

Abstract
Polyglutamine (polyQ) diseases are inherited neurodegenerative disorders that are caused by the expansion of trinucleotide CAG repeats in the causative genes. Spinal and bulbar muscular atrophy (SBMA) is an inherited motor neuron disease that is caused by the expansion of a polyQ tract within the androgen receptor (AR). p62 is a ubiquitin- and light-chain 3-binding protein that is known to regulate the degradation of targeted proteins via autophagy and inclusion formation. In this study, we examined the effects of p62 depletion and overexpression on cultured cells and in a transgenic mouse model that overexpressed the mutant AR. Here, we demonstrate that depletion of p62 significantly exacerbated motor phenotypes and the neuropathological outcome, whereas overexpression of p62 protected against mutant AR toxicity in SBMA mice. Depletion of p62 significantly increased the levels of monomeric mutant AR and mutant AR protein complexes in an SBMA mouse model via the impairment of autophagic degradation. In addition, p62 overexpression improved SBMA mouse phenotypes by inducing cytoprotective inclusion formation. Our results demonstrate that p62 provides two different therapeutic targets in SBMA pathogenesis: (1) autophagy-dependent degradation and (2) benevolent inclusion formation of the mutant AR.
Introduction
Polyglutamine (polyQ) diseases are inherited neurodegenerative disorders that are caused by the expansion of trinucleotide CAG repeats in specific genes (Di Prospero and Fischbeck, 2005). Spinal and bulbar muscular atrophy (SBMA) is a motor neuron disease that is caused by the expansion of a polyQ tract within the androgen receptor (AR) (Adachi et al., 2007). SBMA is characterized by motor neuron loss in the spinal cord and brainstem, along with diffuse nuclear accumulation and nuclear inclusions (NIs) of the mutant AR in residual motor neurons and specific visceral organs (Sobue et al., 1989; Li et al., 1998; Adachi et al., 2005). Nuclear and cytoplasmic inclusions are common pathological features in polyQ diseases and colocalize with many components of the ubiquitin–proteasome system (UPS) and autophagy, which raises the possibility that UPS and autophagy might actively degrade components of these inclusions (Buchberger et al., 2010). Furthermore, molecular chaperones also facilitate the refolding or proteolysis of toxic misfolded proteins and could play a role in protecting neuronal cells against the toxic properties of expanded polyQ (Adachi et al., 2009; Labbadia et al., 2011).
Macroautophagy is a process in which cells form double-membrane vesicles called autophagosomes around a portion of the cytoplasm. The protein p62, which is also known as sequestosome-1 (SQSTM1), is an adaptor protein that binds to ubiquitin and the autophagosome membrane light-chain 3 (LC3) in the autophagic degradation pathway (Ichimura et al., 2008). These autophagosomes ultimately fuse with lysosomes, which results in the degradation of their substrates. Specific phosphorylation of p62 at serine 403 in its ubiquitin-associated (UBA) domain increases the affinity between UBA and the polyubiquitin chain, which results in efficiently targeted polyubiquitinated proteins and stabilization of the sequestosome structure (Matsumoto et al., 2011). Although p62 plays a pivotal role in nuclear and cytoplasmic inclusion formation for disease-causative proteins in neurodegenerative disorders (Kuusisto et al., 2001, 2008; Al-Sarraj et al., 2011; King et al., 2011; Troakes et al., 2012), the role of p62 in the pathogenesis of neurodegenerative disorders, including SBMA, has not yet been fully resolved.
In this study, we examined the effect of p62 depletion and overexpression in cultured cells and a transgenic mouse model that overexpresses mutant AR to explore a potential strategy for SBMA therapy. In the SBMA mouse model, depletion of p62 significantly exacerbated physical, behavioral, and neuropathological outcomes, and overexpression of p62 protected against the toxicity of mutant AR. Depletion of p62 significantly increased the accumulation of monomeric mutant ARs and mutant AR protein complexes in SBMA mice via the impairment of autophagic degradation, whereas overexpression of p62 promoted inclusion body formation that was less toxic in the spinal cord and muscle of SBMA mice. Our results demonstrate that p62 exhibits two different therapeutic aspects that are critical components in the autophagy-dependent degradation of the mutant AR and provides harmless inclusion body formation in SBMA pathogenesis.
Materials and Methods
Plasmid constructs and siRNA.
pIRESpuro3, which encodes 3xFLAG-tagged mouse p62, and the PB1 p62 mutant (K7A and D69A, M0) plasmids were kindly provided by Dr. Masaaki Komatsu (Laboratory of Frontier Science, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). The PB1 p62 mutant (D69A) plasmid with an HA-tag (see Fig. 2D), deletion mutant plasmids for p62 (M1–M7; see Fig. 2E), and a plasmid with ubiquitin-binding-disrupting mutation (M406V) of p62 with a FLAG tag (see Fig. 2G) were generated using a KOD Plus Mutagenesis Kit (Toyobo). The plasmids pCR3.1–AR–24Q and pCR3.1–AR–97Q (Waza et al., 2005) were also used. All of the constructs were verified by DNA sequencing. siRNAs that target p62 were used at a concentration of 10 nm and had the following sequences: 5′-GAAAGTGCATGAGAAGAGATT-3′ (sense) and 5′-TCTCTTCTCATGCACTTTCTT-3′ (antisense) for the PC12 cells and 5′-CTTGTAGTTGCATCACGTATT-3′ (sense) and 5′-TACGTGATGCAACTACAAGTT-3′ (antisense) for the Neuro2A cells.
Figure 2.

p62 interacts with AR. A, Neuro2A cells transfected with wild-type (24Q) and mutant (97Q) AR were treated with bafilomycin A1 (20 nm) for 16 h. Immunoprecipitation was then performed with non-immune rabbit IgG (NI) or an antibody for endogenous p62 (I). B, PB1 mutant p62 (M0) or wild-type p62 (Wt) and AR were cotransfected in Neuro2A cells, followed by immunoprecipitation with FLAG M2 agarose beads. Mutant p62 coprecipitated with AR, whereas the control did not show an interaction. C, PB1 mutant p62 (M0) and AR were cotransfected in Neuro2A cells. Immunoprecipitation was then performed using FLAG M2 agarose beads. D, p62 with an HA tag and ARs were cotransfected in Neuro2A cells. Immunoprecipitation was then performed using an HA antibody. E, A schematic representation of p62 deletion mutants fused with an N-terminal 3xFLAG generated by mutagenesis. F, AR–97Q and p62 mutants (M0–M7) were cotransfected, and immunoprecipitation was performed. G, Mutant p62 (M406V) or wild-type p62 (Wt) and AR–97Q were cotransfected in Neuro2A cells, followed by immunoprecipitation using FLAG M2 agarose beads. IPs, Immunoprecipitations; C, control; K7A D69A, Phox and Bem1p (PB1) domain with mutations for each amino acid; ZZ, ZZ-type zinc finger; LRS, LC3 recognition sequence; Ub, ubiquitin.
Cell culture and DNA transfection.
PC12 Tet-On cells that express human wild-type AR (AR–10Q) or mutant AR (AR–112Q) were kindly provided by Dr. Diane E. Merry (Departments of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA) (Walcott and Merry, 2002). Plasmid DNA and siRNA were transfected into Neuro2A cells, NSC34 cells, or PC12 Tet-On cells using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Cells were plated in 6-cm dishes in 5 ml of DMEM/10% FCS with penicillin and streptomycin, and each dish was transfected with 4 μg of plasmid DNA and p62 or control (mock) siRNA. The PC12 cells were cultured in DMEM that contained 10 ng/ml nerve growth factor (NGF) (Alomone Labs), 1 μg/ml doxycycline, and 1 nm dihydrotestosterone (DHT) for the indicated days, as described previously (Walcott and Merry, 2002). Neuro2A cells were cultured in differentiation medium (DMEM/2% FCS; DMEM supplemented with 2% fetal calf serum) that contained retinoic acid (20 μm) and DHT (1 nm) after transfection. The transfection efficiency was 60–70%. The NSC34 cells were cultured in DMEM/10% FCS for 48 h at 37°C under 5% CO2.
Generation of AR transgenic/p62 knock-out mice and genotyping.
p62 knock-out mice in the C57BL/6J (B6) background were kindly provided by Tetsuro Ishii (Graduate School of Comprehensive Human Sciences, University of Tsukuba, Tsukuba, Japan) (Komatsu et al., 2007). Homozygous p62 knock-out females (p62−/− mice) of the B6 strain were mated with male mice of the BDF1/B6 strain that expressed full-length human AR with 24-polyQ tracts (AR–24Q mice, 5–5 line) or 97-polyQ tracts (AR–97Q mice, 7–8 line) to produce a mixed BDF1 and B6 genetic background. First-generation AR–24Q/p62+/− or AR–97Q/p62+/− mice were mated with p62+/− or p62−/− mice to produce all of the combinations and numbers for each analysis. We screened tail DNA by PCR to detect the deletion of the p62 gene using two primer pairs: 5′-CTGCATGTCTTCTCCCATGAC-3′/5′-TAGATACCTAGGTGAGCTCTG-3′ and 5′-CTTACGGGTCCTTTTCCCAAC-3′/5′-TCCTCCTTGCCCAGAAGATAG-3′.
Transgene construction.
Full-length human p62 cDNA was generated from total RNA that was extracted from SH-SY5Y cells by reverse transcription (RT)-PCR. Full-length human p62 was constructed by subcloning p62 inserts that were derived from the full-length human p62 cDNA into the pCAGGS vector (Niwa et al., 1991) using PCR. The HA-tagged p62 fragments were then subcloned by mutagenesis methods, and the DNA sequence was confirmed. The final plasmids were digested to remove the transgene (see Fig. 6A).
Figure 6.
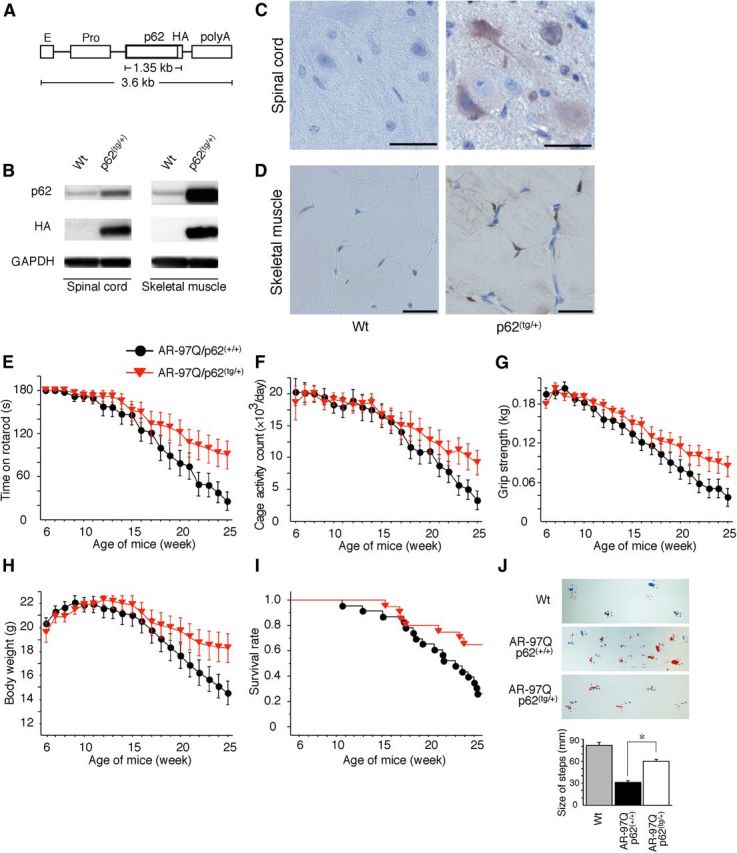
p62 overexpression ameliorates behavioral and visible phenotypes in male AR–97Q mice. A, A schematic view of the transgene construct. The microinjected fragment was composed of a cytomegalovirus enhancer (E), a chicken β-actin promoter (Pro), full-length human p62 with an HA tag, and a rabbit β-globin polyadenylation signal sequence (polyA). B, Western blotting analysis of total spinal cord and muscle protein lysates from wild-type (Wt) and p62(tg/+) mice immunolabeled with antibodies against p62 and HA. C, D, HA immunohistochemistry in the spinal anterior horn (C) and skeletal muscle (D) of 13-week-old wild-type (Wt) and p62(tg/+) mice counterstained with Mayer's hematoxylin. p62 immunoreactivity localized to the nuclei and cytoplasms of anterior horn cells (C) and skeletal muscle (D). HA staining was absent in the wild-type mice. Scale bars, 50 μm. E–I, Rotarod task (E), cage activity (F), grip strength (G), body weight (H), and survival rate (I) of AR–97Q/p62(+/+) (●; n = 25) and AR–97Q/p62(tg/+)(▾; n = 21) mice. J, Footprints of representative 25-week-old AR–97Q/p62(+/+) and AR–97Q/p62(tg/+) mice. The front paws are indicated in blue, and the hindpaws are indicated in red. Values are expressed as the means ± SEM. *p < 0.01.
Generation and maintenance of p62 transgenic mice and genotyping.
We generated mice that overexpressed p62 by microinjection of the transgene into B6 fertilized eggs; we obtained four founders. Heterozygous p62 transgenic females of the B6 strain were mated with male mice of the BDF1/B6 strain that overexpressed AR–24Q mice (5–5 line) or AR–97Q mice (7–8 line) to produce a mixed BDF1 and B6 genetic background. First-generation AR–24Q/p62(tg/+) or AR–97Q/p62(tg/+) mice were used for each analysis. We screened tail DNA by PCR for the presence of the transgene using the following primers: 5′-AGCTGCCTTGTACCCACATC-3′ and 5′-AGCGTAATCTGGAACATCGT-3′.
Neurological and behavioral assessments of SBMA model mice.
AR–24Q and AR–97Q mice were generated and maintained as described previously (Katsuno et al., 2002). All of the animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and under the approval of the Nagoya University Animal Experiment Committee. The AR–97Q male mice exhibited progressive muscular atrophy and weakness in addition to diffuse nuclear staining (DNS) and NIs of the mutant AR. Similar to SBMA patients, these phenotypes were pronounced in male transgenic mice. The mouse rotarod task (Ugo Basile) was performed on a weekly basis, and cage activity was measured weekly using the AB system (Neuroscience), as described previously (Katsuno et al., 2002). Spontaneous motor activity was monitored in 24 h periods; all vertical and horizontal spontaneous movements, including locomotion, rearing, and head movements, were counted and automatically totaled. Grip strength analysis was performed using an MK380M Grip Strength Meter (Muromachi Kikai).
Immunocytochemistry.
PC12 Tet-On cells were plated in a collagen-coated four-chamber slide (BD Biosciences) and were cultured in DMEM that contained 10 ng/ml NGF, 1 μg/ml doxycycline, and 1 nm DHT for the indicated days, as described previously (Walcott and Merry, 2002). The cells were also treated with bafilomycin A1 (11707; Sigma) for 16 h and 10 ng/ml leptomycin B (Enzo Diagnostics) for 2 h before harvest and were fixed with 4% paraformaldehyde for 15 min at room temperature. The cells were then treated in formic acid for 5 min at room temperature and were sequentially incubated with anti-p62 antibody (1:1000; PROGEN Biotechnik), anti-LC3 (1:1000; PM046; Medical and Biological Laboratories), and anti-AR (1:1000; Perseus Proteomics) at 4°C overnight. Next, the sections were incubated with Alexa Fluor 647-conjugated goat anti-guinea pig IgG, Alexa Fluor 546-conjugated goat anti-rabbit IgG, and Alexa Fluor 488-conjugated goat anti-mouse IgG (1:1000; Invitrogen) for 1 h at room temperature. The stained sections were imaged using a confocal laser-scanning microscope (LSM 710; Carl Zeiss).
Patients.
Tissue was obtained from three patients with clinicopathologically and genetically confirmed SBMA (51–77 years of age; mean age, 65.7 years) and three controls without neurological disease (51–78 years of age; mean age, 64.0 years). These patients were hospitalized and evaluated at Nagoya University Hospital and its affiliated hospitals. The collection of human tissues and their use for this study were approved by the Ethics Committee of Nagoya University Graduate School of Medicine. Informed consent was obtained to use these tissues for research purposes. Paraffin-embedded sections of the spinal cord were processed and examined in the same manner as was used for the transgenic mice.
Immunohistochemistry and histopathology.
Mice were deeply anesthetized with pentobarbital sodium and were transcardially perfused with 20 ml of 4% paraformaldehyde fixative in phosphate buffer, pH 7.4. The spinal cord and skeletal muscle tissues were removed, postfixed overnight in 10% phosphate-buffered Formalin, and processed for paraffin embedding. Sections (6 μm thickness) of the tissue samples were deparaffinized, dehydrated with alcohol, and treated in formic acid for 5 min at room temperature. For the immunohistochemical studies, the paraffin sections were preheated in a microwave oven for 10 min and were blocked with normal animal serum (1:20). The sections were then incubated with anti-expanded polyQ antibody (1:10,000; 1C2; Millipore) and anti-p62 antibody (1:1000; PROGEN Biotechnik) for all of the human tissues and anti-p62 antibody (1:1000; PM045; Medical and Biological Laboratories), anti-glial fibrillary acidic protein (GFAP) antibody (1:1000; Roche Diagnostics), anti-HA antibody (1:500; C29F4; Cell Signaling Technology), and anti-ubiquitin antibody (1:1000; catalog #3936; Cell Signaling Technology) for the mouse tissues. Primary antibodies were probed using a biotinylated anti-species-specific IgG (Vector Laboratories), and the immune complexes were visualized using streptavidin–horseradish peroxidase (Dako) and 3,3-diaminobenzidine (Dojindo) as a substrate. Anti-ubiquitin antibody binding was probed with a labeled polymer of the secondary antibody as part of the Envision system, which contained horseradish peroxidase (Dako). The sections were counterstained with Mayer's hematoxylin. Paraffin-embedded sections (6 μm thickness) of the gastrocnemius muscle were air dried and stained with hematoxylin and eosin. For double-immunofluorescence staining, the sections were blocked with TSA Blocking Reagent (PerkinElmer Life and Analytical Sciences) and were then sequentially incubated with anti-p62 antibody (1:1000; PM045; Medical and Biological Laboratories) and 1C2 antibody (1:10,000; Millipore) at 4°C overnight. The sections were then incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:1000; Invitrogen) and Alexa Fluor 546-conjugated goat anti-mouse IgG (1:1300; Invitrogen) for 8 h at 4°C. The stained sections were imaged using a confocal laser-scanning microscope (LSM 710; Carl Zeiss).
Quantification of 1C2-positive cells and ubiquitin-positive NIs.
To assess 1C2-positive cells and ubiquitin-positive NIs in the ventral horn of the spinal cord, 50 consecutive transverse sections of the thoracic spinal cord were prepared from each individual mouse. Cells positive for 1C2 within the ventral horn of every fifth section were counted as described previously (Adachi et al., 2001). Populations of 1C2-positive cells and ubiquitin-positive NIs were expressed in units of number per square millimeters (see Fig. 5G). To examine 1C2-positive cells and ubiquitin-positive NIs in muscle, the number was calculated from counts of 500 fibers in randomly selected areas of individual mice and was expressed as the number per 100 muscle fibers (see Fig. 5H). The ratio of the number of cells with NIs or DNS to that of the total number of 1C2-positive cells was calculated (see Fig. 7B,C). The 1C2 signal intensity of the DNS was analyzed using Image Gauge software version 4.22 (GE Healthcare). The mean signal intensity of DNS was calculated by correcting the signal intensity of all of the 1C2-positive neurons in the spinal cord and all of the 1C2-positive nuclei in the muscle. The relative signal intensity (R.S.I.) of the DNS was computed as the signal intensity of each cell divided by the mean signal intensity of cells without inclusions (see Fig. 7D). The quantitative data for six individual mice are expressed as the mean ± SEM.
Figure 5.
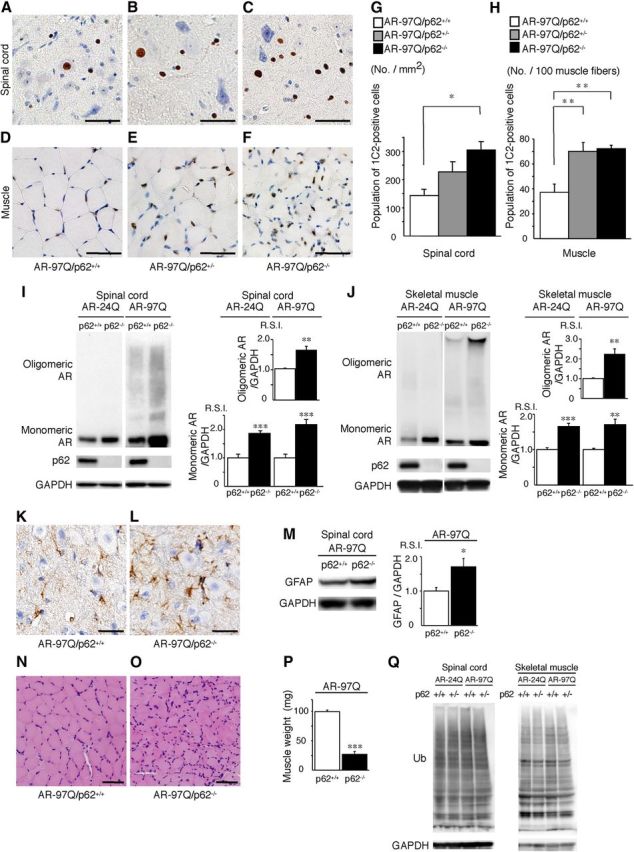
Depletion of p62 induces the accumulation of AR in an SBMA mouse model. A–F, PolyQ immunohistochemistry (1C2) in the spinal anterior horn (A–C) and muscle (D–F) of 13-week-old AR–97Q/p62+/+, AR–97Q/p62+/−, and AR–97Q/p62−/− mice. Scale bars, 50 μm. G, H, Quantitative assessment of 1C2 staining in the spinal ventral horn (G) and muscle (H). Bars represent the density of 1C2-positive cells in AR–97Q/p62+/+, AR–97Q/p62+/−, and AR–97Q/p62−/− mice. The results are expressed as the means ± SEM (n = 6). I, J, Western blotting analysis of the total tissue homogenates from the spinal cord (I) and muscle (J) of AR–24Q and AR–97Q mice (13 weeks of age, n = 6), probed with anti-AR. Values are expressed as the means ± SEM (n = 6). K, L, Immunohistochemical staining with anti-GFAP antibody in the spinal anterior horn. Scale bars, 30 μm. M, Western blotting analysis of the total tissue homogenates from the spinal cord of AR–97Q mice (13 weeks of age, n = 6) probed with anti-GFAP. N, O, Hematoxylin and eosin staining of the muscles. P, The gastrocnemius muscles from AR–97Q/p62+/+ and AR–97Q/p62−/− mice were dissected and weighed. Q, Western blotting analysis of the total tissue homogenates from the spinal cord and muscle of AR–24Q and AR–97Q mice (13 weeks of age) probed with anti-ubiquitin (Ub). Scale bars, 50 μm. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 7.
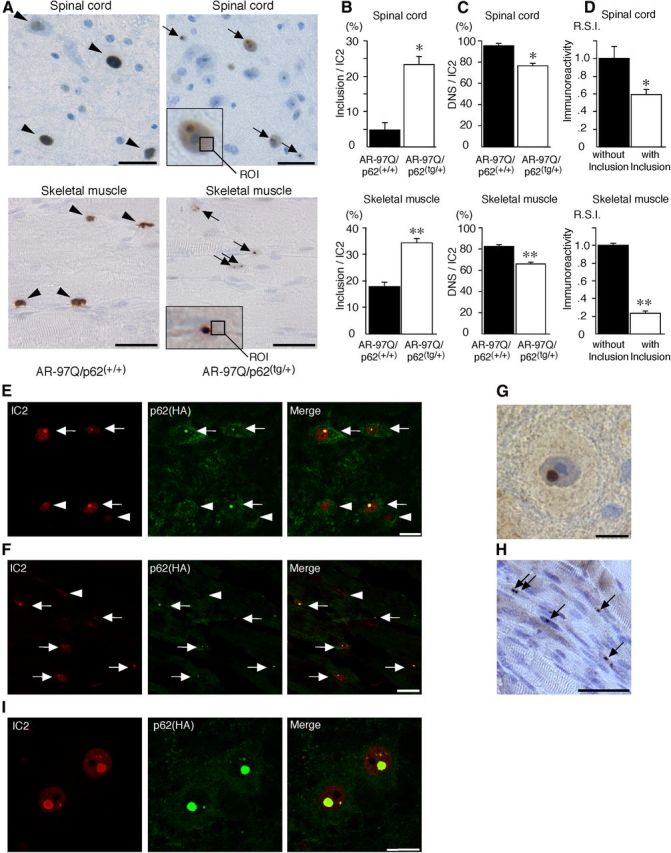
p62 promotes inclusion body formation of ARs in male AR–97Q mice. A, Immunohistochemical staining with 1C2 antibody revealed DNS (arrowhead) in AR–97Q/p62(+/+) mice and many NIs (arrow) in the AR–97Q/p62(tg/+) mice in the spinal anterior horn and skeletal muscle at 25 weeks of age. The region of interest for the DNS is defined as the ROI (inset; open square). Scale bars, 30 μm. B, The ratio of the number of cells with NIs to the total number of IC2-positive cells. C, The ratio of the number of cells with DNS to the total number of IC2-positive cells. D, The R.S.I. of the DNS was significantly reduced in cells with NIs compared with those without NIs in spinal anterior horn neurons and skeletal muscle. E, F, Double-immunofluorescence staining with 1C2 (red) and anti-HA (green) antibodies in the spinal anterior horn (E) and skeletal muscle (F) of AR–97Q/p62(tg/+) mice revealed the complete colocalization of HA-tagged p62 and mutant AR in NIs (arrow), but no colocalization was observed in the DNS (arrowhead). Scale bars, 20 μm. G, H, p62 immunohistochemistry in the spinal anterior horn neurons (G) and skeletal muscle (H) of AR–97Q/p62(tg/+) mice. Arrows indicate NIs. Scale bars: G, 30 μm; H, 50 μm. Values are expressed as the means ± SEM (n = 6). I, 1C2 (red) and anti-HA (green) double immunofluorescence in the spinal cord of 25-week-old AR–97Q/p62(tg/+) mice. *p < 0.01; **p < 0.001.
Protein expression analyses.
Forty-eight hours after transfection, the cells were lysed in TNE buffer (10 mm Tris-HCl, pH 7.5, 1% Nonidet P-40, 150 mm NaCl, and 1 mm EDTA), supplemented with 1 mm PMSF and 6 μg/ml aprotinin and centrifuged at 15,000 × g for 15 min at 4°C. Thirteen- and 25-week-old mice were exsanguinated under pentobarbital sodium anesthesia, and tissues were snap frozen with powdered CO2 in acetone. The tissues were homogenized in CelLytic-M Mammalian Cell Lysis/Extraction Reagent (Sigma) with 1 mm PMSF and 6 μg/ml aprotinin and were centrifuged at 2,500 × g for 15 min at 4°C. Supernatant protein concentrations were determined using the DC protein assay (Bio-Rad). Aliquots of the supernatant fractions were loaded on 5–20% SDS-PAGE gels, in which each lane contained 7 μg of cell proteins and 80 μg of neural and muscle tissue. To prevent carryover, the pellets were washed with CelLytic-M Mammalian Cell Lysis/Extraction Reagent, followed by sonication and centrifugation. The insoluble pellets were lysed in 300 μl of 8 m urea solution (30 mm Tris, pH 8.5, 2 m Thiourea, 4% CHAPS, and 8 m urea), sonicated, and centrifuged at 25,000 ×g for 15 min at 4°C. Aliquots of the supernatant fractions were loaded on 5–20% SDS-PAGE gels. The gels were then transferred to Hybond-P membranes (GE Healthcare) using 25 mm Tris, 192 mm glycine, 0.1% SDS, and 10% methanol as the transfer buffer. Primary antibodies were used at the following concentrations: rabbit anti-AR, 1:1000 (N20; Santa Cruz Biotechnology); rabbit anti-AR, 1:1000 (H280; Santa Cruz Biotechnology); rabbit anti-p62, 1:1000 (PM045; Medical and Biological Laboratories); rabbit anti-LC3 for cells, 1:1000 (PM036; Medical and Biological Laboratories); rabbit anti-LC3 for mouse tissues, 1:2000 (NB600-1384; Novus Biologicals); mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 1:10000 (MAB374; Millipore); rabbit anti-HA, 1:1000 (Y-11; Santa Cruz Biotechnology); and mouse anti-ubiquitin, 1:1000 (catalog #3936; Cell Signaling Technology). Primary antibodies were probed using HRP-conjugated anti-rabbit Ig F(ab′)2 and anti-mouse Ig F(ab′)2 (1:5000; GE Healthcare) secondary antibodies and were detected using the ECL Plus kit (GE Healthcare). An LAS-3000 imaging system was used to produce digital images and to quantify band intensities, which were then analyzed using Image Gauge software version 4.22 (GE Healthcare). Densitometric values of ARs were normalized to those of endogenous GAPDH. The R.S.I. was computed as the signal intensity of each sample divided by that of the nontreated cells and control siRNA-transfected cells (see Figs. 1, 3) or the signal intensities of AR–24Q/p62+/+ or AR–97Q/p62+/+ mice (see Fig. 5) and AR–24Q/p62(+/+) or AR–97Q/p62(+/+) mice (see Fig. 9).
Figure 1.
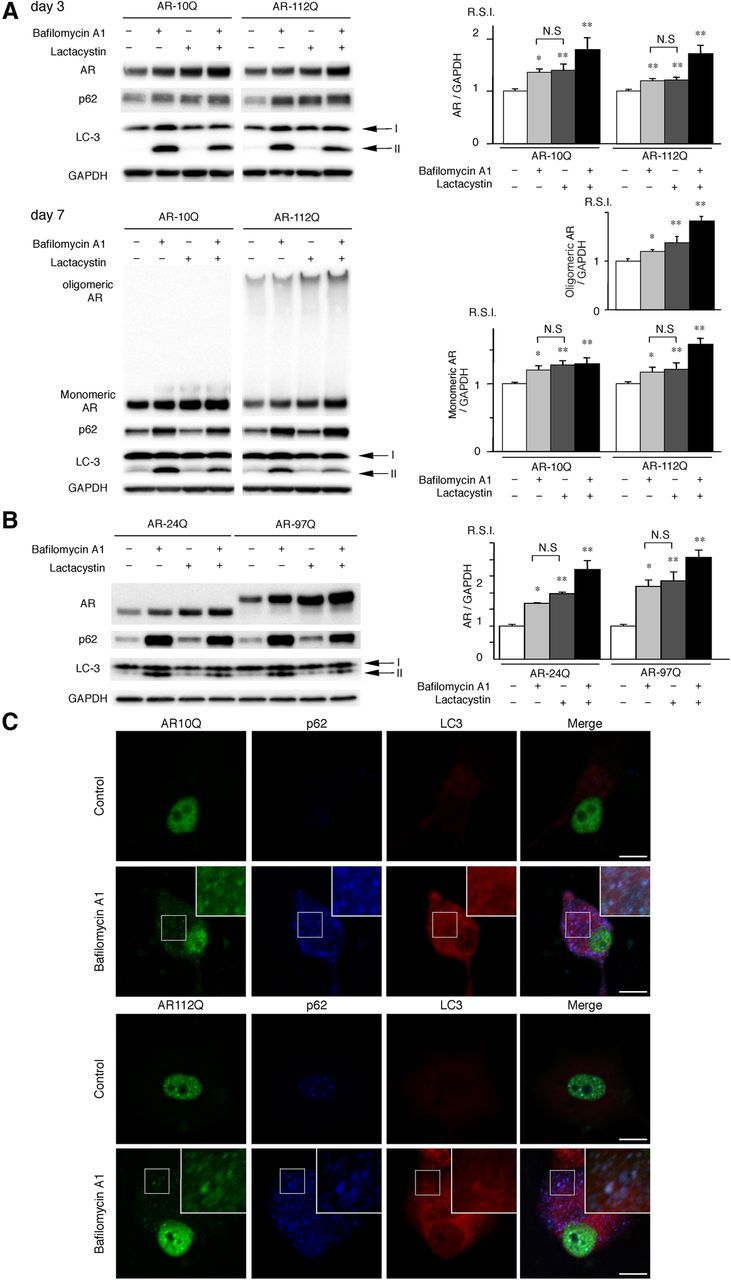
AR is degraded in a p62-dependent autophagic pathway. A, PC12 cells that express wild-type (10Q) and mutant (112Q) AR were treated with bafilomycin A1 (20 nm) and/or lactacystin (2.5 μm) for 16 h after DHT treatment for 3 or 7 d. Immunoblot analysis was performed. B, Neuro2A cells that were transfected with wild-type (24Q) and mutant (97Q) ARs were treated with bafilomycin A1 (20 nm) and lactacystin (2.5 μm) for 16 h. C, Immunofluorescence for AR10Q and 112Q (green), p62 (blue), LC3 (red), and an overlay (white) of the three signals in PC12 cells for no treatment (control) and a treatment of bafilomycin A1 (20 nm). Scale bars, 10 μm. These experiments were repeated in five sets of cells, and equivalent results were obtained. All of the values are expressed as the means ± SEM (n = 5). *p < 0.05; **p < 0.01.
Figure 3.
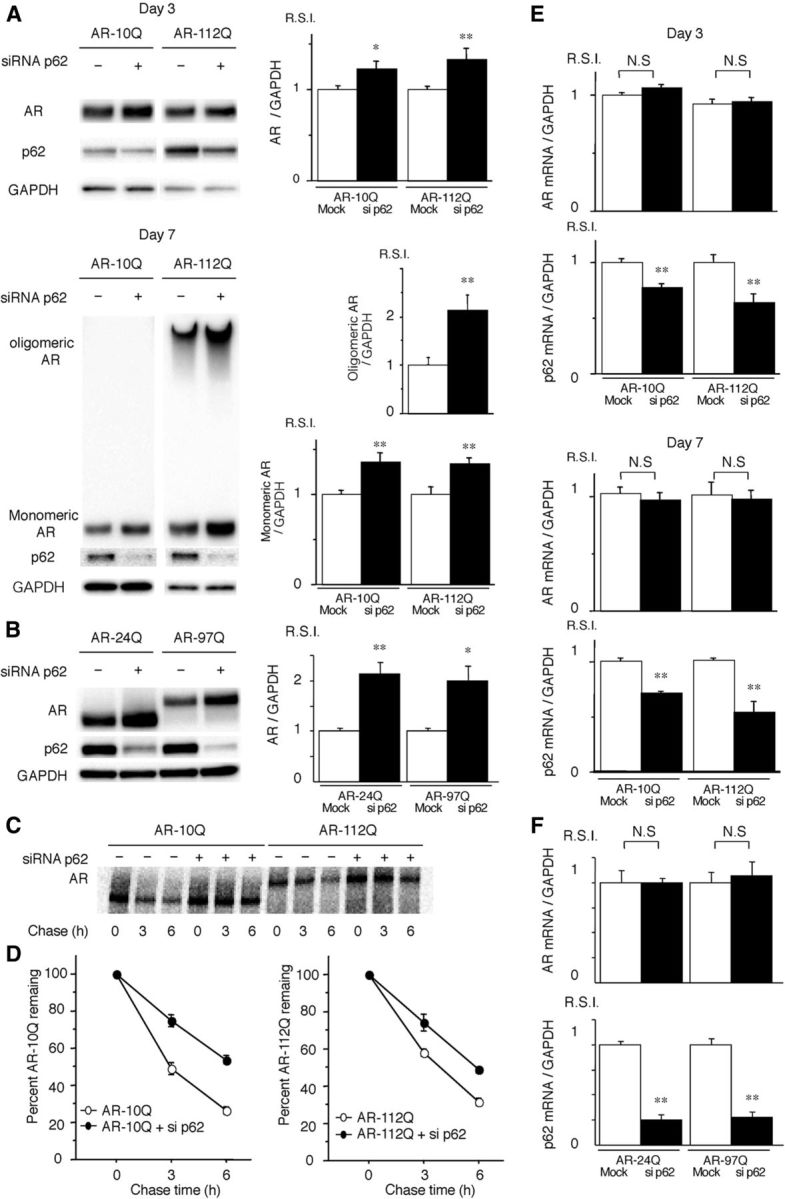
Depletion of p62 promotes the accumulation of AR. A, PC12 cells expressing wild-type (10Q) and mutant (112Q) AR were transfected with either control or p62 siRNA. The cells were treated with DHT for 3 or 7 d. B, Neuro2A cells transfected with wild-type (24Q) and mutant (97Q) AR were cotransfected with control or p62 siRNA. C, Pulse-chase analysis of two forms of AR in PC12 cells. Data from one representative experiment for wild-type and mutant AR. D, Pulse-chase assessment of half-life of the wild-type (left) and mutant AR (right). The percentages of wild-type (10Q) and mutant (112Q) remaining in the presence (●) and absence (○) of p62 siRNA are indicated. E, Real-time RT-PCR for wild-type (10Q) and mutant (112Q) AR mRNA normalized to GAPDH levels in PC12 cells. F, Real-time RT-PCR for wild-type (24Q) and mutant (97Q) AR mRNA normalized to GAPDH levels in Neuro2A cells. These experiments were repeated in five sets of cells, and equivalent results were obtained. All of the values are expressed as the means ± SEM (n = 5). *p < 0.05; **p < 0.01.
Figure 9.
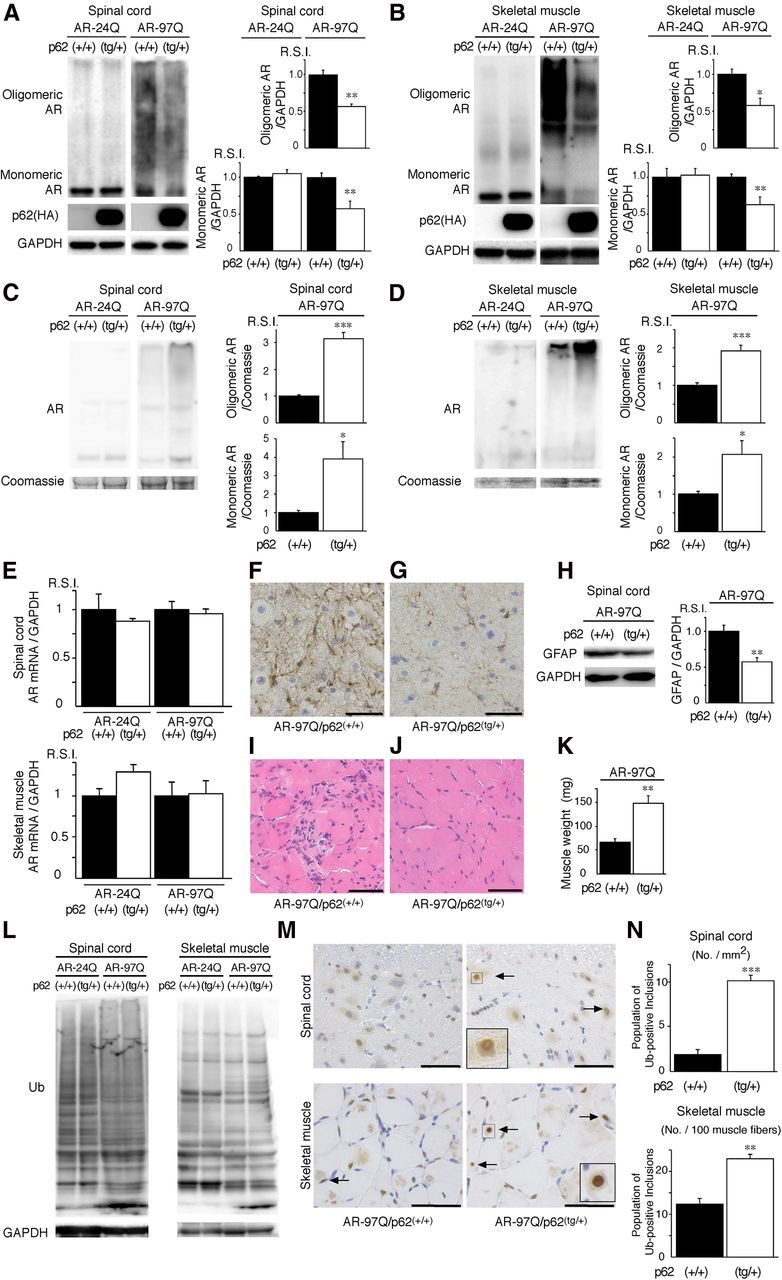
An increase in p62 reduces mutant AR expression. A, B, Western blotting analysis of total tissue homogenates from the spinal cord (A) and muscle (B) of AR–24Q and AR–97Q mice (25 weeks of age) probed with anti-AR. Values are expressed as the means ± SEM (n = 5). C, D, Western blotting analysis of the pellet lysed in 8 m urea solution from the spinal cord (C) and muscle (D) of AR–24Q and AR–97Q mice (25 weeks of age) probed with anti-AR. Values are expressed as the means ± SEM (n = 5). E, Real-time RT-PCR of wild-type and mutant AR mRNA normalized to GAPDH levels. F, G, Immunohistochemical staining with anti-GFAP antibody in the spinal anterior horn. Scale bars, 30 μm. H, Western blotting analysis of the total tissue homogenates from the spinal cord of AR–97Q mice (25 weeks of age, n = 5) probed with anti-GFAP. I, J, Hematoxylin and eosin staining of the muscle. Scale bars, 50 μm. K, The gastrocnemius muscles from AR–97Q/p62(+/+) and AR–97Q/p62(tg/+) mice were dissected and weighed. L, Western blotting analysis of the total tissue homogenates from the spinal cord and muscle of AR–24Q and AR–97Q mice (25 weeks of age) probed with anti-ubiquitin (Ub). M, Ubiquitin immunohistochemistry revealed NIs (arrow) in the spinal anterior horn and muscle of 25-week-old AR–97Q/p62(+/+) and AR–97Q/p62(tg/+) mice. The inset shows a magnified image in the square box. Scale bars, 50 μm. N, Quantitative assessment of ubiquitin-positive NIs in the spinal ventral horn and muscle. Bars represent the density of ubiquitin-positive NIs in AR–97Q/p62(+/+) and AR–97Q/p62(tg/+) mice. The results are expressed as the means ± SEM (n = 6). *p < 0.05; **p < 0.01; ***p < 0.001.
Immunoprecipitation assay.
We constructed p62 deletion mutant plasmids using a mutagenesis method with the appropriate primers. Each construct was designed against the functional domains and exons of the corresponding mouse p62 gene to assess which domain of p62 was required for the interaction with the AR (see Fig. 2E). Neuro2A cells transfected with AR–24Q and/or AR–97Q and the mutant p62 vectors were lysed. The sample, which contained 0.5 mg of total protein in 500 μl of TNE buffer, was rotated with 40 μl of anti-FLAG M2 agarose beads (Sigma) for 8 h at 4°C. The protein was eluted from the beads using 3xFLAG peptide (Sigma) under native conditions. Anti-HA tag monoclonal antibody magnetic beads (M132-9; Medical and Biological Laboratories), rabbit anti-AR (N20; Santa Cruz Biotechnology), and rabbit anti-p62 (PM045; Medical and Biological Laboratories) with 50 μl of protein G Dynabeads (Invitrogen) was also used in an immunoprecipitation assay. The supernatants were then boiled for 5 min in SDS sample buffer supplemented with 2-mercaptoethanol and loaded on to SDS-polyacrylamide gels. The blots were probed as described for Western blots with anti-AR (1:1000; N20; Santa Cruz Biotechnology), anti-DYKDDDK (1:1000; 018-22381; Wako Pure Chemicals), anti-HA tag HRP-DirecT (1:1000; 561-7; Medical and Biological Laboratories), anti-ubiquitin (1:1000; catalog #3936; Cell Signaling Technology), and anti-GAPDH (1:10,000; Millipore).
Pulse-chase labeling assay.
Cells were transfected as described above; they were starved for 1 h in methionine- and cysteine-free DMEM containing 10 ng/ml NGF, 1 μg/ml doxycycline, and 1 nm DHT and were then labeled for 1 h with 150 Ci of Redivue Pro-Mix L- [35S] in vitro cell-labeling mix (GE Healthcare) per milliliter of the culture medium. After being washed in PBS, the cells were chased for the indicated time intervals in complete medium. Immunoprecipitation was performed using equivalent amounts of protein lysates, 50 μl of protein G Dynabeads (Invitrogen), and 5 μl of anti-AR antibody (H280; Santa Cruz Biotechnology) for 10 min at room temperature. Each sample was separated by 5–20% SDS-PAGE and was analyzed by phosphorimaging (Typhoon LFA 9000 PhosphorImager; GE Healthcare) using Image Gauge software version 4.22 (GE Healthcare).
Quantitative real-time RT-PCR.
Total RNA was isolated from the PC12 cells and Neuro2A cells using the RNeasy Mini kit (Qiagen) and from mouse tissues using the PureLink RNA Mini kit (Invitrogen), according to the instructions of the manufacturer. The total RNA from the cells (3 μg) and mouse tissues (2.5 μg) was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen). Real-time RT-PCR was performed in a total volume of 25 μl containing 12.5 μl of 2× QuantiFast SYBR Green PCR Master Mix (Qiagen) and each primer at a concentration of 1 μm. The PCR products were detected using the iCycler system (Bio-Rad). The reaction conditions were 95°C for 5 min and then 40 cycles of 10 s at 95°C and 30 s at 60°C. As an internal standard control, the expression of GAPDH was simultaneously quantified using the following primers: 5′-CCTGGAGAAACCTGCCAAGTAT-3′ and 5′-TGAAGTCGCAGGAGACAACCT-3′. The primers used to detect the AR were designed as described previously (Waza et al., 2005). The primers used for p62 detection were 5′-ATGCTGTCCATGGGTTTCTC-3′ and 5′-GGTGGAGGGTGCTTTGAATA-3′ for PC12 cells and 5′-CCAGTGATGAGGAGCTGACA-3′ and 5′-CATCACAGATCACATTGGGG-3′ for Neuro2A cells. The R.S.I. was computed as the signal intensity of each sample divided by the signal intensity of the control siRNA-transfected cells (see Fig. 3) and AR–24Q/p62(+/+) or AR–97Q/p62(+/+) mice (see Fig. 9).
Statistical analyses.
The data were analyzed using unpaired t tests and post hoc tests (Tukey–Kramer tests) for multiple comparisons, along with the Kaplan–Meier and log-rank tests to assess the survival rate, as calculated using Statview software version 5 (Hulinks).
Results
AR is degraded via the p62-dependent autophagic pathway in cultured cells
p62 is degraded in the autophagic–lysosomal pathway through its interaction with LC3 (Komatsu et al., 2007; Ichimura et al., 2008). In addition, the AR can also be degraded by autophagy (Montie et al., 2009). To determine whether normal and mutant ARs are degraded by autophagy in cells in which the protein forms inclusions in the nucleus, as in motor neurons in SBMA, we treated an inducible PC12 cell model of SBMA that expresses full-length human AR with wild-type (AR–10Q) or mutant (AR–112Q) AR with bafilomycin A1, which is a potent inhibitor of autophagy. In this model, AR–112Q forms NIs in response to DHT, and treatment with DHT for 7 d increased the frequency of inclusions nearly threefold compared with treatment for 3 d (Walcott and Merry, 2002). Immunoblot analysis revealed a significant accumulation of monomeric wild-type (AR–10Q) and mutant (AR–112Q) AR in the PC12 cells that were treated with bafilomycin A1 after DHT treatment for 3 and 7 d; p62 also accumulated in the bafilomycin A1-treated cells (Fig. 1A). Bafilomycin A1 treatment also significantly increased the amount of the high-molecular-weight complex of mutant AR in the AR–112Q cells after DHT treatment for 7 d (Fig. 1A). Similar accumulations of AR–24Q, AR–97Q, and p62 were observed when we transiently transfected wild-type (AR–24Q) or mutant (AR–97Q) AR into Neuro2A cells and treated the cells with bafilomycin A1 (Fig. 1B). Accumulation of wild-type and mutant AR was similarly observed in treatment with lactacystin, which is a potent inhibitor of the ubiquitin–proteasome pathway (Fig. 1A,B). These findings suggest that the AR is degraded not only via the UPS but also via the autophagic–lysosomal pathway, and this effect occurs independently of inclusion formation in the nuclei. To examine whether AR was localized in LC3-positive cytoplasmic puncta, we cultured the PC12 cells with doxycycline, NGF, and DHT for 7 d. Immunofluorescence staining revealed that bafilomycin A1 treatment increased p62- and LC3-positive cytoplasmic puncta, which colocalized with both wild-type and mutant ARs (Fig. 1C). This result suggests that both wild-type and mutant ARs are degraded in the p62-dependent autophagic pathway. These findings are consistent with a previous study that reported that p62 interacted with mutant huntingtin and was degraded by autophagy (Bjørkøy et al., 2005). Nevertheless, p62 colocalized with AR–112Q in the NIs (Fig. 1C).
p62 interacts with the AR
p62 interacts with LC3 via a short domain of its C-terminal portion and is selectively degraded by autophagy (Ichimura et al., 2008; Ichimura and Komatsu, 2010). However, it is unclear whether p62 can interact with the AR protein. Because wild-type p62 was difficult to use in immunoprecipitation assay experiments given its propensity for aggregation, we used p62 with a PB1 (Phox and Bem1p) domain mutation (Fig. 2B–F). The self-oligomerization of p62 has been shown to be severely attenuated by this mutation (Lamark et al., 2003; Ichimura et al., 2008). In immunoprecipitation experiments, AR–24Q and AR–97Q coimmunoprecipitated with both endogenous and transfected p62, which suggests that p62 could either directly or indirectly recognize the AR (Fig. 2A–G). To determine the region of p62 that is required for the interaction with AR, we performed immunoprecipitation assay experiments using a series of p62 deletion mutants. The functional domains and exons of the corresponding mouse p62 gene were preserved in each construct (Fig. 2E). Deletion mutants of p62 (M6 and M7) failed to interact with the AR, whereas other mutants (M0–M5) interacted with the AR, which suggests that amino acid sequences from exon 4 (amino acids 180–225) of p62 are essential for its interaction with the AR (Fig. 2F). This AR–p62 interaction did not change when ubiquitin binding was disrupted using the M406V mutation in the p62 expression vector (Fig. 2G) (Falchetti et al., 2004). These results indicate that p62 can either directly or indirectly recognize the AR without ubiquitination and that the AR is degraded via autophagy.
Depletion of p62 promotes the accumulation of AR in a cell culture model
To determine whether depletion of p62 influences the turnover of AR, we treated the inducible PC12 cells that expressed AR–10Q and AR–112Q with either control siRNA or p62 siRNA. Immunoblot analysis revealed the accumulation of both monomeric wild-type (AR–10Q) and mutant (AR–112Q) AR after DHT treatment for 3 and 7 d and the accumulation of a high-molecular-weight complex of the mutant AR in the AR–112Q cells after DHT treatment for 7 d in the PC12 cells that were depleted of p62 (p < 0.01), which suggests that both wild-type and mutant ARs were degraded via p62-dependent autophagic degradation (Fig. 3A). Similar accumulations of AR–24Q, AR–97Q, and p62 were observed when we transiently transfected wild-type (AR–24Q) or mutant (AR–97Q) AR into Neuro2A cells and treated the cells with p62 siRNA (Fig. 3B). The turnover of wild-type (AR–10Q) and mutant (AR112Q) ARs was then assessed using a pulse-chase labeling assay in PC12 cells to determine whether the enhanced degradation of AR was attributable to protein degradation or to a change in RNA expression. Wild-type and mutant ARs were degraded almost equally in the absence of p62 siRNA, as reported previously (Bailey et al., 2002; Lieberman et al., 2002). In the absence of p62, wild-type and mutant ARs exhibited markedly diminished degradation (Fig. 3C,D), whereas the mRNA levels for both AR–10Q and AR–112Q were quite similar when treated with control or p62 siRNA constructs (Fig. 3E). Similarly, the mRNA levels for both AR–24Q and AR–97Q in Neuro2A cells were approximately equal when treated with mock or p62 siRNA constructs (Fig. 3F). These data indicate that deprivation of p62 influenced the autophagic degradation of both wild-type and mutant proteins without altering the mRNA levels, even in cells with NIs.
Depletion of p62 impairs the phenotypes of male AR–97Q mice
The aforementioned results suggest that endogenous p62 plays an important role in the degradation of misfolded polyQ protein in cells. We hypothesized that, if p62 contributes to the cellular quality control of the mutant polyQ protein, then reducing the expression of p62 should exacerbate polyQ disease. To test this hypothesis in vivo, we crossed an SBMA transgenic mouse that overexpressed the full-length human AR with either 24Q or 97Q mice (AR–24Q or AR–97Q mice) with p62 knock-outs (−/−) (Komatsu et al., 2007). Because SBMA mice (AR–97Q mice) have a small body size, a shorter lifespan, progressive muscle atrophy and weakness, reduced cage activity, and a male preponderance similar to that of human SBMA patients (Katsuno et al., 2002), we used male transgenic mice in this study. The disease progression in the AR–97Q mice that had a homozygous depletion of p62 (AR–97Q/p62−/−) was significantly exacerbated compared with the disease progression in SBMA transgenic mice with normal p62 activity (AR–97Q/p62+/+) and in AR–97Q/p62+/− mice with a heterozygous depletion of p62. Immunohistochemical studies of the spinal anterior horn and muscle using the p62-specific antibody in AR–97Q/p62+/+ and AR–97Q/p62−/− mice confirmed the absence of p62 staining in the nuclei and cytoplasms of knock-out animals (Fig. 4A–D). Western blotting analysis confirmed the decline of p62 expression in the spinal cord and muscle of p62+/− mice and p62−/− mice (Fig. 4E). Mice of all three genotypes appeared to be normal until 6–9 weeks of age. However, by 10–15 weeks, the AR–97Q/p62−/− mice displayed prominent kyphosis, muscle weakness and atrophy, and a foot-dragging walk with foot inversion (Fig. 4F–K). After onset, the phenotype of the AR–97Q/p62−/− mice progressed rapidly to death over a 10–20 week period. By 20 weeks, only 10% of the AR–97Q/p62−/− mice remained alive, in contrast to ∼40% of the AR97Q/p62+/+ mice (Fig. 4J). Furthermore, up to 25 weeks of age, none of the AR–24Q/p62−/− mice showed any neurological phenotypes (data not shown).
Figure 4.
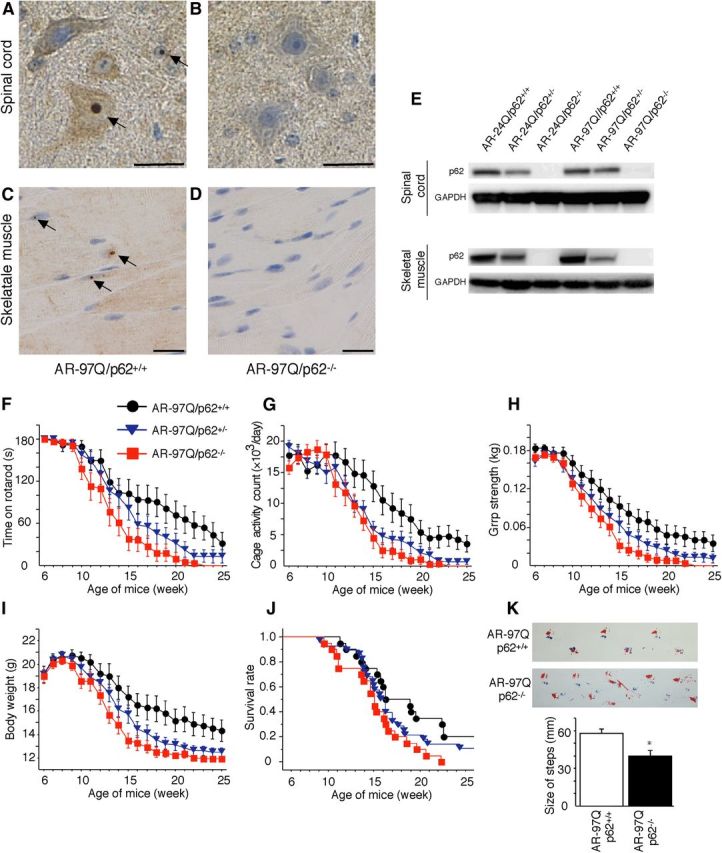
Depletion of p62 impairs behavioral and visible phenotypes in male AR–97Q mice. A–D, p62 immunohistochemistry in the spinal anterior horn (A, B) and skeletal muscle (C, D) of 25-week-old AR–97Q/p62+/+ (A, C) and 13-week-old AR–97Q/p62−/− (B, D) mice counterstained with Mayer's hematoxylin. p62 immunoreactivity was localized to the nuclei and cytoplasm, with NIs (arrow) in the anterior horn cells and skeletal muscle. Scale bars, 20 μm. E, Western blotting analysis of p62 expression in the total spinal cord and muscle protein lysates from the indicated mice immunolabeled with antibodies against p62. F–J, Rotarod task (F), cage activity (G), grip strength (H), body weight (I), and survival rate (J) of the AR–97Q/p62+/+(●; n = 20), AR–97Q/p62+/−(▾; n = 28), and AR–97Q/p62−/− (■; n = 20) mice. Although none of the parameters tested at 15 weeks revealed significant differences between AR–97Q/p62+/− and AR–97Q/p62+/+ mice, the AR–97Q/p62+/− mice performed more poorly than the AR–97Q/p62+/+ mice in all of the parameters. K, Footprints of representative 13-week-old AR–97Q/p62+/+ and AR–97Q/p62−/− mice. The front paws are indicated in blue, and the hindpaws are indicated in red. Values are expressed as the means ± SEM. *p < 0.05.
To further characterize the motor phenotype of these mice, we performed rotarod testing for 15-week-old AR–97Q/p62+/+ and AR–97Q/p62−/− mice. AR–97Q/p62−/− mice exhibited significant motor impairment as assessed by the rotarod task when compared with AR–97Q/p62+/+ mice (p < 0.05; Fig. 4F). Locomotor cage activity for AR–97Q/p62−/− and AR97Q/p62+/− mice was also significantly decreased when compared with that of the AR–97Q/p62+/+ mice (p < 0.01 and 0.05, respectively; Fig. 4G). AR–97Q/p62−/− mice consistently performed more poorly than the other genotypes in tasks for testing grip strength (p < 0.01; Fig. 4H). In addition, none of the genotypes were distinguishable in terms of body weight at birth. However, AR–97Q/p62−/− mice lost weight significantly earlier and more profoundly when compared with AR–97Q/p62+/+ mice (p < 0.01; Fig. 4I). Depletion of p62 also significantly decreased the survival rate of SBMA mice when compared with AR–97Q/p62+/+ mice (AR–97Q/p62+/+ vs AR–97Q/p62+/−, p = 0.247 vs AR–97Q/p62−/−, p = 0.007; Fig. 4J). Moreover, AR–97Q/p62−/− mice exhibited early onset of motor weakness, with dragging legs and shorter steps when compared with AR–97Q/p62+/+ mice (Fig. 4K). Although the AR–97Q/p62+/− and AR–97Q/p62−/− mice both showed phenotypic expression, the AR–97Q/p62+/− mice had better performance than the AR–97Q/p62−/− mice for most of these parameters, which suggests that the affected motor phenotype depended on the expression level of p62 rather than the genetic background.
Depletion of p62 induces the accumulation of ARs in an SBMA mouse model
We immunohistochemically examined mouse tissue samples for mutant ARs using the 1C2 antibody, which specifically recognizes expanded polyQs, and we noted a marked increase in DNS in the spinal cord (Fig. 5A–C) and muscles (Fig. 5D–F) in AR–97Q/p62−/− mice at 13 weeks when compared with the AR–97Q/p62+/− and AR–97Q/p62+/+ mice. Quantitative assessments showed significantly more 1C2-positive cells in the spinal cord (Fig. 5G) and muscles (Fig. 5H) of AR–97Q/p62−/− mice when compared with AR–97Q/ p62+/+ mice. Because wild-type and mutant ARs similarly accumulate in the absence of p62 in vitro, we also examined the expression of ARs in SBMA mice. Western blotting analysis of lysates from the spinal cord and muscle of AR–97Q mice revealed a high-molecular-weight mutant AR protein complex that was retained in the stacking gel as smear-shaped bands in addition to a band that could represent the monomeric mutant AR. Moreover, only the band for the wild-type monomeric AR was visible in the tissue from the AR–24Q mice (Fig. 5I,J). Depletion of p62 significantly increased the amount of the high-molecular-weight complex, the monomeric form of the mutant AR in the spinal cord and muscle of AR–97Q mice, and the amount of wild-type monomeric AR in AR–24Q mice (Fig. 5I,J). These observations indicate that p62 affects p62-dependent degradation of wild-type and mutant AR proteins via autophagy. Anti-GFAP antibody staining showed an apparent increase in reactive astrogliosis in AR–97Q/p62−/− mice when compared with AR–97Q/p62+/+ mice in the spinal anterior horn (Fig. 5K,L). Western blotting analyses using an antibody against GFAP revealed that depletion of p62 upregulated GFAP, which indicates that the depletion of p62 exacerbated the neurodegenerative changes (Fig. 5M). Muscle histology and weight measurement also revealed significant exacerbation of muscle atrophy in AR–97Q/p62−/− mice compared with AR–97Q/p62+/+ mice (Fig. 5N–P). Depletion of p62 did not influence the expression of total ubiquitinated proteins in the spinal cord and muscle of AR–24Q and AR–97Q mice, as determined by ubiquitin staining of Western blots (Fig. 5Q) and histology (data not shown).
p62 overexpression ameliorates phenotypes in male AR–97Q mice
To test whether increased expression of p62 provides protective effects in AR–97Q mice, we generated transgenic mice that overexpressed full-length human p62 with a C-terminal HA tag under the control of a cytomegalovirus enhancer and a chicken β-actin promoter (Fig. 6A). The promoter used for this transgenic mouse was the same as that used in the SBMA model mice. Among 10 available mouse lines, we established four that expressed p62 in the spinal cord and skeletal muscle, and we examined the effects of the overexpressed p62 on mouse phenotypes. Western blotting analysis revealed that p62 expression in the spinal cords of p62(tg/+) mice was fourfold higher than the expression of endogenous p62 in wild-type mice. In muscle, p62 expression was 12-fold higher in p62(tg/+) mice compared with wild-type mice (Fig. 6B). Immunohistochemical studies performed on p62(tg/+) mouse tissue using an anti-HA tag antibody confirmed that spinal anterior horn neurons and muscle cells expressed p62. Moreover, p62 protein was distributed in the cytoplasms and nuclei of the spinal anterior horn neurons, glial cells, and muscles (Fig. 6C,D). Up to 35 weeks of age, none of the hemizygous transgenic mice that overexpressed p62 presented any neurological phenotypes, as assessed by the rotarod task, cage activity, grip strength, and body weight (data not shown). Histological examination at 35 weeks did not reveal any detectable effects on neuronal cell morphology, neuronal cell number, or muscle structure (data not shown). These studies indicate that overexpression of human p62 alone does not impair neuronal development or motor function.
We crossed the AR–24Q and AR–97Q mice with mice that overexpress human p62 (p62 1–1 line) to generate hemizygotic AR–24Q/p62(tg/+) and AR–97Q/p62(tg/+) mice and AR–24Q/p62(+/+) and AR–97Q/p62(+/+) mice for use as controls. The AR–97Q mice that overexpressed p62 (AR–97Q/p62(tg/+)) demonstrated amelioration of the disease course after 18 weeks of age (Fig. 6E–I). In addition, AR–97Q/p62(tg/+) mice showed improvements in their motor impairment, as assessed by the rotarod task at 25 weeks (p < 0.05; Fig. 6E). Locomotor cage activity of the AR–97Q/p62(tg/+) mice was also significantly increased at 25 weeks when compared with that of the AR–97Q/p62(+/+) mice (p < 0.05; Fig. 6F). Furthermore, grip strength was stronger in the AR–97Q/p62(tg/+) mice than in the control mice (p < 0.05; Fig. 6G), and AR–97Q/p62(+/+) mice lost weight significantly earlier and more profoundly than the AR–97Q/p62(tg/+) mice (p < 0.05; Fig. 6H). An increase in p62 also significantly increased the survival rate of AR–97Q/p62(tg/+) mice compared with AR–97Q/p62(+/+) mice (p = 0.008; Fig. 6I). Although the AR–97Q/p62(tg/+) mice took relatively shorter steps than the wild-type mice, they walked with significantly longer steps than the AR–97Q/p62(+/+) mice (p < 0.01; Fig. 6J).
p62 overexpression promotes NI body formation and reduces DNS of mutant AR in an SBMA mouse model
We performed immunohistochemical staining for mutant AR using the 1C2 antibody in the spinal cord and skeletal muscle of 25-week-old mice. Intriguingly, numerous NIs of the mutant AR were observed in AR–97Q/p62(tg/+) mice but not in AR–97Q/p62(+/+) mice (Fig. 7A). The intensity of the DNS was reduced in cells with NIs (Fig. 7A, arrow) compared with cells without NIs (Fig. 7A, arrowhead). The ratio of the number of cells with NIs to the number of total 1C2-positive cells significantly increased in AR–97Q/p62(tg/+) mice (Fig. 7B), and the number of cells with DNS was significantly decreased in AR–97Q/p62(tg/+) mice (Fig. 7C). The R.S.I. of the DNS was significantly reduced in cells with NIs compared with those without NIs in the spinal anterior horn and skeletal muscle (Fig. 7D). We evaluated the colocalization of overexpressed human p62 and mutant AR in AR–97Q/p62(tg/+) mice. Intriguingly, double-immunofluorescence staining with anti-HA and 1C2 antibodies and immunohistochemistry with an anti-HA antibody demonstrated that p62–HA and mutant AR colocalized in all of the NIs but not in the DNS in the spinal anterior horn cells and skeletal muscles of AR–97Q mice (Fig. 7E–H). In addition, p62 localized in the core of NIs of mutant AR (Fig. 7I). Double-immunofluorescence staining with anti-p62 and 1C2 antibodies also revealed that endogenous p62 and mutant AR colocalized in the NIs in the spinal anterior horn cells and skeletal muscles of AR–97Q mice (Fig. 8A,B) and in the spinal anterior horn neurons of human SBMA patients (Fig. 8C,D), which suggests that endogenous p62 coexists with mutant AR and can exert a cell-protective function in both AR–97Q mice and SBMA patients. There were no NIs in the spinal cord and skeletal muscle of the AR–24Q/p62(tg/+) mice (data not shown).
Figure 8.
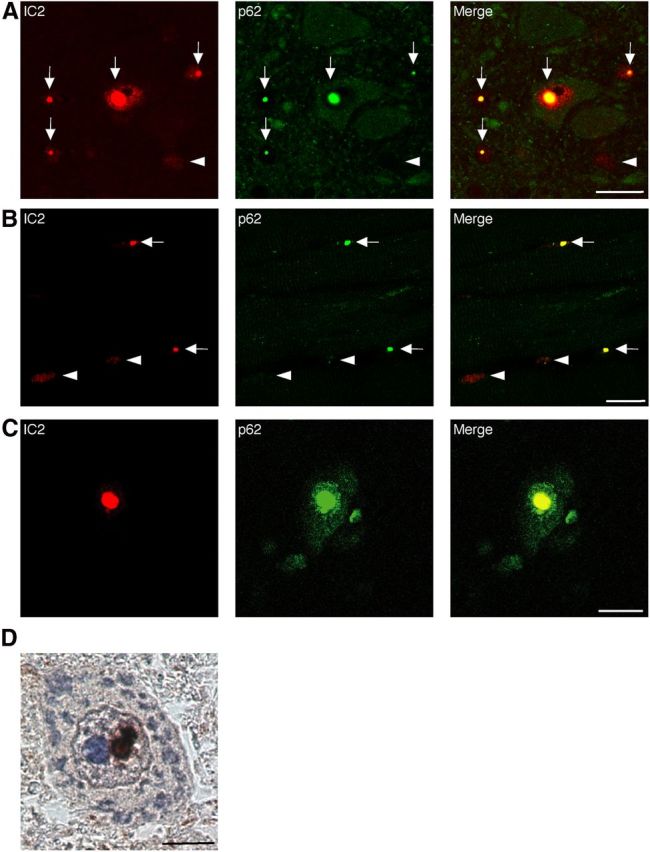
Colocalization of nuclear-localized p62 with mutant AR. A–C, 1C2 (red) and anti-HA (green) double immunofluorescence in the spinal cord (A) and skeletal muscle (B) of 16-week-old AR–97Q mice and in the spinal anterior horn cells (C) of SBMA patients. Double-immunofluorescence staining revealed p62 and mutant AR colocalization in NIs (shown in yellow, arrow), but no colocalization was observed in the DNS (arrowhead) in AR–97Q mice and SBMA patients. Scale bars, 20 μm. D, Immunohistochemistry for the anti-p62 antibody in SBMA patients. p62 was localized in the NIs. Scale bar, 10 μm.
p62 overexpression reduces the monomeric and oligomeric protein expression of mutant AR in an SBMA mouse model
Overexpression of p62 significantly decreased the amount of the high-molecular-weight mutant AR protein complex and the monomeric form of the mutant AR (AR–97Q) in the spinal cord and skeletal muscle of AR–97Q mice, whereas it did not decrease the expression of wild-type monomeric AR (AR–24Q) (Fig. 9A,B). To address whether nuclear ARs shift from a more soluble oligomer to a less soluble and aggregated state in AR–97Q/p62(tg/+) mice, we lysed the pellet in 8 m urea solution and performed Western blotting. Overexpression of p62 significantly increased AR protein in the pellet of the spinal cord and skeletal muscle of AR–97Q/p62(tg/+) mice, suggesting that increasing p62 shifts from soluble oligomeric AR to insoluble AR (Fig. 9C,D). The respective levels of wild-type and mutant AR mRNA were similar in AR–24Q and AR–97Q mice overexpressing p62 (Fig. 9E). Anti-GFAP staining showed an apparent decrease in reactive astrogliosis in AR–97Q/p62(tg/+) mice when compared with AR–97Q/p62(+/+) mice in the spinal anterior horn (Fig. 9F,G). Western blotting analyses with an antibody against GFAP revealed that overexpression of p62 downregulated GFAP, which indicates that the overexpression of p62 mitigated the neurodegenerative changes (Fig. 9H). Muscle histology and weight measurements also demonstrated a significant amelioration of muscle atrophy in AR–97Q/p62(tg/+) mice compared with AR–97Q/p62(+/+) mice (Fig. 9I,J). The AR–97Q/p62(tg/+) muscles were 2.2 times heavier than the AR–97Q/p62(+/+) muscles (Fig. 9K). Overexpression of p62 did not influence the expression of the total amount of ubiquitinated proteins in the spinal cord and muscle of AR–24Q and AR–97Q mice, as determined by Western blots (Fig. 9L), whereas more ubiquitin-positive NIs were identified in the AR–97Q/p62(tg/+) mice than in the AR–97Q/p62(+/+) mice (Fig. 9M,N). The expression of the autophagic marker LC3 (Mizushima et al., 2010) was slightly elevated in the spinal cord and muscle of AR–24Q/p62(tg/+) and AR–97Q/p62(tg/+) mice compared with AR–24Q/p62(+/+) and AR–97Q/p62(+/+) mice (Fig. 10A,B), which suggests that high expression of p62 induced autophagosome formation to some extent. However, the induction of autophagosome formation could be insufficient to affect the degradation of the AR protein because p62 overexpression did not decrease the expression of wild-type AR in the spinal cord and skeletal muscle of AR–24Q mice (Fig. 9A,B). Overexpression of p62 did not influence the expression of wild-type and mutant AR in two SBMA cellular models (Fig. 11A,B). In this culture system, we detected a band that represented a monomeric mutant AR in the separating gel. However, it was difficult to detect the high-molecular-weight mutant AR protein complex and NIs. Thus, this cell culture model is more appropriate for estimating changes in monomeric mutant AR expression. These observations suggest that an increase in p62 promotes the downregulation of soluble-toxic mutant AR species via harmless inclusion body formation rather than by autophagic degradation and exerts therapeutic effects in AR–97Q mice. Our results were consistent with those of a previous study that showed that NI formation exhibits protective effects in a cellular model of Huntington's disease (Arrasate et al., 2004).
Figure 10.

LC3-II expression in AR–97Q/p62(tg/+) mice. A, Western blotting analysis of total spinal cord protein lysates from the AR–97Q/p62(+/+) and AR–97Q/p62(tg/+) mice immunolabeled with antibodies against LC3. B, Quantitative analysis for the amounts of LC3-II. LC3-II levels were slightly elevated in the AR–97Q/p62(tg/+) mice. This experiment was repeated in four sets of mice with equivalent results. All of the values are expressed as the means ± SEM (n = 5). *p < 0.05; **p < 0.01.
Figure 11.

p62 overexpression does not affect AR expression. A, Full-length wild-type (24Q) or mutant (97Q) AR and HA-tagged p62 (pCAGGS–p62–HA) were cotransfected into Neuro2A cells for 48 h. B, Full-length wild-type (24Q) or mutant (97Q) AR and FLAG-tagged p62 (pIRESpuro3–3XFLAG–p62) were cotransfected into NSC34 cells for 48 h. All of the cells were cultured in DMEM/10% FCS at 37°C and with 5% CO2. Immunoblots revealed that the expression of AR was similar in the presence or absence of HA–p62 or FLAG–p62.
Discussion
The protein p62 is a ubiquitously expressed cellular protein, and it has been shown recently to be a selective substrate for autophagy (Bjørkøy et al., 2005; Pankiv et al., 2007; Ichimura et al., 2008). This protein is localized at the autophagosome formation site (Itakura and Mizushima, 2011) and directly interacts with LC3, which is an autophagosome-localizing protein (Pankiv et al., 2007; Ichimura et al., 2008). Subsequently, p62 incorporates into the autophagosome and is then degraded (Pankiv et al., 2007; Ichimura et al., 2008). Thus, impaired autophagy is accompanied by the accumulation of p62, followed by the formation of p62- and ubiquitin-positive aggregates because of the nature of self-oligomerization and ubiquitin binding by p62 (Mizushima and Komatsu, 2011). In contrast, inclusion bodies that are positive for both ubiquitin and p62 have been identified in various neurodegenerative disorders (Kuusisto et al., 2001, 2008), which suggests that p62 contributes to inclusion formation. Furthermore, p62-positive aggregates observed in hepatocytes and neurons of liver- and brain-specific Atg7-deficient mice are completely dispersed by the additional loss of p62 (Komatsu et al., 2007).
We generated an SBMA transgenic mouse model (AR–97Q) that displayed progressive muscular atrophy, weakness, and DNS and NIs of the mutant AR in neuronal and non-neuronal tissues (Katsuno et al., 2002). Because AR has a specific ligand (i.e., testosterone), the pathogenesis of SBMA is unique among the polyQ diseases (Katsuno et al., 2003). The success of androgen deprivation therapy in SBMA mouse models has been translated into clinical trials (Katsuno et al., 2010; Fernández-Rhodes et al., 2011). In addition, the elucidation of SBMA pathophysiology using animal models has led to the development of other therapeutics, including chaperone-related disease-modifying therapy (Waza et al., 2005; Tokui et al., 2009), posttranslational modification of the AR (Palazzolo et al., 2009; Montie et al., 2011), AR mRNA stability (Miyazaki et al., 2012), and modulation of the NH2−/carboxyl-terminal interaction of the AR (Orr et al., 2010), which inhibit the pathogenic process of neuronal degeneration. Thus, there is increasing evidence that AR ligands, molecular chaperones–UPS, and autophagy play crucial roles in the pathogenesis of SBMA. Furthermore, mutant AR that bears an expanded polyQ repeat adopts an altered conformation that results in protein aggregation and inclusion formation; however, recent studies have suggested that soluble causative protein species that include protein aggregates, rather than protein inclusions, could be toxic; thus, they represent targets in the treatment of neurodegenerative disorders (Bauer and Nukina, 2009; Naiki and Nagai, 2009; Hands and Wyttenbach, 2010). In this study, we addressed the questions of whether depletion of p62 exacerbates neuropathological outcomes and whether overexpression of p62 can protect against mutant AR toxicity and exert therapeutic effects on the SBMA phenotype. Depletion of p62 significantly increased the accumulation of monomeric mutant AR and mutant AR protein complexes in AR–97Q mice via an impairment of autophagic degradation, whereas helpful inclusion body formation was promoted in the spinal cord and muscle of the AR–97Q/p62 double transgenic mice.
In a neuronal cell culture model of SBMA, we demonstrated that decreasing levels of p62 inhibited the degradation of monomeric wild-type and mutant ARs even in cells with NIs, which suggests that p62 plays a pivotal role in AR protein turnover. Growing evidence indicates that p62, together with ubiquitinated proteins, is transported into autophagosomes, which suggests that p62 is a receptor for ubiquitinated proteins that is necessary for their degradation in lysosomes (Johansen and Lamark, 2011). As a receptor, p62 would also contribute to autophagic degradation of Parkin-mediated ubiquitinated mitochondria (Geisler et al., 2010). Furthermore, p62 is localized at the autophagosome formation site and directly interacts with LC3, which facilitates the delivery of its polyubiquitinated protein for lysosomal degradation (Ichimura et al., 2008). Subsequently, p62 is also incorporated into the autophagosome and is then degraded. In addition, p62 has been shown to directly interact with mutant copper–zinc superoxide dismutase (SOD1) protein (Gal et al., 2007), which indicates that p62 directly or indirectly recognizes the mutant SOD1 protein and is degraded via autophagy. p62 also has the capacity to sequester aggregate-prone proteins that might otherwise undergo proteasomal degradation (Korolchuk et al., 2009). In our study, both wild-type and mutant ARs colocalized with endogenous p62- and LC3-positive puncta (Fig. 1C). In addition, ARs were recognized directly by p62 (Fig. 2), which suggests that mutant AR is degraded in a p62-dependent autophagic pathway. Furthermore, our Western blotting analysis revealed that monomeric mutant AR and the high-molecular-weight form of mutant AR protein complexes retained in the stacking gel were upregulated in the spinal cord and muscle of AR–97Q transgenic/p62 knock-out mice, which suggests that degradation of mutant AR was inhibited by the deletion of p62.
In addition, the overexpression of p62 did not influence the expression of wild-type and mutant AR in a cellular model of SBMA or the expression of wild-type AR in a transgenic mouse model, which suggests that p62 overexpression was not sufficient to promote the degradation of both wild-type and mutant AR (Figs. 9A,B, 11A,B). However, p62 overexpression enhanced the inclusion body formation of mutant ARs, which resulted in a reduction of expression in a transgenic mouse model (Figs. 7, 9), and this effect can occur independently of the degradation in lysosomes. More importantly, overexpressed p62 existed in all of the mutant AR NIs in the spinal cord and skeletal muscle of AR–97Q/p62 double transgenic mice. Furthermore, p62 also colocalized with mutant AR NIs that were present in the anterior horn cells of postmortem tissues obtained from SBMA patients. Inclusion bodies that were positive for both ubiquitin and p62 have been identified in various neurodegenerative diseases, which suggests that p62 is involved in the formation of disease-related inclusion bodies (Kuusisto et al., 2001, 2008; Labbadia et al., 2012). Intriguingly, the overexpressed p62 and mutant AR colocalized in all of the NIs but not in the DNS, and p62 was localized in the core of NIs of mutant AR (Fig. 7I). p62 can either directly or indirectly interact with the AR without its ubiquitination (Fig. 2G) and can also function with the misfolded AR, as indicated by the fact that we did not observe NI formation in the AR–24Q/ p62(tg/+) mice. These results suggest that p62 is capable of self-oligomerization and could provide an opportunity for inclusion formation to generate the seeds of NIs.
However, the extent of diffuse nuclear accumulation of mutant AR in motor and sensory neurons of the spinal cord of autopsied SBMA patients was strongly correlated with the CAG repeat length but not with the number of NIs (Adachi et al., 2005). Accumulating evidence suggests that NIs are not harmful polyQ species (Arrasate et al., 2004) and instead that an oligomeric form of the mutant protein could be the major pathogenic species (Nagai et al., 2007; Sathasivam et al., 2010). Several studies have suggested that inclusion formation could be a cellular response against the toxicity of abnormal polyQ proteins (Arrasate et al., 2004; Bowman et al., 2005), whereas the nuclear localization or accumulation of abnormal proteins can be decisive in inducing neuronal cell dysfunction and degeneration in polyQ diseases, including SBMA (Klement et al., 1998; Saudou et al., 1998; Adachi et al., 2005). In view of the time course of SBMA, diffuse nuclear accumulation of mutant proteins with an expanded polyQ tract might be an early event that occurs before NI formation, which is closely related to the manifestation of neuronal dysfunction (Watase et al., 2002; Yoo et al., 2003). The molecular pathogenetic process by which diffuse nuclear mutant AR accumulation induces neuronal dysfunction and death remains unclear; however, it is notable that overexpression of p62 can promote NI formation and the packaging of accumulated soluble and toxic nuclear mutant AR.
Accumulation of misfolded proteins is causally related to many age-related neurodegenerative diseases (Bates, 2006). Prompt removal and/or refolding could be required more in aged or damaged cells than in young healthy cells, in which protein quality control systems function appropriately. In SBMA patients, diffuse nuclear accumulation of mutant AR is frequent and extensive, and it occurs in a wide array of CNS nuclei and visceral organs. The precise mechanism used by the mutant protein must be determined in future studies; however, we have used cellular and mouse models of SBMA and demonstrated that p62 plays a beneficial role in autophagic degradation and NI formation of the mutant AR protein. These findings provide new insights into the cytoprotective functions of p62; thus, they could have important implications for the development of therapeutic strategies for the treatment of SBMA and other polyQ diseases.
Footnotes
This work was supported by a Center-of-Excellence grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and grants from the Mochida Memorial Foundation for Medical and Pharmaceutical Research. We also thank Dr. Masaaki Komatsu (Protein Metabolism Project, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) for kindly providing the vectors containing p62 with mutations and Dr. Diane E. Merry (Departments of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA) for kindly providing the inducible PC12 cells that express wild-type or mutant AR. We thank Miwa Ito and Kazuko Matsuba (Department of Neurology, Nagoya University, Nagoya, Japan) and Noboru Ogiso, Yasutaka Ohya, and Kumiko Yano (Division for Research of Laboratory Animals at the Center for Research of Laboratory Animals and Medical Research Engineering, Nagoya University, Nagoya, Japan) for their technical assistance.
The authors declare no competing financial interests.
References
- Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Do J, Sang C, Kobayashi Y, Doyu M, Sobue G. Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet. 2001;10:1039–1048. doi: 10.1093/hmg/10.10.1039. [DOI] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Yoshida M, Hashizume Y, Sobue G. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128:659–670. doi: 10.1093/brain/awh381. [DOI] [PubMed] [Google Scholar]
- Adachi H, Waza M, Katsuno M, Tanaka F, Doyu M, Sobue G. Pathogenesis and molecular targeted therapy of spinal and bulbar muscular atrophy. Neuropathol Appl Neurobiol. 2007;33:135–151. doi: 10.1111/j.1365-2990.2007.00830.x. [DOI] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Waza M, Minamiyama M, Tanaka F, Sobue G. Heat shock proteins in neurodegenerative diseases: pathogenic roles and therapeutic implications. Int J Hyperthermia. 2009;25:647–654. doi: 10.3109/02656730903315823. [DOI] [PubMed] [Google Scholar]
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobágyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11:515–523. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- Bates GP. BIOMEDICINE: one misfolded protein allows others to sneak by. Science. 2006;311:1385–1386. doi: 10.1126/science.1125246. [DOI] [PubMed] [Google Scholar]
- Bauer PO, Nukina N. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem. 2009;110:1737–1765. doi: 10.1111/j.1471-4159.2009.06302.x. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet. 2005;14:679–691. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Di Prospero NA, Fischbeck KH. Therapeutics development for triplet repeat expansion diseases. Nat Rev Genet. 2005;6:756–765. doi: 10.1038/nrg1690. [DOI] [PubMed] [Google Scholar]
- Falchetti A, Di Stefano M, Marini F, Del Monte F, Mavilia C, Strigoli D, De Feo ML, Isaia G, Masi L, Amedei A, Cioppi F, Ghinoi V, Bongi SM, Di Fede G, Sferrazza C, Rini GB, Melchiorre D, Matucci-Cerinic M, Brandi ML. Two novel mutations at exon 8 of the Sequestosome 1 (SQSTM1) gene in an Italian series of patients affected by Paget's disease of bone (PDB) J Bone Miner Res. 2004;19:1013–1017. doi: 10.1359/JBMR.040203. [DOI] [PubMed] [Google Scholar]
- Fernández-Rhodes LE, Kokkinis AD, White MJ, Watts CA, Auh S, Jeffries NO, Shrader JA, Lehky TJ, Li L, Ryder JE, Levy EW, Solomon BI, Harris-Love MO, La Pean A, Schindler AB, Chen C, Di Prospero NA, Fischbeck KH. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol. 2011;10:140–147. doi: 10.1016/S1474-4422(10)70321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal J, Ström AL, Kilty R, Zhang F, Zhu H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J Biol Chem. 2007;282:11068–11077. doi: 10.1074/jbc.M608787200. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Hands SL, Wyttenbach A. Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta Neuropathol. 2010;120:419–437. doi: 10.1007/s00401-010-0703-0. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol. 2010;32:431–436. doi: 10.1007/s00281-010-0220-1. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192:17–27. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Yabe I, Sasaki H, Aoki M, Morita M, Nakano I, Kanai K, Ito S, Ishikawa K, Mizusawa H, Yamamoto T, Tsuji S, Hasegawa K, Shimohata T, Nishizawa M, Miyajima H, et al. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:875–884. doi: 10.1016/S1474-4422(10)70182-4. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/S0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, Inukai A, Sobue G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med. 2003;9:768–773. doi: 10.1038/nm878. [DOI] [PubMed] [Google Scholar]
- King A, Maekawa S, Bodi I, Troakes C, Al-Sarraj S. Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathology. 2011;31:239–249. doi: 10.1111/j.1440-1789.2010.01171.x. [DOI] [PubMed] [Google Scholar]
- Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/S0092-8674(00)81781-X. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001;12:2085–2090. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- Kuusisto E, Kauppinen T, Alafuzoff I. Use of p62/SQSTM1 antibodies for neuropathological diagnosis. Neuropathol Appl Neurobiol. 2008;34:169–180. doi: 10.1111/j.1365-2990.2007.00884.x. [DOI] [PubMed] [Google Scholar]
- Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, Woodman B, Moussaoui S, Frentzel S, Luthi-Carter R, Paganetti P, Bates GP. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest. 2011;121:3306–3319. doi: 10.1172/JCI57413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Novoselov SS, Bett JS, Weiss A, Paganetti P, Bates GP, Cheetham ME. Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain. 2012;135:1180–1196. doi: 10.1093/brain/aws022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Perander M, Outzen H, Kristiansen K, Øvervatn A, Michaelsen E, Bjørkøy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Harmison G, Strand AD, Olson JM, Fischbeck KH. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet. 2002;11:1967–1976. doi: 10.1093/hmg/11.17.1967. [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Adachi H, Katsuno M, Minamiyama M, Jiang YM, Huang Z, Doi H, Matsumoto S, Kondo N, Iida M, Tohnai G, Tanaka F, Muramatsu S, Sobue G. Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat Med. 2012;18:1136–1141. doi: 10.1038/nm.2791. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montie HL, Cho MS, Holder L, Liu Y, Tsvetkov AS, Finkbeiner S, Merry DE. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2009;18:1937–1950. doi: 10.1093/hmg/ddp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montie HL, Pestell RG, Merry DE. SIRT1 modulates aggregation and toxicity through deacetylation of the androgen receptor in cell models of SBMA. J Neurosci. 2011;31:17425–17436. doi: 10.1523/JNEUROSCI.3958-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, Goto Y, Naiki H, Toda T. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- Naiki H, Nagai Y. Molecular pathogenesis of protein misfolding diseases: pathological molecular environments versus quality control systems against misfolded proteins. J Biochem. 2009;146:751–756. doi: 10.1093/jb/mvp119. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- Orr CR, Montie HL, Liu Y, Bolzoni E, Jenkins SC, Wilson EM, Joseph JD, McDonnell DP, Merry DE. An interdomain interaction of the androgen receptor is required for its aggregation and toxicity in spinal and bulbar muscular atrophy. J Biol Chem. 2010;285:35567–35577. doi: 10.1074/jbc.M110.146845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, Sobue G, Taylor JP, Sumner CJ, Fischbeck KH, Pennuto M. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–328. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, Paganetti P, Muchowski PJ, Wilson S, Bates GP. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington's disease. Hum Mol Genet. 2010;19:65–78. doi: 10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/S0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain. 1989;112:209–232. doi: 10.1093/brain/112.1.209. [DOI] [PubMed] [Google Scholar]
- Tokui K, Adachi H, Waza M, Katsuno M, Minamiyama M, Doi H, Tanaka K, Hamazaki J, Murata S, Tanaka F, Sobue G. 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an SBMA model mouse. Hum Mol Genet. 2009;18:898–910. doi: 10.1093/hmg/ddn419. [DOI] [PubMed] [Google Scholar]
- Troakes C, Maekawa S, Wijesekera L, Rogelj B, Siklós L, Bell C, Smith B, Newhouse S, Vance C, Johnson L, Hortobágyi T, Shatunov A, Al-Chalabi A, Leigh N, Shaw CE, King A, Al-Sarraj S. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2012;32:505–514. doi: 10.1111/j.1440-1789.2011.01286.x. [DOI] [PubMed] [Google Scholar]
- Walcott JL, Merry DE. Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J Biol Chem. 2002;277:50855–50859. doi: 10.1074/jbc.M209466200. [DOI] [PubMed] [Google Scholar]
- Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, Kano M, Atkinson R, Sun Y, Armstrong DL, Sweatt JD, Orr HT, Paylor R, Zoghbi HY. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34:905–919. doi: 10.1016/S0896-6273(02)00733-X. [DOI] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, Armstrong DL, Wu SM, Sweatt JD, Zoghbi HY. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. 2003;37:383–401. doi: 10.1016/S0896-6273(02)01190-X. [DOI] [PubMed] [Google Scholar]