

Abstract
Enteric neural crest-derived cells (ENCCs) migrate from the anterior foregut in a rostrocaudal direction to colonize the entire gastrointestinal tract and to form the enteric nervous system. Genetic approaches have identified many signaling molecules regulating the migration of ENCCs; however, it remains elusive how the activities of the signaling molecules are regulated spatiotemporally during migration. In this study, transgenic mice expressing biosensors based on Förster resonance energy transfer were generated to video the activity changes of the signaling molecules in migrating ENCCs. In an organ culture of embryonic day 11.25 (E11.25) to E13 guts, ENCCs at the rostral wavefront migrated as a cellular chain faster than the following ENCCs that formed a network. The faster-migrating cells at the wavefront exhibited lower protein kinase A (PKA) activity than did the slower-migrating trailing cells. The activities of Rac1 and Cdc42 exhibited an inverse correlation with the PKA activity, and PKA activation decreased the Rac1 activity and migration velocity. PKA activity in ENCCs was correlated positively with the distribution of GDNF and inversely with the distribution of endothelin 3 (ET-3). Accordingly, PKA was activated by GDNF and inhibited by ET-3 in cultured ENCCs. Finally, although the JNK and ERK pathways were previously reported to control the migration of ENCCs, we did not find any correlation of JNK or ERK activity with the migration velocities. These results suggest that external cues regulate the migration of ENCCs by controlling PKA activity, but not ERK or JNK activity, and argue for the importance of live imaging of signaling molecule activities in developing organs.
Introduction
Cells of the enteric nervous system (ENS) are originated from enteric neural crest-derived cells (ENCCs), which undergo migration, proliferation, differentiation, and colonization during embryonic development (Yntema and Hammond, 1954; Le Douarin and Teillet, 1973). Shortly after emigration from the neural tube, ENCCs invade into the anterior foregut at embryonic day 9 (E9)–E9.5 and migrate in a rostrocaudal direction, approach the termination of the bowel at E13.5, and colonize the entire gastrointestinal tract by E14.5 (Durbec et al., 1996; Druckenbrod and Epstein, 2005; Anderson et al., 2006; Burns and Thapar, 2006). During migration, some ENCCs proliferate (Young et al., 2005) or differentiate into neuronal or glial cells (Conner et al., 2003; Young et al., 2004). This process is elaborately regulated by intrinsic programs and also by extrinsic cues, and aberrance therein causes serious disorders such as in Hirschsprung's disease (Heanue and Pachnis, 2007; Gershon, 2010).
A number of molecules have been shown to play critical roles in the development of the ENS in vertebrates (Young, 2008; Burzynski et al., 2009). In addition to proteins that are expressed specifically in ENCCs, including Ret and endothelin receptor B (EDNRB), ubiquitously expressed signaling molecules have also been shown to affect the fate of ENCCs. For example, protein kinase A (PKA) is a key component in the integration of signals from Ret and EDNRB. Inhibition of PKA abrogates GDNF-mediated chemotaxis of ENCC in vitro (Barlow et al., 2003). Furthermore, mutation of a putative PKA phosphorylation site of Ret has been shown to cause ENCC migration defects and aganglionosis (Asai et al., 2006). Meanwhile, Rho-family GTPases, including RhoA, Rac1, and Cdc42, are molecular switches that control actin dynamics (Heasman and Ridley, 2008). They play central roles in neuronal morphogenesis (Luo, 2000; Rupp and Kulesa, 2007), and also play important roles in neuronal migration and proliferation of ENCCs (Stewart et al., 2007; Fuchs et al., 2009).
Detailed analysis of the signaling molecules in ENS has been hampered by two problems. First, methods to monitor the activities of signaling molecules in situ are limited. Second, identification of signaling molecules requires fixation or homogenization of the cells. A solution to these problems is the use of biosensors based on the principle of Förster resonance energy transfer (FRET). For a long period, however, the use of FRET biosensors was limited to tissue culture cells due to a lack of methods to establish stable expression in mice (Aoki et al., 2012). Recently, we and others have reported the establishment of transgenic mice expressing FRET biosensors (Yamaguchi et al., 2011; Kamioka et al., 2012). With these mice in hand we are now able to see the activities of signaling molecules in living animals.
In this study we performed live imaging of the activities of signaling molecules in ENCCs by two-photon excitation microscopy. We found clear antagonistic actions of PKA activity and Rac1 activity in a stage- and region-dependent manner. Furthermore, JNK and ERK activities did not show detectable correlation with the velocity of ENCC migration, although a JNK inhibitor did abrogate the migration of ENCCs.
Materials and Methods
Chemicals.
PD184352 (2-(2-chloro-4-iodophenylamino)-N-cyclopropylmethoxy-3,4-difluorobenzamide), PI-103 (3-[4-(4-morpholinyl)pyrido[3′,2′:4,5]furo[3,2-d]pyrimidin-2-yl]-phenol), and 554340 (1,3-dihydro-5,6-dimethoxy-3-((4-hydroxyphenyl)methylene)-2H-indol-2-one) were purchased from Calbiochem. SP-8-Br-cAMPS (8-bromoadenosine-3′,5′-cyclic monophosphorothioate, Sp− isomer) was purchased from BioLog. SP600125 (anthra[1–9-cd]pyrazol-6(2H)-one) and BQ788 (2,6-dimethylpiperidinecarbonyl-γ-Methyl-Leu-Nin-(Methoxycarbonyl)-d-Trp-d-Nle, N-[N-[N-[(2,6-dimethyl-1-piperidinyl)carbonyl]-4-methyl-l-leucyl]-1-(methoxycarbonyl)-d-tryptophyl]-d-norleucine sodium salt) were purchased from Sigma-Aldrich.
FRET biosensors and generation of transgenic mice.
Transgenic mice expressing a PKA biosensor AKAR3EV, a negative control FRET biosensor AKAR3EV-NC, and an ERK biosensor EKAREV-NLS were reported previously (Kamioka et al., 2012). FRET biosensors for Rac1, Cdc42, and JNK named RaichuEV-Rac1, RaichuEV-Cdc42, and JNKAR1EV-NLS were reported previously (Komatsu et al., 2011), and the methods for producing transgenic mice expressing FRET biosensors for Rac1, Cdc42, and JNK were described previously (Sumiyama et al., 2010; Kamioka et al., 2012). Founder animals were bred with Jcl:ICR mice to produce stable lines. Newborn mice were illuminated with a blue flashlight LEDGFP-3W (Optocode) and inspected for green fluorescence through yellow-colored glasses. The animal protocols were reviewed and approved by the Animal Care and Use Committee of Kyoto University Graduate School of Medicine (No. 10584).
Generation of Ret-mCherry mice.
Transgenic mice expressing mCherry under a Ret promoter, referred to as Ret-mCherry mice hereafter, were produced by the same strategy as Ret-TGM mice (Enomoto et al., 2001): a cDNA encoding GFP inserted into the first coding exon of Ret was exchanged for cDNA encoding mCherry. Male mice heterozygous for the Ret-mCherry mutation (Ret-mCherry/+ mice) were mated to wild-type (C57BL6) females. The genotype of Ret-mCherry mice was determined by PCR using the primers P1 (5′-CGAGACCCGCCTGCTCCTCAACCGC-3′), P2 (5′-AGCGCTAACTTCACCCCGGCCCCTACCGTC-3′), and P3 (5′-GTTATCCTCCTCGCCCTTGCTCAC-3′) (35 cycles of 94°C for 30 s, 65°C for 30 s, and 72°C for 60 s).
Organ culture.
FRET mice and Ret-mCherry/+ mice were mated and pregnant mice were killed by cervical dislocation. Live imaging of embryonic guts was performed essentially as described previously (Aoki and Matsuda, 2009). Embryos were illuminated with a blue flashlight LEDGFP-3W and inspected for green fluorescence through yellow-colored glasses. Guts were dissected from each embryo and transferred into PBS in 24-well plates (Nunc). Each gut was inspected for RFP fluorescence by an epifluorescence microscope. Guts with RFP+ cells were then placed on a piece of filter paper (Millipore Corporation). The preparation was placed in tissue culture medium (DMEM/F12) containing 10% Knockout Serum Replacement (KSR) (Invitrogen), and penicillin/streptomycin sulfate solution in a 35 mm dish (Nunc).
In vitro explant assay.
For the in vitro explant assay, E11.5 or E12.5 guts in which ENCCs expressed both FRET biosensor and ret-mCherry were prepared as described previously (Uesaka and Enomoto, 2010). Guts were cut into short segments and transferred onto 35 mm glass-base dishes (Asahi Techno Glass) coated with human fibronectin (Biomedical Technologies), and cultured in DMEM/F12 supplemented with 10% KSR and 50 ng/ml GDNF (R&D Systems) for a day before imaging with an epifluorescence microscope.
Two-photon excitation microscopy and confocal laser scanning microscopy.
The protocol for FRET imaging of ENCCs in explant assay with an epifluorescence microscope has been described previously (Aoki and Matsuda, 2009). The organ-cultured gut was maintained in an incubation chamber (Tokai Hit) and imaged using a 20× water-immersion objective lens (XLUMPLFL20; Olympus) on a BX61WI/FV1000 upright microscope, which was connected to a Mai Tai DeepSee HP Ti:Sapphire Laser (Spectra Physics), 440 nm laser diode (Olympus), and 543 nm laser diode (Melles Griot). For FRET imaging with a two photon excitation microscope, the excitation wavelength for CFP was 840 nm. We used an IR-cut filter, RDM650, a DM505 dichroic mirror, and a band-path filter BA460-510 for CFP or BA510-560 for YFP (Olympus). For FRET imaging and mCherry imaging with a confocal laser scanning unit, we used a 440 nm laser diode for CFP excitation, a 543 nm laser diode for mCherry excitation, and the following filter sets purchased from Olympus: an excitation dichroic mirror, BS/20/80, a second dichroic mirror, DM510, a third dichroic mirror, SDM560, and three emission filters, BA465-495 for CFP, BA520-550 for FRET, and BA560-660 for mCherry, respectively.
Immunostaining of phospho-CREB.
Embryonic guts were dissected at E12 and fixed overnight in 4% formaldehyde in PBS at 4°C. After washing with PBS, whole-mount preparations were processed for immunohistochemistry as described previously (Uesaka and Enomoto, 2010). Phospho-CREB (Ser133) antibody (#9191, Cell Signaling Technology) and Alexa Fluor 488 goat anti-rabbit IgG (Life Technologies) were used as the primary and secondary antibodies, respectively.
Image processing.
Acquired images were analyzed with MetaMorph software (Universal Imaging). For calculation of the migration speed of individual ENCCs expressing a FRET biosensor in the nucleus in a 3D environment, the original confocal images were analyzed with Imaris Software (Bitplane AG). For calculation of the migration speed of individual ENCCs expressing a FRET biosensor in the cytosol, the total path length that the cell moved was determined by measuring the change of center position of the nucleus of each cell using MetaMorph software, and divided by the time. For quantification of phospho-CREB, fluorescent intensity of >20 ENCCs was quantified at the rostral or caudal regions of three E12 mouse embryos.
Expression of GDNF and endothelin 3.
Expression of endothelin 3 (ET-3) was examined by in situ hybridization according to the manufacturer's protocol (Roche Diagnostics Corp.). A cDNA fragment encompassing from nt 217 to 804 of ET-3 cDNA (Gene ID: 13616, 2963 bp) was used as the probe. GDNF expression was examined in GDNFlacZ/+ mice as described previously (Moore et al., 1996; Uesaka et al., 2007).
Results
Activity maps of signaling molecules in ENS by means of ex vivo imaging of transgenic mice expressing FRET biosensors
We have recently developed a method to efficiently generate transgenic mice expressing a FRET biosensor by the Tol2 transposase-mediated gene transfer method (Sumiyama et al., 2010; Kamioka et al., 2012). As a proof of concept, we reported transgenic mice expressing FRET biosensors for ERK and PKA. Here, to examine the spatiotemporal dynamics of the activities of Rho-family GTPases and JNK in ENCCs, we added transgenic mouse lines for Rac1, Cdc42, and JNK. The FRET biosensors in these mice, collectively called FRET mice, are expressed ubiquitously under the CAG promoter (Kamioka et al., 2012). To distinguish ENCCs from morphologically indistinguishable mesenchymal cells, we used transgenic mice expressing the mCherry fluorescent protein under the Ret promoter, which is active specifically in ENCCs. These Ret-mCherry mice were crossed with the FRET mice (Fig. 1A,B). The embryonic mouse gut in organ culture was time lapse-imaged for CFP, FRET, and mCherry by either two-photon excitation microscopy or confocal microscopy. ENCCs were identified on the mCherry images (Fig. 1C), which were binarized to generate clipping masks. The mCherry masks were used to identify ENCCs on the FRET/CFP ratio images. Outlines of the gut wall were determined on the unprocessed FRET/CFP ratio images. In this way, the intercellular activity variations of Rac1, Cdc42, PKA, ERK, and JNK were visualized in ENCCs. Notably, in the FRET mice stained for NADPH we did not find any anomaly in myenteric plexuses, excluding potential interference on the development of ENCCs by the expression of FRET biosensors.
Figure 1.
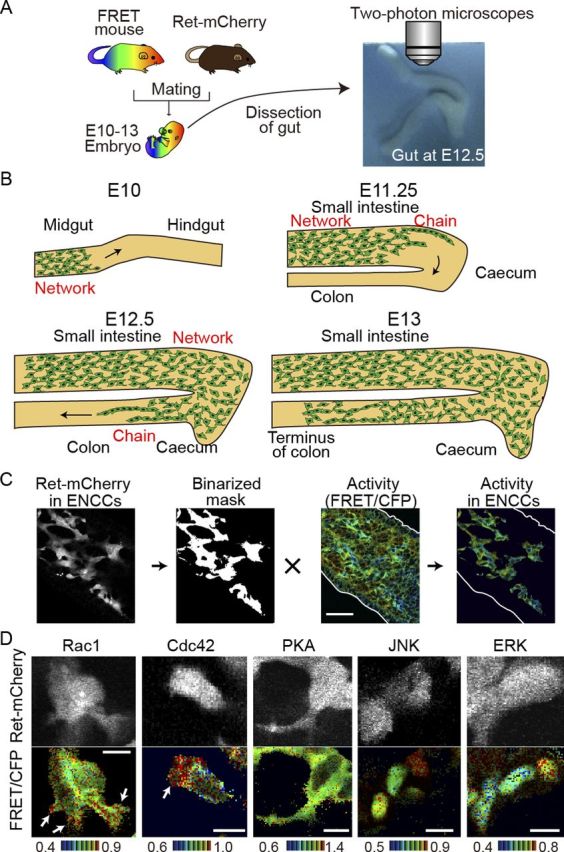
Activities of signaling molecules in ENCCs by means of ex vivo imaging of transgenic mice expressing FRET biosensors. A, A FRET mouse was crossed with a Ret-mCherry mouse. The gut was dissected from an embryo and cultured on a piece of sterile paper. B, A schematic illustration showing the migration of ENCCs in the developing gut at E10–E13. C, Image processing for the preparation of activity maps of signaling molecules in ENCCs. ENCCs are identified by the Ret-driven expression of mCherry. The mCherry images are binarized to generate masks, which are used to crop ENCCs from the FRET/CFP ratio image. The FRET/CFP ratio images are presented in the intensity-modulated display (IMD) mode. In the IMD mode, eight colors from red to blue are used to represent the FRET/CFP ratio, with the light intensity of each color indicating the mean intensity of FRET and CFP. D, Activities of signaling molecules in single ENCCs. The FRET/CFP ratio images and Ret-mCherry images are shown. The ratio ranges are shown at the bottom of the panel. The FRET biosensors of Rac1 and Cdc42 are localized at the plasma membrane, that of PKA is localized in the cytoplasm, and those of JNK and ERK are localized in the nucleus. Scale bars, 10 μm.
Under higher magnifications the subcellular activity maps of Rac1 and Cdc42 could also be drawn in a single ENCC (Fig. 1D). The ENCCs extended the lamellipodia in the direction of their migration, wherein high activities of Rac1 and Cdc42 were observed as reported in 2D cultures (Kraynov et al., 2000; Itoh et al., 2002). Note that the FRET biosensors for Rac1 and Cdc42 are localized on the plasma membrane, that the FRET biosensor for PKA is localized at the cytoplasm, and that the FRET biosensors for JNK and ERK are localized in the nucleus.
Inverse correlation of PKA activity with Rac1 and Cdc42 activities and migration velocity of ENCCs at E12.5
During the course of development, ENCCs derived from the transgenic mice expressing both FRET biosensors and mCherry did not show any defects in migration or differentiation. ENCCs unidirectionally migrated along the small intestine, passed the cecum, and reached the terminus of the colon by E13.5 as reported previously (Fig. 1B) (Young and Newgreen, 2001). We examined the activities of PKA, Rac1, Cdc42, JNK, and ERK in ENCCs of the colon at E12.5, when the caudal wavefront of ENCCs reached the hindgut. The ENCCs at the wavefront invaded the uncolonized mesenchyme in the form of cellular chains. On the other hand, cells in the rostral region formed a network that presages the reticulated arrangement characteristic of the ENS (Fig. 2A,B). These observations were again consistent with previous reports (Young et al., 2004; Druckenbrod and Epstein, 2007).
Figure 2.
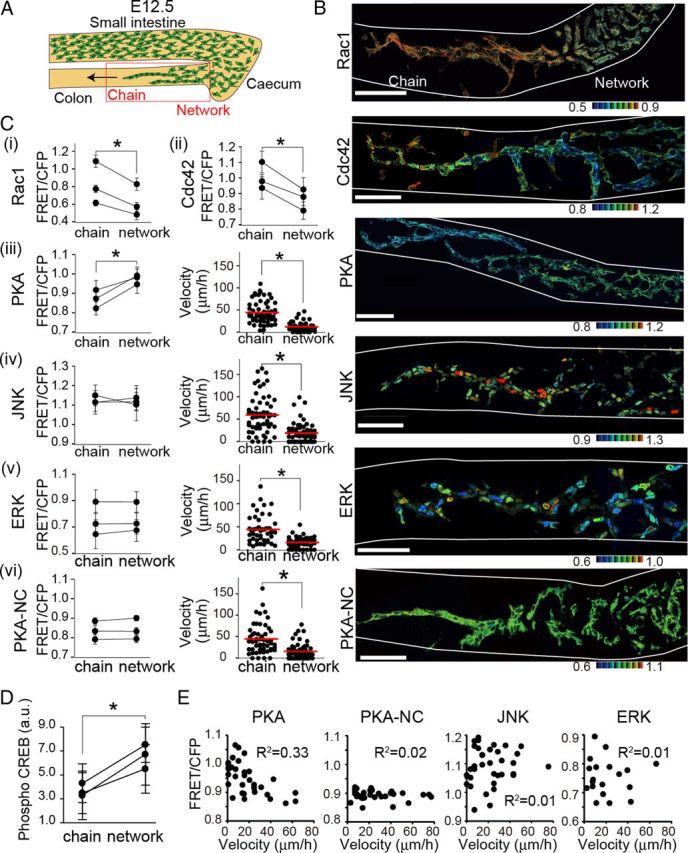
Activity maps of signaling molecules in ENCCs at E12.5. A, A schematic illustration of migrating ENCCs at E12.5 in the embryonic gut organ culture. The leading ENCCs migrate as chains, whereas the trailing ENCCs form networks. ENCCs within the red box were observed ex vivo. B, Representative FRET images of ENCCs expressing a FRET biosensor for Rac1, Cdc42, PKA, JNK or ERK. PKA-NC is a negative control biosensor, wherein the PKA phosphorylation site is mutated. Scale bars, 50 μm. The white lines show the contours of the guts. C, In each transgenic mouse, >20 ENCCs migrating as a chain or forming a network were analyzed to obtain the mean FRET/CFP ratio. Three mice were analyzed for each biosensor. Error bars show the SD. Asterisks indicate a significant difference by the paired t test analysis (p < 0.05). iii–vi, Migration velocities of single ENCCs in the caudal chain or the rostral network are plotted by dots. The numbers of analyzed cells are as follows: AKAR3EV, n = 60 (chain), n = 60 (network); AKAR3EV-NC, n = 55 (chain), n = 57 (network); JNKAR1EV-NLS, n = 59 (chain), n = 56 network); EKAREV-NLS, n = 49 (chain), n = 50 (network). Red lines and asterisks represent mean velocities and the statistical significance by t test (p < 0.05). D, Whole embryonic guts at E12 were immunostained with an anti-phospho-CREB antibody and an secondary antibody conjugated with Alexa Fluor 488. Fluorescent intensity of ENCCs at the rostral and caudal regions was quantified. Error bars show the SD. Asterisks indicate a significant difference by the paired t test analysis (p < 0.05). E, Each dot shows the FRET/CFP ratio and velocity of a single cell. R2 represents the correlation coefficient. The numbers of analyzed cells are as follows: AKAR3EV, n = 34; AKAR3EV-NC, n = 33; JNKAR1EV-NLS, n = 34; EKAREV-NLS, n = 22.
ENCCs chain-migrating in the caudal region exhibited lower PKA activity and higher Rac1 and Cdc42 activities compared with ENCCs in the rostral region (Fig. 2B,C). We did not find any significant difference in the averaged activities of ERK and JNK between the caudal and rostral ENCCs, although there was significant intercellular variance. PKA_NC is a negative control biosensor, wherein the phosphorylation site in the FRET biosensor of PKA was ablated. As expected, the FRET/CFP values were almost homogenous in all ENCCs of PKA-NC mice. To validate the finding obtained by the PKA FRET biosensor, the phosphorylation of CREB, a PKA substrate, was compared quantitatively by immunohistochemistry with anti-phospho-CREB antibody (Fig. 2D). As expected, the phosphorylation of CREB was lower in the chain-migrating ENCCs in the caudal region than those in the rostral region.
In the GFP-expressing transgenic mice used in the previous studies, individual ENCCs could not be easily distinguished from each other due to the homogenous localization of GFP between the nucleus and the cytoplasm, and also among ENCCs (Young et al., 2004). In contrast, in the transgenic mice used in this study, the FRET biosensors were expressed at the nuclei (ERK, JNK), cytoplasm (PKA, PKA-NC), or plasma membrane (Rac1, Cdc42). Except for cells expressing plasma membrane-localized FRET biosensors, the nucleus could be readily distinguished, allowing us to trace the migration of ENCCs during time-lapse imaging. We found that ENCCs at the wavefront migrated rapidly, whereas ENCCs in the rostral region migrated slowly and stochastically into various directions (Fig. 2C). To confirm the inverse correlation of PKA activity with migration velocity, the FRET/CFP ratio of each ENCC was plotted against the average velocity (Fig. 2E).
Regulation of Rac1-dependent ENCC migration by PKA and by PI3K
To untangle the signaling network regulating ENCC migration, ENCCs at E12.5 were treated with a cAMP analog (SP-8-Br-cAMPS), PI3K inhibitor (PI-103), MEK inhibitor (PD184352), or JNK inhibitor (SP600125) (Fig. 3A,B). The cAMP analog robustly activated PKA as expected, but neither the PI3K inhibitor nor the MEK inhibitor affected PKA activity to a detectable level (Fig. 3A). Rac1 activity was decreased by the cAMP analog and the PI3K inhibitor, but not by the MEK inhibitor (Fig. 3B). Thus, Rac1 was regulated positively by PI3K and negatively by PKA in ENCCs (Fig. 3B). ERK was inhibited both by the PI3K inhibitor and the MEK inhibitor (Fig. 3B). Treatment of the cAMP analog had no effect on ERK activity. Neither the cAMP analog, PI3K inhibitor, nor the MEK inhibitor affected JNK activity. We also examined the effect of PKA inhibitors such as H89, but none of them changed the PKA activity. We speculated that PKA inhibitors might not permeate through the serosa.
Figure 3.
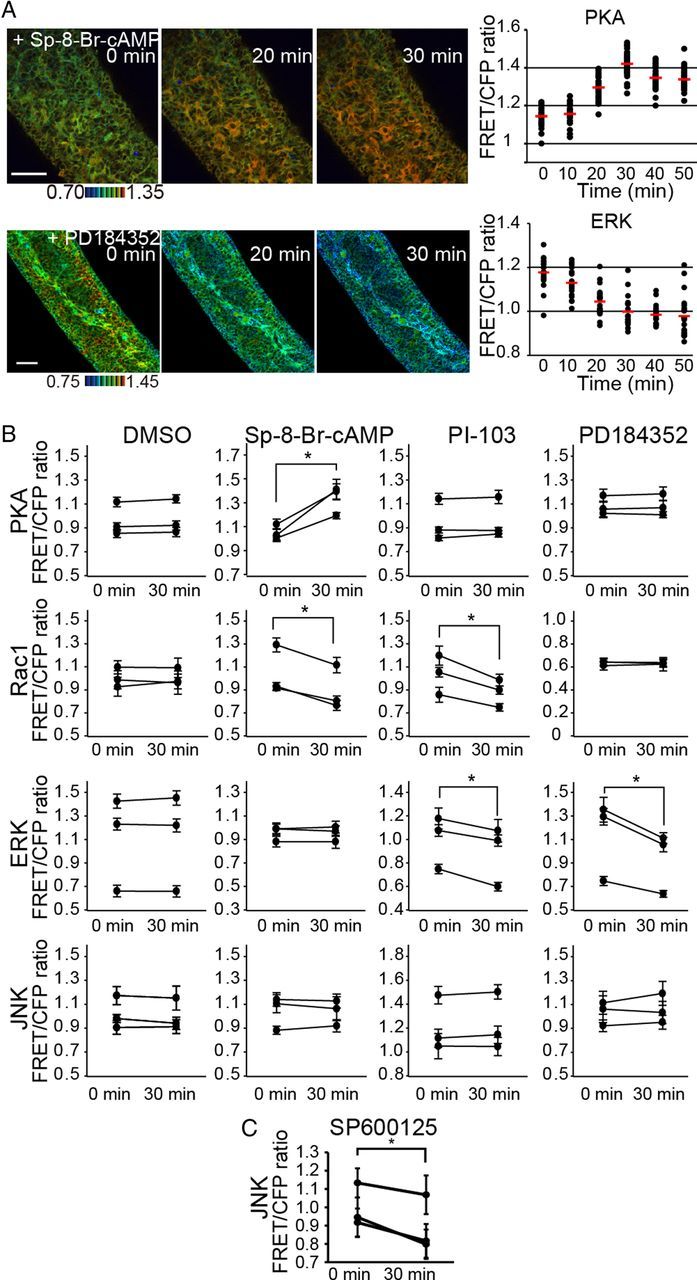
Regulation of Rac1 activity by PKA and PI3K, but not by ERK, in ENCCs. A, Embryonic gut organ cultures prepared from transgenic mice expressing a biosensor for PKA and ERK were time-lapse imaged and examined for the effect of 120 μm SP-8-Br-cAMP and 10 μm PD184352, respectively. Shown here are representative FRET/CFP ratio images in IMD mode and of FRET/CFP at the indicated time points after drug treatment. Scale bars, 50 μm. Data points represent the FRET/CFP ratio of each ENCC at the indicated time points. The mean FRET/CFP ratio at each time point is represented by a red line. B, ENCCs in organ culture were treated with DMSO, 120 μm SP-8-Br-cAMP, 30 μm PI-103 and 10 μm PD184352 for 30 min. The FRET/CFP ratios of >20 cells were measured for each organ culture before and after drug treatment. Data obtained from three independent organ cultures are shown. The asterisks indicate statistical significance by the paired t test analysis (p < 0.05). Error bars show the SD. C, ENCCs in organ culture were treated with 30 μm SP600125 for 30 min. The FRET/CFP ratios of >20 cells were measured for each organ culture before and after drug treatment. Data obtained from three independent organ cultures are shown. The asterisks indicate statistical significance by the paired t test analysis (p < 0.05). Error bars show the SD.
We next examined the effect of these reagents on the migration of ENCCs. The cAMP analog, the PI3K inhibitor, and the JNK inhibitor perturbed the migration of ENCCs, but not the MEK inhibitor (Fig. 4A,B). These results suggested that Rac1-dependent migration of ENCCs was regulated negatively by PKA and positively by PI3K. Although we did not find any biased spatial distribution of JNK activity, the JNK inhibitor suppressed JNK activity (Fig. 3C) and migration of ENCCs (Fig. 4A,B). In contrast, ERK did not perturb migration of ENCCs.
Figure 4.
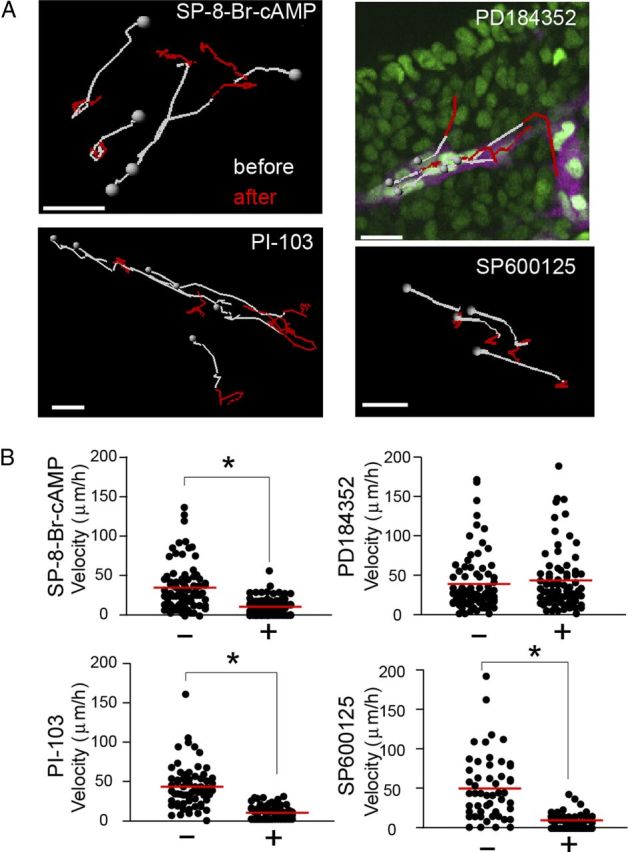
Regulation of ENCC motility by PKA, PI3K, and JNK, but not by ERK. A, Transgenic mice expressing JNKAR1EV-NLS and Ret-mCherry were used for the tracking of ENCCs, because the nuclear localization of the biosensor allowed automatic tracking by the software. During time-lapse imaging, the following reagents were added (in μm): 120 SP-8-Br-cAMP, 30 PI-103, 10 PD184352, and 30 SP600125. The nuclei of ENCCs were tracked for 30 min before drug treatment (white) and for 30 min after drug treatment (red). Gray spheres represent the start position of the trajectories. Scale bars, 20 μm. B, Migration velocities before and 30 min after the drug treatment. Data points represent individual cells compiled from three independent experiments. The red lines represent mean migration velocities of ENCCs. The asterisks indicate the results of t test analysis; *p < 0.05.
Activities of Rac1 and PKA in ENCCs at E10 and E11.5
It has been shown that the mode of ENCC migration changes during development of the intestine (Burns and Thapar, 2006). Thus, we next examined the activities of signaling molecules at earlier stages. As reported previously, ENCCs colonized the stomach and the proximal small intestine at E10, and the cecum at E11.25 (Fig. 5A). Notably, the migration of ENCCs at E10 was significantly slower than the migration of ENCCs at E12.5. At this time, ENCCs at the wavefront and the trailing regions exhibited similar levels of PKA and Rac1 activities (Fig. 5B,C). At E11.25 ENCCs at the wavefront started migrating fast as a chain and moving forward to the cecum. At this time ENCCs in the cecum showed lower PKA activity and higher Rac1 activity than did ENCCs in the rostral region (Fig. 5D,E). When the migration velocities of ENCCs were quantified, ENCCs in the caudal region and ENCCs in the rostral region migrated with similar velocities at E10, but ENCCs in the cecum, or caudal region, migrated faster than ENCCs in the rostral region at E11.25, as was observed at E12.5 (Fig. 5F). It was reported previously that chains of cells were observed at E10.5, when ENCCs enter the midgut (Druckenbrod and Epstein, 2005). Therefore, PKA inactivation and the subsequent Rac1 activation presumably occur at E10.5 in the midgut, which drives ENCCs to migrate quickly in the form of cellular chains.
Figure 5.

Migration velocity positively correlated with Rac1 activity and negatively correlated with PKA activity in the early stages of ENCCs. A, A schematic illustration of migrating ENCCs at E10 and E11.25. B, Representative FRET images of ENCCs expressing the FRET biosensors for PKA and Rac1 at E10. Scale bars, 50 μm. C, Mean FRET/CFP ratios of ENCCs in the rostral or caudal region at E10. FRET/CFP ratios of >15 cells were averaged for each organ culture and the results of three independent experiments are shown. Error bars show the SD. D, Representative FRET images of ENCCs at E11.25. Scale bars, 50 μm. E, Mean FRET/CFP ratios of ENCCs migrating as chains or forming a network at E11.25. FRET/CFP ratios of >20 cells were averaged for each organ culture and the results of three independent experiments are shown. The asterisks indicate statistical significance by paired t test analysis (p < 0.05). Error bars indicate SEM. F, Migration velocity of ENCCs expressing the biosensor for PKA at E10, E11.25, and E12.5. Data points represent individual cells compiled from three independent experiments. The red line represents the mean migration velocities of ENCCs. The asterisks indicate statistical significance by t test analysis (p < 0.05).
Activities of Rac1 and PKA in ENCCs at E13
Next, we observed Rac1 and PKA activity at E13, when the caudal region of ENCCs approached the terminus of the colon (Fig. 6A). In clear contrast to ENCCs at E12.5, ENCCs at the caudal region (Fig. 6B, left region) showed higher PKA activity than the following ENCCs (Fig. 6B, central region). Importantly, these ENCCs with low PKA activity continued to ENCCs with high PKA at the rostral region (Fig. 6B, right region). A mirror image of this PKA activity map was obtained for Rac1 (Fig. 6B). As in the other regions, the migration velocity correlated inversely with PKA activity and positively with Rac1 activity (Fig. 6C,D). Thus, ENCCs ceased migration when PKA activity increased and Rac1 activity decreased at the terminus of the colon. This observation strongly suggested that PKA controls the migration of ENCCs during the development of the intestine via Rac1.
Figure 6.
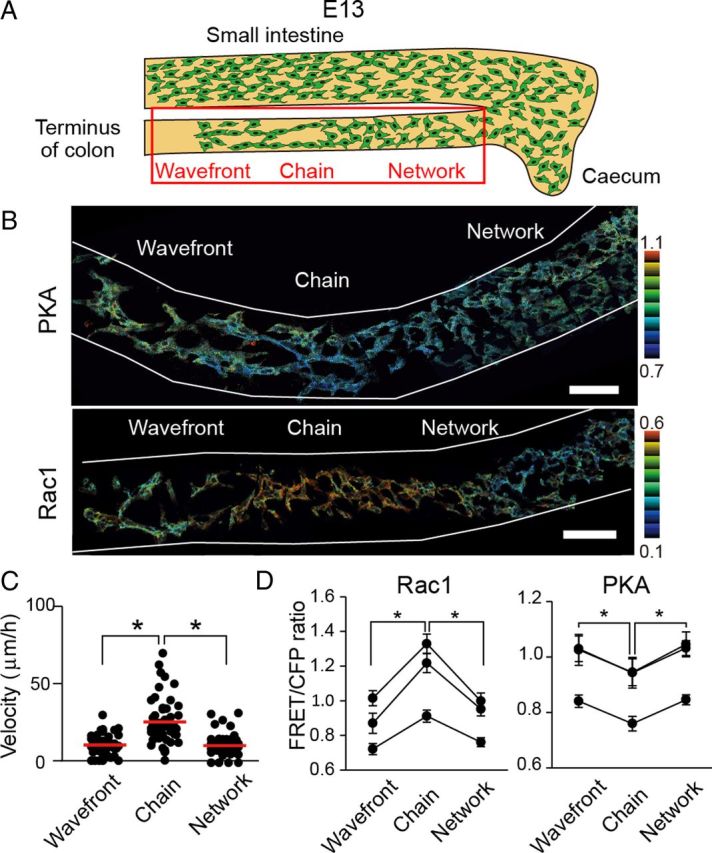
Increased PKA activity and decreased Rac1 activity in the ENCCs that reached the end of the colon at E13. A, A schematic illustration of ENCCs at E13. B, Representative FRET images of ENCCs expressing the FRET biosensor for PKA or Rac1 at E13. Scale bars, 50 μm. C, Migration velocity of ENCCs at the end, chain, or network region. Data points represent individual cells compiled from three independent experiments (n = 58 for end, n = 45 for chain, n = 45 for network). The red line represents the mean of average velocities of ENCCs. The asterisks indicate statistical significance by t test analysis (p < 0.05). D, Mean FRET/CFP ratios of ENCCs that halted at the end of colon, were migrating as a chain, or were forming a network at E13. The FRET/CFP ratios of >20 cells were averaged for each organ culture and the results of three independent experiments are shown. Error bars indicate SEM.
Regulation of PKA and Rac1 by GDNF and ET-3
Finally we searched for extrinsic cues that could modulate the activities of PKA and Rac1. The candidate molecules are GDNF and ET-3, which have been shown to be key players of ENCC development (Burns and Thapar, 2006; Heanue and Pachnis, 2007). Bath application of GDNF and ET-3 to the organ culture of the intestine did not work, probably due to inefficient permeation across the serosa. Therefore, we examined the effect of GDNF and ET-3 by the time-lapse FRET imaging of ENCC explants (Fig. 7A). GDNF activated Rac1 and inhibited PKA in ENCCs but not in the surrounding mesenchymal cells (Fig. 7B,C,E). The GDNF-induced Rac1 activation and PKA inhibition were cancelled by the addition of 554340, an inhibitor of Ret (Fig. 7C,E). In contrast to GDNF, ET-3 activated PKA and inhibited Rac1 in 30 min (Fig. 7B,D,F). The ET-3-induced Rac1 inhibition and PKA activation were abrogated by BQ788, a specific agonist of EDNRB (Fig. 7D). Notably, ET-3 transiently activated Rac1 within 5 min in a BQ788-independent manner, the significance of which should be evaluated in a future study. Neither GDNF nor ET-3 exhibited any effect on JNK activity in ENCCs (Fig. 7G). These results strongly suggest that PKA serves as a molecular switch of Rac1-dependent migration that is controlled negatively by GDNF and positively by ET-3.
Figure 7.
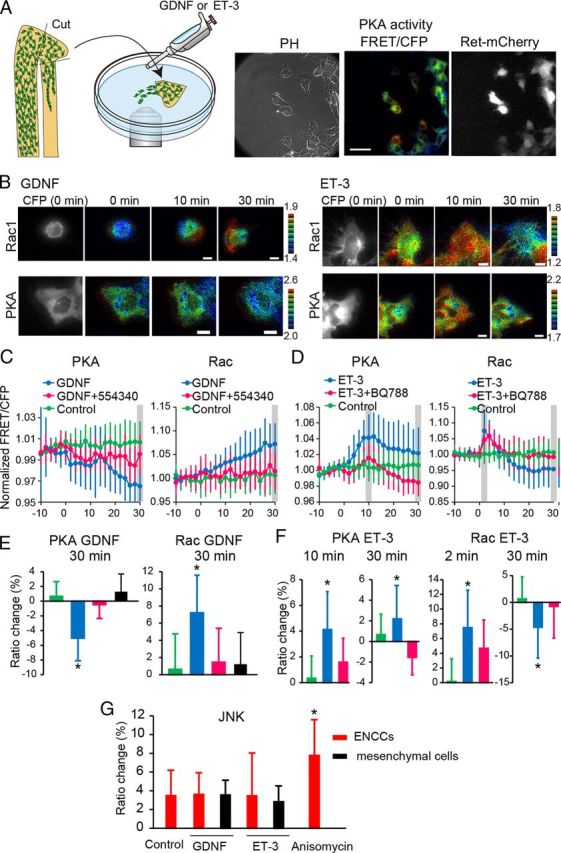
Activities of PKA and Rac1 regulated by GDNF and ET-3. A, A schematic illustration of the explant assay. Representative phase contrast, PKA activity (FRET/CFP), and ret-mCherry images of ENCCs expressing the FRET biosensor for PKA. Scale bars, 50 μm. The ret-mCherry image was used to mark ENCCs. B, ENCCs expressing the FRET biosensor for Rac1 or PKA were stimulated with 100 ng/ml GDNF or 100 nm ET-3 at time point zero. Images were acquired every 2 min for 40 min. Representative FRET/CFP images in the IMD mode and Ret-mCherry images at the indicated time points are shown. Scale bars, 10 μm. C, FRET/CFP values of each cell were normalized to the average FRET/CFP value between −10 and 0 min. Cells were mock-stimulated or stimulated with GDNF in the presence or absence of the Ret kinase inhibitor 554340. In ENCCs expressing the biosensor for PKA, the numbers of analyzed cells were as follows: GDNF, n = 17; GDNF+554340, n = 15; control, n = 30. In ENCCs expressing the biosensor for Rac1, the numbers of analyzed cells were as follows: GDNF, n = 21; GDNF+554340, n = 21; control, n = 26. Error bars are the SD. D, Cells were analyzed similarly to those in C, except that cells were stimulated with ET-3 and the ET-3 inhibitor BQ788. In ENCCs expressing the biosensor for PKA, the numbers of analyzed cells were as follows: ET-3, n = 25; ET-3+BQ788, n = 20; no treatment, n = 30. In ENCCs expressing the biosensor for Rac1, the numbers of analyzed cells were as follows: ET-3, n = 35; ET-3+BQ788, n = 24; no treatment, n = 26. Error bars are the SD. E, Cells expressing the FRET biosensor for PKA or Rac1 were mock-stimulated or stimulated with 100 ng/ml GDNF in the presence or absence of the Ret kinase inhibitor 554340 (1 μm). Bar graphs represent PKA and Rac1 activities 30 min after treatment. Expression of Ret-mCherry was used to discriminate ENCCs from mesenchymal cells. The numbers of experiments were as follows: PKA (Control, n = 30; GDNF, n = 40; GDNF+554340, n = 15; GDNF on mesenchymal cells, n = 12), Rac1 (Control, n = 30; GDNF, n = 40; GDNF+554340, n = 15; GDNF on mesenchymal cells, n = 12). F, Cells expressing the FRET biosensor for PKA or Rac1 were mock-stimulated or stimulated with 100 nm ET-3 in the presence or absence of ET-3 inhibitor BQ788. The numbers of experiments were as follows: PKA (ET-3, n = 27; ET-3+BQ788, n = 27; no treatment, n = 33), Rac1 (ET-3, n = 36; ET-3+BQ788, n = 24; no treatment, n = 26). G, Cells expressing the FRET biosensor for JNK were mock-stimulated or stimulated with 100 ng/ml GDNF, 100 nm ET-3, or 1 μg/ml Anisomycin. The numbers of experiments were as follows: Control, n = 12; GDNF, n = 12; GDNF on mesenchymal cells, n = 13; ET-3, n = 13; ET-3 on mesenchymal cells, n = 12; Anisomycin, n = 12. Error bars show the SD.
To examine the role of GDNF and ET-3 in ENCC migration, expression of GDNF and ET-3 was examined at E13. Both GDNF and ET-3 were expressed most abundantly at cecum and decreased gradually toward the caudal end (Fig. 8A).
Figure 8.
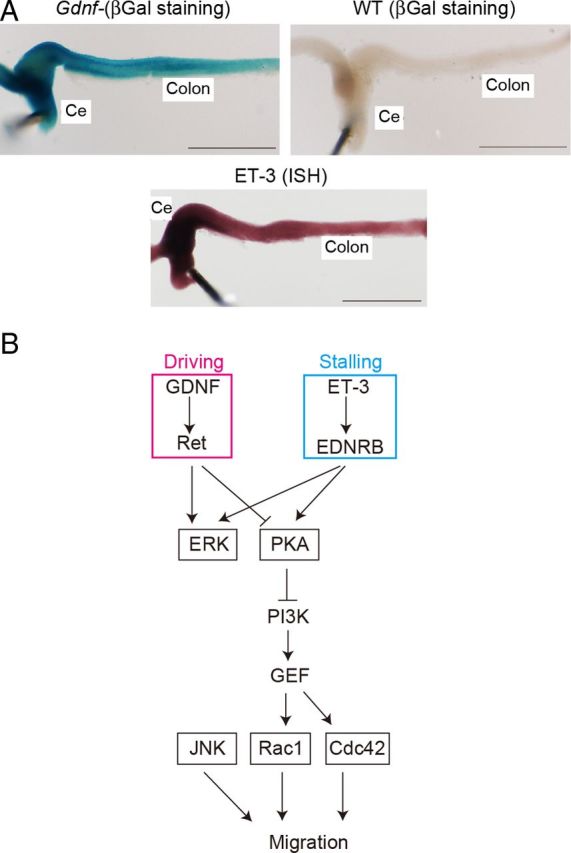
Mechanism of migration of ENCCs. A, Expression of GDNF and ET-3 in ENCCs at E13. Expression of GDNF was examined with a GDNFLacZ/+ mouse. Expression of ET-3 was examined by in situ hybridization. B, A regulatory model of ENCC migration. GDNF serves as a chemoattractant during chain migration, and ET-3 at the end of the colon stalls migration. Both signals merge at PKA. JNK regulates ENCC migration independently of the PKA-Rac1 pathway.
Discussion
The genetic and histochemical work by Barlow et al. (2003) has shown that ENCCs are coordinately controlled by the ET-3-EDNRB and GDNF-Ret pathways. This work elegantly demonstrated that the two pathways function cooperatively for the replication, but antagonistically for the chemotaxis of ENCCs. However, due to a lack of methods to monitor the activities of signaling molecules in situ, the mechanism of the spatiotemporal control of the signaling molecules remains elusive. For example, based on reports that the activation of EDNRA or EDNRB receptors inhibits PKA activity in certain cell types (James et al., 1994; Ono et al., 1994), it has been proposed that PKA is a positive regulator of ENCC migration. This observation would seem to contradict our proposal that ET-3 activates PKA and thereby inhibits the chain migration of ENCCs. EDNRB is primarily coupled to Gq and the effect of ET-3 on PKA can be either positive or negative, depending on cell types (Sokolovsky, 1995): The activation of EDNRB receptors can activate PKA in neuronal cells (Jaureguiberry et al., 2004) and melanocytes (Sato-Jin et al., 2008). Thus, measurement of the activity in situ is critical for determining the contribution of PKA or other signaling molecules, for which our approach with FRET mice will be applicable in a versatile way.
GDNF, which is expressed in regions that precede the wavefront of ENCC migration (i.e., the stomach and cecum), drives ENCC migration (Natarajan et al., 2002). Meanwhile, ET-3, which is expressed at the cecum and colon, serves to halt migration of ENCCs (Barlow et al., 2003). We obtained similar results at E13 and did not find higher expression of ET-3 at the terminus of the colon than in the rostral regions. Therefore, the antagonistic action of GDNF versus ET-3 alone could not explain the mechanism underlying the stalling of ENCC migration at the terminus of the colon.
We have found that the migration velocity of ENCCs correlates inversely with PKA activity (Fig. 2) and that, in line with this observation, PKA activation by a cAMP analog inhibits ENCC migration (Fig. 4). The mechanism by which PKA inhibits migration has not been studied in ENCCs, but in other cell types two mechanisms have been proposed to link PKA activity to cell migration. In endothelial cells PKA inhibits cell migration in an Src-dependent manner (Jin et al., 2010). In a mouse lymphoid cell line, PKA inhibits guanine nucleotide exchange of RhoA, thereby perturbing integrin-mediated lymphocyte adhesion (Laudanna et al., 1997). However, a link from PKA to Rac1 was not established in either study.
We have shown that PKA activity is inversely correlated with Rac1 activity (Fig. 2), which naturally leads to speculation that PKA inhibits Rac1 and ENCC migration (Fig. 8B). Reflecting the pleiotropic roles of PKA in signal transduction, PKA can either inhibit or activate Rac1 in a cell context-dependent manner. PKA phosphorylates Rac1 at Serine 71 and thereby downregulates Rac1 upon bacterial infection of epithelial cells (Brandt et al., 2009). PKA also downregulates Rac1 indirectly by means of phosphorylation of guanine nucleotide exchange factors of Rac1. For example, PKA binds to, phosphorylates, and inhibits P-Rex1, a Rac1 GEF (guanine nucleotide exchange factor), which regulates chemotaxis of leukocytes (Zhao et al., 2007). In contrast, PKA is required for a GDNF-mediated increase in Rac1 GEF activity and lamellipodia formation in SH-SY5Y neuroblastoma cells (Fukuda et al., 2002). In PC12 rat pheochromocytoma cells, PKA phosphorylates and activates STEF/Tiam2, another Rac1 GEF, and induces neurite extension (Goto et al., 2011). Meanwhile, many Rac1 GEFs are regulated by PI3K. In agreement with this observation, we found that the migration of ENCCs was inhibited by a PI3K inhibitor (Fig. 4). Thus, we could postulate a signaling cascade comprised of PKA, PI3K, Rac1 GEF, and Rac1 (Fig. 8B). Further studies of potential PKA-regulated GEFs and/or GTPase-activating proteins will be needed to untangle the signaling pathway from PKA to Rac1 in ENCCs.
Asai et al. (2006) previously reported that PKA regulates Ret and JNK and promotes the migration of ENCCs in the developing gut. We also confirmed that JNK is required for ENCC migration by the use of a specific JNK inhibitor (Fig. 4). However, in contradiction to the previous proposal that JNK is activated by GDNF in dorsal root ganglion cells, we failed to observe JNK activation in GDNF-stimulated ENCCs (Fig. 7G). Furthermore, we also failed to observe any correlation between migration velocity and JNK activity in ENCCs (Fig. 2). Because the FRET biosensor for JNK monitors the activity balance between JNK and phosphatases, we might have failed to detect JNK activation, if both JNK and phosphatases had been activated by GDNF. But in this case the signal from JNK is counteracted by the phosphatases and not transmitted to the downstream molecules. Thus, JNK activity does not play a regulatory role in ENCC migration, but probably serves to maintain the ability for ENCCs to migrate as reported previously in epithelial cells (Wang et al., 2010).
Natarajan et al. (2002) reported that ERK is phosphorylated upon GDNF stimulation and required for the GDNF-induced migratory response of ENCCs. In another study, however, the MEK1 inhibitor PD98059 (2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) did not affect ENCC migration in the colon (Asai et al., 2006). Colonization of ENCCs is an integrated process of migration, survival, proliferation, and differentiation. A reduction in the number of ENCCs by cell death or another mechanism leads to failure of complete colonization and results in aganglionosis (Burns et al., 2000; Uesaka and Enomoto, 2010). In the previous experimental protocols, it was difficult to distinguish in which processes of migration, survival, proliferation, or differentiation ERK is required. In this study, we failed to observe any correlation between ERK activity and migration velocity in the timescale of minutes or between ERK activity and the location of each ENCC (Fig. 2). It has been reported that proliferation rates are equivalent in all regions of the ENS, regardless of the position relative to the migratory wavefront (Young et al., 2005). Thus, ERK may play a critical role in the regulation of ENCC replication, which may affect the overall rate of ENCC migration, but ERK may not directly control the velocity or direction of ENCC migration.
In conclusion, we have shown the activity changes of small GTPases and protein kinases in migrating ENCCs, and found that PKA serves as a critical node of external cues and negatively regulates Rac1 and, thereby, ENCC migration. Furthermore, against previous predictions, JNK and ERK do not spatiotemporally regulate ENCC migration, despite their essential roles in the migration of ENCCs. These observations argue for the importance of the visualization of signaling molecule activities in living animals.
Footnotes
This work was supported by a Grant-in-Aid for Scientific Research on the Innovative Area “Fluorescence Live imaging” (No. 22113002), by a Research Program of Innovative Cell Biology “Cell Innovation,” and by an Innovative Techno-Hub for the Integrated Medical Bio-imaging Project of the Special Coordination Funds for Promoting Science and Technology by Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. A.G. was supported by a Grant-in-Aid for Japan Society for the Promotion of Science Fellows by MEXT, Japan. We thank Kyoko Hirano, Yoshie Inaoka, Akiko Sakurai-Katsumata, and Akiko Kawagishi for technical assistance and members of the Matsuda Laboratory for their input.
Michiyuki Matsuda received a research fund from Olympus Corporation.
References
- Anderson RB, Stewart AL, Young HM. Phenotypes of neural-crest-derived cells in vagal and sacral pathways. Cell Tissue Res. 2006;323:11–25. doi: 10.1007/s00441-005-0047-6. [DOI] [PubMed] [Google Scholar]
- Aoki K, Matsuda M. Visualization of small GTPase activity with fluorescence resonance energy transfer-based biosensors. Nat Protoc. 2009;4:1623–1631. doi: 10.1038/nprot.2009.175. [DOI] [PubMed] [Google Scholar]
- Aoki K, Komatsu N, Hirata E, Kamioka Y, Matsuda M. Stable expression of FRET biosensors: a new light in cancer research. Cancer Sci. 2012;103:614–619. doi: 10.1111/j.1349-7006.2011.02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai N, Fukuda T, Wu Z, Enomoto A, Pachnis V, Takahashi M, Costantini F. Targeted mutation of serine 697 in the Ret tyrosine kinase causes migration defect of enteric neural crest cells. Development. 2006;133:4507–4516. doi: 10.1242/dev.02616. [DOI] [PubMed] [Google Scholar]
- Barlow A, de Graaff E, Pachnis V. Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron. 2003;40:905–916. doi: 10.1016/s0896-6273(03)00730-x. [DOI] [PubMed] [Google Scholar]
- Brandt S, Kenny B, Rohde M, Martinez-Quiles N, Backert S. Dual infection system identifies a crucial role for PKA-mediated serine phosphorylation of the EPEC-Tir-injected effector protein in regulating Rac1 function. Cell Microbiol. 2009;11:1254–1271. doi: 10.1111/j.1462-5822.2009.01330.x. [DOI] [PubMed] [Google Scholar]
- Burns AJ, Thapar N. Advances in ontogeny of the enteric nervous system. Neurogastroenterol Motil. 2006;18:876–887. doi: 10.1111/j.1365-2982.2006.00806.x. [DOI] [PubMed] [Google Scholar]
- Burns AJ, Champeval D, Le Douarin NM. Sacral neural crest cells colonise aganglionic hindgut in vivo but fail to compensate for lack of enteric ganglia. Dev Biol. 2000;219:30–43. doi: 10.1006/dbio.1999.9592. [DOI] [PubMed] [Google Scholar]
- Burzynski G, Shepherd IT, Enomoto H. Genetic model system studies of the development of the enteric nervous system, gut motility and Hirschsprung's disease. Neurogastroenterol Motil. 2009;21:113–127. doi: 10.1111/j.1365-2982.2008.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner PJ, Focke PJ, Noden DM, Epstein ML. Appearance of neurons and glia with respect to the wavefront during colonization of the avian gut by neural crest cells. Dev Dyn. 2003;226:91–98. doi: 10.1002/dvdy.10219. [DOI] [PubMed] [Google Scholar]
- Druckenbrod NR, Epstein ML. The pattern of neural crest advance in the cecum and colon. Dev Biol. 2005;287:125–133. doi: 10.1016/j.ydbio.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Druckenbrod NR, Epstein ML. Behavior of enteric neural crest-derived cells varies with respect to the migratory wavefront. Dev Dyn. 2007;236:84–92. doi: 10.1002/dvdy.20974. [DOI] [PubMed] [Google Scholar]
- Durbec PL, Larsson-Blomberg LB, Schuchardt A, Costantini F, Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development. 1996;122:349–358. doi: 10.1242/dev.122.1.349. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Crawford PA, Gorodinsky A, Heuckeroth RO, Johnson EM, Jr, Milbrandt J. RET signaling is essential for migration, axonal growth and axon guidance of developing sympathetic neurons. Development. 2001;128:3963–3974. doi: 10.1242/dev.128.20.3963. [DOI] [PubMed] [Google Scholar]
- Fuchs S, Herzog D, Sumara G, Büchmann-Møller S, Civenni G, Wu X, Chrostek-Grashoff A, Suter U, Ricci R, Relvas JB, Brakebusch C, Sommer L. Stage-specific control of neural crest stem cell proliferation by the small rho GTPases Cdc42 and Rac1. Cell Stem Cell. 2009;4:236–247. doi: 10.1016/j.stem.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Kiuchi K, Takahashi M. Novel mechanism of regulation of Rac activity and lamellipodia formation by RET tyrosine kinase. J Biol Chem. 2002;277:19114–19121. doi: 10.1074/jbc.M200643200. [DOI] [PubMed] [Google Scholar]
- Gershon MD. Developmental determinants of the independence and complexity of the enteric nervous system. Trends Neurosci. 2010;33:446–456. doi: 10.1016/j.tins.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Goto A, Hoshino M, Matsuda M, Nakamura T. Phosphorylation of STEF/Tiam2 by protein kinase A is critical for Rac1 activation and neurite outgrowth in dibutyryl cAMP-treated PC12D cells. Mol Biol Cell. 2011;22:1780–1790. doi: 10.1091/mbc.E10-09-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heanue TA, Pachnis V. Enteric nervous system development and Hirschsprung's disease: advances in genetic and stem cell studies. Nat Rev Neurosci. 2007;8:466–479. doi: 10.1038/nrn2137. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Itoh RE, Kurokawa K, Ohba Y, Yoshizaki H, Mochizuki N, Matsuda M. Activation of rac and cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Mol Cell Biol. 2002;22:6582–6591. doi: 10.1128/MCB.22.18.6582-6591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AF, Xie LH, Fujitani Y, Hayashi S, Horie M. Inhibition of the cardiac protein kinase A-dependent chloride conductance by endothelin-1. Nature. 1994;370:297–300. doi: 10.1038/370297a0. [DOI] [PubMed] [Google Scholar]
- Jaureguiberry MS, di Nunzio AS, Dattilo MA, Bianciotti LG, Vatta MS. Endothelin 1 and 3 enhance neuronal nitric oxide synthase activity through ETB receptors involving multiple signaling pathways in the rat anterior hypothalamus. Peptides. 2004;25:1133–1138. doi: 10.1016/j.peptides.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Jin H, Garmy-Susini B, Avraamides CJ, Stoletov K, Klemke RL, Varner JA. A PKA-Csk-pp60Src signaling pathway regulates the switch between endothelial cell invasion and cell-cell adhesion during vascular sprouting. Blood. 2010;116:5773–5783. doi: 10.1182/blood-2010-07-296210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamioka Y, Sumiyama K, Mizuno R, Sakai Y, Hirata E, Kiyokawa E, Matsuda M. Live imaging of protein kinase activities in transgenic mice expressing FRET biosensors. Cell Struct Funct. 2012;37:65–73. doi: 10.1247/csf.11045. [DOI] [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, Matsuda M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22:4647–4656. doi: 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac activation dynamics visualized in living cells. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- Laudanna C, Campbell JJ, Butcher EC. Elevation of intracellular cAMP inhibits RhoA activation and integrin-dependent leukocyte adhesion induced by chemoattractants. J Biol Chem. 1997;272:24141–24144. doi: 10.1074/jbc.272.39.24141. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Teillet MA. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J Embryol Exp Morphol. 1973;30:31–48. [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Moore MW, Klein RD, Fariñas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- Natarajan D, Marcos-Gutierrez C, Pachnis V, de Graaff E. Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development. 2002;129:5151–5160. doi: 10.1242/dev.129.22.5151. [DOI] [PubMed] [Google Scholar]
- Ono K, Tsujimoto G, Sakamoto A, Eto K, Masaki T, Ozaki Y, Satake M. Endothelin-A receptor mediates cardiac inhibition by regulating calcium and potassium currents. Nature. 1994;370:301–304. doi: 10.1038/370301a0. [DOI] [PubMed] [Google Scholar]
- Rupp PA, Kulesa PM. A role for RhoA in the two-phase migratory pattern of post-otic neural crest cells. Dev Biol. 2007;311:159–171. doi: 10.1016/j.ydbio.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Sato-Jin K, Nishimura EK, Akasaka E, Huber W, Nakano H, Miller A, Du J, Wu M, Hanada K, Sawamura D, Fisher DE, Imokawa G. Epistatic connections between microphthalmia-associated transcription factor and endothelin signaling in Waardenburg syndrome and other pigmentary disorders. FASEB J. 2008;22:1155–1168. doi: 10.1096/fj.07-9080com. [DOI] [PubMed] [Google Scholar]
- Sokolovsky M. Endothelin receptor heterogeneity, G-proteins, and signaling via cAMP and cGMP cascades. Cell Mol Neurobiol. 1995;15:561–571. doi: 10.1007/BF02071317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AL, Young HM, Popoff M, Anderson RB. Effects of pharmacological inhibition of small GTPases on axon extension and migration of enteric neural crest-derived cells. Dev Biol. 2007;307:92–104. doi: 10.1016/j.ydbio.2007.04.024. [DOI] [PubMed] [Google Scholar]
- Sumiyama K, Kawakami K, Yagita K. A simple and highly efficient transgenesis method in mice with the Tol2 transposon system and cytoplasmic microinjection. Genomics. 2010;95:306–311. doi: 10.1016/j.ygeno.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Uesaka T, Enomoto H. Neural precursor death is central to the pathogenesis of intestinal aganglionosis in Ret hypomorphic mice. J Neurosci. 2010;30:5211–5218. doi: 10.1523/JNEUROSCI.6244-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesaka T, Jain S, Yonemura S, Uchiyama Y, Milbrandt J, Enomoto H. Conditional ablation of GFRalpha1 in postmigratory enteric neurons triggers unconventional neuronal death in the colon and causes a Hirschsprung's disease phenotype. Development. 2007;134:2171–2181. doi: 10.1242/dev.001388. [DOI] [PubMed] [Google Scholar]
- Wang X, He L, Wu YI, Hahn KM, Montell DJ. Light-mediated activation reveals a key role for Rac in collective guidance of cell movement in vivo. Nat Cell Biol. 2010;12:591–597. doi: 10.1038/ncb2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Shinotsuka N, Nonomura K, Takemoto K, Kuida K, Yosida H, Miura M. Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. J Cell Biol. 2011;195:1047–1060. doi: 10.1083/jcb.201104057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yntema CL, Hammond WS. The origin of intrinsic ganglia of trunk viscera from vagal neural crest in the chick embryo. J Comp Neurol. 1954;101:515–541. doi: 10.1002/cne.901010212. [DOI] [PubMed] [Google Scholar]
- Young HM. Functional development of the enteric nervous system—from migration to motility. Neurogastroenterol Motil 20 Suppl. 2008;1:20–31. doi: 10.1111/j.1365-2982.2008.01098.x. [DOI] [PubMed] [Google Scholar]
- Young HM, Newgreen D. Enteric neural crest-derived cells: origin, identification, migration, and differentiation. Anat Rec. 2001;262:1–15. doi: 10.1002/1097-0185(20010101)262:1<1::AID-AR1006>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Young HM, Bergner AJ, Anderson RB, Enomoto H, Milbrandt J, Newgreen DF, Whitington PM. Dynamics of neural crest-derived cell migration in the embryonic mouse gut. Dev Biol. 2004;270:455–473. doi: 10.1016/j.ydbio.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Young HM, Turner KN, Bergner AJ. The location and phenotype of proliferating neural-crest-derived cells in the developing mouse gut. Cell Tissue Res. 2005;320:1–9. doi: 10.1007/s00441-004-1057-5. [DOI] [PubMed] [Google Scholar]
- Zhao T, Nalbant P, Hoshino M, Dong X, Wu D, Bokoch GM. Signaling requirements for translocation of P-Rex1, a key Rac2 exchange factor involved in chemoattractant-stimulated human neutrophil function. J Leukoc Biol. 2007;81:1127–1136. doi: 10.1189/jlb.0406251. [DOI] [PubMed] [Google Scholar]