every 20 s. The current responses observed during the falling phase from –30 mV to –120 mV were used for I–V relationship analyses. Changes in net currents caused by a 2 min bath application of CRF or NPY were determined by subtracting the current responses to the voltage ramp in the periods of 0–3 min before drug application from those of 13–16 min after drug application. In some experiments, effects of NPY on I–V relationships were examined in the presence of ZD7288.
To measure holding currents, a window with a 1 s duration was placed every 20 s, and the average holding current within the window was determined. To monitor the changes in input resistance, input resistance was calculated by measuring the change in the holding current evoked by voltage steps (–5 mV, 30 ms) every 20 s. Holding currents and input resistance were measured in the periods of 0–3 min before and 13–16 min after drug application. The effects of drugs were evaluated by comparing the average values observed at these two periods.
Measurement of firing activity.
To measure changes in firing activity, current-clamp recordings without current injection were carried out. To inhibit GABAA receptor-mediated and ionotropic glutamate receptor-mediated synaptic transmission, 10 μm SR95531 and 1 mm kynurenic acid, respectively, were added to superfused Ringer's solution. To analyze firing activity, a window with 2.5 s duration was placed every 5 s, and the numbers of spikes in 36 windows were counted during 3 min. After counting basal firing activities, 1 μm CRF was perfused through the recording chamber for 2 min in the presence or absence of 1 μm NPY. NPY was perfused from 7.5 to 11 min before CRF application to the end of the experiments. Firing activities were measured during a period of 0–3 min before the application of peptides and 17–20 min after CRF application. The latter period was chosen because maximum firing activities were observed in this period in most of the neurons tested. The effects of drugs were evaluated by comparing the average values observed during these periods.
Statistical analyses.
Data are expressed as means ± SEM. In vivo microdialysis data were assessed using one-way repeated-measures ANOVA followed by the Newman–Keuls post hoc test. Time spent in the pain- or drug-paired compartment during preconditioning and test sessions in the CPA test was analyzed using within-group paired t tests. CPA scores were analyzed using one-way ANOVA followed by the Newman–Keuls post hoc test or Student's t test for comparisons among more than two groups or between two groups, respectively. Two-way repeated-measures ANOVA was used for the data from measurements of nociceptive behaviors. Electrophysiological data were analyzed using the paired t test, Student's t test, or one-way repeated-measures ANOVA followed by the Newman–Keuls post hoc test. Statistical analyses were performed using IBM SPSS statistics v.20.0.0 or using GraphPad Prism v.6.00 (GraphPad Software); p values < 0.05 were considered to indicate statistically significant differences.
Results
Histology
After the in vivo microdialysis experiments and behavioral tests, histological analyses were performed. Data from rats with correct placements of the microdialysis probe (Fig. 1A) and of the bilateral microinjection cannulae (Fig. 1B–E) were used for the statistical analyses.
Figure 1.

The placements of the tips of the microdialysis probes (A) and microinjection cannulae (B–E). B, NBI27914 1.0 nmol, closed circles; 0.3 nmol, open circles; AS-30 1.0 nmol, closed triangles; 0.3 nmol, open triangles; vehicle, open rhomboids. C, NPY 0.3 nmol, closed squares; 0.1 nmol, open squares; vehicle, open rhomboids. D, CRF 0.3 nmol, closed circles; 0.1 nmol, open circles; vehicle, open rhomboids. E, CRF, closed circles; CRF+NPY, closed squares; CRF+NPY+BIBP3226, open reversed triangles; CRF+NPY+ L-152,804, open triangles. The illustrations of coronal sections were taken from the atlas of Paxinos and Watson (1998); 0.2, –0.26, –0.4, and –0.8 indicate distances (mm) from bregma.
Pain-induced CRF release within the dlBNST
The averaged baseline concentration of CRF in the dialysates was 6.8 ± 0.5 pg/15 μl. Intraplantar injection of formalin produced a transient increase in extracellular CRF levels within the dlBNST (Fig. 2). One-way repeated-measures ANOVA demonstrated a significant effect of the formalin injection (F(10,76) = 4.50, p < 0.001, n = 7). A significant increase in the CRF level was observed at 15–30 min after the formalin injection (186 ± 14%; p < 0.01 compared with the last baseline sample (–15 to 0 min), Newman–Keuls post hoc test).
Figure 2.

Effects of intraplantar injection of formalin (n = 7) on extracellular CRF levels in the dlBNST were examined. The arrow indicates the time point of intraplantar injection. Data are expressed as means ± SEM the percentage baseline control value, calculated as an average of three consecutive dialysates before intraplantar injection. ##p < 0.01 compared with the value just before intraplantar injection (Newman–Keuls post hoc test).
Effects of intra-dlBNST injection of CRF receptor antagonists on formalin-induced CPA and nociceptive behaviors
To determine the role of CRF-mediated neurotransmission within the dlBNST in the affective component of pain, the effects of bilateral intra-dlBNST injection of NBI27914 (CRF1 receptor antagonist) and AS-30 (CRF2 receptor antagonist) on formalin-induced CPA were examined. In the intra-dlBNST vehicle-injected rats, the time spent in the pain-paired compartment during the test session (374 ± 33 and 391 ± 18 s in Fig. 3A,B, respectively) was significantly shorter (t = 4.55 (df = 6), p < 0.01 and t = 4.91 (df = 5), p < 0.01, respectively, paired t test) than the time during the preconditioning session (529 ± 19 and 538 ± 29 s). In the intra-dlBNST NBI27914 (0.3 and 1 nmol/side)-injected rats, no significant difference (t = 2.19 (df = 7), p > 0.05 and t = 1.82 (df = 6), p > 0.05, respectively, paired t test) was observed in the time spent in the pain-paired compartment between the test (480 ± 50 and 508 ± 41 s, respectively) and the preconditioning (551 ± 28 and 546 ± 32 s, respectively) sessions. Also, in the intra-dlBNST AS-30 (0.3 and 1 nmol/side)-injected groups, no significant difference (t = 1.56 (df = 5), p > 0.05 and t = 1.51 (df = 6), p > 0.05, respectively, paired t test) was observed in the time spent in the pain-paired compartment between the test (475 ± 20 and 502 ± 24 s, respectively) and the preconditioning (533 ± 32 and 535 ± 24 s, respectively) sessions. CPA scores showed dose-dependent attenuation of formalin-induced CPA by intra-dlBNST injection of these antagonists. As shown in Figure 3, C and D, one-way ANOVA indicated a significant difference among groups (NBI27914, F(2,19) = 3.87, p < 0.05 and AS-30, F(2,16) = 5.96, p < 0.05). Post hoc comparisons revealed that NBI27914 at a dose of 1 nmol/side (37.6 ± 20.7 s; p < 0.05), but not 0.3 nmol/side (71.3 ± 32.5 s; p > 0.05), and AS-30 at doses of 0.3 and 1 nmol/side (58.3 ± 20.8 s, p < 0.05 and 33.3 ± 22.1 s, p < 0.05, respectively) attenuated formalin-induced CPA significantly compared with the vehicle-injected group (155 ± 34 s and 147 ± 30, respectively).
Figure 3.

A–D, Effects of intra-dlBNST injection of NBI27914 (A, C, vehicle, n = 7; 0.3 nmol, n = 8; 1.0 nmol, n = 7) or AS-30 (B, D, vehicle, n = 6; 0.3 nmol, n = 6; 1.0 nmol, n = 7) on formalin-induced CPA. Data are expressed as means ± SEM. The columns show the time spent in the pain-paired compartment in the preconditioning (white columns) and test (black columns) sessions (A, B). ##p < 0.01 compared with the preconditioning session (paired t test). The columns show the CPA scores (C, D). #p < 0.05 compared with vehicle-injected rats (Newman–Keuls post hoc test). E, Effects of intra-dlBNST injection of vehicle (n = 11), NBI27914 (0.3 nmol, n = 6; 1.0 nmol, n = 7), or AS-30 (0.3 nmol, n = 6; 1.0 nmol, n = 6) on formalin-induced nociceptive behaviors. Data are expressed as means ± SEM.
Because the dlBNST is located close to the lateral ventricle, it was possible that drugs leaking to the lateral ventricle might act on brain regions other than the dlBNST and suppress formalin-induced CPA. Thus, off-site control experiments were performed by injecting these antagonists into the lateral ventricle at a dose of 1 nmol/side. In the intracerebroventricularly injected NBI27914 and AS-30 groups (n = 7 each), the time spent in the pain-paired compartment during the test session (382 ± 23 and 452 ± 29 s, respectively) was significantly shorter (t = 4.91 (df = 6), p < 0.01 and t = 3.61 (df = 6), p < 0.05, respectively, paired t test) than the time during the preconditioning session (482 ± 8 and 535 ± 20 s, respectively). Thus, these results showed no suppressing effects of intracerebroventricularly administered NBI27914 and AS-30 on formalin-induced CPA, suggesting that the dlBNST was the likely site of action of these antagonists in suppressing formalin-induced CPA.
To examine whether intra-dlBNST injection of NBI27914 or AS-30 per se produced conditioned place preference (CPP) or CPA, these drugs (1 nmol/side) were injected into the bilateral dlBNST in the absence of intraplantar formalin injection. In both the intra-dlBNST NBI27914- and AS-30-injected groups (n = 6 and n = 7, respectively), no significant difference (t = 1.06 (df = 5), p > 0.05 and t = 0.12 (df = 6), p > 0.05, respectively, paired t test) was observed in the time spent in the drug-paired compartment between the test (491 ± 22 and 487 ± 33 s, respectively) and preconditioning (509 ± 28 and 491 ± 10 s, respectively) sessions. CPA scores were 18.3 ± 17.3 and 4.14 ± 33.5 s, respectively, which were not significantly different (t = 0.21 (df = 9), p > 0.05 and t = 0.51 (df = 11), p > 0.05, respectively, Student's t test) from the CPA score of the vehicle-injected group (23.8 ± 19.2 s, n = 5 and 28.7 ± 34.8 s, n = 6, respectively). These data showed that neither CPP nor CPA was induced by the intra-dlBNST injection of these antagonists, indicating that these drugs have no motivational effect by themselves when injected into the dlBNST at these doses.
As shown in Figure 3E, intra-dlBNST injection of NBI27914 (0.3 nmol/side or 1 nmol/side) or AS-30 (0.3 nmol/side or 1 nmol/side) did not affect formalin-induced nociceptive behaviors compared with the vehicle-injected group. Two-way repeated-measures ANOVA revealed no significant effect of these drugs (F(4,31) = 1.05; p > 0.05) and no significant interaction between the drugs and time (F(44,341) = 0.89; p > 0.05). These results showed that intra-dlBNST injection of neither NBI27914 nor AS-30 affected the sensory component of pain.
Effects of intra-dlBNST injection of NPY on formalin-induced CPA and nociceptive behaviors
The effects of intra-dlBNST injection of NPY (0.1 and 0.3 nmol/side) on formalin-induced CPA were examined. In the intra-dlBNST vehicle-injected and NPY (0.1 nmol/side)-injected groups, the time spent in pain-paired compartment during the test session (408 ± 23 and 438 ± 33 s, respectively) was significantly shorter (t = 3.81 (df = 7), p < 0.01 and t = 4.20 (df = 6), p < 0.01, respectively, paired t test) than the time during the preconditioning session (553 ± 29 and 527 ± 22 s, respectively; Fig. 4A). In the intra-dlBNST NPY (0.3 nmol/side)-injected group, no significant difference (t = 0.57 (df = 6), p > 0.05, paired t test) was observed in the time spent in the pain-paired compartment between the test (504 ± 24 s) and preconditioning (520 ± 13 s) sessions. CPA scores showed dose-dependent attenuation of formalin-induced CPA by intra-dlBNST injection of NPY (Fig. 4B). One-way ANOVA indicated a significant difference among groups (F(2,19) = 4.51, p < 0.05). Post hoc comparisons revealed that NPY at a dose of 0.3 nmol/side (15.7 ± 27.3 s, p < 0.05), but not 0.1 nmol/side (89.0 ± 21.2 s), attenuated formalin-induced CPA significantly compared with the vehicle-injected group (145 ± 38.0 s).
Figure 4.
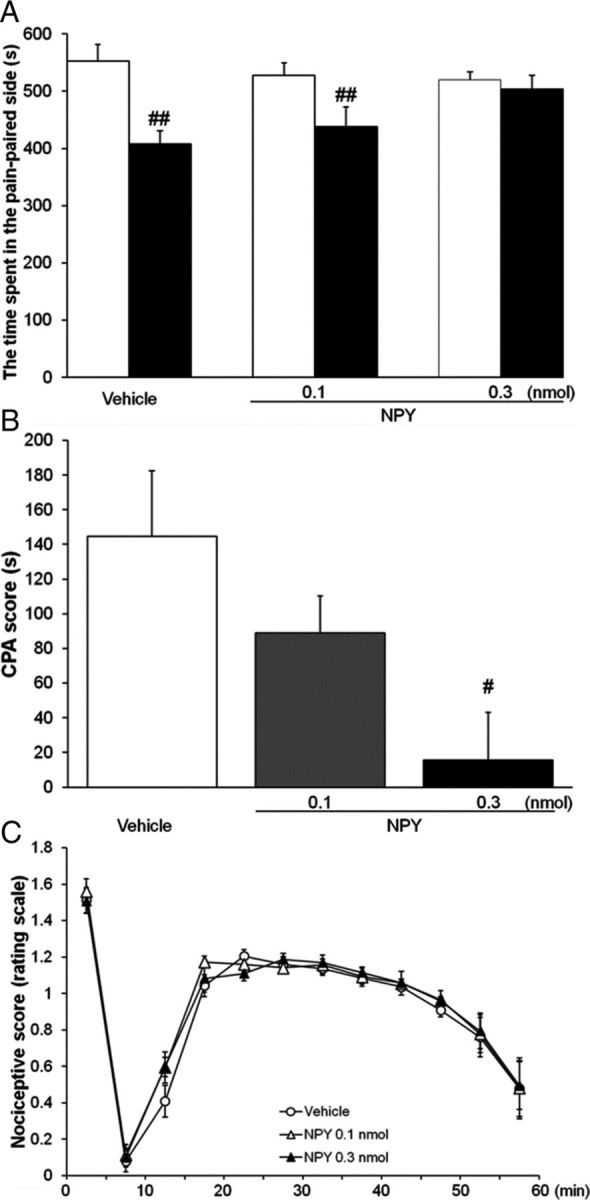
A, B, Effects of intra-dlBNST injection of vehicle (n = 8) or NPY (0.1 nmol, n = 7; 0.3 nmol, n = 7) on formalin-induced CPA. Data are expressed as means ± SEM. The columns show the time spent in the pain-paired compartment in the preconditioning (white columns) and test (black columns) sessions (A). ##p < 0.01 compared with the preconditioning session (paired t test). The columns show CPA scores (B). #p < 0.05 compared with vehicle-injected rats (Newman–Keuls post hoc test). C, Effects of intra-dlBNST injection of vehicle (n = 6) or NPY (0.1 nmol, n = 5; 0.3 nmol, n = 7) on formalin-induced nociceptive behaviors. Data are expressed as means ± SEM.
The effects of off-site control injections of NPY (0.3 nmol/side) into the lateral ventricle on formalin-induced CPA were examined. In the intracerebroventricular NPY-injected group (n = 7), the time spent in the pain-paired compartment during the test session was 448 ± 30 s, which was significantly shorter (t = 3.13 (df = 6), p < 0.05, paired t test) than the time during the preconditioning session (553 ± 25 s). This result showed no suppressing effect of intracerebroventricularly administered NPY on formalin-induced CPA, suggesting that the dlBNST was the likely site of action of this peptide in suppressing formalin-induced CPA.
To examine whether intra-dlBNST injection of NPY per se produced CPP or CPA, NPY (0.3 nmol/side) was injected into the bilateral dlBNST in the absence of intraplantar formalin injection. In the intra-dlBNST NPY-injected group (n = 5), no significant difference (t = 0.42 (df = 4), p > 0.05, paired t test) was observed in the time spent in the drug-paired compartment between the test (501 ± 40 s) and preconditioning (488 ± 10 s) sessions. The CPA score (–13.4 ± 32.3 s) was not significantly different (t = 0.09 (df = 7), p > 0.05, Student's t test) from that of the vehicle-injected group (–18.3 ± 41.5 s, n = 4). These findings showed that neither CPP nor CPA was induced by the intra-dlBNST injection of NPY, indicating that this peptide has no motivational effect by itself when injected into the dlBNST at this dose.
As shown in Figure 4C, intra-dlBNST injection of NPY (0.1 nmol/side or 0.3 nmol/side) did not affect formalin-induced nociceptive behaviors compared with the vehicle-injected group. Two-way repeated-measures ANOVA revealed no significant effect of intra-dlBNST NPY (F(2,15) = 0.22; p > 0.05) and no significant interaction between the drugs and time (F(22,165) = 0.48; p > 0.05). These results showed that intra-dlBNST injection of NPY did not affect the sensory component of pain.
Effect of intra-dlBNST injection of NPY on CRF-induced CPA
Sahuque et al. (2006) reported that intra-BNST administration of CRF produced a dose-dependent CPA. First, we confirmed the induction of CPA by the intra-dlBNST injection of CRF. As shown in Figure 5A, in rats injected with CRF at doses of 0.1 and 0.3 nmol/side, the time spent in the drug-paired compartment during the test session (434 ± 49 and 399 ± 34 s, respectively) was significantly shorter (t = 2.64 (df = 9), p < 0.05 and t = 6.50 (df = 10), p < 0.001, respectively, paired t test) than the time during the preconditioning session (534 ± 21 and 558 ± 13 s, respectively). CPA scores revealed a dose-dependent induction of CPA by intra-dlBNST injection of CRF (Fig. 5B). One-way ANOVA indicated a significant difference among groups (F(2,28) = 4.15, p < 0.05). Post hoc comparisons showed that CRF at a dose of 0.3 nmol/side (160 ± 25 s, p < 0.05), but not 0.1 nmol/side (99.4 ± 38.2 s, p > 0.05), significantly induced CPA compared with the vehicle-treated group (15.1 ± 44.1 s).
Figure 5.

Effect of intra-dlBNST injection of NPY on CRF-induced CPA. A, B, Effects of intra-dlBNST injection of vehicle (n = 10) or CRF (0.1 nmol, n = 10; 0.3 nmol, n = 11) were examined using a place conditioning paradigm. Data are expressed as means ± SEM. A, The columns show the time spent in the drug-paired compartment in the preconditioning (white columns) and test (black columns) sessions. #, ###p < 0.05, 0.001 compared with the preconditioning session (paired t test). B, The columns show CPA scores. #p < 0.05 compared with vehicle-injected rats (Newman–Keuls post hoc test). C, D, Effects of intra-dlBNST injection of NPY in the presence or absence of subtype-selective NPY antagonists on CRF-induced CPA (CRF, n = 10; CRF + NPY, n = 10; CRF + NPY + BIBP3226, n = 11; CRF +N PY + L-152,804, n = 12). C, The columns show the time spent in the drug-paired compartment in the preconditioning (white columns) and test (black columns) sessions. ##, ###p < 0.01, 0.001 compared with the preconditioning session (paired t test). D, The columns show CPA scores. ##p < 0.01 compared with CRF alone-injected rats; *p < 0.05 compared with CRF + NPY-injected rats (Newman–Keuls post hoc test).
Next, the effect of coadministration of NPY on CRF-induced CPA was examined. In the rats simultaneously injected with CRF (0.3 nmol/side) and NPY (0.3 nmol/side), no significant difference (t = 0.40 (df = 9), p > 0.05, paired t test) was observed in the time spent in the drug-paired compartment between the test (496 ± 34 s) and preconditioning (510 ± 18 s) sessions (Fig. 5C). Coadministration of NPY with CRF significantly (t = 3.43 (df = 18), p < 0.01, Student's t test) reduced the CPA score (14.0 ± 35.3 s) compared with the CRF alone-injected group (167.7 ± 87.2 s; Fig. 5D).
To examine whether intra-dlBNST repeated injections of NPY per se produced CPP or CPA, NPY (0.3 nmol/side; n = 6) was administered into the bilateral dlBNST in the absence of CRF over 3 d. No significant difference (t = 0.48 (df = 5), p > 0.05, paired t test) was observed in the time spent in the drug-paired compartment between the test (524 ± 19 s) and preconditioning (516 ± 21 s) sessions. The CPA score (–8.00 ± 16.8 s) was not significantly different (t = 0.39 (df = 14), p > 0.05, Student's t test) from that of the vehicle-injected group (15.1 ± 44.1 s, n = 10). These data showed that neither CPP nor CPA was induced by intra-dlBNST repeated injections of NPY alone, indicating that NPY had no motivational effect by itself when repeatedly injected into the dlBNST at this dose.
To identify the receptor subtype(s) involved in the inhibitory effect of NPY on CRF-induced CPA, the effects of coadministration of BIBP3226 (Y1 selective antagonist) or L-152,804 (Y5 selective antagonist) were examined. In the groups coadministered BIBP3226 (3.0 nmol/side) or L-152,804 (3.0 nmol/side) in addition to CRF and NPY, significant differences (t = 4.68 (df = 10), p < 0.001 or t = 3.71 (df = 11), p < 0.01, respectively, paired t test) were observed in the time spent in the drug-paired compartment between the test (412 ± 35 or 407 ± 35 s, respectively) and preconditioning (546 ± 16 or 521 ± 16 s, respectively) sessions (Fig. 5C). CPA scores of these groups (134 ± 29 or 114 ± 31 s, respectively) increased significantly (t = 2.67 (df = 19), p < 0.05 or t = 2.15 (df = 20), p < 0.05, respectively, Student's t test) compared with the score of the group injected with CRF and NPY (14.0 ± 35.3 s) (Fig. 5D).
Effects of CRF and NPY on membrane potentials in dlBNST neurons
Effects of CRF and NPY on the membrane potentials in dlBNST neurons were examined using a whole-cell patch-clamp technique in slice preparations. First, we examined the effects of a 2 min bath application of 1 μm CRF on membrane potentials in dlBNST neurons (Fig. 6). In type I neurons, CRF did not affect the membrane potential (–69.74 ± 0.55 and –70.76 ± 0.99 mV in the periods before and after CRF application, respectively; t = 0.86 (df = 4), p > 0.05, paired t test; Fig. 6B, top), and the input resistance was not changed (301.0 ± 29.5 and 311.4 ± 40.2 MΩ in the periods before and after CRF application, respectively; t = 0.50 (df = 4), p > 0.05, paired t test; Fig. 6C, top). On the other hand, CRF depolarized membrane potentials gradually in all of the type II neurons tested (n = 5). CRF application significantly changed the membrane potential from –69.97 ± 0.90 mV to –65.33 ± 0.56 mV (t = 7.59 (df = 4), p < 0.01, paired t test; Fig. 6B, second row). The input resistance in the postdrug period (327.0 ± 26.8 MΩ) was significantly higher than that in the predrug period (277.4 ± 26.0 MΩ; t = 2.79 (df = 4), p < 0.05, paired t test; Fig. 6C, second row). The CRF-induced depolarization and increase in input resistance were not observed in the presence of CRF receptor antagonists (300 nm NBI27914 + 300 nm AS-30), indicating that these changes were mediated by activation of CRF receptors (–69.59 ± 0.71 and –70.99 ± 1.10 mV before and after CRF application, respectively; t = 1.66 (df = 4), p > 0.05, paired t test; Fig. 6B, bottom; 228.9 ± 18.5 and 234.5 ± 14.3 MΩ before and after CRF application, respectively; t = 0.41 (df = 4), p > 0.05, paired t test; Figure 6C, bottom). Although CRF increased the input resistance in type III neurons slightly, but significantly (270.6 ± 67.0 and 300.5 ± 73.6 MΩ in the periods before and after CRF application, respectively; t = 3.71 (df = 4), p < 0.05, paired t test; Fig. 6C, third row), the membrane potential was not affected by CRF (–70.14 ± 0.95 and –69.05 ± 1.35 mV in the periods before and after CRF application, respectively; t = 1.20 (df = 4), p > 0.05, paired t test; Fig. 6A,B, third row) in this type of neurons.
Figure 6.

Effects of CRF on membrane potential and input resistance in three types of dlBNST neurons. A, Representative traces from current-clamp recordings in type I (top), type II (second row), and type III (third row) dlBNST neurons, and type II dlBNST neurons in the presence of NBI27914 (300 nm) + AS-30 (300 nm) (bottom). B, C, Effects of CRF (1 μm, 2 min) on membrane potential (B) and input resistance (C) in type I (n = 5; top), type II (n = 5; second row), and type III (n = 5; third row) dlBNST neurons, and type II dlBNST neurons in the presence of NBI27914 + AS-30 (n = 5; bottom). Left, Time courses of membrane potential (B) and input resistance (C). Right, Averaged membrane potential (B) and input resistance (C) in the period of pre-CRF (0–3 min) and post-CRF (16–19 min) application. Gray symbols and lines show data obtained from individual neurons, and black symbols and lines show averaged data obtained from five neurons. Data are expressed as means ± SEM. *p < 0.05, **p < 0.01 compared with pre-CRF application (paired t test).
Next, we investigated the effects of a 2 min bath application of 1 μm NPY on the membrane potentials in dlBNST neurons (Fig. 7). In type I neurons, NPY did not affect either the membrane potential (–69.89 ± 0.66 and –69.78 ± 1.44 mV in the periods before and after NPY application, respectively; t = 0.06 (df = 5), p > 0.05, paired t test; Fig. 7A,B, top), and the input resistance (270.2 ± 10.1 and 288.5 ± 16.4 MΩ in the periods before and after NPY application, respectively; t = 1.82 (df = 5), p > 0.05, paired t test; Fig. 7C, top). On the other hand, NPY hyperpolarized membrane potentials gradually in all of the type II neurons tested (n = 8). NPY application significantly changed membrane potential from –68.91 ± 0.59 to –73.05 ± 1.06 mV (t = 5.39 (df = 7), p < 0.01, paired t test; Fig. 7B, second row). The input resistance in the postdrug period (254.9 ± 28.5 MΩ) was significantly larger than that in the predrug period (223.7 ± 19.5 MΩ; t = 3.15 (df = 7), p < 0.05, paired t test; Fig. 7C; second row). In the presence of NPY receptor antagonists (1 μm BIBP3226 + 1 μm L-152,804), NPY did not affect either the membrane potentials (–68.75 ± 0.56 and –69.35 ± 0.56 mV before and after NPY application, respectively; t = 0.82 (df = 5), p > 0.05, paired t test; Fig. 7B, bottom) or the input resistance (287.5 ± 16.9 and 307.9 ± 28.9 MΩ before and after NPY application, respectively; t = 1.41 (df = 5), p > 0.05, paired t test; Fig. 7C, bottom), suggesting that the effects of NPY were mediated via NPY receptors. In type III neurons, NPY did not affect either the membrane potential (–69.94 ± 0.96 and –69.61 ± 1.77 mV in the periods before and after NPY application, respectively; t = 0.29 (df = 4), p > 0.05, paired t test; Fig. 7B, third row) or the input resistance (387.8 ± 49.2 and 376.2 ± 52.5 MΩ in the periods before and after NPY application, respectively; t = 0.68 (df = 4), p > 0.05, paired t test; Fig. 7C, third row).
Figure 7.

Effects of NPY on membrane potential and input resistance in three types of dlBNST neurons. A, Representative traces from current-clamp recordings in type I (top), type II (second row), and type III (third row) dlBNST neurons, and type II dlBNST neurons in the presence of BIBP3226 (1 μm) + L-152,804 (1 μm) (bottom). B, C, Effects of NPY (1 μm, 2 min) on membrane potential (B) and input resistance (C) in type I (n = 6; top), type II (n = 8; second row), and type III (n = 5; third row) dlBNST neurons, and type II dlBNST neurons in the presence of BIBP3226 + L-152,804 (n = 6; bottom). Left, Time courses of membrane potential (B) and input resistance (C). Right, Averaged membrane potential (B) and input resistance (C) in the period of pre-NPY (0–3 min) and post-NPY (16–19 min) application. Gray symbols and lines show data obtained from individual neurons, and black symbols and lines show averaged data obtained from 5–8 neurons. Data are expressed as means ± SEM. *p < 0.05, **p < 0.01 compared with pre-NPY application (paired t test).
The opposing effects of CRF and NPY on the membrane potential in type II dlBNST neurons allowed us to examine the effects of NPY on CRF-induced depolarization (Fig. 8). As seen in Figure 8, A and B, in type II neurons, significant hyperpolarization was observed after NPY application (–68.20 ± 0.43 and –70.46 ± 0.75 mV in the periods before and after NPY application, respectively; p < 0.05, compared with the pre-application of drugs (Control), one-way repeated measured ANOVA (F(2,8) = 4.83, p < 0.05) with Bonferroni's post hoc test, n = 5;Figure 8B). In the presence of NPY, CRF did not depolarize the membrane potential in type II neurons (–70.46 ± 0.75 and –71.06 ± 0.83 mV in the periods before and after CRF application, respectively; Fig. 8B). We occasionally observed a transient excitation after CRF application in the presence of NPY (Fig. 8A). However, the depolarization was very small and did not persist over ∼16 min, where CRF usually exhibits maximal depolarization in the absence of NPY. The input resistance was also unaltered by CRF in the presence of NPY (316.1 ± 59.8 and 328.0 ± 54.3 MΩ in the periods before and after CRF application, respectively; Fig. 8C).
Conductances associated with the actions of CRF and NPY in type II dlBNST neurons
To investigate the conductances underlying the actions of CRF and NPY, we studied the effects of these peptides on the steady-state I–V relationships in type II neurons by applying a voltage ramp protocol. As shown in Figure 9, A and B, CRF-induced net currents in individual neurons were heterogeneous: CRF decreased net currents in some neurons (n = 4, Fig. 9A, left, B, black traces), while the increase in net currents was observed in other neurons (n = 6; Fig. 9A, right, B, gray traces). Thus, we classified these neurons into two groups and designated them as type IIa and type IIb. The averaged current trace of type IIa neurons revealed that the reversal potential of this current was approximately –95 mV (–94.4 ± 3.9 mV, n = 4), which is close to the potassium equilibrium potential (–109.3 mV) calculated from the Nernst equation under our experimental conditions (Fig. 9C, left). On the other hand, the CRF-induced increased currents reversed the polarity at approximately –50 mV (–55.9 ± 6.1 mV, n = 6) with a clear outward rectification in type IIb neurons, suggesting a contribution of nonselective cationic conductance (Takano et al., 1996; Yang and Ferguson, 2002; Murai and Akaike, 2005; Kaneko et al., 2008) (Fig. 9C, right). CRF significantly increased negative holding currents (–58.92 ± 8.54 and –67.77 ± 9.63 pA before and after CRF application, respectively; t = 2.80 (df = 9), p < 0.05, paired t test; Fig. 9D, upper), whereas the input resistance was not affected by CRF (339.9 ± 36.3 and 342.5 ± 39.1 MΩ before and after CRF application, respectively; t = 0.31 (df = 9), p > 0.05, paired t test; Fig. 9D, lower). These results suggest that at least two conductances might be associated with the CRF-induced depolarization. We observed the different effects of CRF on changes in input resistance between voltage- and current-clamp recordings. Voltage-clamp recordings were obtained from four type IIa and six type IIb neurons, which exhibited increased and decreased input resistance after CRF application, respectively. Thus, the apparent net change in input resistance seemed to be unchanged. On the other hand, in current-clamp recordings (Fig. 6), averaged input resistance was increased following CRF application. Although we did not determine the cell classes (IIa or IIb) in the current-clamp recording experiment, based on the changes in input resistance observed in individual neurons, it might be possible that the recorded neurons included four type IIa and one type IIb neurons.
Figure 9.

Conductances associated with the action of CRF in the type II dlBNST neurons. A, Representative steady-state current responses to voltage ramp pulses from –30 to –120 mV recorded from type IIa (left) and type IIb (right) neurons in the periods of pre-CRF (gray) and post-CRF (black) application (1 μm, 2 min). The insets show the subtraction of the currents (CRF-induced net currents). Axis titles in A apply in the insets. B, Traces of CRF-induced net currents in four type IIa (black) and six type IIb (gray) neurons. C, Average traces of CRF-induced currents in type IIa (n = 4, left) and type IIb (n = 6, right) neurons. Gray shadows represent SEM. Vertical dashed lines show the reversal potential. D, Effects of CRF on the holding currents at –70 mV (upper) and the input resistance (lower). Gray lines represent the data obtained from individual neurons (n = 10), and black symbols and lines show the averaged data obtained from 10 neurons. *p < 0.05 compared with pre-CRF application (paired t test).
On the other hand, NPY-induced net currents in individual neurons were similar in all of the neurons tested: NPY decreased net currents in all recorded neurons (n = 8, Fig. 10A,B). This result suggests that type II neurons cannot be classified according to the response to NPY. In other words, both type IIa and IIb neurons may respond to NPY in the same manner. NPY-induced change in the net currents was accompanied by significantly reduced negative holding currents (–62.64 ± 10.06 and –54.81 ± 10.12 pA before and after NPY application, respectively; t = 4.05 (df = 7), p < 0.01, paired t test; Fig. 10C, upper) and increased input resistance (304.3 ± 30.3 and 359.9 ± 48.6 MΩ before and after NPY application, respectively; t = 2.37 (df = 7), p < 0.05, paired t test; Fig. 10C, lower). The averaged trace exhibited that the current associated with NPY reversed the polarity approximately –60 mV (–60.4 ± 6.0 mV, n = 8; Fig. 10B), suggesting a contribution of multiple conductances to the action of NPY. One of the candidate conductances is hyperpolarization-activated current (Ih), the suppression of which has been shown to be critical for NPY-induced hyperpolarization in the BLA (Giesbrecht et al., 2010). Thus, we tested this hypothesis by using Ih blocker ZD7288 (Fig. 10D–F). Bath application of 10 μm ZD7288 alone elicited significant hyperpolarizing currents (–31.72 ± 8.62 and –11.43 ± 6.70 pA in the periods 0–3 min before and 7–10 min after ZD7288 application, respectively; t = 3.66 (df = 7), p < 0.01, paired t test, n = 8) and increased input resistance (482.21 ± 36.7 and 744.9 ± 124.6 MΩ in the periods 0–3 min before and 7–10 min after ZD7288 application, respectively; t = 2.75 (df = 7), p < 0.05, paired t test, n = 8), indicating that Ih constitutively depolarizes membrane potentials in type II neurons. In the presence of ZD7288, NPY did not affect either the holding currents (–11.07 ± 8.92 and –16.54 ± 7.40 pA before and after NPY application, respectively; t = 1.47 (df = 7), p > 0.05, paired t test; Fig. 10F, upper) or the input resistance (662.9 ± 95.8 and 791.8 ± 183.9 MΩ before and after NPY application, respectively; t = 2.05 (df = 7), p > 0.05, paired t test; Fig. 10F, lower). In the presence of ZD7288, the reversal potential of NPY-associated currents shifted to a more negative potential (–85.3 ± 6.2 mV, n = 8; Fig. 10E) compared with the reversal potential in the absence of ZD7288 (–60.4 ± 6.0 mV). These results indicate that NPY-induced hyperpolarization was mediated primarily by suppression of Ih in type II dlBNST neurons. The remaining ZD7288-resistant component, which showed a negative slope conductance with the reversal potential of approximately –85 mV, might be mediated, at least in part, by closing potassium channels.
Figure 10.

Conductances associated with the action of NPY in the type II dlBNST neurons. A, D, Representative steady-state current responses to voltage ramp pulses from –30 to –120 mV recorded from type II neurons in the periods of pre-NPY (gray) and post-NPY (black) (1 μm, 2 min) application in the absence (A) or presence (D) of ZD7288 (10 μm). The insets show the subtraction of the currents (NPY-induced net currents). Axis titles in A and D apply in the insets. B, E, Average traces of NPY-induced currents in type II neurons in the absence (B; n = 8) or presence (E; n = 8) of ZD7288. Gray shadows represent SEM. Vertical dashed lines show the reversal potential. C, F, Effects of NPY on the holding currents at –70 mV (upper) and the input resistance (lower). Gray lines represent the data obtained from individual neurons, and black symbols and lines show the averaged data obtained from eight neurons. *p < 0.05, **p < 0.01 compared with pre-NPY application (paired t test).
Effects of CRF and NPY on the firing activity in type II dlBNST neurons
The effects of CRF and NPY on the firing activity in type II dlBNST neurons were examined. A 2 min bath application of 1 μm CRF gradually depolarized membrane potentials, as observed in the above experiments, and then induced spikes (Fig. 11A), which persisted over 30 min. The number of spikes significantly increased from 12.2 ± 7.1 spikes/1.5 min in the period before CRF application to 206.8 ± 68.1 spikes/1.5 min in the period after CRF application (t = 3.35 (df = 5), p < 0.05, n = 6, paired t test; Fig. 11B). On the other hand, NPY tended to suppress the firing activity (Fig. 11C). The number of spikes decreased from 28.5 ± 26.5 in the period before NPY application to 15.0 ± 12.3 spikes/1.5 min in the period during NPY application (Fig. 11D). In the presence of NPY, no significant increase in the spikes was observed after the application of 1 μm CRF (35.8 ± 30.8 spikes/1.5 min; t = 0.21 (df = 5), p > 0.05 vs control (before NPY application) and t = 0.62 (df = 5), p > 0.05 vs NPY alone (during NPY application), n = 6, paired t test; Figure 11D).
Figure 11.

Effects of CRF on firing activity of type II dlBNST neurons in the absence (A, B; n = 6) and presence (C, D; n = 6) of NPY. A, C, Representative traces (36 overlaid) from current-clamp recordings in type II dlBNST neurons before and after application of peptides. B, D, Effects of CRF and NPY on the number of spikes in type II dlBNST neurons. Gray symbols and lines show data obtained from individual neurons, and black symbols and lines show averaged data obtained from six neurons. *p < 0.05 compared with pre-CRF application control (paired t test).
Discussion
In the current study, using a conditioned place paradigm and an in vivo microdialysis technique, we demonstrated that enhanced neurotransmission via CRF receptors within the dlBNST plays a key role in the negative affective component of pain. Additionally, we showed that intra-dlBNST injection of NPY suppressed pain-induced CPA. To our knowledge, these findings are the first reported providing evidence of the opposing roles of CRF and NPY within the dlBNST in the negative affective component of pain. We also demonstrated that intra-dlBNST CRF injection produced CPA even in the absence of formalin-evoked noxious stimulation and that coadministration of NPY suppressed CRF-induced CPA. These results suggest that NPY injected into the dlBNST may suppress formalin-induced CPA through inhibition of CRF-mediated neurotransmission.
Behavioral studies using a CPA test have successfully elucidated the neural substrates and mechanisms underlying the affective component of pain (Johansen et al., 2001; Tanimoto et al., 2003; Deyama et al., 2008). Because this test was based on associative learning between a noxious stimulus-induced aversive affect and a neutral environmental context, whether the attenuation of CPA was due to impairment of associative learning or suppression of the primary aversive affect is difficult to determine. In the present study, we demonstrated that intra-dlBNST CRF injection produced CPA even in the absence of formalin-induced noxious stimulus. This indicates that the activation of CRF receptors within the dlBNST is sufficient to produce the negative affective states. Therefore, the attenuation of CPA by inhibition of intra-dlBNST CRF-mediated neurotransmission may be due to the reduction of the primary aversive affect.
To investigate the cellular mechanisms of CRF-induced CPA and the counteracting effect of NPY, we examined the effects of CRF and NPY on neuronal excitability in dlBNST neurons. Interestingly, we found that only type II, not type I or III, neurons responded to both CRF and NPY in the dlBNST. Bath application of CRF depolarized membrane potentials significantly in type II neurons. In contrast, NPY-induced hyperpolarization in type II neurons. Additionally, coapplication of NPY extinguished the CRF-induced depolarization. Similar results were observed in firing activities of type II dlBNST neurons. Specifically, CRF increased firing activities of type II neurons significantly, and coapplication of NPY suppressed them. These results demonstrated opposing effects of CRF and NPY at the cellular level, as observed at the behavioral level. These counteracting effects of CRF and NPY on neuronal excitability in type II dlBNST neurons may explain the opposing roles of these peptides in the negative affective component of pain.
Analyses of I–V relationships in the absence and presence of CRF demonstrate that there may be at least two classes of type II dlBNST neurons. The steady-state current in type IIa neurons was reduced by CRF and reversed its polarity at approximately –95 mV, suggesting the involvement of potassium channels. Jedema and Grace (2004) reported the direct action of CRF on locus ceruleus neurons, increasing neuronal excitability by decreasing a potassium conductance in a cAMP-dependent manner. A similar mechanism may be involved in the depolarizing effect of CRF on type IIa dlBNST neurons. On the other hand, CRF increased steady-state current and its reversal potential was approximately –50 mV in type IIb neurons, implying the contribution of nonselective cation current (Takano et al., 1996; Yang and Ferguson, 2002; Kaneko et al., 2008). In support of this hypothesis, CRF has been reported to activate nonselective cationic conductances in the pituitary grand (Takano et al., 1996). The reversal potentials observed in the type IIa and IIb neurons were slightly deviated from the predicted (–109.3 mV for potassium current) or reported (>–45 mV for nonselective cation current) (Takano et al., 1996; Ito and Dulon, 2002; Yang and Ferguson, 2002; Kaneko et al., 2008) values, suggesting that both channels express in both type IIa and IIb neurons with different levels, rather each channel exclusively expresses in either type IIa or IIb neurons. Further studies are necessary to characterize the detailed physiological properties of these two types of neurons.
NPY hyperpolarized type II dlBNST neurons with an increase in input resistance. I–V relationship analyses revealed that NPY reduced steady-state currents, the reversal potential of which was approximately –60 mV, indicating suppression of cationic conductance(s). Consistent with this finding, ZD7288, an Ih blocker, inhibited the hyperpolarizing effect of NPY and shifted the reversal potential of NPY-associated currents toward a more negative potential, demonstrating that NPY-induced hyperpolarization was mediated by blocking Ih channels. Similar hyperpolarizing effect of NPY via inhibition of Ih channels has been reported in BLA pyramidal neurons (Giesbrecht et al., 2010).
A previous study suggested that the majority of type II BNST neurons were GABAergic interneurons, which may innervate output neurons within the BNST (Hammack et al., 2007). Thus, CRF-induced activation of type II neurons could result in the inhibition of output neurons. Indeed, in the ventrolateral BNST, Dumont and Williams (2004) demonstrated that activation of neurons exhibiting a depolarizing sag (type I or II neurons) enhanced GABAA-IPSC in the output neurons projecting to the ventral tegmental area (VTA). Georges and Aston-Jones (2001, 2002) have reported that the BNST sends excitatory drive to VTA dopaminergic neurons. Recently, we have demonstrated that this excitatory drive from the BNST to VTA dopaminergic neurons is mainly composed of two GABAergic neurons (Kudo et al., 2012). Specifically, most VTA-projecting BNST neurons are GABAergic neurons, which preferentially innervate VTA GABAergic neurons. Therefore, activation of VTA-projecting BNST output neurons is predicted to promote VTA dopaminergic neuron activity through a disinhibition mechanism. Pain-induced increase in CRF release within the BNST may activate the type II BNST neurons, which could suppress VTA-projecting BNST output neurons, thereby attenuating the excitatory drive from the BNST to the VTA dopaminergic neurons. The attenuation of VTA dopaminergic neuron activity through this mechanism may be involved in pain-induced aversion. In support of this hypothesis, it has recently been reported that optogenetic inhibition of VTA dopaminergic neurons causes CPA (Tan et al., 2012).
Although several lines of evidence suggest the critical role of CRF1 receptors in CRF-induced negative emotion such as anxiety and fear, there is still continued controversy over the role of CRF2 receptors in these emotional states (Takahashi, 2001; Bale and Vale, 2004). Regarding the BNST, Sahuque et al. (2006) reported that anxiety-like behaviors induced by the intra-BNST CRF injection were prevented by coadministration of a CRF1, but not a CRF2, receptor antagonist. On the other hand, either CRF1 or CRF2 receptor antagonist prevented CPA induced by intra-BNST CRF injection, consistent with our current results showing that both CRF1 and CRF2 receptor antagonists suppressed pain-induced CPA. These findings suggest that distinct neuronal pathways may be involved in different negative emotional states such as anxiety and aversion. Further studies using neuronal pathway-specific methods such as optogenetic approaches are necessary to address this issue.
A large body of literature suggests that anxiolytic and anti-aversive effects of NPY are mediated primarily by Y1 receptors (Heilig, 1995; Kask et al., 1997; Nakajima et al., 1998; Primeaux et al., 2005). Additionally, there are reports that Y5 receptors also mediate the suppression of negative affect (Sørensen et al., 2004). In contrast, Y2 receptors have been reported to mediate negative emotions, such as anxiety (Nakajima et al., 1998; Sajdyk et al., 2002). Thus, in the present study, we examined the involvement of Y1 and Y5 subtypes in the inhibitory effect of intra-dlBNST NPY on CRF-induced CPA and found that both Y1 and Y5 receptors play important roles in this effect of NPY. In this study, we revealed the heterogeneity of type II dlBNST neurons. Further electrophysiological experiments using Y1- and Y5-specific agonists are needed to clarify the neuronal populations expressing these receptors.
Increasing data have demonstrated opposing effects of CRF and NPY. Intracerebroventricular injection of Y1 receptor antagonist BIBP3226 induced an anxiogenic effect in the elevated plus maze test in rats, which was blocked by pretreatment with a nonselective CRF receptor antagonist (Kask et al., 1997). Sajdyk et al. (2006) reported that urocortin I, a CRF receptor agonist, injected into the BLA-induced CPA, which was reversed by coadministration of NPY. Electrophysiological analysis revealed opposing actions of CRF and NPY on the excitability of BLA neurons (Giesbrecht et al., 2010). In the ventrolateral BNST, it has been reported that NPY and CRF have opposite modulating effects on GABAA-IPSC (Kash and Winder, 2006). The present study has added pain-induced aversion to the list of negative emotional states in which opposing effects of CRF and NPY play important roles.
Further studies are necessary to determine the neuronal circuits involved in the induction of negative affect associated with pain stimulation, and the molecular mechanisms operating the ion channels involved in the opposing actions of CRF and NPY on dlBNST neuronal excitability. Nevertheless, we have uncovered some of the neuronal mechanisms underlying the negative affective component of pain by showing the opposing roles of intra-dlBNST CRF and NPY in pain-induced aversive behaviors and the opposing actions of these peptides on neuronal excitability converging on the same target, type II neurons, within the dlBNST.
Footnotes
This study was supported by a grant for Interdisciplinary Project for Psychosomatological Research in Hokkaido University; a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS) (M.M., 23300130); a Grant-in-Aid for Young Scientists (B) from JSPS (S.I., 22790239); Health Labour Sciences Research Grant (research on chronic pain) from the Ministry of Health, Labour, and Welfare (M.M.); and grants from the Hoansha Foundation (M.M.) and Naito Foundation (M.M.).
The authors declare no competing financial interests.
References
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Deyama S, Nakagawa T, Kaneko S, Uehara T, Minami M. Involvement of the bed nucleus of the stria terminalis in the negative affective component of visceral and somatic pain in rats. Behav Brain Res. 2007;176:367–371. doi: 10.1016/j.bbr.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Deyama S, Katayama T, Ohno A, Nakagawa T, Kaneko S, Yamaguchi T, Yoshioka M, Minami M. Activation of the β-adrenoceptor-protein kinase A signaling pathway within the ventral bed nucleus of the stria terminalis mediates the negative affective component of pain in rats. J Neurosci. 2008;28:7728–7736. doi: 10.1523/JNEUROSCI.1480-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyama S, Katayama T, Kondoh N, Nakagawa T, Kaneko S, Yamaguchi T, Yoshioka M, Minami M. Role of enhanced noradrenergic transmission within the ventral bed nucleus of the stria terminalis in visceral pain-induced aversion in rats. Behav Brain Res. 2009;197:279–283. doi: 10.1016/j.bbr.2008.08.024. [DOI] [PubMed] [Google Scholar]
- Deyama S, Ide S, Kondoh N, Yamaguchi T, Yoshioka M, Minami M. Inhibition of noradrenaline release by clonidine in the ventral bed nucleus of the stria terminalis attenuates pain-induced aversion in rats. Neuropharmacology. 2011;61:156–160. doi: 10.1016/j.neuropharm.2011.03.023. [DOI] [PubMed] [Google Scholar]
- Dumont EC, Williams JT. Noradrenaline triggers GABAA inhibition of bed nucleus of the stria terminalis neurons projecting to the ventral tegmental area. J Neurosci. 2004;24:8198–8204. doi: 10.1523/JNEUROSCI.0425-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges F, Aston-Jones G. Potent regulation of midbrain dopamine neurons by the bed nucleus of the stria terminalis. J Neurosci. 2001;21:RC160. doi: 10.1523/JNEUROSCI.21-16-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges F, Aston-Jones G. Activation of ventral tegmental area cells by the bed nucleus of the stria terminalis: a novel excitatory amino acid input to midbrain dopamine neurons. J Neurosci. 2002;22:5173–5187. doi: 10.1523/JNEUROSCI.22-12-05173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesbrecht CJ, Mackay JP, Silveira HB, Urban JH, Colmers WF. Countervailing modulation of Ih by neuropeptide Y and corticotrophin-releasing factor in basolateral amygdala as a possible mechanism for their effects on stress-related behaviors. J Neurosci. 2010;30:16970–16982. doi: 10.1523/JNEUROSCI.2306-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman AR, Yang Y, Ressler KJ, Davis M. The role of neuropeptide Y in the expression and extinction of fear-potentiated startle. J Neurosci. 2008;28:12682–12690. doi: 10.1523/JNEUROSCI.2305-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammack SE, Mania I, Rainnie DG. Differential expression of intrinsic membrane currents in defined cell types of the anterolateral bed nucleus of the stria terminalis. J Neurophysiol. 2007;98:638–656. doi: 10.1152/jn.00382.2007. [DOI] [PubMed] [Google Scholar]
- Hara Y, Nakaya H. Dual effects of extracellular ATP on the muscarinic acetylcholine receptor-operated K+ current in guinea-pig atrial cells. Eur J Pharmacol. 1997;324:295–303. doi: 10.1016/s0014-2999(97)00088-5. [DOI] [PubMed] [Google Scholar]
- Heilig M. Antisense inhibition of neuropeptide Y (NPY)-Y1 receptor expression blocks the anxiolytic-like action of NPY in amygdala and paradoxically increases feeding. Regul Pept. 1995;59:201–205. doi: 10.1016/0167-0115(95)00103-i. [DOI] [PubMed] [Google Scholar]
- Ito K, Dulon D. Nonselective cation conductance activated by muscarinic and purinergic receptors in rat spiral ganglion neurons. Am J Physiol Cell Physiol. 2002;282:C1121–1135. doi: 10.1152/ajpcell.00364.2001. [DOI] [PubMed] [Google Scholar]
- Jedema HP, Grace AA. Corticotropin-releasing hormone directly activates noradrenergic neurons of the locus ceruleus recorded in vitro. J Neurosci. 2004;24:9703–9713. doi: 10.1523/JNEUROSCI.2830-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Fields HL. Glutamatergic activation of anterior cingulate cortex produces an aversive teaching signal. Nat Neurosci. 2004;7:398–403. doi: 10.1038/nn1207. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Fields HL, Manning BH. The affective component of pain in rodents: direct evidence for a contribution of the anterior cingulate cortex. Proc Natl Acad Sci U S A. 2001;98:8077–8082. doi: 10.1073/pnas.141218998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K, Tamamaki N, Owada H, Kakizaki T, Kume N, Totsuka M, Yamamoto T, Yawo H, Yagi T, Obata K, Yanagawa Y. Noradrenergic excitation of a subpopulation of GABAergic cells in the basolateral amygdala via both activation of nonselective cationic conductance and suppression of resting K+ conductance: a study using glutamate decarboxylase 67-green fluorescent protein knock-in mice. Neuroscience. 2008;157:781–797. doi: 10.1016/j.neuroscience.2008.09.029. [DOI] [PubMed] [Google Scholar]
- Kash TL, Winder DG. Neuropeptide Y and corticotropin-releasing factor bi-directionally modulate inhibitory synaptic transmission in the bed nucleus of the stria terminalis. Neuropharmacology. 2006;51:1013–1022. doi: 10.1016/j.neuropharm.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Kask A, Rägo L, Harro J. Alpha-helical CRF(9–41) prevents anxiogenic-like effect of NPY Y1 receptor antagonist BIBP3226 in rats. Neuroreport. 1997;8:3645–3647. doi: 10.1097/00001756-199711100-00044. [DOI] [PubMed] [Google Scholar]
- Kosten TA. Clonidine attenuates conditioned aversion produced by naloxone-precipitated opiate withdrawal. Eur J Pharmacol. 1994;254:59–63. doi: 10.1016/0014-2999(94)90370-0. [DOI] [PubMed] [Google Scholar]
- Kudo T, Uchigashima M, Miyazaki T, Konno K, Yamasaki M, Yanagawa Y, Minami M, Watanabe M. Three types of neurochemical projection from the bed nucleus of the stria terminalis to the ventral tegmental area in adult mice. J Neurosci. 2012;32:18035–18046. doi: 10.1523/JNEUROSCI.4057-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Davis M. Role of the hippocampus, the bed nucleus of the stria terminalis, and the amygdala in the excitatory effect of corticotropin-releasing hormone on the acoustic startle reflex. J Neurosci. 1997;17:6434–6446. doi: 10.1523/JNEUROSCI.17-16-06434.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin SM, Ling N, Liu XJ, Kahl SD, Gehlert DR. Differential distribution of urocortin- and corticotropin-releasing factor-like immunoreactivities in the rat brain. Neuroscience. 1999;92:281–291. doi: 10.1016/s0306-4522(98)00732-5. [DOI] [PubMed] [Google Scholar]
- Murai Y, Akaike T. Orexins cause depolarization via nonselective cationic and K+ channels in isolated locus coeruleus neurons. Neurosci Res. 2005;51:55–65. doi: 10.1016/j.neures.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yamamoto R, Fujio M, Suzuki Y, Minami M, Satoh M, Kaneko S. Involvement of the bed nucleus of the stria terminalis activated by the central nucleus of the amygdala in the negative affective component of morphine withdrawal in rats. Neuroscience. 2005;134:9–19. doi: 10.1016/j.neuroscience.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Inui A, Asakawa A, Momose K, Ueno N, Teranishi A, Baba S, Kasuga M. Neuropeptide Y produces anxiety via Y2-type receptors. Peptides. 1998;19:359–363. doi: 10.1016/s0196-9781(97)00298-2. [DOI] [PubMed] [Google Scholar]
- Parker RM, Herzog H. Regional distribution of Y-receptor subtype mRNAs in rat brain. Eur J Neurosci. 1999;11:1431–1448. doi: 10.1046/j.1460-9568.1999.00553.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego, CA: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Primeaux SD, Wilson SP, Cusick MC, York DA, Wilson MA. Effects of altered amygdalar neuropeptide Y expression on anxiety-related behaviors. Neuropsychopharmacology. 2005;30:1589–1597. doi: 10.1038/sj.npp.1300705. [DOI] [PubMed] [Google Scholar]
- Rafieian-Kopaei M, Gray AM, Spencer PS, Sewell RD. Contrasting actions of acute or chronic paroxetine and fluvoxamine on morphine withdrawal-induced place conditioning. Eur J Pharmacol. 1995;275:185–189. doi: 10.1016/0014-2999(94)00770-8. [DOI] [PubMed] [Google Scholar]
- Sahuque LL, Kullberg EF, Mcgeehan AJ, Kinder JR, Hicks MP, Blanton MG, Janak PH, Olive MF. Anxiogenic and aversive effects of corticotropin-releasing factor (CRF) in the bed nucleus of the stria terminalis in the rat: role of CRF receptor subtypes. Psychopharmacology. 2006;186:122–132. doi: 10.1007/s00213-006-0362-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajdyk TJ, Schober DA, Smiley DL, Gehlert DR. Neuropeptide Y-Y2 receptors mediate anxiety in the amygdala. Pharmacol Biochem Behav. 2002;71:419–423. doi: 10.1016/s0091-3057(01)00679-7. [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Fitz SD, Shekhar A. The role of neuropeptide Y in the amygdala on corticotropin-releasing factor receptor-mediated behavioral stress responses in the rat. Stress. 2006;9:21–28. doi: 10.1080/10253890600557315. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Shibasaki T, Lederis K. Distribution and efferent projections of corticotropin-releasing factor-like immunoreactivity in the rat amygdaloid complex. Brain Res. 1986;382:213–238. doi: 10.1016/0006-8993(86)91332-6. [DOI] [PubMed] [Google Scholar]
- Sørensen G, Lindberg C, Wörtwein G, Bolwig TG, Woldbye DP. Differential roles for neuropeptide Y Y1 and Y5 receptors in anxiety and sedation. J Neurosci Res. 2004;77:723–729. doi: 10.1002/jnr.20200. [DOI] [PubMed] [Google Scholar]
- Takahashi LK. Role of CRF1 and CRF2 receptors in fear and anxiety. Neurosci Biobehav Rev. 2001;25:627–636. doi: 10.1016/s0149-7634(01)00046-x. [DOI] [PubMed] [Google Scholar]
- Takano K, Yasufuku-Takano J, Teramoto A, Fujita T. Corticotropin-releasing hormone excites adrenocorticotropin-secreting human pituitary adenoma cells by activating a nonselective cation current. J Clin Invest. 1996;98:2033–2041. doi: 10.1172/JCI119008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouèbe G, Deisseroth K, Tye KM, Lüscher C. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012;73:1173–1183. doi: 10.1016/j.neuron.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto S, Nakagawa T, Yamauchi Y, Minami M, Satoh M. Differential contributions of the basolateral and central nuclei of the amygdala in the negative affective component of chemical somatic and visceral pains in rats. Eur J Neurosci. 2003;18:2343–2350. doi: 10.1046/j.1460-9568.2003.02952.x. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol. 1998;56:613–672. doi: 10.1016/s0301-0082(98)00060-4. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Walter A, Mai JK, Lanta L, Görcs T. Differential distribution of immunohistochemical markers in the bed nucleus of the stria terminalis in the human brain. J Chem Neuroanat. 1991;4:281–298. doi: 10.1016/0891-0618(91)90019-9. [DOI] [PubMed] [Google Scholar]
- Yang B, Ferguson AV. Orexin-A depolarizes dissociated rat area postrema neurons through activation of a nonselective cationic conductance. J Neurosci. 2002;22:6303–6308. doi: 10.1523/JNEUROSCI.22-15-06303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]