

Abstract
Huntingtin (HTT), the protein mutated in Huntington's disease (HD), controls transport of the neurotrophin, brain-derived neurotrophic factor (BDNF), within corticostriatal neurons. Transport and delivery of BDNF to the striatum are reduced in disease, which contributes to striatal neuron degeneration. BDNF released by cortical neurons activates TrkB receptors at striatal dendrites to promote striatum survival. However, it remains to be determined whether transport of TrkB, the BDNF receptor, depends on HTT and whether such transport is altered in mutant situation. Here we show that TrkB binds to and colocalizes with HTT and dynein. Silencing HTT reduces vesicular transport of TrkB in striatal neurons. In HD, the polyQ expansion in HTT alters the binding of TrkB-containing vesicles to microtubules and reduces transport. Using a combination of microfluidic devices that isolate dendrites from cell bodies and BDNF coupled to quantum dots, we selectively analyzed TrkB retrograde transport in response to BDNF stimulation at dendrite terminals. We show that the retrograde transport of TrkB vesicles within striatal dendrites and the BDNF/TrkB-induced signaling through ERK phosphorylation and c-fos induction are decreased in neurons from an HD mouse model. Together, our findings demonstrate that HTT is a crucial regulator of TrkB trafficking. Transport defects in HD are not restricted to BDNF transport in cortical neurons but also affect trafficking of its ligand-bound receptor in the striatal neurons. This transport alteration may further impair BDNF-TrkB survival signaling within the corticostriatal connection that is most affected in HD.
Introduction
The neurodegenerative Huntington's disease (HD) is caused by an abnormal polyglutamine (polyQ) expansion in huntingtin protein (HTT). HD is characterized by the loss of striatal and cortical neurons. The exact pathogenic mechanisms remain elusive but involve transcriptional deregulation, excitotoxicity, energy metabolism deficits, and mitochondrial injury (Borrell-Pages et al., 2006a). Increasing evidence indicates that vesicular transport defects, especially of BDNF, play a key role in HD (Gunawardena et al., 2003; Szebenyi et al., 2003; Gauthier et al., 2004; Trushina et al., 2004; Her and Goldstein, 2008) as BDNF is not produced by striatal neurons but is synthesized in the cortex and transported to the striatum by corticostriatal projecting neurons (Altar et al., 1997). Decreasing BDNF cortical production results in striatal degeneration in health and HD (Altar et al., 1997; Ivkovic and Ehrlich, 1999; Baquet et al., 2004; Canals et al., 2004; Rauskolb et al., 2010; Bosse et al., 2012). Conversely, restoring axonal transport or secretion of BDNF increases BDNF release and neuronal survival (Borrell-Pages et al., 2006b; Dompierre et al., 2007; Gharami et al., 2008; Zala et al., 2008; Pineda et al., 2009).
BDNF is a member of the neurotrophin family that binds to TrkB receptors leading to their dimerization and internalization into endosomes (Cosker et al., 2008). TrkB-positive endosomes undergo dynein-dependent retrograde transport along microtubules (MTs) to the cell body where TrkB induces survival signals (Watson et al., 1999; Yano et al., 2001; Heerssen et al., 2004; Valdez et al., 2005; Deinhardt et al., 2006; Ha et al., 2008). However, retrograde transport of Trk receptors in response to trophic factors has been studied primarily in axons. Compartmentalized culture chambers, such as Campenot chambers (Campenot, 1977), or more recently microfluidic chambers (Taylor et al., 2005) allow to separate axons from cell bodies and allowed the study of axonal specific events (Cosker et al., 2008). In contrast, physical separation of dendrites from cell bodies is technically more challenging. Only recently have some of the signaling pathways elicited in dendrites upon BDNF treatment been described (Cohen et al., 2011). Therefore, the mechanisms responsible for retrograde transport of TrkB receptors from postsynaptic sites into the cell body remain largely unknown.
In HD, dendritic retrograde transport of TrkB receptors could be of particular importance as TrkB receptor activation participates in the development and survival of striatal neurons, the most severely affected neurons. Selectively invalidating TrkB receptors in striatum leads to loss of striatal neurons in mouse brains (Baydyuk et al., 2011). In HD, TrkB receptor density may be low and its signaling impaired (Gines et al., 2006; Zuccato et al., 2008; Gines et al., 2010). However, the primary mechanisms responsible for this reduced signaling remain to be elucidated. No studies have investigated TrkB receptor dynamics in HD striatal dendrites. Given the role of HTT in MT-dependent transport in axons, we selectively analyzed its role in the retrograde transport and signaling of TrkB receptors within striatal dendrites using complementary approaches, such as TrkB receptor imaging, microfluidic devices to selectively isolate dendrites, and BDNF-coupled quantum dots.
Materials and Methods
Reagents and antibodies.
The following antibodies were used: mouse anti-dynein (1:1000 for Western blotting and 1:100 for immunocytochemistry; Millipore Bioscience Research Reagents), mouse anti-HTT (1:4000 for Western blotting; mAb 4C8, Euromedex), rabbit anti-HTT (1:1000 for Western blotting; D7F7 mAb #5656, Cell Signaling Technology), mouse Anti-TrkB (1:500 for Western blotting; Transduction Laboratories), rabbit anti-TrkB (1:100 for immunocytochemistry; Santa Cruz Biotechnology), mouse anti-α-tubulin (1:3000 for Western blotting and 1:500 for immunocytochemistry; Sigma), mouse anti-GFP (1:1000 for Western blotting; Roche), rabbit anti-DsRed (1:1000 for Western blotting; Clontech), rabbit anti-phospho-ERK1/2 (1:100 for immunocytochemistry; Cell Signaling Technology), rabbit anti-c-fos (1:200 for immunocytochemistry; Cell Signaling Technology), and rabbit anti-phospho-CREB (1:100 for immunochemistry; Cell Signaling Technology). The anti-rabbit or anti-mouse HRP-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories. Alexa-conjugated secondary antibodies were purchased from Invitrogen. The following plasmids were previously described elsewhere: TrkB-RFP, TrkB-GFP (Watson et al., 1999), dynein-IC1B-RFP and dynein-IC2C-RFP (Ha et al., 2008), and MAP2-GFP (Kaech and Banker, 2006). Streptavidin-quantum dots-705 were purchased from Invitrogen. Amine-PEG3-biotin, EDC (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) are from Pierce. siRNA are from Eurogentec, and shRNA-HTT (AGCTTTGATGGATTCTAA-TCTCTTGAA-ATTAGAATCCATCAAAGCT) and shRNA-Luciferase (CGTACGCGGAATACTTCGA-TGACAGGAAG-TCGAAGTATTCCGCGTACG) plasmids contain a separate expression cassette encoding the GFP reporter gene under the control of the mouse PGK promoter (Drouet et al., 2009). All other reagents were obtained from Sigma.
Cultures and transfection.
Primary striatal neurons were prepared from E17 rat embryos or E15 mouse embryos as previously described (Saudou et al., 1998). Briefly, ganglionic eminence was dissected in cold DM buffer, pH 7.4 (82 mm Na2SO4, 30 mm K2SO4, 5.8 mm MgCl2, 0.25 mm CaCl2, 1 mm HEPES, 20 mm glucose, 0.5% Phenol red) supplemented with kynurenic acid (1 mm) and MgCl2 (10 mm). Tissue digestion was performed with 2 incubations of 10 min in a solution of papain and cysteine followed by 2 incubations of 7 min and 30 s with a solution of trypsin inhibitor. Tissues were then dissociated mechanically in optiMEM-1 (Invitrogen) supplemented with glucose. Neurons in suspension (5 millions) were electroporated with Amaxa Nucleofector kit for rat neurons (Lonza) according to the supplier's manual. Neurons electroporated were plated on glass coverslips coated with poly-l-lysine (1 mg/ml). After 3 h, medium was replaced with fresh Neurobasal supplemented with 2% B27, 2 mm glutamax, 1% penicillin/streptomycin, 10 μm forskolin, and 100 μm IBMX.
Striatal cell lines (STHdh+/+) or their HD mutant counterparts (STHdhQ111/Q111 cells) (Trettel et al., 2000) were cultured in DMEM supplemented with 10% bovine calf serum, 2 mm glutamine, and 0.4 mg/ml G418 (Invitrogen). These cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. HEK cells were transfected with calcium phosphate method. HeLa cells and COS7 cells were transfected with Lipofectamine 2000. These 2 cell lines were cultured in DMEM supplemented with 10% bovine calf serum and 2 mm glutamine. All primary neurons and cell lines were maintained at 37°C in a humidified 5% CO2 atmosphere, except for striatal cell lines, which were kept at 33°C.
TrkB-RFP vesicles and EB3-GFP imaging.
Cells were grown on glass coverslip mounted in a Ludin chamber. TrkB-RFP videomicroscopy experiments on STHdh cells were performed 2–3 d after transfection. For trafficking studies of TrkB-RFP and EB3-GFP on primary neurons, videomicroscopy experiments were performed 6–9 d after electroporation. The microscope and the chamber were kept at 37°C (33°C for striatal cells). Images were recorded every 2 s with a 100× PlanApo NA 1.4 oil-immersion objective coupled to a piezo device (z-step of 0.3 μm). Images were collected in stream mode using a Micromax camera (Ropper Scientific) set at 2 × 2 binning with an exposure time of 100–150 ms. Projections, animations, and analyses were generated with ImageJ software (http://rsb.info.nih.gov/ij/; National Institutes of Health).
Immunocytochemistry.
Striatal rat primary neurons were stimulated with 50 ng/ml BDNF (PreproTech) for 10 min at 37°C and fixed with PFA 4% 20 min at room temperature (RT) and then washed 2 times with PBS. Blocking was performed with PBS + 3% BSA + 0.3% Triton X-100. Primary antibodies were incubated 1 h at RT followed by 3 washes in PBS and incubated with Alexa-conjugated secondary antibodies during 1 h at RT. For vesicle localization on microtubules, HeLa or COS7 cells transfected with TrkB-RFP and wild-type (1301–17Q-YFP) or mutant HTT (1301–73Q-YFP) were washed once with PHEM buffer (PIPES 60 mm, pH 6.9, HEPES 25 mm, EGTA 10 mm, Mg acetate 2 mm). Prelysis was performed by incubating cells with saponin (0.02%) in PHEM buffer for 4 min at 37°C. Cells were then fixed using PFA 4% and glutaraldehyde 0.02% in PHEM buffer (20 min at 37°C). After 2 washes with PBS, a quenching was performed with sodium borohydride 1% in PBS during 10 min at RT. Cells were washed twice with PBS, incubated with PBS + BSA 3% for 30 min, and then with an antibody raised against α-tubulin (1 h at RT). Cells were washed three times with PBS and incubated for 1 h at RT with the secondary antibody coupled to Alexa-488. For phospho-CREB immunostaining, neurons were stimulated with 100 ng/ml BDNF or biotinylated BDNF for 10 min at 37°C and then fixed in PFA 4% during 20 min at RT. After 2 washes with PBS, blocking was performed with PBS + 3% BSA + 0.1% Triton X-100. Primary antibody was incubated 1 h at RT followed by 3 washes in PBS, and cells were incubated with Alexa-555-conjugated secondary antibodies during 1 h at RT.
Western blotting.
Cells were homogenized in lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% NP40, 1:100 protease inhibitor mixture, 1:50 phosphate inhibitor mixture, 1 mm PMSF) on ice. After a 10 min centrifugation at 15,000 × g at 4°C, supernatants were collected and protein concentration was assessed with the BCA kit (Pierce). A total of 20 μg of total proteins was denaturated 5 min at 95°C in SDS loading buffer and loaded on a 8% SDS-acrylamide/bis-acrylamide gel. Proteins were transferred on a nitrocellulose membrane. Membranes were blocked in TBS buffer (10 mm Tris, 200 mm NaCl) + 0.1% Tween 20 + 5% nonfat dry milk for 30 min at RT. Primary antibodies were prepared in TBS-0.1% Tween 20 + 5% nonfat dry milk, and membranes were incubated 2 h at RT. After 3 washes with TBS + 0.1% Tween 20, membranes were incubated with HRP-conjugated secondary antibodies prepared in TBS + 0.1% Tween 20 for 1 h at RT, and washed again 3 times with TBS-Tween 20. Detection of proteins was performed with the ECL chemiluminescent kit (Pierce).
Coimmunoprecipitation.
HEK cells were lysed in lysis buffer on ice. Proteins at the concentration of 1 mg/ml were incubated 1 h at 4°C with a preformed complex: beads protein G Sepharose (25 μl) + antibody (1 μg). Then beads were washed 3 times with lysis buffer and boiled 5 min at 95°C in SDS loading buffer to be denaturated, and Western blot was performed.
Cell fractionation.
Vesicular fractionation involved the following: cells were scraped in buffer A (150 mm NaCl, 1 mm EGTA, 1 mm MgCl2, 10 mm HEPES, pH 7.4) and centrifuged 5 min at 150 × g. Pellet was lysed in buffer A supplemented with protease inhibitor mixture (1:100), phosphatase inhibitor mixture (1:50), and PMSF (1 mm). A first 10 min centrifugation at 1000 × g produced the P1 and S1 fractions. S1 fraction was centrifuged 30 min at 12,000 × g to obtain P2 and S2 fractions. S2 fraction was centrifuged 2 h 30 min at 100,000 × g to give P3, the “small vesicles” fraction, and S3, the cytosolic fraction. Pellets were lysed in buffer A supplemented with 1% NP40. All samples were sonicated.
Microtubule fractionation.
Cells were treated with 1 μm taxol for 1 h at 37°C before being scraped in PBS. Cells were centrifuged 5 min at 1000 × g, and the pellet was dissociated in PM2G buffer (0.1 m PIPES, pH 6.9, 2 m glycerol, 5 mm MgCl2, 2 mm EGTA). An equivalent volume of PM2G supplemented with 0.4% NP40 was added to the cell pellet and incubated at 37°C during 15 min. A 5 min centrifugation at 1000 × g at RT was performed. Supernatant was centrifuged at 10,000 × g at RT during 30 min. Pellet was washed with PM2G buffer and resuspended in SDS buffer (31 mm Tris, pH 6.8, 1% SDS, 5% glycerol) and heated at 95°C during 5 min, whereas supernatant was diluted in SDS buffer 2×. All samples were sonicated.
Mice were killed by cervical dislocation. Brains were removed, and striata were quickly dissected. All the following procedures were performed at temperatures <4°C. Tissues were homogenized with glass douncer and then lysed by passing the samples 10 times in a 15-G syringe in ice-cold BRB80 buffer (PIPES 80 mm, MgCl2 1 mm, and EGTA 1 mm) containing a mixture of protease inhibitor and PMSF 1 mm. Samples were clarified at 10,000 × g for 30 min at 4°C. Supernatant (total) was incubated at 37°C with 1 mm GTP for 15 min, and then taxol 50 μm was added to stabilized microtubules for 15 min at 37°C. Samples were centrifuged at 10,000 × g for 30 min at 30°C, and supernatant and pellet-containing microtubules and associated proteins were collected. Pellet was resuspended in one-fifth of the supernatant volume.
BDNF biotinylation.
BDNF (PreproTech) was prepared in MES buffer (100 mm, pH 5.5) and mixed with amine-PEG3-biotin (50 mm) and EDC (100 mm) during 2 h at RT. Next, biotinylated BDNF was dialyzed against PBS to exchange MES buffer and remove unbound biotin.
Microchambers and neuron plating.
Fabrication of the polydimethylsiloxane microfluidic devices with 100 μm microgrooves was adapted from Taylor et al. (2005). Master was made with SU-8 resin on silicon wafer using photolithography. Fabrication was done in 2 steps: a first layer with SU-8 2002 for the microchannels (3 μm high) being crossed by neuronal processes and a second layer with SU-8 3050 for the chamber (50 μm high) where cell bodies are seeded. Silicon wafers were placed in a Petri dish, and a mixture of silicon elastomer (PDMS, Sylgard 184, Dow Corning) and its curing agent (ratio 10:1) was poured on the masters. This PDMS piece served as negative mold to replicate the master with epoxy resin. Silicon or epoxy masters were filled with the PDMS mixture. Air bubbles were removed by incubation in a dessicator under vacuum during 1 h, and polymerization was performed by incubating 3 h at 60°C. Finalized PDMS microchambers were cut and washed with ethanol and distilled water and placed, with Iwaki boxes with a glass bottom coverslip (27 mm diameter), into a plasma cleaner under vacuum during 30 s for surface activation. After a rapid passage in an oven at 60°C, PDMS pieces and Iwaki boxes were brought together to form an irreversible tight seal. The microfluidic devices were coated with a mixture of poly-d-lysin (0.5 mg/ml) + laminin (10 μg/ml) and incubated overnight at 4°C and then washed 3 times with Neurobasal medium. One million neurons were plated in the upper chambers in Neurobasal supplemented with 2% B27, 1% Glutamax, and 1% penicillin/streptomycin. After 1 week, neuronal processes crossed the microchannels, dendrites were submitted to a short exposure (2 min) to biotinylated BDNF, paying attention to have a difference in volume between compartments to maintain a pressure and avoid BDNF diffusing in the cell body compartment. Neurons were quickly washed with PBS, fixed at RT with 3.7% formaldehyde + 5% sucrose in PBS (30 min), and washed again with PBS. After permeabilization with PBS + 0.1% Triton X-100, cells were incubated with quantum dot-705 at 20 nm in PBS during 30 min at RT followed by 3 washes with PBS. Quantum dots showed no or limited binding to BDNF-untreated neurons in the microfluidic device (see Fig. 6D). Acquisition of images was done on confocal SP5 microscope (Leica). For signaling experiments, one-week old striatal dendrites of wild-type (Hdh+/+) and mutant (HdhQ111/+ or HdhQ111/Q111) neurons were stimulated with BDNF (PreproTech) during 10 min. Neurons were next fixed and immunostained as described above. We first counted immunopositive neurons for phospho-ERK or c-fos that correspond to those activated in response to BDNF. This number ranges from 700 to 1500 for phospho-ERK and from 400 to 1100 for c-fos. We normalized these numbers to the number of dendrites crossing the 100 μm channels (an average of 1100). We next compared the number of neurons activated by BDNF versus control and then calculated the fold increase in BDNF-induced phospho-ERK activation in Hdh+/+ and HdhQ111/+ or HdhQ111/Q111 neurons. Both HdhQ111/+ and HdhQ111/Q111 neurons showed the same defect in their signaling responses to BDNF for c-fos and phospho-ERK. These findings are in agreement with the fact that HD is a pure dominant disorder. Values were pooled and data referred as HdhQ111/Q111 in Figure 7. The neurite outgrowth and viability of Hdh+/+ and HdhQ111/Q111 neurons in microchambers were similar in the experiments analyzing TrkB retrograde transport (see Figs. 5 and 6) and signaling (see Fig. 7). A total of 6 and 4 independent experiments were performed for phospho-ERK and c-fos analyses, respectively.
Figure 6.
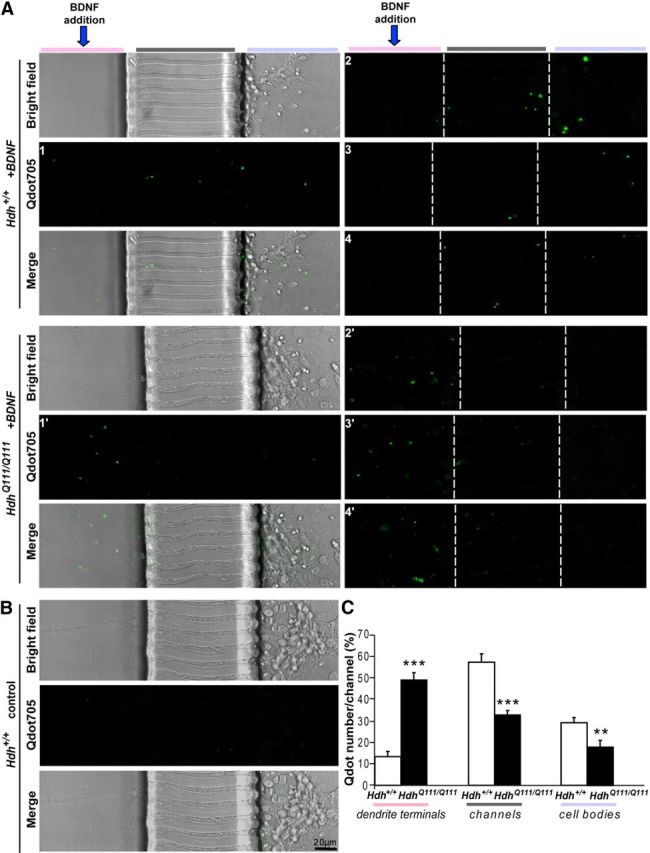
Mutant polyQ huntingtin specifically alters retrograde transport of TrkB vesicles in striatal dendrites. A, Representative images (4 different images of the Qdot705 channel): A1–A4, for Hdh+/+. A1′–A4′, for HdhQ111/Q111. Images show the position of Qdots within the microfluidic devices after 2 min incubation of the primary striatal neurons from Hdh+/+ (top) or HdhQ111/Q111 (bottom) mice with biotinylated BDNF. Left, Microchannels in bright field and merge. The other representative images only show the Qdot positions. The Qdots are mostly located in the channels and cell body compartment for wild-type cells, whereas they are mostly in the dendrite compartment in mutant situation. B, Only very few Qdots could be detected in chambers containing dendrites that were not treated with BDNF. C, Quantification of >200 microchannels reveals a significant reduction in the number of TrkB-containing vesicles that crossed the channels to reach the cell bodies of striatal neurons in HD neurons. Error bars indicate SEM. **p < 0.01 (unpaired, one-tailed t test). ***p < 0.001 (unpaired, one-tailed t test).
Figure 7.
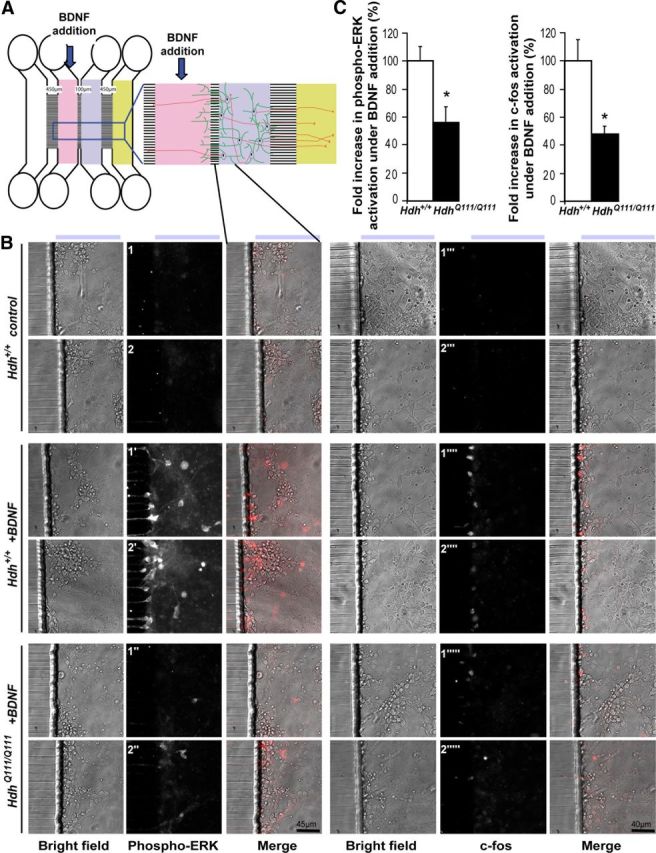
Mutant polyQ huntingtin alters BDNF-TrkB-induced signaling in striatal neurons. A, Scheme of the microfluidic device. B, Representative images showing the compartment of the microfluidic device shown in A after 10 min (for phospho-ERK experiment) or 1 h (for c-fos experiment) incubation of primary striatal neurons from Hdh+/+ (middle) or HdhQ111/Q111 (bottom) mice with BDNF. Immunostainings of neurons with anti-phospho-ERK (left) or anti-c-fos (right) antibodies were performed in control condition (Hdh+/+ neurons, no treatment, top), BDNF-treated Hdh+/+ neurons (middle), or BDNF-treated HdhQ111/Q111 neurons (bottom). Two representative images are presented for each condition. C, Quantification of the fold increases in phospho-ERK (n = 6) or c-fos (n = 4) activation after BDNF addition reveals a significant decrease in both phospho-ERK and c-fos in neurons from HdhQ111/Q111 mutant mice. Error bars indicate SEM. *p < 0.05 (unpaired, one-tailed t test).
Figure 5.
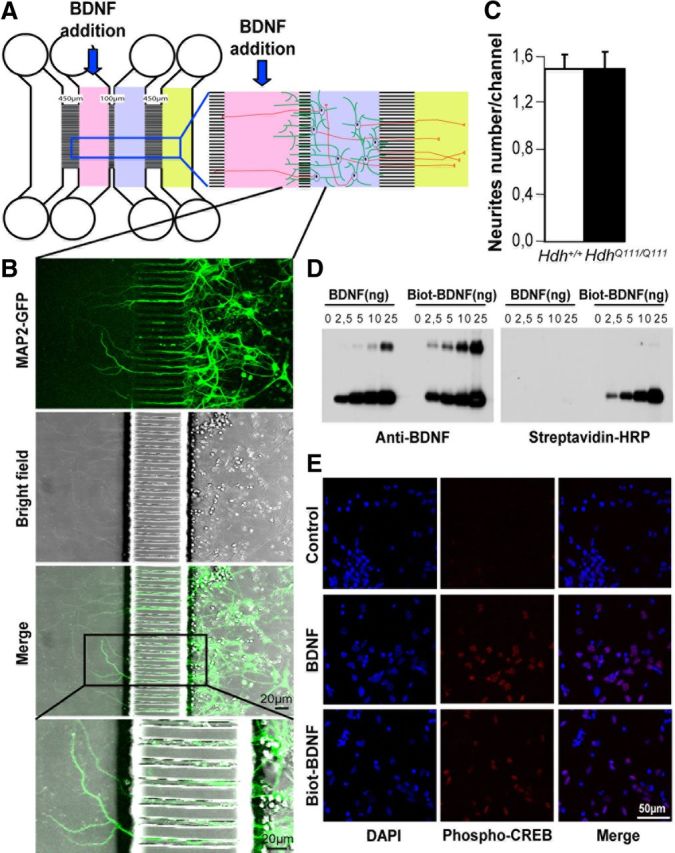
Microfluidic devices to separate dendrites from cell bodies. A, Scheme of the microfluidic device allowing to physically separate dendrites (green processes) from axons (red processes). B, Rat primary striatal neurons were electroporated with MAP2-GFP and plated in the cell body compartment (A, blue compartment) of the microfluidic device. Most of the 100 μm microchannels are crossed by striatal dendrites that extend to the distal compartment (pink). C, Quantification 1 week after plating of neurites from Hdh+/+ and HdhQ111/Q111 striatal neurons revealed no difference in neurite outgrowth. D, Western blotting of BDNF and biotinylated BDNF using an anti-BDNF antibody or a streptavidin-HRP solution shows that BDNF is efficiently biotinylated. E, Rat primary striatal neurons untreated (control) or treated with BDNF or with biotinylated BDNF were stained with an antibody directed against phospho-CREB (in red) and counterstained with DAPI (blue). The staining shows that biotinylated BDNF is biologically active.
Mice.
HdhQ111 mice were previously described (Wheeler et al., 2002). Animals were maintained with access to food and water ad libitum in a colony room kept at a constant temperature (19°C-22°C) and humidity (40–50%) on a 12:12 h light/dark cycle. All experimental procedures were performed in strict accordance with the recommendations of the European Community (86/609/EEC) and the French National Committee (87/848) for care and use of laboratory animals. Experiments in Figures 3D and 4E, F were performed on males.
Figure 3.
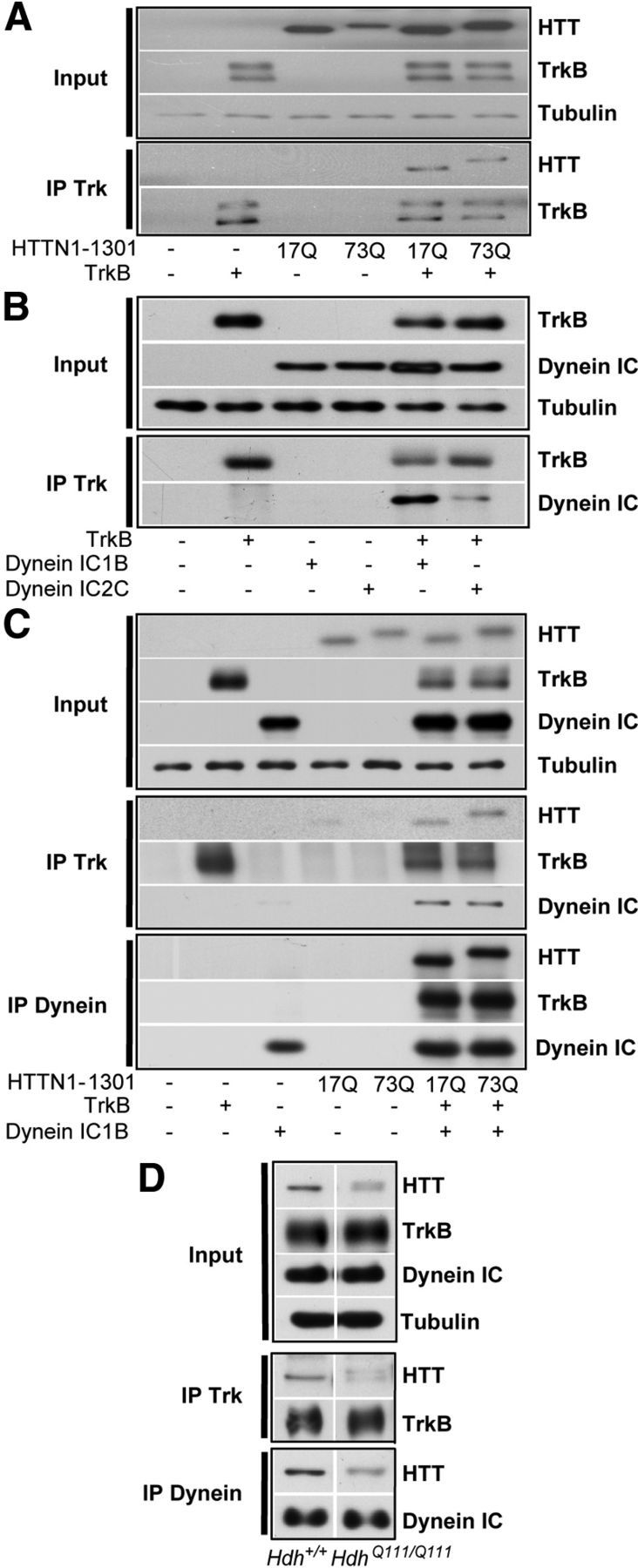
Huntingtin interacts with dynein and TrkB. A, Coimmunoprecipitation of HTT with TrkB from HEK cells transfected with TrkB and HTTN-1301–17Q or HTTN-1301–73Q show no interaction differences upon polyQ expansion in HTT. B, Immunoprecipitation of TrkB from HEK cells transfected with TrkB and dynein IC-1B or IC-2C reveals a preferential interaction of TrkB with dynein IC-1B compared with dynein IC-2C. C, HEK cells were transfected with TrkB, dynein IC-1B, and HTTN-1301–17Q or HTTN-1301–73Q. Anti-TrkB or dynein antibodies specifically coimmunoprecipitate wild-type and mutant HTT with the same efficacy. D, Trk or dynein immunoprecipitations were performed from Hdh+/+ and HdhQ111/Q111 mouse brain extracts. HD mutation does not affect HTT coimmunoprecipitation.
Figure 4.
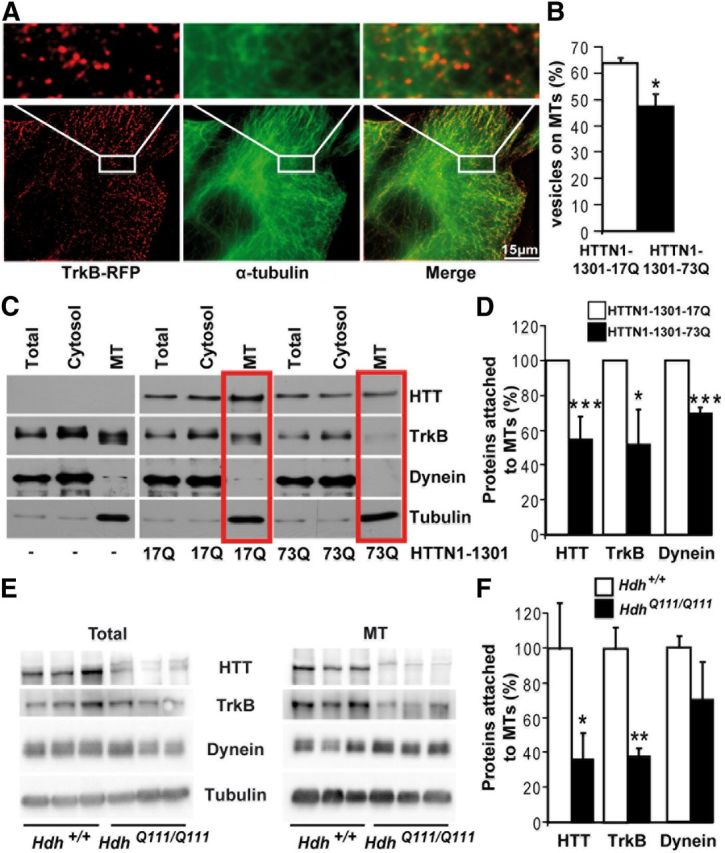
Mutant polyQ huntingtin disrupts TrkB vesicle attachment to microtubules. A, Immunostaining of Cos7 cells expressing TrkB-RFP (red) for α-tubulin (green) reveals the localization of TrkB on MTs. B, Quantification of TrkB vesicles on MTs in three independent experiments (HTTN1–1301-17Q, n = 33 cells; HTTN1–1301-73Q, n = 38 cells) shows a reduction of TrkB localization on MTs upon abnormal polyQ expansion in HTT. C, MT fractionation of cells expressing either HTTN-1301–17Q or HTTN-1301–73Q reveals a reduced association of HTT, TrkB, and dynein in pathological conditions with quantification (n = 5) in D. E, MT fractionation of Hdh+/+ and HdhQ111/Q111 mouse brain extracts shows a decreased association of TrkB and HTT to MTs in mutant situation with quantification of three independent experiments in F. Error bars indicate SEM. *p < 0.05 (unpaired, one-tailed t test). **p < 0.01 (unpaired, one-tailed t test). ***p < 0.001 (unpaired, one-tailed t test).
Statistics.
Statview 4.5 software (SAS Institute) was used for statistical analysis. Groups were compared by ANOVA followed by Fisher's PLSD post hoc analyses. The criterion for statistical significance was set at p < 0.05. Error bars indicate SEM.
Results
Huntingtin facilitates TrkB vesicular transport
To investigate whether HTT is involved in the retrograde transport of TrkB-containing vesicles, we used TrkB fused to RFP (Fig. 1A). As a rationale for this investigation, we immunostained primary cultures of rat striatal neurons with antibodies against HTT, TrkB, and dynein (dynein IC). Dynein is the main molecular motor that retrogradely transports TrkB (Yano et al., 2001; Bhattacharyya et al., 2002; Heerssen et al., 2004; Ha et al., 2008). We analyzed their colocalization in neurites by spinning disk confocal microscopy and z-axis projection. We observed a significant colocalization of HTT with TrkB along neurites (Fig. 1B). As expected, we also observed a strong colocalization of TrkB with dynein along neurites. Our findings agree with the reported interactions of HTT with dynein (Caviston et al., 2007) and their colocalization in neurites of striatal neurons (data not shown).
Figure 1.
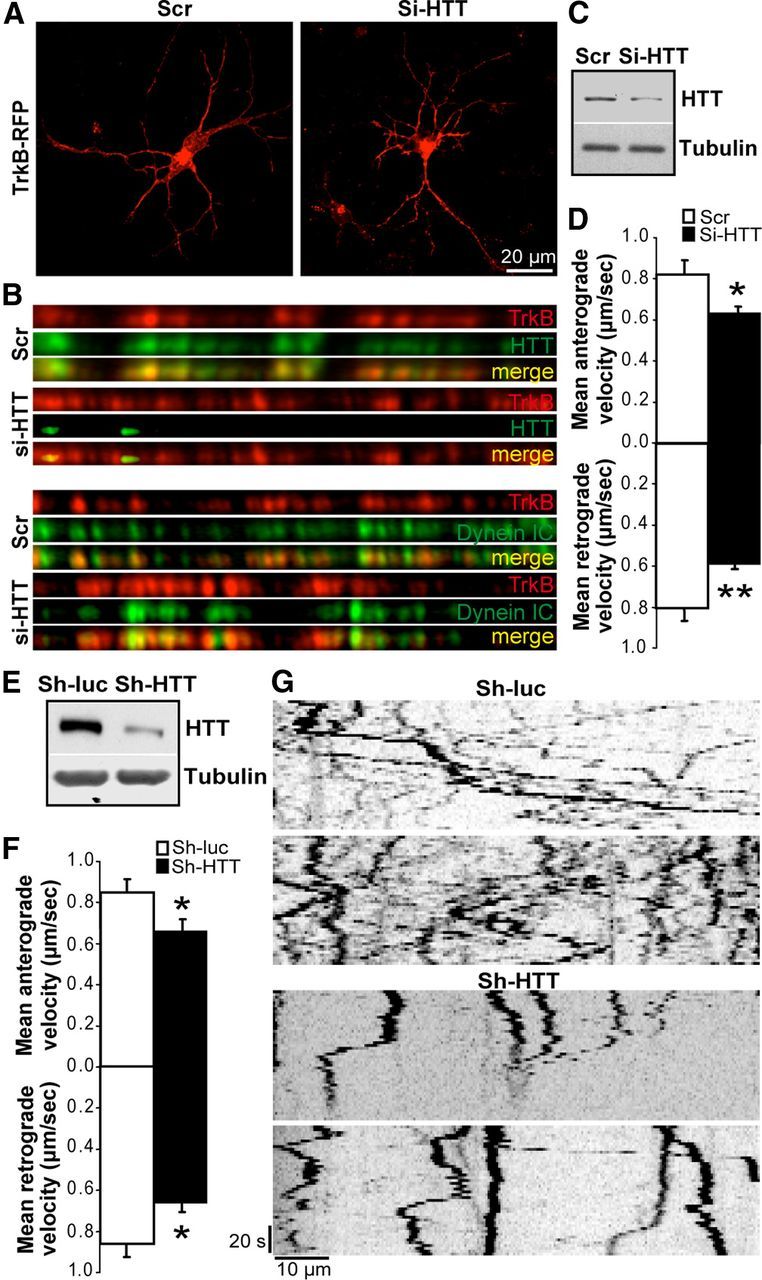
TrkB transport depends on huntingtin. A, Morphology and survival of striatal neurons are not altered by TrkB-RFP expression or HTT silencing (Si-HTT, right) compared with scramble situation (Scr, left). B, Immunostainings of TrkB and HTT (top) or dynein intermediate chain (Dynein IC, bottom) in neurites of rat primary striatal neurons reveal no marked difference of TrkB-DIC colocalization upon HTT silencing (Si-HTT) compared with the Scramble situation. C, Silencing of HTT was verified by Western blot. D, Anterograde and retrograde velocities were determined by videomicroscopy and were significantly reduced by Si-HTT. E–G, Rat primary striatal neurons were electroporated with TrkB-RFP and an anti-luciferase shRNA (Sh-luc) or anti-htt shRNA (Sh-HTT). E, Silencing of HTT was verified by Western blot. F, Anterograde and retrograde velocities were significantly reduced by Sh-HTT as determined by videomicroscopy. G, Representative independent kymographs show a reduced movement of TrkB particles upon Sh-HTT electroporation. Error bars indicate SEM. *p < 0.05 (unpaired, one-tailed t test). **p < 0.01 (unpaired, one-tailed t test).
We next electroporated primary striatal neurons with a TrkB-RFP and GFP expression vectors and either an anti-HTT siRNA (Si-HTT) or an siRNA with a scrambled sequence (Scr). At the time of the videomicroscopy experiments, we did not find any toxic effect of TrkB overexpression (percentage of healthy GFP neurons: pcDNA, 95.3 ± 1.6, n = 276; TrkB-RFP, 93.8 ± 3.0, n = 194 in 2 independent experiments) or of the Scr and Si-HTT siRNAs (Zala et al., 2008), and we did not observe a significant effect of Si-HTT on neuronal morphology (Fig. 1A). For each videomicroscopy experiment, we performed immunoblotting on electroporated striatal neuron extracts to verify that HTT protein levels were reduced by the Si-HTT (Fig. 1C). Quantification of four independent experiments revealed a significant reduction in HTT levels within the whole neuronal extract of 39.30 ± 8.14%. Next, we measured TrkB vesicular velocity in the anterograde and retrograde directions using fast videomicroscopy as described previously (Colin et al., 2008; Zala et al., 2008). The velocities of the TrkB-containing vesicles were similar to previously published values (Ha et al., 2008). The anterograde and retrograde velocities of TrkB-positive vesicle were significantly reduced in neurons silenced for HTT compared with neurons treated with the Scr RNA (Fig. 1D). To control for any possible off-target effects, we extended the experiment by coelectroporating the TrkB-RFP plasmid with a plasmid that coexpressed GFP and an anti-HTT shRNA (Sh-HTT) that targeted another region of the HTT mRNA sequence or a shRNA control targeting luciferase (sh-luc). HTT protein levels were decreased by the Sh-HTT as shown by immunoblotting of the whole neuronal extract (Fig. 1E) and the quantification of 3 independent experiments (28.52 ± 8.49%). We next specifically measured TrkB trafficking in the sh-targeted neurons as they are GFP-positive. The silencing of HTT induced a significant reduction in TrkB dynamics as shown for two representative kymographs in both control and Sh-HTT conditions (Fig. 1G). As with Si-HTT, the mean velocities of both anterograde and retrograde vesicle transport decreased in Sh-HTT-expressing neurons compared with control neurons expressing an anti-Luciferase shRNA (Sh-Luc) (Fig. 1F). Our findings that reducing HTT levels decreases, but does not completely abolish, transport are in agreement with previous experiments (Gauthier et al., 2004; Colin et al., 2008; Zala et al., 2008). It agrees with the observation that HTT is a facilitator/regulator of axonal transport but is not absolutely required for transport, unlike molecular motors, such as dynein or kinesin, whose inhibition leads to an almost complete blockade of transport (Colin et al., 2008; Caviston and Holzbaur, 2009; Hirokawa et al., 2010). To start investigating the mechanism that leads to reduced transport, we analyzed the consequences of silencing HTT on the colocalization of TrkB with dynein. Although silencing HTT in neurons was effective as shown by the almost complete absence of HTT within neurites (Fig. 1B, top), we found no significant loss of TrkB colocalization with dynein. These findings are consistent with HTT facilitating the activity of dynein but not regulating its attachment to vesicles (Gauthier et al., 2004; Caviston et al., 2007; Colin et al., 2008; Zala et al., 2008). Together, these results suggest a role for wild-type HTT in TrkB vesicular transport via a dynein-dependent mechanism.
Dendritic TrkB retrograde transport is altered in an HD model
We next investigated the consequences of the polyQ expansion in the HTT protein on TrkB trafficking. We first used immortalized striatal cell lines prepared from embryonic striatal neurons dissected from either wild-type mice or knock-in mice harboring an Htt gene that encoded a polyQ tract of 111 glutamines (Wheeler et al., 2002). We expressed TrkB-RFP in the wild-type striatal cells (STHdh+/+) and in their HD mutant counterparts (referred as STHdhQ111/Q111 cells) (Trettel et al., 2000) and followed TrkB vesicles by videomicroscopy. TrkB vesicular dynamics were significantly altered in the HD mutant cells as vesicles exhibited a lower mean velocity in these cells compared with that in wild-type cells (Fig. 2A).
Figure 2.
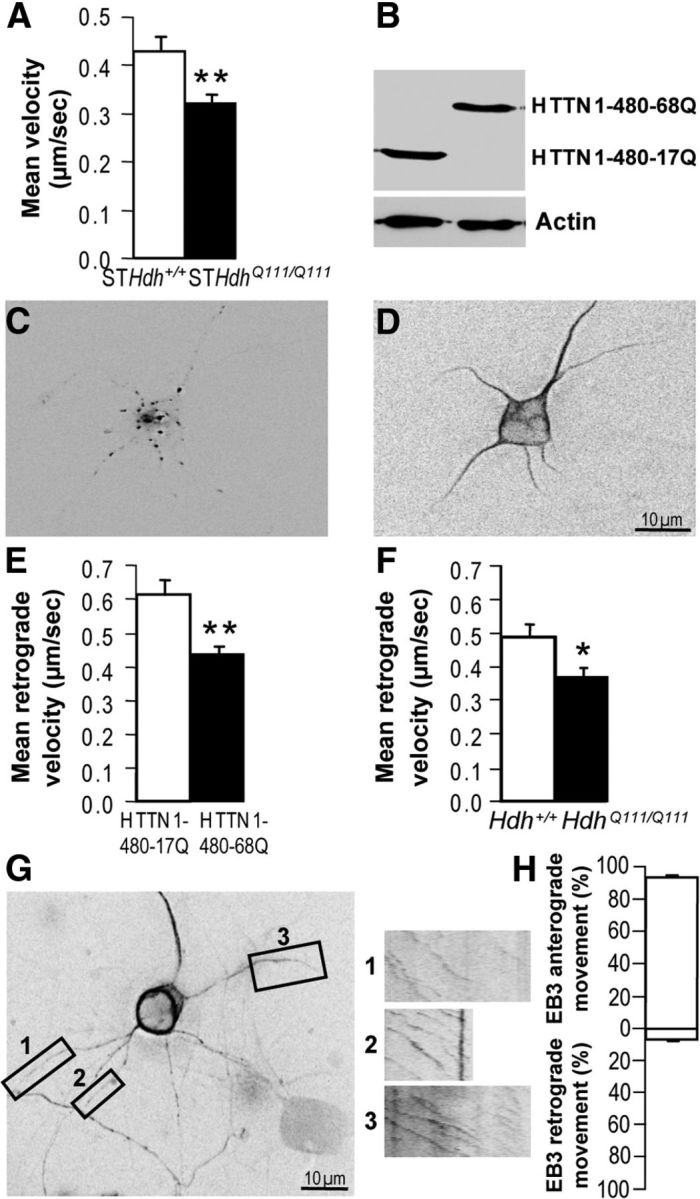
TrkB retrograde transport is altered in mutant HTT expressing neurons. A, The mean velocity of TrkB vesicles is reduced in immortalized mutant (STHdhQ111/Q111) cells compared with wild-type (STHdh+/+) cells as determined by videomicroscopy. B, Western blotting analyses show that HTTN1–480-17Q and HTTN1–480-68Q constructs are expressed at similar levels in primary striatal neurons. C, D, Fluorescence image of a rat primary striatal neuron expressing TrkB-RFP (C) and MAP2-GFP (D). E, Compared with HTTN1–480-17Q, HTTN1–480-68Q induces a reduction in the retrograde velocity of TrkB vesicles in dendrites of rat primary striatal neurons electroporated with TrkB-RFP and MAP2-GFP. F, Retrograde velocity of TrkB-RFP vesicles in dendrites of mouse primary striatal neurons from HdhQ111/Q111 mice is reduced compared with neurons from Hdh+/+ mice. G, Fluorescence image of a rat primary striatal neuron expressing EB3-GFP and kymographs of EB3-GFP movements in three distal dendrites are depicted on boxes 1–3. H, EB3 particle movements are mostly toward dendrite tips as shown by quantification of the movement of EB3 particles toward dendrite tips (anterograde) or toward the cell bodies (retrograde). Error bars indicate SEM. *p < 0.05 (unpaired, one-tailed t test). **p < 0.01 (unpaired, one-tailed t test).
To further investigate TrkB trafficking in HD pathological situation, we used primary cultures of rat striatal neurons that were coelectroporated with the TrkB-RFP plasmid (Fig. 2C) and either a wild-type or a mutant polyQ-HTT expression construct encoding the first 480 N-terminal amino acids of HTT (HTTN1–480-17Q or HTTN1–480-68Q, respectively) containing the polyQ expansion. This fragment is sufficient for axonal transport in the wild-type situation, and transport is altered when this N-terminal fragment contains the abnormal polyQ expansion (Colin et al., 2008; Zala et al., 2008). Both constructs were expressed at the same levels as shown by Western blotting analysis (Fig. 2B). To selectively assess retrograde transport in dendrites, we also included a plasmid expressing MAP2-GFP (Fig. 2D). The mean retrograde velocity of TrkB-containing vesicles was significantly lower (Fig. 2E) in dendrites from neurons expressing the mutant HTT construct than in dendrites from neurons expressing the wild-type HTT construct.
To unequivocally demonstrate that TrkB vesicle trafficking is altered in HD neurons and to avoid any artifacts caused by the overexpression mutant HTT, we investigated TrkB trafficking in mouse primary striatal cultures from knock-in HdhQ111/Q111 mice (Wheeler et al., 2002). We electroporated these neurons with the TrkB-RFP plasmid and recorded the movement of TrkB vesicles. Again, retrograde transport of TrkB vesicles was significantly lower (Fig. 2F) in neurons from HdhQ111/Q111 mice than in neurons from control mice Hdh+/+. Together, these results demonstrate that TrkB vesicle dynamics were significantly altered in HD striatal dendrites.
As a first step toward understanding the role of molecular motors within this process, we investigated MT polarity within our assay. Within axons, MTs orientate with their plus-end at the nerve terminals. In dendrites, MTs have a mixed polarity in the proximal part of dendrites but show a plus-end polarity toward dendrite terminals in their distal part (Stepanova et al., 2003; Kapitein et al., 2010; Kapitein and Hoogenraad, 2011). To test whether the retrograde transport in striatal distal dendrites depends on dynein, we analyzed MT orientation in our culture condition by transfecting striatal neurons with EB3-GFP, a neuronal plus-end tip protein fused to GFP (Stepanova et al., 2003). As shown in three independent kymographs, we found that EB3 trafficking was orientated toward dendrite terminals (Fig. 2G, boxes 1–3). Quantification of 671 EB3 particles showed that in 95% of the EB3 observed movements, EB3 traffics toward the neurite tip (Fig. 2H). These results suggest that, in our culture conditions, MTs in the distal part of striatal neurons are orientated with their plus-end toward dendrite terminals and that retrograde transport in such dendrites is likely to depend on the retrograde molecular motor dynein.
Huntingtin interacts with TrkB and dynein
Our experiments showing a colocalization of TrkB with HTT and dynein, and that the dynein-dependent retrograde transport of TrkB depends on HTT and is altered in HD pathogenic situation, led to us to investigate the interaction between HTT, dynein, and TrkB. We cotransfected HEK cells with a full-length TrkB construct and either wild-type or polyQ constructs encoding the first 1301 amino acids of the N-terminal domain of HTT (HTTN1–1301), which contains the dynein-interacting domain (Caviston and Holzbaur, 2006). Immunoprecipitation of TrkB from the cotransfected cells led to the coimmunoprecipitation of HTT, whether or not it contained an expanded polyQ tract (Fig. 3A). To verify the specificity of these immunoprecipitations, we investigated the interaction between TrkB and dynein. Two different isoforms of dynein intermediate chain, dynein IC-1B and IC-2C, were previously shown to interact specifically with TrkB and TrkA, respectively (Ha et al., 2008). Accordingly, we found that TrkB interacted more strongly with dynein IC-1B than with dynein IC-2C (Fig. 3B). Therefore, we coexpressed wild-type or polyQ-HTT with dynein IC-1B and TrkB in cells and analyzed their respective interactions. HTT and dynein IC-1B efficiently coimmunoprecipitated with TrkB. Conversely, HTT and TrkB efficiently coimmunoprecipitated with dynein (Fig. 3C). Again, we found that an expanded polyQ stretch within HTT had no obvious effect on the nature of this complex. Finally, we investigated these interactions in vivo by performing immunoprecipitations of TrkB or dynein from mouse brains extracts from wild-type Hdh+/+ and mutant HdhQ111/Q111 mice. As previously reported in HdhQ111/Q111 mice (Keryer et al., 2011), the levels of endogenous mutant HTT protein are lower compared with those of wild-type HTT. Nevertheless, we could detect similar interactions between HTT and TrkB or dynein in both wild-type and mutant mice (Fig. 3D), suggesting as for the in vitro experiments (Fig. 3A–C) that polyQ expansion in HTT alters TrkB transport via a mechanism distinct from disrupting interactions among TrkB, dynein, and HTT.
Mutant polyQ huntingtin alters attachment of TrkB vesicles to microtubules
TrkB-vesicular transport is MT-dependent (Watson et al., 1999; Ginty and Segal, 2002; Ha et al., 2008). Therefore, we investigated whether an abnormal polyQ expansion in HTT could affect the interactions between TrkB vesicles and MTs. We expressed TrkB-RFP in COS7 cells, then prelysed the cells to remove the cytosolic components, and then fixed and immunostained the cells for α-tubulin. We found high numbers of TrkB-containing vesicles associated with MTs (Fig. 4A). We next coexpressed TrkB-RFP with wild-type (HTTN1–1301-17Q) or mutant (HTTN1–1301-73Q) HTT and quantified the number of TrkB vesicles present on MTs. We found that the percentage of vesicles attached to MTs was significantly lower in cells expressing mutant HTT than in cells expressing wild-type HTT (Fig. 4B). Next, we fractionated the subcellular components of COS7 cells coexpressing TrkB-GFP with either HTTN1–1301-17Q (wild-type) or HTTN1–1301-73Q (mutant) and obtained MT and cytosolic fractions. The same protein amounts from total, cytosolic, and MT fractions were analyzed by Western blotting (Fig. 4C). As previously reported (Gauthier et al., 2004; Zala et al., 2008), levels of the HTT and dynein proteins were lower in the MT fraction containing the pathogenic polyQ-HTT than in MT fractions containing wild-type HTT (Fig. 4C,D). We especially found the MT-associated levels of TrkB to be significantly reduced, indicating that HTT, TrkB receptors, and dynein detached from MTs in mutant condition. As the MT fraction corresponds in protein content to <10% of the cytosolic proteins, detachment of TrkB receptors from MTs, as expected, was not followed by an increase in TrkB content in the cytosolic fraction. We also noticed a slight shift in TrkB migration within the MT fraction. This could result from post-translational modifications, such as glycosylation that could depend on TrkB association with MTs. Nevertheless, it is unlikely that this migration shift could account for a difference in TrkB trafficking as these low molecular weight species are specific to the MT fraction and were found in both wild-type and mutant conditions. To further confirm our findings, we performed a similar fractionation on brains from wild-type Hdh+/+ and mutant HdhQ111/Q111 mice. As also observed in Figure 3D, total levels of mutant HTT were reduced compared with wild-type HTT (Fig. 4E). In addition, we observed the presence of an additional band in striatal extracts from HdhQ111/Q111 mice that could correspond to proteolytic cleavage that occurs preferentially in the striatum from old animals (13 months) as previously reported (Mende-Mueller et al., 2001). In support, cortical extracts from the same mice did not show such cleavage (data not shown). We next measured the level of proteins associated with MTs and observed reduced levels of both HTT and, importantly, of TrkB in the microtubule fraction purified from HdhQ111/Q111 mouse brains (Fig. 4E,F). We did not find decreased attachment of endogenous dynein to MTs. This may be the result of the experimental conditions in which we are analyzing the distribution of TrkB and HTT endogenous proteins. Therefore, most of the dynein analyzed may correspond to dynein associated with other cargos than TrkB and that are not specifically regulated by HTT.
Together, these results suggest that the abnormal polyQ expansion in HTT does not impair interactions between the dynein retrograde motor and TrkB vesicles but instead alters the attachment of these vesicles to MTs.
Retrograde transport of TrkB-BDNF vesicles is altered in HD striatal dendrites leading to a decreased striatal signaling
Although we demonstrated that retrograde transport of TrkB in dendrites is altered, the TrkB vesicles we observed might have corresponded to vesicles moving bidirectionally within dendrites rather than TrkB-BDNF-containing endosomes undergoing retrograde transport after BDNF-mediated receptor activation and internalization. Although axonal retrograde transport of receptors has been previously reported (Heerssen et al., 2004; Kuruvilla et al., 2004), the specific retrograde transport of Trk receptors within dendrites has been difficult to achieve.
We therefore took advantage of the recent development of microfluidic devices consisting of microfluidic chambers containing two compartments to separate neuronal cell bodies and dendrites from axons (Taylor et al., 2005) with channels that are 5 μm wide, 450 μm long, and 3 μm deep. This configuration allows CNS axons to grow into a fluidically isolated environment free of any chemoattractants or trophic factors. Embryonic rat striatal neurons can extend long processes into the channels, but only axons can reach the distal chamber located 450 μm away from the proximal one, which contains the cell bodies and most of the dendrites (Taylor et al., 2005). As we wanted to specifically study dendrites, and not axons, we generated an equivalent microfluidic device with four compartments with two microgroove parts (one with microchannels 450 μm long and one with microchannels 100 μm long) to more precisely discriminate axons from dendrites (Fig. 5A). This microfluidic device is similar to one that was recently developed, which uses a microchannel of 75 μm to selectively isolate dendrites from cell bodies (Cohen et al., 2011). In agreement with previous findings, we found that most of the neurites in the 100-μm-long channel corresponded to dendrites, as they were MAP2-GFP (Fig. 5B). We plated striatal neurons prepared from the ganglionic eminence of Hdh+/+ or HdhQ111/Q111 E15 mouse embryos in the cell body compartment of the microfluidic device (Fig. 5A). We observed no major effect of the polyQ expansion on the survival of the neurons within chambers (data not shown). We also investigated whether dendrite growth is altered by the HD mutation and quantified the number of neurites that cross the channels. We found no difference between neurons from Hdh+/+ and HdhQ111/Q111 mice (Fig. 5C). To selectively label TrkB receptors that would be internalized and retrogradely transported within dendrites, we prepared biotinylated BDNF as was done previously for NGF to track single NGF receptors (Cui et al., 2007). We verified that BDNF is biotinylated (Fig. 5D) and is biologically active as shown by the phosphorylation of CREB upon addition of biotinylated BDNF to primary striatal neurons (Fig. 5E).
One week after plating of the primary neurons in the microfluidic device, we added biotinylated BDNF to the dendritic compartment (distal compartment) during 2 min. We found this time to be sufficient to allow receptor internalization and significant to complete retrograde trafficking of endosomes to the cell body compartment in the wild-type situation (Fig. 6A). To guarantee the exclusive stimulation of dendrites and not cell bodies, we generated a pressure difference between the compartments containing the cell body and the dendrites by maintaining a volume difference between the two compartments. After BDNF stimulation, microchambers were washed with PBS to remove the noninternalized BDNF, and the neurons were then fixed, permeabilized and incubated with streptavidin-Qdots. In neurons from wild-type mice, Qdots were found localized primarily in the microchannels that contained dendrites and in the compartment containing the cell bodies (Fig. 6A,C). In contrast, in neurons from HdhQ111/Q111 mice, Qdots were mostly found in dendrite terminals compartment and in the channels containing dendrites (Fig. 6A,C). To make sure that all Qdots observed in the microchambers corresponded to internalized receptors, we also incubated Qdots in chambers that did not received BDNF. Only very few Qdots could be detected in this condition (Fig. 6B). We quantified Qdot localization by analyzing close to 200 microchannels per condition in two independent experiments. We found significantly more Qdots in the dendrite terminals of HdhQ111/Q111 neurons than in those of Hdh+/+ neurons (Fig. 6C). Conversely, the channels and the cell bodies compartments of chambers with Hdh+/+ neurons contained significantly more Qdots than the corresponding compartments of chambers with HdhQ111/Q111 neurons. This demonstrates that the retrograde transport of endogenous TrkB in the dendrites of striatal neurons in response to BDNF is impaired.
Finally, we investigated the physiological consequences of such a decrease in TrkB retrograde transport on downstream signaling. As in Figure 6, we plated striatal neurons prepared from the ganglionic eminence of Hdh+/+ or HdhQ111/Q111 E15 mouse embryos, waited 1 week, then added BDNF on the dendrites of the striatal neurons during 10 min (for phospho-ERK experiment) or 1 h (for c-fos experiment) (Fig. 7A). We extended the time compared with Figure 6 to allow the retrograde transport of endosomes to the cell body and the subsequent activation of downstream signalings. Cultures were fixed and immunostained for phospho-ERK or c-fos, two downstream targets of BDNF that were recently shown to be required for the BDNF-TrkB-induced dendrite-to-nucleus signaling (Cosker et al., 2008; Cohen et al., 2011). We observed a reduction in the number of phospho-ERK and c-fos-positive neurons from HdhQ111/Q111 mice compared with neurons from Hdh+/+ mice in the striatal cell body compartment (Fig. 7B). We quantified the number of phospho-ERK and c-fos-positive neurons after BDNF treatment and normalized it to the number of dendrites crossing the channels. We then calculated the difference in fold activation in phospho-ERK or c-fos between Hdh+/+ and HdhQ111/Q111 neurons by establishing the ratio between BDNF addition and control conditions. Analysis of 4–6 independent experiments revealed a respective decrease in BDNF-induced phospho-ERK and c-fos activation in neurons from HdhQ111/Q111 mice of 44% and 52% (Fig. 7C).
Together, these results provide evidence that mutant HTT alters TrkB vesicular transport in striatal dendrites and leads to physiological defects by impacting on downstream signaling.
Discussion
The consequences of the abnormal polyQ expansion in mutant HTT on BDNF trafficking in cortical neurons have been well characterized. However, whether the selective retrograde trafficking of TrkB receptors in striatal neurons depends on HTT and whether such transport is altered in HD have not been elucidated. As shown by videomicroscopy and coimmunoprecipitation experiments, our findings provide evidence that HTT is involved in the retrograde transport of TrkB vesicles in neurons. In support, we show that retrograde transport of TrkB is altered when HTT is silenced or when it contains the polyQ expansion. The retrograde transport of TrkB in distal dendrites depends on dynein as shown by the selective orientation of MTs using EB3-GFP videomicroscopy. Moreover, HTT interacts with dynein IC-1B, which is specifically involved in the transport of TrkB, but does not interact with dynein IC-2C (Ha et al., 2008). Therefore, in addition to BDNF, APP, and Ti-VAMP, TrkB can be added to the list of cargos that are transported by HTT.
By selectively exposing striatal dendrites to BDNF and its subsequent coupling using quantum dots, we found that retrograde transport of TrkB in the dendrites of striatal neurons is impaired in HD. How does BDNF signal in striatal dendrites? In contrast to axonal retrograde neurotrophin signaling, which has been extensively studied, it is only recently that some of the mechanisms involved in dendrite-to-nucleus signaling have been reported (Cohen et al., 2011). Indeed, until this study, it has been impossible to address this question because of the difficulty of separating dendrites from cell bodies. As for our study, the microfluidic compartmentalization of dendrites was essential for revealing dendrite-specific mechanisms. This approach led to the demonstration that BDNF applied to dendrites activates TrkB receptors in the cell body and that this signaling requires the ERK/MEK1/2 pathway (Cohen et al., 2011). Our findings that the retrograde trafficking of TrkB-BDNF vesicles is impaired in the dendrites of striatal neurons from HD mice and that this leads to a decrease induction of the ERK pathway in HD context is consistent with this study. These results warrant further studies that would recapitulate the corticostriatal projections using microfluidics and in which TrkB retrograde transport in striatal neurons would be studied in response to a lower BDNF transport from cortical afferences.
Our results show that HTT transports TrkB vesicles and that the dynein retrograde trafficking of TrkB-BDNF vesicles is impaired in the dendrites of HD striatal neurons. Consistent with the notion that dynein-dependent retrograde transport is defective in HD striatum, mice that contain point mutations in dynein heavy chain 1 gene exhibit striatal dysfunction and neuritic abnormalities (Braunstein et al., 2010). Furthermore, HTT regulates endosome positioning via a dynein-dependent mechanism (Caviston et al., 2011) and through Rab5 (Pal et al., 2006), a small GTPase involved in the early endosomal trafficking of Trk receptors (Deinhardt et al., 2006). Together, our study reveals that HTT is a key protein in dynein-dependent retrograde dendritic signaling in striatal neurons.
Our finding that dendritic transport of TrkB is altered in HD neurons is consistent with the crucial role played by TrkB in the survival of striatal neurons, which is exemplified by the extensive loss of medium spiny neurons that accompanies selective ablation of striatal TrkB (Baydyuk et al., 2011). These findings are also supported by reports of defective TrkB signaling in HD (Gines et al., 2010) and by the observations that sustained application of BDNF on striatal neurons reduces cell death in vitro and in vivo (Saudou et al., 1998; Borrell-Pages et al., 2006b; Gharami et al., 2008; Xie et al., 2011). Interestingly, we found that, in addition to a defect in the selective retrograde transport of activated TrkB-BDNF-bound receptors, the anterograde transport of TrkB in dendrites is also reduced (data not shown). This suggests that the secretion and transport of de novo TrkB receptors is impaired in HD and could also result in a reduction in the TrkB receptor density at postsynaptic sites. In support, a decrease in TrkB receptors has been previously reported in HD (Gines et al., 2006; Zuccato et al., 2008; Gines et al., 2010).
In conclusion, an essential role for HTT in BDNF and TrkB trafficking supports the notion that cell and non–cell-autonomous mechanisms are responsible for the dysfunction and death of striatal neurons in HD. Indeed, HTT not only controls the neurotrophic support of striatal neurons via its role in the transport of BDNF through corticostriatal afferences, but also the efficacy of the TrkB neurotrophic retrograde signaling pathway in striatal dendrites. Therefore, the alteration in the transport of both BDNF and of its receptor in HD situation is likely to have a major impact on the survival of the connected neurons.
Footnotes
This work was supported by Agence Nationale pour la Recherche Grant ANR-08-MNP-039 to F.S., Fondation pour la Recherche Médicale (équipe labellisée) to F.S., CNRS, INSERM, and the Institut Curie (F.S.). Fellowship support was provided by the following: Région Ile de France (G.L.), Institut Curie (D.Z.), CHDI Foundation Inc (D.Z., G.L.), and Fondation Pierre-Gilles de Gennes pour la Recherche (D.Z., G.L.). We thank S. Humbert for support and discussions; E. Jaumain, C. Benstaali, G. Poizat, and H. Yu for culture preparations; C. Lemercier-Neuillet for microchambers; A. Lubin for experiments; and R. Segal, K. Pfister, N. Déglon, G. Banker, M. Chao, and H. Yano for reagents.
The authors declare no competing financial interests.
References
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydyuk M, Russell T, Liao GY, Zang K, An JJ, Reichardt LF, Xu B. TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc Natl Acad Sci U S A. 2011;108:1669–1674. doi: 10.1073/pnas.1004744108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Watson FL, Pomeroy SL, Zhang YZ, Stiles CD, Segal RA. High-resolution imaging demonstrates dynein-based vesicular transport of activated Trk receptors. J Neurobiol. 2002;51:302–312. doi: 10.1002/neu.10062. [DOI] [PubMed] [Google Scholar]
- Borrell-Pagès M, Zala D, Humbert S, Saudou F. Huntington's disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. 2006a;63:2642–2660. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell-Pagès M, Canals JM, Cordelières FP, Parker JA, Pineda JR, Grange G, Bryson EA, Guillermier M, Hirsch E, Hantraye P, Cheetham ME, Néri C, Alberch J, Brouillet E, Saudou F, Humbert S. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J Clin Invest. 2006b;116:1410–1424. doi: 10.1172/JCI27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse KE, Maina FK, Birbeck JA, France MM, Roberts JJ, Colombo ML, Mathews TA. Aberrant striatal dopamine transmitter dynamics in brain-derived neurotrophic factor-deficient mice. J Neurochem. 2012;120:385–395. doi: 10.1111/j.1471-4159.2011.07531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein KE, Eschbach J, Ròna-Vörös K, Soylu R, Mikrouli E, Larmet Y, René F, Gonzalez De Aguilar JL, Loeffler JP, Müller HP, Bucher S, Kaulisch T, Niessen HG, Tillmanns J, Fischer K, Schwalenstöcker B, Kassubek J, Pichler B, Stiller D, Petersen A, et al. A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons. Hum Mol Genet. 2010;19:4385–4398. doi: 10.1093/hmg/ddq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci U S A. 1977;74:4516–4519. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martín-Ibañez R, Muñoz MT, Mengod G, Ernfors P, Alberch J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci. 2004;24:7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Holzbaur EL. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006;16:530–537. doi: 10.1016/j.tcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Caviston JP, Holzbaur EL. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009;19:147–155. doi: 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Ross JL, Antony SM, Tokito M, Holzbaur EL. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc Natl Acad Sci U S A. 2007;104:10045–10050. doi: 10.1073/pnas.0610628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Zajac AL, Tokito M, Holzbaur EL. Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Mol Biol Cell. 2011;22:478–492. doi: 10.1091/mbc.E10-03-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Bas Orth C, Kim HJ, Jeon NL, Jaffrey SR. Neurotrophin-mediated dendrite-to-nucleus signaling revealed by microfluidic compartmentalization of dendrites. Proc Natl Acad Sci U S A. 2011;108:11246–11251. doi: 10.1073/pnas.1012401108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin E, Zala D, Liot G, Rangone H, Borrell-Pagès M, Li XJ, Saudou F, Humbert S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27:2124–2134. doi: 10.1038/emboj.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosker KE, Courchesne SL, Segal RA. Action in the axon: generation and transport of signaling endosomes. Curr Opin Neurobiol. 2008;18:270–275. doi: 10.1016/j.conb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui B, Wu C, Chen L, Ramirez A, Bearer EL, Li WP, Mobley WC, Chu S. One at a time, live tracking of NGF axonal transport using quantum dots. Proc Natl Acad Sci U S A. 2007;104:13666–13671. doi: 10.1073/pnas.0706192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouet V, Perrin V, Hassig R, Dufour N, Auregan G, Alves S, Bonvento G, Brouillet E, Luthi-Carter R, Hantraye P, Déglon N. Sustained effects of nonallele-specific Huntingtin silencing. Ann Neurol. 2009;65:276–285. doi: 10.1002/ana.21569. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gharami K, Xie Y, An JJ, Tonegawa S, Xu B. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington's disease phenotypes in mice. J Neurochem. 2008;105:369–379. doi: 10.1111/j.1471-4159.2007.05137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginés S, Bosch M, Marco S, Gavaldà N, Díaz-Hernández M, Lucas JJ, Canals JM, Alberch J. Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur J Neurosci. 2006;23:649–658. doi: 10.1111/j.1460-9568.2006.04590.x. [DOI] [PubMed] [Google Scholar]
- Ginés S, Paoletti P, Alberch J. Impaired TrkB-mediated ERK1/2 activation in huntington disease knock-in striatal cells involves reduced p52/p46 Shc expression. J Biol Chem. 2010;285:21537–21548. doi: 10.1074/jbc.M109.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol. 2002;12:268–274. doi: 10.1016/S0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/S0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Ha J, Lo KW, Myers KR, Carr TM, Humsi MK, Rasoul BA, Segal RA, Pfister KK. A neuron-specific cytoplasmic dynein isoform preferentially transports TrkB signaling endosomes. J Cell Biol. 2008;181:1027–1039. doi: 10.1083/jcb.200803150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerssen HM, Pazyra MF, Segal RA. Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci. 2004;7:596–604. doi: 10.1038/nn1242. [DOI] [PubMed] [Google Scholar]
- Her LS, Goldstein LS. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J Neurosci. 2008;28:13662–13672. doi: 10.1523/JNEUROSCI.4144-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Ehrlich ME. Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J Neurosci. 1999;19:5409–5419. doi: 10.1523/JNEUROSCI.19-13-05409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Hoogenraad CC. Which way to go? Cytoskeletal organization and polarized transport in neurons. Mol Cell Neurosci. 2011;46:9–20. doi: 10.1016/j.mcn.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Schlager MA, Kuijpers M, Wulf PS, van Spronsen M, MacKintosh FC, Hoogenraad CC. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol. 2010;20:290–299. doi: 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
- Keryer G, Pineda JR, Liot G, Kim J, Dietrich P, Benstaali C, Smith K, Cordelières FP, Spassky N, Ferrante RJ, Dragatsis I, Saudou F. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J Clin Invest. 2011;121:4372–4382. doi: 10.1172/JCI57552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell. 2004;118:243–255. doi: 10.1016/j.cell.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Mende-Mueller LM, Toneff T, Hwang SR, Chesselet MF, Hook VY. Tissue-specific proteolysis of Huntingtin (htt) in human brain: evidence of enhanced levels of N- and C-terminal htt fragments in Huntington's disease striatum. J Neurosci. 2001;21:1830–1837. doi: 10.1523/JNEUROSCI.21-06-01830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington's disease. J Cell Biol. 2006;172:605–618. doi: 10.1083/jcb.200509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington's disease. Mol Brain. 2009;2:33. doi: 10.1186/1756-6606-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/S0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/S0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, 2nd, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Akmentin W, Philippidou P, Kuruvilla R, Ginty DD, Halegoua S. Pincher-mediated macroendocytosis underlies retrograde signaling by neurotrophin receptors. J Neurosci. 2005;25:5236–5247. doi: 10.1523/JNEUROSCI.5104-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson FL, Heerssen HM, Moheban DB, Lin MZ, Sauvageot CM, Bhattacharyya A, Pomeroy SL, Segal RA. Rapide nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J Neurosci. 1999;19:7889–7900. doi: 10.1523/JNEUROSCI.19-18-07889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, Gutekunst CA, Vrbanac V, Lebel LA, Schilling G, Hersch S, Friedlander RM, Gusella JF, Vonsattel JP, Borchelt DR, MacDonald ME. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum Mol Genet. 2002;11:633–640. doi: 10.1093/hmg/11.6.633. [DOI] [PubMed] [Google Scholar]
- Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington's disease phenotypes in YAC128 mice. J Neurosci. 2010;30:14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Lee FS, Kong H, Chuang J, Arevalo J, Perez P, Sung C, Chao MV. Association of Trk neurotrophin receptors with components of the cytoplasmic dynein motor. J Neurosci. 2001;21:RC125. doi: 10.1523/JNEUROSCI.21-03-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zala D, Colin E, Rangone H, Liot G, Humbert S, Saudou F. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet. 2008;17:3837–3846. doi: 10.1093/hmg/ddn281. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Marullo M, Conforti P, MacDonald ME, Tartari M, Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington's disease. Brain Pathol. 2008;18:225–238. doi: 10.1111/j.1750-3639.2007.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]