

Abstract
Panglial networks are essential for normal physiology in the CNS, and the function of distinct connexins participating in these networks is not well understood. We generated Connexin32 (Cx32)-deficient mice with additional deletion of astrocytic Cx43 to explore the role of both connexins in panglial networks. Cx43/Cx32 double knock-out (dKO) mice revealed strong microglial activation in corpus callosum and cingulum along with severe astrogliosis and scar formation. In addition, most of the fine myelinated fibers projecting from the corpus callosum into the cortex were lost. Myelin loss was caused by a strong decrease of oligodendrocytes in the cingulum of Cx43/Cx32dKO mice. Immunoblot analyses using newly generated specific Cx47 antibodies revealed that oligodendrocytic Cx47 is phosphorylated in vivo depending on astrocytic Cx43 expression. In Cx43-deficient mice, Cx47 protein levels were strongly decreased, whereas Cx47 mRNA levels were not altered. Using Cx43G138R/Cx30KO mice, we show that Cx47 expression depends on the presence of astrocytic Cx43 protein and that its gap junctional channel function is not necessary for Cx47 stabilization. In consequence, Cx43/Cx32dKO mice additionally lack Cx47 expression and therefore cannot form oligodendrocytic gap junctions, which explains the phenotypic similarities to Cx32/Cx47dKO mice. Our findings provide strong evidence that phosphorylation and stability of oligodendrocytic Cx47 proteins is dependent on astrocytic Cx43 expression. These results further unravel the complexity of panglial networks and show that results of previous studies using astrocytic Cx43-deficient mice have to be reconsidered.
Introduction
Complex multicellular organisms are dependent on direct cell–cell communication through gap junction (GJ) channels. GJ channels are characterized by their ability to allow diffusion of ions and metabolites (<1200 Da) (Bruzzone et al., 1996). They are composed of two connexons (hemichannels), whereby each connexon is provided by one adjacent cell. Each connexon is composed of six connexin protein subunits. Identical connexons form homotypic channels, whereas heterotypic channels are composed of two different hemichannels. Neurons and macroglial cells express distinct sets of connexin isoforms. Astrocytes express Connexin 30 (Cx30) and Cx43. In addition, Cx26 is expressed in a small subpopulation of gray matter astrocytes (Nagy et al., 2011). Oligodendrocytes express Cx47, Cx32, and Cx29, but Cx29 does not form functional GJ channels in vitro (Kleopa et al., 2004). Expression of oligodendrocytic and astrocytic connexins results in functional intra-astroglial (A:A), intraoligodendroglial (O:O), and interastro-oligodendroglial (A:O) coupling (Wallraff et al., 2006; Maglione et al., 2010; Wasseff and Scherer, 2011).
Several connexin single and double knock-out mice (dKO) have been investigated to understand the function of glial connexins participating in panglial networks. Single KOs did not result in gross morphological alterations (Sutor et al., 2000; Odermatt et al., 2003; Teubner et al., 2003; Theis et al., 2003; Eiberger et al., 2006; Nagy et al., 2011). Loss of both astrocytic connexins (Cx30 and Cx43) leads to dismyelination and vacuolization of gray as well as white matter regions (Lutz et al., 2009), whereas mice deficient for both oligodendrocytic connexins, Cx32 and Cx47, show severe myelin abnormalities and die at approximately postnatal day 42 (P42) (Menichella et al., 2003; Odermatt et al., 2003). In addition, mice deficient for one astrocytic (Cx30) and one oligodendrocytic (Cx47) connexin show complete loss of A:O coupling, early onset myelin pathology, motor impairments, and 40% of Cx30/Cx47dKO animals die between P42 and P90 (Tress et al., 2012).
Altogether, these studies indicate that A:O coupling is essential for myelin maintenance and are in line with a theoretical model of the coupling situation in the CNS based on cell culture experiments, in which A:O coupling can be established by Cx30:Cx32, Cx30:Cx47, and Cx43:Cx47 heterotypic channels, whereas combinations of Cx43 and Cx32 cannot form functional channels (Orthmann-Murphy et al., 2007; Magnotti et al., 2011a).
Recently, a study on Cx43 and Cx32 double-deficient mice revealed severe phenotypic abnormalities with astrocyte loss, white matter vacuolization, and early death at ∼16 weeks after birth (Magnotti et al., 2011b). This was surprising because Cx30:Cx47 channels should maintain functional coupling among astrocytes and oligodendrocytes in Cx43/Cx32dKO mice, whereas O:O and A:A coupling should be maintained by Cx47 and Cx30 expression, respectively.
With the present study, we aimed to identify the mechanism underlying the unexpected phenotype of Cx43/Cx32dKO mice and provide new insights into the function and contribution of distinct oligodendrocytic and astrocytic connexins. We show that Cx47 is phosphorylated in vivo and that Cx47 expression in oligodendrocytes and phosphorylation of Cx47 depend on astrocytic Cx43 expression in the mouse brain. Therefore, the phenotype of Cx43/Cx32dKO mice results from loss of both oligodendrocytic connexins Cx32 and Cx47.
Materials and Methods
Animals.
All mice used were kept under standard housing conditions with a 12 h dark/light cycle and with food and water ad libitum. All experiments were performed in accordance with local and state regulations for research with animals. hGFAP–Cre mice used by Magnotti et al. (2011b) are based on the human glial fibrillary acidic protein (GFAP) promoter and suffer from a variable mosaic expression and therefore unreliable excision of floxed Cx43 (Requardt et al., 2009). We decided to choose the mGFAP–Cre mice, which show a more reliable excision using the mouse GFAP promoter as driver for the Cre recombinase (Garcia et al., 2004) to generate Cx43flox/Cx32KO:mGFAP–Cre mice. For simplification, we named the Cx43flox/flox:mGFAP–Cre/Cx32y/− mice as Cx43/Cx32dKOmGFAP mice. This means that, in the so-called Cx43/Cx32dKOmGFAP mice, the floxed Cx43 allele is deleted by a Cre recombinase, which is driven by the mouse GFAP promoter, whereas Cx32 represents a nonconditional KO. Cx43/Cx32dKOmGFAP mice and control littermates were obtained by breeding Cx43flox/flox:mGFAP–Cre/Cx32y/+ males and Cx43flox/flox:mGFAP–Cre/Cx32+/− females. Only male animals were used for our experiments. Cx43flox/flox and Cx43flox/flox:mGFAP–Cre mice were used as control littermates. Cx43floxG138R/floxG138R:Nestin–Cre/Cx30−/− mice in the text are designated as Cx43ODDD mice. In addition, we generated Cx43flox/flox/Cx32y/−:Nestin–Cre mice, which are termed Cx43/Cx32dKONestin in this study. Cx43cKONestin mice are Cx43 conditional (single) deficient after Nestin–Cre-mediated deletion of Cx43. Cx43cKOmGFAP are Cx43 conditional (single) deficient mice after mGFAP–Cre-mediated deletion of Cx43. Genotyping of the mice was performed as described previously (Cx30, Teubner et al., 2003; Cx32, Nelles et al., 1996; Cx43, Theis et al., 2001; mGFAP–Cre, Lutz et al., 2009; and Nestin–Cre, Tronche et al., 1999).
Semiquantitative RT-PCR.
Tissue samples were isolated from the cerebellum, DNA-free mRNA was prepared, and semiquantitative (sq) RT-PCR was performed as described previously (Tress et al., 2012). For β-actin and Cx47 detection and quantification, a Taqman probe/primer mix was used (Applied Biosystems). Water and mRNA from Cx47-deficient cerebella were used as negative controls. A gene expression ratio XCx47/Xβ-actin was determined (Tress et al., 2012). The amplification efficiency, E, was 1.98 for Cx47 and 1.97 for β-actin.
Immunohistochemical staining on vibratome sections.
At different ages, mice were killed by injecting an anesthetic solution (0.3 mg of ketamine and 0.03 mg of xylazine per gram body weight) intraperitoneally. Mice were transcardially perfused with 30 ml of PBS followed by 30 ml of phosphate-buffered 4% formaldehyde solution (Roti-Histofix; Roth). Brains were rapidly prepared and postfixed in 2% phosphate-buffered formaldehyde solution for at least 48 h at 4°C. Vibratome sections (25 μm) were obtained (VT 1200 S; Leica), and free-floating slices were incubated in blocking solution [5% normal goat serum (NGS), 4% bovine serum albumin (BSA), and 0.5% Triton X-100] for 1 h at room temperature to avoid unspecific cross-reactivity. Primary antibodies diluted in blocking solution were applied overnight at room temperature. After washing with PBS, sections were incubated with corresponding biotin-conjugated secondary antibodies for 2 h at room temperature and washed again. Vectastain Peroxidase ABC reagent (Vector Laboratories) was applied according to the instructions of the manufacturers. After 30 min incubation in working solution, free-floating sections were washed in PBS for 60 min and then transferred to distilled water for at least 5 min before NovaRed (Vector Laboratories) staining. Sections were mounted on glass slides, air dried at 42°C on a slide warmer, and coverslipped with Entellan (Merck Chemicals). Rat primary antibodies directed against myelin basic protein (MBP) (1:1000; Millipore Bioscience Research Reagents) were used for myelin staining; rabbit polyclonal antibodies against ionized calcium binding adaptor molecule 1 (Iba1) (1:500; Wako Chemicals) were used for staining of microglia. Rabbit polyclonal GFAP antibodies (1:2000; Dako) were used for labeling of astrocytes, and rabbit polyclonal anti-glutathione S-transferase π (GST-π) (1:250; Millipore) was used for labeling myelinating oligodendrocytes. Cells of the oligodendroglial lineage were stained by rabbit polyclonal anti-oligodendrocyte transcription factor 2 (Olig2) (1:500; Millipore Bioscience Research Reagents).
Quantification of Olig2-positive cells.
The numbers of Olig2- and GST-π-positive cells were quantified in an area of 202,500 μm2 located directly above the cingulum in 25 μm vibratome sections of at least three mice per genotype and age. Regions were cut out from pictures with identical magnification levels by manual selection using Photoshop software (Adobe Photoshop CS4; Adobe Systems). Cell counting was performed using MBF-ImageJ software (NIH ImageJ).
Immunofluorescence staining on vibratome sections.
For immunofluorescence labeling, vibratome sections (25 μm) were blocked with PBS containing 0.5% Triton X-100, 5% NGS, and 4% BSA for 1 h at room temperature. Sections were incubated with primary antibodies: rabbit polyclonal anti-Cx43 [1:2500 (Wilgenbus et al., 1992)], mouse monoclonal anti-2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) (1:200; Sigma-Aldrich), and polyclonal rabbit anti-Cx47 (1:2000, newly generated) at room temperature for 2 h in blocking solution, washed with PBS-T (0.1% Triton X-100), and incubated for 1 h with corresponding goat polyclonal secondary antibodies conjugated to Alexa Fluor-488, Alexa Fluor-594, or Alexa Fluor-647 (1:1000; Invitrogen) diluted in blocking solution. After PBS washes, sections were mounted with PermaFluor (Thermo Fisher Scientific), and images were taken with a Laser Scanning Microscope (LSM 710; Carl Zeiss). The dye Hoechst33258 was used for labeling DNA.
Immunoblotting.
Mice were killed by cervical dislocation, and cerebella were quickly dissected out and flash frozen in liquid nitrogen. Tissues were stored at −80°C until extraction in modified RIPA buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1 mm NaF, 1% NP-40, and 0.25% Na-deoxycholate, pH 7.4) containing protease inhibitors (Complete Mini; Roche Diagnostics). The samples were homogenized using a Precellys24 tissue homogenizer (Peqlab). After centrifugation for 15 min at 10,000 × g and 4°C, the supernatant was retrieved in new 1.5 ml tubes and kept at −80°C. HeLa–Cx47–eGFP cell lysates were prepared as described previously (Tress et al., 2011). Before proteins were separated by electrophoresis on a 8–12% polyacrylamide gel, samples were prepared by mixing lysates with Laemmli's buffer (Laemmli, 1970). After SDS-gel separation, proteins were transferred to Hybond ECL membrane (GE Healthcare). Blots were preincubated in a blocking solution of 5% milk powder (MP) in TBST (50 mm Tris, 150 mm NaCl, pH 7.5, and 0.1% Tween 20) for 1 h at room temperature, incubated with primary antibodies overnight at 4°C, and after three washing steps, with horseradish peroxidase (HRP)-conjugated antibodies (1:5000 to 1:20,000; Dianova). Primary antibodies were polyclonal rabbit Cx43 [1:25,000 in 5% MP (Wilgenbus et al., 1992)], polyclonal rabbit Cx47 (1:5000 in 5% MP, newly generated), monoclonal mouse anti-tubulin (1:20,000 in 5% MP; Millipore Bioscience Research Reagents), and monoclonal mouse anti-GAPDH. Protein bands were detected by incubation of the membranes with enhanced chemiluminescence reagents (GE Healthcare) and development on x-ray films. Densitometric analyses were performed with the MacBiophotonics ImageJ software and by normalizing the band intensities to tubulin or GAPDH values.
Generation of new antibodies.
The 22 aa peptide (CVGEQSRPGAQEQLATKPRAGS) corresponding to a part of the C terminus of mouse Cx47 was synthesized by Prof. Maarten Egmond (University of Utrecht, Utrecht, The Netherlands), coupled, and injected into two guinea pigs and one rabbit by the Pineda Antibody Services. After six boosts, final bleeding was performed, and the serum was further processed in our laboratory. The serum was affinity purified with the same peptide using a HiTrap affinity column (GE Healthcare). Glycine at 0.2 m, pH 2.5, was used for elution in combination with Tris-HCl, pH 8, in the receiver tube. Directly after elution, the antibody solution was dialyzed against PBS overnight at 4°C, and antibodies were concentrated by Amicon Ultra-4 centrifugal filter devices with Ultracel-100 membrane (Millipore) and finally stored in PBS with 1% BSA and 0.01% sodium azide.
Statistical analyses.
Data are expressed as means ± SEM and analyzed by Microsoft Excel software. Differences between groups of mice in quantitative immunoblot, sqRT-PCR, and immunohistochemical staining were examined with the Student's t test. p values <0.001 were considered statistically highly significant.
Results
In this study, we used several different connexin mutated mice. We decided to abbreviate the correct genetic designations for easier readability. Instead of Cx43flox/flox:mGFAP–Cre/Cx32y/−, these mice are named Cx43/Cx32dKOmGFAP mice. Cx43floxG138R/floxG138R:Nestin–Cre/Cx30−/− mice in the text are termed Cx43ODDD mice, and Cx43flox/flox:Nestin–Cre/Cx32y/− mice are named Cx43/Cx32dKONestin. Conditional Cx43 (single) deficient mice are named Cx43cKO with the promoter of the Cre-recombinase used in superscript.
Activated microglia and reactive gliosis in Cx43/Cx32dKOmGFAP mice
To investigate the phenotype of the Cx43 and Cx32 double-deficient mice, we crossbred Cx43flox mice (Theis et al., 2001) with Cx32KO mice (Nelles et al., 1996) and mGFAP–Cre mice (Garcia et al., 2004). mGFAP–Cre-driven deletion of the Cx43flox allele resulted mostly in Cx43-deficient astrocytes.
Our initial experiments addressed the question whether there is any inflammation or reactive gliosis in the CNS of Cx43/Cx32dKOmGFAP mice. We immunohistochemically stained for Iba1 and GFAP. Iba1 is expressed by microglia, the resident immune cells of the CNS. Under normal conditions, these cells have a small cell body and cover the whole brain with their fine protrusions. During activation, microglial cells change their shape toward macrophage-like morphology. Normal microglial morphology can be seen in Figure 1, A, C, E, and G, which shows sections of control mouse forebrain and cerebellum stained for Iba1 on P30 and P90. In contrast, highly activated microglia were present in corpus callosum and cingulum of Cx43/Cx32dKOmGFAP mice on P30 (Fig. 1B). Remarkably, activated microglia align with white matter tracts and myelinated fibers in dKO mice. However, we did not find microglial activation in cerebellum of 30-d-old dKO mice (Fig. 1D). Because myelination in the mouse brain mainly takes place between P10 and P60 (Baumann and Pham-Dinh, 2001), we chose P90 as the time point of completed myelin development in our study. We found persistent microglial activation on P90 in corpus callosum and cingulum, although microglial activation appeared less focused on myelin tracts (Fig. 1F) compared with P30. In addition, microglial activation was present in cerebellar white matter on P90 (Fig. 1H).
Figure 1.

Activated microglia and reactive astroglia in the CNS of Cx43/Cx32dKOmGFAP mice. Immunohistochemical analysis on 25 μm brain slices from 30- and 90-d-old mice were stained for Iba1, a microglia marker. Control mice showed normal distribution of ramified microglial cells in all time points and regions tested (A, C, E, G). In Cx43/Cx32dKOmGFAP mice, a strong microglial activation along the corpus callosum and in the cingulum was detected at P30 (B), whereas the cerebellum was not affected (D). With advancing age, the intensity of the microglial activation in the cingulum decreased (F), but on P90, microglial activation occurred in the cerebellar white matter (H). Immunohistochemical analysis of GFAP on brain slices of 30- and 90-d-old controls showed the typical distribution of GFAP-positive cells in cortex and cerebellum (I, K, M, O). GFAP is only weakly expressed in the corpus callosum and in some astrocytes surrounding blood vessels in the cortex, whereas it is prominent in the hippocampus. In contrast, GFAP expression was highly increased in corpus callosum and especially cortex of Cx43/Cx32dKO mice on P30 (J). There were no differences between control (K) and dKO (L) mice in cerebellar GFAP expression. During maturation, astrogliosis in the cortex of Cx43/Cx32dKO mice increased. Nearly the entire cortical area was covered by strongly GFAP-positive cells on P90 (N), and astrogliosis was additionally detected in the cerebellum of dKO mice on P90 (P). Scale bars: 500 μm; insets, 100 μm.
Astrogliosis is a natural reaction to any harm on the CNS, e.g., inflammation, ischemia, or neurodegenerative diseases (Sofroniew, 2009). To detect harmed tissue, we performed immunostainings against GFAP. Under normal conditions, only some GFAP-positive astrocytes are located in the cortex, as shown for P30 in Figure 1, I and K, as well as for P90 control mice (Fig. 1M,O). Severe astrogliosis occurred in cingulum and cortex of 30-d-old Cx43/Cx32dKOmGFAP mice (Fig. 1J), whereas GFAP staining in the cerebellar sections did not reveal any signs of astrogliosis (Fig. 1L). The intensity of the astrogliosis increased during development and resulted in glial scar formation in the cortex of dKO mice on P90 (Fig. 1N). Furthermore, reactive astrocytes could also be detected in cerebellar white matter at this time point (Fig. 1P).
The extent of microglial activation seems to peak during myelination, whereas astrogliosis shows a progressive course of events.
Disturbed myelin maintenance in Cx43/Cx32dKOmGFAP mice
The Iba1 staining revealed that activated microglial cells were mainly located at white matter tracts. To clarify whether myelin is affected by these inflammatory responses, we performed immunohistochemical stainings against MBP using 25 μm vibratome sections of 30- and 90-d-old mice. The cingulum as well as the fine fibers projecting from the cingulum to the cortex consistently expressed MBP in control mice on P30 (Fig. 2A). In contrast, Cx43/Cx32dKOmGFAP mice showed an irregular MBP expression pattern in the cortex (Fig. 2B). At higher magnifications, the strictly organized structure of the fine myelinated fibers was visible in control mice (Fig. 2C), whereas the staining in dKO mice looked patchy and revealed a partial loss of MBP-positive fibers (Fig. 2D).
Figure 2.
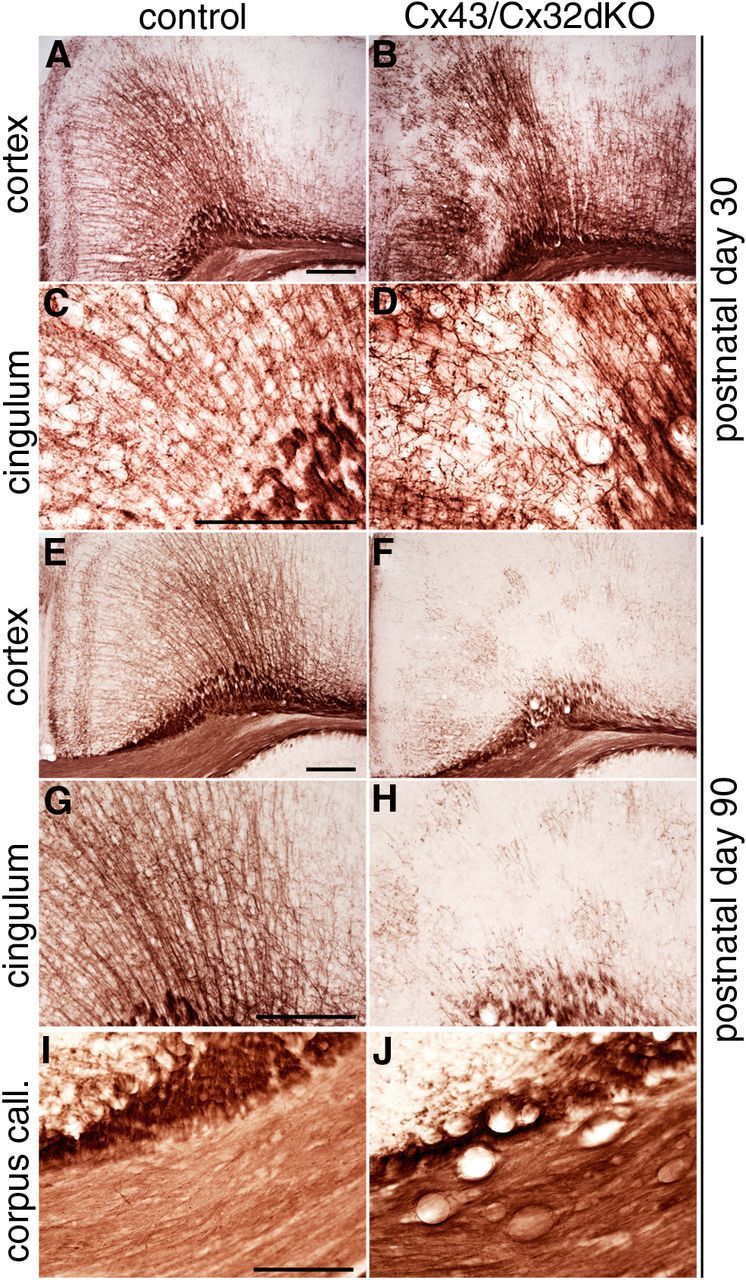
Formation of vacuoles in the corpus callosum and loss of fine myelinated fibers in the cingulum of Cx43/Cx32dKOmGFAP mice. Immunohistochemical staining against MPB was performed on brain slices of 30- and 90-d-old mice. In control mice on P30 (A, C), the myelin looks normal. Fine myelinated fibers spread out from the corpus callosum to the cortical layers. In Cx43/Cx32dKO mice, this pattern is disturbed (B, D). At higher magnifications, the tightly organized structure built up by fine myelinated fibers of the cingulum can be seen (C), whereas the organization is partially lost in Cx43/Cx32dKO brain (D). When comparing 90-d-old controls (E, G, I) with 90-d-old dKOs (F, H, J), a strong loss of myelinated fibers in the cingulum can be observed. In addition, there is a formation of vacuoles in the corpus callosum (J) of dKO animals. At higher magnification, only some islets of myelinated fibers are left in the cingulum of dKOs (H), whereas in control mice, a robust myelination is maintained (G). Scale bars, 200 μm.
In the cortex of 90-d-old mice, MBP staining yielded the typical pattern of fine fibers pervading the cortex (Fig. 2E). In contrast, MBP staining in Cx43/Cx32dKOs revealed a mosaic pattern of myelinated fibers and unstained tissue (Fig. 2F). Some remaining little islets of myelinated fibers were visible at a higher magnification, whereas most of the fine myelinated fibers were lost (Fig. 2H). Additionally, vacuole formation was evident in the corpus callosum of Cx43/Cx32dKOmGFAP (Fig. 2J) but not in control mice (Fig. 2I).
Oligodendrocyte population is decreased in the cingulum of Cx43/Cx32dKOmGFAP mice
Next we evaluated oligodendrocyte numbers in demyelinated cortical areas of Cx43/Cx32dKOmGFAP mice. We performed immunohistochemical stainings for two oligodendrocyte marker proteins. First, we used antibodies to Olig2, which is expressed by adult oligodendrocytes and oligodendrocyte precursor cells. We quantified the number of Olig2-positive cells in an area of 202,500 μm2 located above the cingulum (Fig. 3A,B, indicated by the box). Olig2-positive cells were significantly decreased in the cortex of 60-d-old Cx43/Cx32dKOmGFAP mice (Fig. 3F) compared with control mice (Fig. 3E). Subsequently, we performed immunostainings on brain sections of 30- and 90-d-old mice. Quantification revealed that the number of Olig2-positive cells in Cx43/Cx32dKOmGFAP mice (103 ± 2; 4 mice; 25 areas quantified) was decreased by 20% compared with control mice (129 ± 2; 6 mice; 56 areas quantified) on P30. The number of Olig2-positive cells in the cortex of Cx43/Cx32dKOmGFAP mice (68 ± 3; 4 mice; 28 areas quantified) was further decreased by 60% in total compared with control littermates on P60 (169 ± 4; 3 mice; 20 areas quantified). Cx43/Cx32dKOmGFAP mice showed the most vigorous decrease by 76% in Olig2-positive cell numbers (51 ± 2; 4 mice; 25 areas quantified) compared with control mice (212 ± 4; 5 mice; 34 areas quantified) on P90. All differences were statistically highly significant (p < 0.001).
Figure 3.

Number of oligodendrocytes is decreased in cingulum of Cx43/Cx32dKOmGFAP mice. Olig2-positive cells have been stained by immunohistochemistry in brain slices of 60-d-old mice. Normal number and distribution of Olig2-positive cells can be seen in brain slices of WT animals (A), whereas there were less Olig2-positive cells in the cingulum of Cx43/Cx32dKO mice (B). For better visualization, we provide higher magnifications of WT (E) and Cx43/Cx32dKO (F) mice. To quantify the number of myelinating oligodendrocytes, immunohistochemical staining against GST-π was performed on brain slices of 60-d-old mice. By comparing WT mice (C, G) with Cx43/Cx32dKO mice (D, H), a dramatic decrease of myelinating cells in the dKO animals becomes obvious. The number of Olig2-positive cells per 202,500 μm2 has been quantified for mice aged P30, P60, and P90 (I), whereas the number of GST-π-positive cells has only been quantified on P60 (J). Scale bars: A–D, 200 μm; E–H, 100 μm. ***p < 0.001.
To address the question whether the reduction in cell numbers is attributable to loss of early oligodendrocytes or loss of myelinating oligodendrocytes, we performed immunohistochemical staining for GST-π, a marker for myelinating cells. Immunostainings revealed a strong decrease in GST-π-positive cells in Cx43/Cx32dKOmGFAP mice (Fig. 3D,H) compared with control mice (Fig. 3C,G). Quantification ascertained a decrease by 87% from control mice (129 ± 6; 4 mice; 22 areas quantified) to Cx43/Cx32dKOmGFAP mice (17 ± 1; 5 mice; 36 areas quantified; p < 0.001) showing that Cx43/Cx32 deficiency results in a striking loss of myelinating cells in the cortex of 60-d-old mice.
Generation and characterization of new polyclonal Cx47 antibodies
To quantify the amount of Cx47 protein in the different transgenic connexin mouse mutants, we had to generate new Cx47 antibodies because commercially available Cx47 antibodies were found to give a false-positive signal at ∼47 kDa in immunoblot analyses of Cx47KO tissues. We used a synthetic peptide corresponding to a Cx47 C-terminal region as well as a GST-fusion protein carrying the whole Cx47 C terminus (amino acid residues 332–436) as antigens. Both were independently used to immunize rabbits. The newly generated antibodies were tested by immunofluorescence and immunoblot analysis. GST-fusion protein derived antibodies showed similar false-positive bands just like commercially available Cx47 antibodies and therefore were disposed. The peptide-derived antibodies were used for immunofluorescence analysis on wild-type (WT) mouse brain sections, in which we obtained a typical Cx47-staining pattern (Fig. 4A), whereas labeling of Cx47KO brain slices (Fig. 4B) showed no signals at all. Furthermore, we verified the antibodies by immunostainings of Cx47-expressing HeLa cells as a positive control (Fig. 4C). Subsequently, we performed immunoblot analyses using cerebellar tissue lysates and HeLa control lysates (Fig. 4D). The immunoblots showed a strong signal for Cx47 in lysates of WT mice at 47 kDa, whereas no signal was detected in Cx47KO lysates. The HeLa–Cx47 cell lysates, which were used as positive control, yielded a signal at 47 kDa, whereas HeLa WT controls did not give any immunosignal. GAPDH immunoblots were used as loading controls, indicating that WT and Cx47KO cerebellum contain similar amounts of protein. HeLa–Cx47 lysates showed no signal for GAPDH because of the low amount of total protein loaded to prevent superimposing of Cx47-positive signals.
Figure 4.

Validation of newly generated Cx47 antibodies by immunofluorescence and immunoblot analysis. Immunofluorescence analysis using our newly generated rabbit polyclonal Cx47 antibodies in the cingulum of WT (A) and Cx47-deficient (B) mouse brain slices. Cx47 signals are shown in red, and nuclear staining by Hoechst33258 is shown in blue. In addition, anti-Cx47 reactivity was confirmed using Cx47-expressing HeLa cells (C). Immunoblot analyses (D) using 50 μg of cerebellar tissue lysate show strong Cx47 expression in WT but no Cx47 expression in Cx47KO cerebellum. HeLa WT and HeLa–Cx47 cells were used as controls, whereas GAPDH was used for standardization of immunoblots. Scale bars, 100 μm.
Nestin–Cre mediated deletion of Cx43 and the effects on Cx47 protein expression
In a previous study, Lutz et al. (2009) reported that mGFAP–Cre-mediated deletion of the floxed Cx43 allele resulted in an incomplete KO of Cx43 in the CNS. To investigate the extent of remaining Cx43 expression in the forebrain, we performed immunofluorescence analyses, which revealed a nearly complete loss of Cx43 in cortex, hippocampus, and corpus callosum, whereas in the thalamus, many Cx43-positive signals remained (see Fig. 9C,D). To achieve deletion of Cx43 in all astrocytes, we crossbred Cx43flox mice with mice expressing the Cre recombinase under control of the Nestin promoter (Tronche et al., 1999). To verify the deletion efficacy of the Nestin promoter-driven Cre recombinase, immunofluorescence analyses were performed as shown in Figure 5. Anti-Cx43 immunostainings on WT brain sections resulted in robust staining in all brain regions except for white matter tracts including the corpus callosum, in which only moderate staining was observed (Fig. 5A,E). After Cre-mediated deletion, Cx43-positive signals were only found in meninges and some large blood vessels, in which Cx43 is expressed in smooth muscle cells (Little et al., 1995) (Fig. 5B,F). Our study shows that Nestin–Cre-mediated deletion of Cx43 is more efficient than mGFAP–Cre-mediated deletion and more reliable than hGFAP–Cre-mediated deletion, which was used in previous studies (Requardt et al., 2009).
Figure 9.

Immunohistochemical analyses of Cx43/Cx32dKONestin mice and comparison with Cx43/Cx32dKOmGFAP mice. Immunohistochemical staining for GFAP on brain slices of 60-d-old Cx43/Cx32dKOmGFAP mice revealed normal distribution of GFAP-positive astrocytes in the thalamus (A, B). mGFAP–Cre-driven deletion of Cx43 is incomplete; immunostainings for Cx43 show several Cx43-positive puncta remaining in the thalamus of Cx43/Cx32dKOmGFAP mice (C, D). Analyses of the phenotypic alterations in Cx43/Cx32dKONestin mice were performed by immunohistochemical stainings for GFAP, Iba1, and MBP on brain slices of 60-d-old-mice. Control mice exhibit normal distribution of GFAP-positive astrocytes in the cortex (E) and thalamus (G), whereas Cx43/Cx32dKONestin mice suffer from strong astrogliosis in the cortex (F) and thalamus (H). This is in contrast to Cx43/Cx32dKOmGFAP mice, which show no astrogliosis in the thalamus. Control mice show normal morphology of microglial cells (I, K), but in Cx43/Cx32dKONestin mice, microglial cells are activated throughout myelinated areas (J, L). Myelin staining shows strong loss of fine myelinated fibers in the cortex of Cx43/Cx32dKONestin mice (N) compared with controls (M). In the thalamus, the myelin appears less dense in Cx43/Cx32dKONestin mice (P) compared with controls (O). Scale bars, 100 μm.
Figure 5.

Verification of Nestin–Cre-mediated Cx43 deletion and loss of Cx47 in Cx43-deficient brain sections. Immunostaining of Cx43 (green) in 25 μm brain slices of 60-d-old mice shows that the Cx43 protein is abundantly expressed in the cortex (A) and thalamus (E) of control mice. After Nestin–Cre-mediated deletion, the exhaustive distribution of Cx43-positive immunosignals is lost (B, F). The remaining Cx43-positive signals are located in the meninges and endothelium of blood vessels. Immunostaining of Cx47 (red) in control mice shows prominent signals in cingulum/corpus callosum (C) and thalamus (G). Nestin–Cre-mediated deletion of Cx43 results in strong loss of Cx47-positive signals in all CNS areas tested (D, H). For better visualization of Cx47 immunosignals, immunofluorescence analyses of higher magnification were performed. Typical somatic Cx47 localization can be seen in brainstem slices obtained from WT mice (J, N), whereas a strong decrease in Cx47 immunosignals could be observed in Cx43cKO mice (R, V). Additionally, nuclear staining (I, M, Q, U) and staining for CNPase (K, O, S, W), an oligodendrocyte and myelin marker, were performed. Merged stainings of nuclei, Cx47, and CNPase (L, P, T, X). Scale bars, 100 μm.
Deletion of one glial connexin can influence the expression pattern or levels of the other glial connexins. For example, deletion of Cx47 leads to a relocalization and removal of Cx43, Cx30, and Cx32 from astrocyte to oligodendrocyte GJ plaques (Li et al., 2008). Because GJ channels formed by Cx43 and Cx47 play a role in panglial networks, we investigated whether Cx43 deletion affects Cx47 expression pattern and level. Thus, we performed immunostainings for Cx47 using Cx43flox:Nestin–Cre mice and Cx43flox mice as controls. Immunostainings on control mouse brain slices showed strong signals for Cx47, especially in the cingulum and the cortical area surrounding the cingulum (Fig. 5C) as well as in the thalamus (Fig. 5G). Furthermore, double immunostainings for CNPase and Cx47 were performed. Cx47-positive immunosignals were located at the cell somata and at the processes contiguous to the soma as described previously (Fig. 5J,N) (Li et al., 2004). Immunostainings in Cx43-deficient mice revealed strong loss of Cx47-positive signals (Fig. 5R,V). CNPase was used as oligodendrocyte marker. No differences in CNPase staining intensities were found between control mice (Fig. 5K,O) and Cx43cKO mice (Fig 5S,W).
Cx47 protein and mRNA levels after Cre-mediated deletion of Cx43
In view of the fact that the occurrence of Cx47 immunosignals on oligodendrocytes (presumed to represent gap junctional plaques) was strongly decreased in Cx43-deficient mice, we performed immunoblot analyses using newly generated Cx47 antibodies. Surprisingly, the banding pattern showed three specific bands for Cx47 in control lysates, whereas no signal was detected in Cx47KO lysates (Fig. 6A). Because certain connexin isoforms may be phosphorylated (for review, see Johnstone et al., 2012), we performed a dephosphorylation assay using λ protein phosphatase (λPP). After dephosphorylation of WT lysates, the immunoblots showed an altered banding pattern for Cx47 (Fig. 6C). We conclude that the Cx47 protein is phosphorylated in vivo and shows two phosphorylated forms in immunoblot analyses.
Figure 6.

Immunoblots showing Cx47 expression in control animals, dephosphorylation assay, and Cx47 expression in Cx43cKONestin mice as well as quantification of protein decrease and mRNA levels. Immunoblot analyses were performed with cerebellar protein lysates obtained from 60-d-old control mice, Cx43cKONestin mice, and Cx47KO mice. Using our newly generated Cx47 antibodies, we found that there are three distinct specific bands detectable for Cx47 in control mice, whereas there are no bands for Cx47 in Cx47KO mouse lysates (A). Interestingly, in Cx43cKONestin mice relative to control mice, there is only one band instead of three bands for Cx47 and the total amount of Cx47 is decreased (B). The household protein GAPDH was used as a loading control. Dephosphorylation assays were performed using 50 μg of crude WT protein lysate. After dephosphorylation by λPP, the Cx47 banding pattern changes and only the lowest band corresponding to 47 kDa remained on the immunoblot (C). Quantification of decreased Cx47 protein in Cx43cKONestin mice resulted in a reduction by 94% compared with control mice (D). Cerebellar RNA was used for sqRT-PCR analyses, which revealed no alterations in Cx47 mRNA expression between Cx43cKOmGFAP mice and control mice (E). ***p < 0.001.
Next, we determined whether the loss of Cx47 immunosignals is accompanied by changes in the Cx47 protein levels in mice deficient for astrocytic Cx43. We performed immunoblot analyses comparing WT and Cx43cKONestin cerebellar lysates of 60-d-old mice. As shown in Figure 6B, the protein levels in Cx43-deficient mice were strongly reduced. Quantitative immunoblot analysis of cerebellar lysates and subsequent Student's t test analysis revealed that Cx47 levels were significantly (p < 0.001; n = 4 for each genotype) reduced to 6% in Cx43cKONestin mice (Fig. 6D). Furthermore, the banding pattern of Cx47 was changed in Cx43-deficient mice. The typical banding pattern for Cx43cKONestin mice is illustrated in Figure 6B, showing only the lower band for Cx47. Loss of Cx43 leads to an altered Cx47 phosphorylation and protein amount in the cerebellum.
Cx47 immunosignals were unexpectedly lost and oligodendrocytic Cx47 protein level was strongly decreased after Cre-mediated deletion of astrocytic Cx43. To evaluate Cx47 mRNA expression levels, we performed sqRT-PCR analysis with RNA obtained from cerebella of 60-d-old mice. Expression levels were normalized to Cx47 levels in cerebellum of Cx43flox mice, and β-actin expression levels were used as internal standard. Cx43cKOmGFAP mice (1.15 ± 0.11; n = 3) showed no significant difference from control mice (1 ± 0.08; n = 3) (Fig. 6E). Specificity of the sqRT-PCR analysis was verified using RNA obtained from Cx47KO cerebellum as negative control.
Our results indicate that Cx47 immunosignals and protein levels are significantly decreased independent of mRNA expression levels in mice deficient for astrocytic Cx43. This suggests a posttranslational mechanism regulating the level of oligodendrocytic Cx47 protein via its dependence on Cx43 expression in contacting astrocytes.
Cx47 immunosignals and protein phosphorylation depend on the presence of Cx43 but not its GJ channel function
Oligodendrocytic Cx47 immunosignals and phosphorylation were impaired after Cre-mediated deletion of astrocytic Cx43. For the following experiments, we used Cx43floxG138R:Nestin–Cre/Cx30KO mice (Cx43ODDD) to assess whether the presence of the Cx43 protein in the plasma membrane is sufficient to maintain Cx47 protein expression, phosphorylation, and plaque formation or whether Cx47 immunosignals and protein phosphorylation depend on functional GJ channels. It had been shown that Cx43G138R GJ channels are not functional (Dobrowolski et al., 2007) and Cx30KO leads to loss of Cx26 in astrocytes (Lynn et al., 2011). As a result, the astrocytes in the Cx43floxG138R:Nestin–Cre/Cx30KO mice only express the nonfunctional Cx43G138R. Thus these mice represent a useful biological tool for our study. First, we performed immunofluorescence analyses to check whether Cx43 is normally expressed in the brain of these mice. The expression pattern of the mutated Cx43G138R in the cingulum of mouse brain (Fig. 7A) is similar to Cx43 in WT mice. Subsequently, immunostainings for Cx47 were performed. Cx47 immunosignals were normally distributed in sections of the CNS, e.g., in the cingulum (Fig. 7B), in which Cx47-positive puncta were mainly localized at somata as expected. Furthermore, immunoblot analyses using cerebellar tissue lysates of these mice were performed. We confirmed previous findings that Cx43G138R is not phosphorylated in vivo. Cx47 phosphorylation pattern in immunoblots (Fig. 7C) was similar to WT controls. We conclude that the Cx43 GJ channel function is not necessary for Cx47 protein stability and phosphorylation.
Figure 7.

Cx43 and Cx47 expression and phosphorylation in Cx43floxG138R/Cx30KO Nestin–Cre mice. Immunostainings for Cx43G138R (A; green) and Cx47 (B; red) were performed on 25 μm brain slices of 50-d-old Cx43ODDD mice. Both connexins are expressed at the cingulum of Cx43ODDD mice. Immunoblot analysis (C) shows that Cx43G138R is not phosphorylated. Similar to WT (Fig. 3), Cx47 protein is phosphorylated in Cx43ODDD mice. Fifty micrograms of protein per lane were loaded onto the SDS-gel. Tubulin was used as loading control. Scale bar, 100 μm.
Maintained myelination and lack of microglial activation in Cx43G138R/Cx30KO mice
In Cx43G138R/Cx30KO mice astrocytes should be completely uncoupled from each other as well as from oligodendrocytes, whereas oligodendrocytes should remain coupled among each other by Cx32 and Cx47. We performed immunostainings for GFAP, Iba1, Olig2, and MBP to clarify whether uncoupling of astrocytes results in inflammation or dismyelination. Immunostainings for MBP revealed no obvious deficits regarding myelin maintenance (Fig. 8A) in 50-d-old mice. Subsequent analysis of microglial cells by Iba1 immunostainings showed no inflammatory responses (Fig. 8B) in contrast to Cx43/Cx32dKOmGFAP mice (Fig. 1B). Microglial cells showed the normal ramified morphology in cingulum (Fig. 8B, inset). Interestingly, strong vacuolization of white matter tracts was visible in sections stained for MBP or Iba1 expression (Fig. 8A,B). Immunostainings for GFAP revealed astrogliosis in the cortex (Fig. 8D) and cerebellum (Fig. 8F, arrows) of Cx43G138R/Cx30KO mice. Distribution and number of Olig2-positive cells in the forebrain of Cx43G138R/Cx30KO mice was normal, as shown by Olig2 immunohistochemical analyses (Fig. 8G,H). Loss of A:A and A:O coupling resulted in severe vacuole formation in white matter tracts as well as astrogliosis in the CNS of Cx43G138R/Cx30KO mice.
Figure 8.

Myelin, microglia, astrocytes and oligodendrocytes in Cx43ODDD mice. Immunostainings on brain slices of 50-d-old mice show normal myelination (A) and non-activated microglial cells (B). Strong astrogliosis is visible in cortex and cerebellum (arrows) of Cx43ODDD mice (D, F) compared with control mice (C, E). Normal distribution of Olig2-positive oligodendrocytes in cingulum and cortex of Cx43ODDD mice (H) compared with controls (G). Vacuoles are mostly found in white matter regions across the brain of Cx43ODDD mice (A, B, D, F, H). Scale bars, 100 μm.
Phenotypical alterations in Cx43/Cx32dKONestin mice are more extensive compared with Cx43/Cx32dKOmGFAP mice
Phenotypic alterations in Cx43/Cx32dKOmGFAP mice were mainly restricted to the cingulum, the cortex, and the corpus callosum (Fig. 1). Immunostainings for GFAP in sections of control mice (Fig. 9A) as well as Cx43/Cx32dKOmGFAP mice (Fig. 9B) yielded only sparse GFAP-positive astrocytes in the thalamus. The regional restriction of the phenotypic alterations might result from an incomplete deletion of Cx43 (Fig. 9C,D) in the thalamus of Cx43/Cx32dKOmGFAP mice. Thus, we generated Cx43/Cx32dKONestin mice to achieve complete deletion of astrocytic Cx43. GFAP immunostainings revealed strong astrogliosis in the cortex of Cx43/Cx32dKONestin mice (Fig. 9F) compared with control mice (Fig. 9E), similar to Cx43/Cx32dKOmGFAP mice. In addition, we detected strong astrogliosis in the thalamus of Cx43/Cx32dKONestin mice (Fig. 9H) in contrast to control mice (Fig. 9G). Additional immunohistochemical analyses of Iba1 (Fig. 9I–L) and MBP (Fig. 9M–P) showed activated microglial cells and mild changes in myelination in the thalamus in addition to the severe phenotypical alterations in the cortex. Together, the Cx43/Cx32dKONestin mice revealed similar phenotypical alterations as the Cx43/Cx32dKOmGFAP mice, but in the CNS of Cx43/Cx32dKONestin mice, additional subcortical brain regions were affected.
Discussion
The common model of panglial networks is based on cell culture experiments, which revealed functional and nonfunctional combinations of glial connexins (Orthmann-Murphy et al., 2007; Magnotti et al., 2011a). In WT mice, Cx30 and Cx43 GJ channels allow for A:A coupling, and O:O coupling is maintained by Cx32 and Cx47 GJ channels. Furthermore, A:O coupling may be mediated by Cx30:Cx32, Cx30:Cx47, and Cx43:Cx47 heterotypic GJs (Fig. 10A). Impaired panglial coupling attributable to dKO of astrocytic connexins (Wallraff et al., 2006; Lutz et al., 2009), oligodendrocytic connexins (Menichella et al., 2003; Odermatt et al., 2003), or Cx30/Cx47 double deficiency (Tress et al., 2012) results in severe phenotypical alterations. Recently, astrocyte loss, white matter vacuolization, and early death at ∼16 weeks of age were reported for mice deficient in Cx43 and Cx32, but the underlying mechanism remained elusive (Magnotti et al., 2011b). The strong phenotypic abnormalities in Cx43/Cx32dKO mice are surprising because panglial networks should still be formed according to previous studies (Orthmann-Murphy et al., 2007; Magnotti et al., 2011a). Thus, A:A coupling should be maintained by Cx30, O:O coupling should be maintained by Cx47, and A:O coupling should be maintained by Cx30:Cx47 (Fig. 10B) heterotypic channels.
Figure 10.
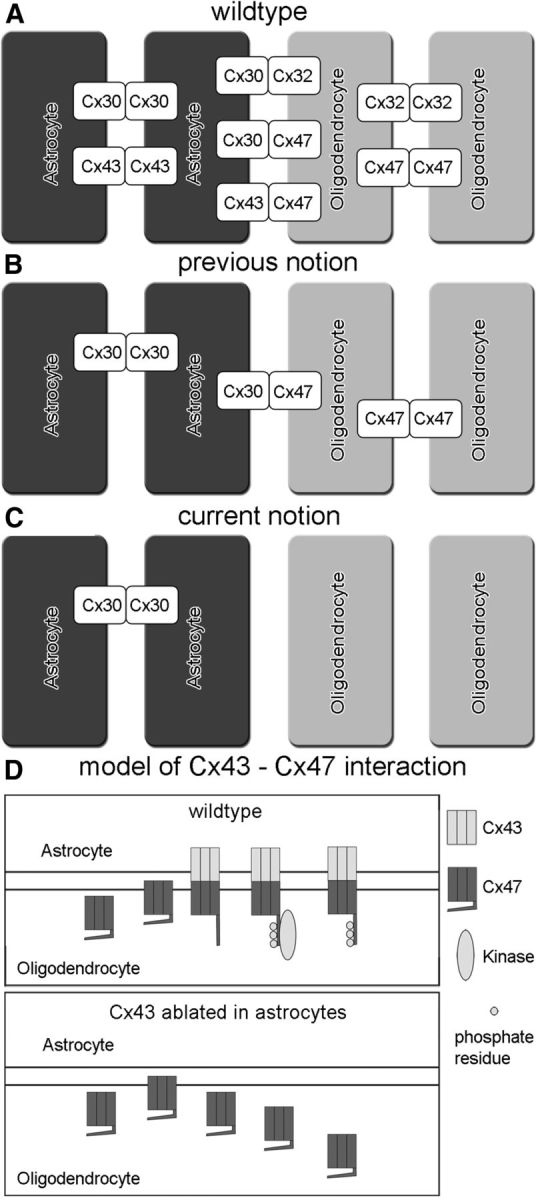
Scheme of panglial network coupling (A) and hypothetical model for Cx47 stabilization by docking to Cx43 hemichannels (B). A, In WT mice, A:A coupling is established by homotypic Cx30 and Cx43 channels, whereas O:O coupling is maintained by homotypic Cx32 and Cx47 channels. For the coupling of astrocytes to oligodendrocytes, three possible heterotypic connexin channels can be formed: Cx30:Cx32, Cx30:Cx47, and Cx43:Cx47. B, The previous notion for mice lacking Cx43 and Cx32 considered Cx30:Cx47 heterotypic channels for coupling between astrocytes and oligodendrocytes, whereas A:A coupling could be maintained by Cx30 and O:O coupling could be maintained by Cx47. C, Based on our new results, connexin expression pattern in Cx43/Cx32dKO mice differs from the previous notion. Because of the loss of Cx47, A:O and O:O coupling should not be possible. Only A:A coupling should occur via Cx30 channels. D, In WT mice, Cx47 is transported as well as integrated into the plasma membrane and can dock to Cx43. This may induce a conformational change that allows phosphorylation (inset) at the C terminus and may stabilize the protein. Internalization and degradation may take place at the normal rate. In mice lacking Cx43 in astrocytes, Cx47 is presumably also transported and integrated into the membrane, but no Cx43 hemichannels are present as docking partner. Therefore, a conformational change is not induced and phosphorylation sites cannot be accessed by kinase(s). Cx47 protein is not stabilized (inset), presumably resulting in higher internalization and protein degradation.
To clarify the mechanism causing the surprisingly strong phenotype, we generated Cx43/Cx32dKO mice in our laboratory. Although Cx43/Cx32dKOmGFAP mice showed strong phenotypic abnormalities, the mGFAP–Cre-driven deletion of Cx43 remained incomplete among astrocytes. The regional deletion of Cx43 correlates to the GFAP expression pattern in mice, but not all astrocytes express GFAP. To achieve a complete KO of astrocytic Cx43, we used Nestin–Cre mice, which express Cre in the entire CNS under control of the rat Nestin promoter (Tronche et al., 1999). Nestin–Cre-driven deletion resulted in complete astrocytic Cx43 deficiency, whereas Cx43 immunosignals remained in meninges and large blood vessels. Although the Nestin promoter is active in neural precursor cells and Cx43 expression in radial glia has been shown to be important for neuronal precursor migration (Elias et al., 2007), Nestin–Cre-mediated Cx43 deficiency did not result in obvious migration defects or any phenotypic abnormalities. Our data provide new insights into the extent of Nestin- and mGFAP-driven Cre-mediated deletion in the brain, which should be considered in future studies.
The dKO of Cx43 and Cx32 resulted in strong microglial activation, astrogliosis, and loss of myelin in the forebrain, whereas the cerebellum was moderately affected in adult mice. Furthermore, we found a strong decrease in the number of oligodendrocytes, especially myelinating oligodendrocytes, in the cingulum of the Cx43/Cx32dKOmGFAP mice in accordance with glial scar formation and most vigorous inflammation. Thus, we hypothesize that loss of oligodendrocytes may be a result of an inflammatory process attributable to a disturbed oligodendrocyte physiology. We carried out TUNEL assays to identify apoptotic cells, but no differences between Cx43/Cx32dKOmGFAP mice and controls were detected (data not shown). Our results suggest that activated microglia are involved in oligodendrocyte loss by internalization of either damaged oligodendrocytes or cell debris resulting from oligodendrocyte necrosis.
Surprisingly, immunostainings revealed loss of Cx47 immunosignals in Cx43-deficient brain sections, suggesting dependency of oligodendrocytic Cx47 GJ channel stability on astrocytic Cx43 expression. Loss of Cx43 effects the expression of several astrocytic genes in cell culture experiments (Iacobas et al., 2003). Transcellular gene regulation of Cx47 in oligodendrocytes after loss of Cx43 in astrocytes cannot be excluded but appears unlikely. Accordingly, we found unchanged Cx47 mRNA levels, indicating posttranslational regulation of Cx47 protein levels in Cx43-deficient mice. By λPP assays, we identified the absence of Cx47 protein phosphorylation in cerebellar lysates obtained from Cx43-deficient mice. Our results confirm a filter-assisted sample preparation method-based brain phosphoproteome study that showed that Cx47 is phosphorylated in vivo (Wiśniewski et al., 2010). Furthermore, we found that in vivo phosphorylation of Cx47 is dependent on Cx43 expression in contacting astrocytes.
To clarify whether functional Cx43 channels are necessary for phosphorylation and stabilization of Cx47 or whether the mere presence of Cx43 protein is sufficient for Cx47 GJ plaque formation, we used Cx43G138R/Cx30KO mice. The mutated Cx43G138R is transported to the plasma membrane, but the presumably formed GJ channels are not functional (Dobrowolski et al., 2007). Immunofluorescence and immunoblot analyses showed that Cx47 is normally expressed and phosphorylated in Cx43G138R/Cx30KO mice. Therefore, Cx47 phosphorylation is dependent on the presence of the Cx43 protein in the membrane regardless of Cx43 GJ channel function. We hypothesize that direct interaction after docking of an astrocytic Cx43 connexon to an oligodendrocytic Cx47 connexon leads to a conformational change in the C-terminal region of Cx47, allowing access for kinase(s). This phosphorylation may then stabilize the Cx47 protein, and the Cx43/Cx47 heterotypic channels remain in the plasma membrane (Fig. 10D). The formation of homotypic Cx47 channels, whose importance for O:O coupling has been discussed previously (Maglione et al., 2010; Wasseff and Scherer, 2011), may not be covered by this model. We could not identify strong Cx47-positive immunosignals in brain slices of Cx43cKO mice, which corresponds to the strong reduction of protein levels observed by immunoblot analyses. However, we cannot exclude that the remaining amount of Cx47, which may not be detected by immunofluorescence analysis, may still form homotypic O:O GJs. Nevertheless, in Cx43/Cx32dKO mice, these low amounts of Cx47 cannot prevent the occurrence of phenotypical alterations. The role and the formation of homotypic Cx47 O:O GJs in the brain needs to be further clarified in the future.
The unexpected impact of Cx43 on phosphorylation and stability of Cx47 dramatically changes the coupling situation in the Cx43/Cx32dKO mice, in which remaining Cx30 channels only provide A:A coupling whereas A:O and O:O coupling are interrupted (Fig. 10C). This results in the phenotypic abnormalities of the Cx43/Cx32dKO mice described here, which are similar to the phenotype of Cx32/Cx47dKO mice published previously (Menichella et al., 2003; Odermatt et al., 2003). The lack in early mortality as described for Cx32/Cx47 dKO mice in Cx43/Cx32dKOmGFAP mice could be attributable to incomplete deletion of Cx43 and subsequently Cx47. Inflammation in the Cx43/Cx32dKOmGFAP mice was restricted to GFAP-positive areas, whereas thalamus, brainstem, and deep cerebellar white matter of the cerebellum were not affected in accordance with the incomplete deletion of Cx43. The phenotypic alterations observed by Magnotti et al. (2011b) are partly consistent with our results, although we did not observe astrocyte loss and early mortality in the mGFAP–Cre:Cx43/Cx32dKO mice. Differences in the observed phenotypical alterations may be based on the different Cre recombinases used. Magnotti et al. (2011b) used the hGFAP–Cre recombinase, driven by the human GFAP promoter, which is active from embryonic day 12.5 onward, and may have a different onset and pattern of expression than the mouse GFAP–Cre recombinase (Zhuo et al., 2001; Garcia et al., 2004).
The complete deletion of astrocytic Cx43 in Cx43/Cx32dKONestin mice resulted in astrogliosis and inflammation in all regions in which myelinating oligodendrocytes were located. In addition, progressive demyelination was taking place in the whole cortex, and the myelin in the thalamus appeared to be less dense. We could not record early mortality because all mice were used for experiments.
Based on the newly identified dependence of Cx47 on the presence of Cx43 protein, results of several previous studies have to be readdressed. Lutz et al. (2009) also used Cx43flox:mGFAP–Cre mice and claimed that loss of the astrocytic connexins leads to a structural phenotype in astrocytes and oligodendrocytes. Here, we show that mGFAP–Cre:Cx43fl/fl mice are essentially deficient in Cx47 protein. Therefore, it is likely that loss of A:O and additionally decreased O:O coupling contributes to the phenotypical alterations observed in mGFAP–Cre:Cx43fl/fl/Cx30−/− mice.
Cx43G138R/Cx30KO mice show Cx47 expression similar to WT levels, but lack of A:A and A:O coupling makes them a useful tool to study the impact of A:O coupling on myelination and oligodendrocyte survival. We observed a great number of vacuoles located in CNS white matter tracts accompanied by strong astrogliosis. Surprisingly, there were no signs for microglial activation and myelination appeared unaffected, including the fine myelinated fibers in the cingulum. Therefore, we hypothesize that A:O coupling is not necessary for myelination and oligodendrocyte survival in the presence of Cx32- and Cx47-mediated O:O coupling. Conversely, vacuoles are caused by loss of A:A and A:O coupling, possibly by insufficient potassium buffering capacity. As a consequence, we assume that O:O coupling is sufficient for myelin formation and maintenance under normal physiological conditions. However, after loss of Cx47 function, A:O coupling becomes essential for the myelin maintenance as shown by Tress et al. 2012.
Taken together, we showed that Cx43 is essential for the stabilization and phosphorylation of Cx47 in vivo. Because Cx43 is expressed in astrocytes and Cx47 is expressed by oligodendrocytes, this is a so far unknown regulation of connexins from one cell type to another. Based on this finding, we further clarified that phenotypic abnormalities of Cx43/Cx32 double-deficient mice are caused by the loss of oligodendrocytic connexins Cx32 and Cx47. Thus, our results provide new insights into the complexity of panglial networks and the regulation of connexins involved.
Footnotes
This work was supported by German Research Foundation Grants Wi270/32-1, SFB645 (B2), and SFB-TR3 (C1), and European Union Grant FP7-202167 (NeuroGlia). We thank Jiong Zhang for providing the Cx43G138R/Cx30KO mice, Christine Siegmund for her excellent technical assistance, and Prof. Maarten Egmond (University of Utrecht, Utrecht, The Netherlands) for synthesis and purification of the immunogenic Cx47 peptide.
The authors declare no competing financial interests.
References
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- Dobrowolski R, Sommershof A, Willecke K. Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J Membr Biol. 2007;219:9–17. doi: 10.1007/s00232-007-9055-7. [DOI] [PubMed] [Google Scholar]
- Eiberger J, Kibschull M, Strenzke N, Schober A, Büssow H, Wessig C, Djahed S, Reucher H, Koch DA, Lautermann J, Moser T, Winterhager E, Willecke K. Expression pattern and functional characterization of connexin29 in transgenic mice. Glia. 2006;53:601–611. doi: 10.1002/glia.20315. [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Iacobas DA, Urban-Maldonado M, Iacobas S, Scemes E, Spray DC. Array analysis of gene expression in connexin-43 null astrocytes. Physiol Genomics. 2003;15:177–190. doi: 10.1152/physiolgenomics.00062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone SR, Billaud M, Lohman AW, Taddeo EP, Isakson BE. Posttranslational modifications in connexins and pannexins. J Membr Biol. 2012;245:319–332. doi: 10.1007/s00232-012-9453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleopa KA, Orthmann JL, Enriquez A, Paul DL, Scherer SS. Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia. 2004;47:346–357. doi: 10.1002/glia.20043. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Li X, Ionescu AV, Lynn BD, Lu S, Kamasawa N, Morita M, Davidson KG, Yasumura T, Rash JE, Nagy JI. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience. 2004;126:611–630. doi: 10.1016/j.neuroscience.2004.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Penes M, Odermatt B, Willecke K, Nagy JI. Ablation of Cx47 in transgenic mice leads to the loss of MUPP1, ZONAB and multiple connexins at oligodendrocyte-astrocyte gap junctions. Eur J Neurosci. 2008;28:1503–1517. doi: 10.1111/j.1460-9568.2008.06431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little TL, Beyer EC, Duling BR. Connexin 43 and connexin 40 gap junctional proteins are present in arteriolar smooth muscle and endothelium in vivo. Am J Physiol. 1995;268:H729–H739. doi: 10.1152/ajpheart.1995.268.2.H729. [DOI] [PubMed] [Google Scholar]
- Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci. 2009;29:7743–7752. doi: 10.1523/JNEUROSCI.0341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn BD, Tress O, May D, Willecke K, Nagy JI. Ablation of connexin30 in transgenic mice alters expression patterns of connexin26 and connexin32 in glial cells and leptomeninges. Eur J Neurosci. 2011;34:1783–1793. doi: 10.1111/j.1460-9568.2011.07900.x. [DOI] [PubMed] [Google Scholar]
- Maglione M, Tress O, Haas B, Karram K, Trotter J, Willecke K, Kettenmann H. Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32. Glia. 2010;58:1104–1117. doi: 10.1002/glia.20991. [DOI] [PubMed] [Google Scholar]
- Magnotti LM, Goodenough DA, Paul DL. Functional heterotypic interactions between astrocyte and oligodendrocyte connexins. Glia. 2011a;59:26–34. doi: 10.1002/glia.21073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnotti LM, Goodenough DA, Paul DL. Deletion of oligodendrocyte Cx32 and astrocyte Cx43 causes white matter vacuolation, astrocyte loss and early mortality. Glia. 2011b;59:1064–1074. doi: 10.1002/glia.21179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Lynn BD, Tress O, Willecke K, Rash JE. Connexin26 expression in brain parenchymal cells demonstrated by targeted connexin ablation in transgenic mice. Eur J Neurosci. 2011;34:263–271. doi: 10.1111/j.1460-9568.2011.07741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelles E, Bützler C, Jung D, Temme A, Gabriel HD, Dahl U, Traub O, Stümpel F, Jungermann K, Zielasek J, Toyka KV, Dermietzel R, Willecke K. Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9565–9570. doi: 10.1073/pnas.93.18.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Büssow H, Schilling K, Steinhäuser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS, Abrams CK. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J Neurosci. 2007;27:13949–13957. doi: 10.1523/JNEUROSCI.3395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requardt RP, Kaczmarczyk L, Dublin P, Wallraff-Beck A, Mikeska T, Degen J, Waha A, Steinhäuser C, Willecke K, Theis M. Quality control of astrocyte-directed Cre transgenic mice: the benefits of a direct link between loss of gene expression and reporter activation. Glia. 2009;57:680–692. doi: 10.1002/glia.20796. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutor B, Schmolke C, Teubner B, Schirmer C, Willecke K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb Cortex. 2000;10:684–697. doi: 10.1093/cercor/10.7.684. [DOI] [PubMed] [Google Scholar]
- Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Söhl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- Theis M, de Wit C, Schlaeger TM, Eckardt D, Krüger O, Döring B, Risau W, Deutsch U, Pohl U, Willecke K. Endothelium-specific replacement of the connexin43 coding region by a lacZ reporter gene. Genesis. 2001;29:1–13. doi: 10.1002/1526-968X(200101)29:1<1::AID-GENE1000>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Döring B, Frisch C, Söhl G, Teubner B, Euwens C, Huston J, Steinhäuser C, Messing A, Heinemann U, Willecke K. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–776. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress O, Maglione M, Zlomuzica A, May D, Dicke N, Degen J, Dere E, Kettenmann H, Hartmann D, Willecke K. Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes Pelizaeus-Merzbacher-like disease in humans. PLoS Genet. 2011;7:e1002146. doi: 10.1371/journal.pgen.1002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress O, Maglione M, May D, Pivneva T, Richter N, Seyfarth J, Binder S, Zlomuzica A, Seifert G, Theis M, Dere E, Kettenmann H, Willecke K. Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J Neurosci. 2012;32:7499–7518. doi: 10.1523/JNEUROSCI.0392-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasseff SK, Scherer SS. Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol Dis. 2011;42:506–513. doi: 10.1016/j.nbd.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilgenbus KK, Kirkpatrick CJ, Knuechel R, Willecke K, Traub O. Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues. Int J Cancer. 1992;51:522–529. doi: 10.1002/ijc.2910510404. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Nagaraj N, Zougman A, Gnad F, Mann M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res. 2010;9:3280–3289. doi: 10.1021/pr1002214. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]