

Abstract
The eukaryotic initiation factor 4E-binding protein-2 (4E-BP2) is a repressor of cap-dependent mRNA translation and a major downstream effector of the mammalian target of rapamycin (mTOR) implicated in hippocampal long-term synaptic plasticity and memory. Yet, synaptic mechanisms regulated by 4E-BP2 translational repression remain unknown. Combining knock-out mice, whole-cell recordings, spine analysis, and translation profiling, we found that 4E-BP2 deletion selectively upregulated synthesis of glutamate receptor subunits GluA1 and GluA2, facilitating AMPA receptor (AMPAR)-mediated synaptic transmission and affecting translation-dependent chemically induced late long-term potentiation (cL-LTP). In 4E-BP2 knock-out (4E-BP2−/−) mice, evoked and miniature EPSCs were increased, an effect mimicked by short-hairpin RNA knockdown of 4E-BP2 in wild-type mice, indicating that 4E-BP2 level regulates basal transmission at mature hippocampal AMPAR-containing synapses. Remarkably, in 4E-BP2−/− mice, the AMPA to NMDA receptor (NMDAR) EPSC ratio was increased, without affecting NMDAR-mediated EPSCs. The enhanced AMPAR function concurred with increased spine density and decreased length resulting from greater proportion of regular spines and less filopodia in 4E-BP2−/− mice. Polysome profiling revealed that translation of GluA1 and GluA2 subunits, but not GluN1 or GluN2A/B, was selectively increased in 4E-BP2−/− hippocampi, consistent with unaltered I–V relation of EPSCs mediated by GluA1/GluA2 heteromers. Finally, translation-dependent cL-LTP of unitary EPSCs was also affected in 4E-BP2−/− mice, lowering induction threshold and removing mTOR signaling requirement while impairing induction by normal stimulation. Thus, translational control through 4E-BP2 represents a unique mechanism for selective regulation of AMPAR synthesis, synaptic function, and long-term plasticity, important for hippocampal-dependent memory processes.
Introduction
Protein synthesis, also called translation, is a finely regulated fundamental biological process (Sonenberg and Hinnebusch, 2009), which is essential for establishing persistent forms of synaptic plasticity and memory (Sutton and Schuman, 2006; Costa-Mattioli et al., 2009). In eukaryotes, translation is regulated primarily at the rate-limiting initiation step, which involves recruiting the ribosome to the mRNA (Sonenberg and Hinnebusch, 2009). A critical interaction in the process involves proper assembly of the eukaryotic initiation factor 4F (eIF4F) complex, which is comprised of the cap-binding protein eIF4E, eIF4A, and the scaffolding protein eIF4G, linking the mRNA to the ribosome (Sonenberg and Hinnebusch, 2009). Translation initiation is regulated via eIF4E-binding proteins (4E-BPs), which act as translation repressors by sequestering eIF4E, thus preventing formation of eIF4F complex and precluding ribosome association with mRNA.
The protein kinase mammalian target of rapamycin (mTOR) is a key regulator of translation initiation via 4E-BP phosphorylation (Hay and Sonenberg, 2004). Depending on its interacting partners, mTOR assembles into two distinct protein complexes: mTORC1, which is inhibited by rapamycin; and mTORC2, which is not (Hay and Sonenberg, 2004; Laplante and Sabatini, 2012). Activated mTORC1 triggers translation initiation by phosphorylating several sites on 4E-BPs, resulting in reduced binding affinity and dissociation from eIF4E (Hay and Sonenberg, 2004). Studies targeting signaling pathways upstream of mTOR suggest that mTOR activation positively regulates excitatory synaptic function (Tang et al., 2002; Schratt et al., 2004; Jaworski et al., 2005; Kumar et al., 2005; Tavazoie et al., 2005; Lee et al., 2011), although mTOR activation has also been linked to depression of excitatory synaptic function (Hou and Klann, 2004). However, the respective role of the multiple downstream effectors of mTOR, 4E-BPs, S6 kinase (S6K), or 5′ TOP-mRNA (Hay and Sonenberg, 2004; Gobert et al., 2008; Laplante and Sabatini, 2012) in the regulation of synaptic function remains unclear.
Of the three known mammalian 4E-BPs (4E-BP1, 2, and 3), 4E-BP2 is the major form in the mammalian CNS and is critical for hippocampal synaptic plasticity and memory (Banko et al., 2005, 2006, 2007). Late-phase long-term potentiation (L-LTP), which is induced by trains of high-frequency electrical stimulation, is inhibited in slices from 4E-BP2 knock-out (4E-BP2−/−) mice (Banko et al., 2005), whereas spatial and contextual memory is impaired in these mice (Banko et al., 2005, 2007). However, L-LTP threshold is facilitated in 4E-BP2−/− mice: a single train of stimulation, subthreshold for L-LTP induction in wild-type slices, effectively induces L-LTP. In addition, metabotropic glutamate receptor (mGluR)-mediated LTD is enhanced in these transgenic mice (Banko et al., 2006). Although basal excitatory synaptic transmission was initially reported intact in field potential studies (Banko et al., 2005), miniature excitatory synaptic activity was recently found to be facilitated in hippocampal pyramidal cells of 4E-BP2−/− mice (Bidinosti et al., 2010), indicating that 4E-BP2-dependent translation regulates excitatory synapse function as well as plasticity. Interestingly, the increased excitatory synaptic activity in 4E-BP2−/− mice was recently found to produce an excitation–inhibition imbalance by means of dysregulated eIF4E-dependent translation of neuroligins and result in autistic-like behaviors in these mice (Gkogkas et al., 2013). However, the mechanisms by which 4E-BP2 translation inhibition regulates excitatory synapse function remain largely unknown.
Here, we use a combination of transgenic mice, whole-cell recordings, spine analysis, and polysome profile analysis to examine how 4E-BP2 translation repression regulates excitatory synaptic transmission and plasticity in CA1 pyramidal neurons. We provide evidence that removal of 4E-BP2 translation inhibition selectively upregulates the synthesis of GluA1 and GluA2 subunits of glutamate receptors, independent of GluN subunits or scaffolding protein PSD95, to specifically control AMPAR-mediated synaptic transmission and translation-dependent long-term plasticity at the single-synapse level.
Materials and Methods
Hippocampal slice cultures
All experiments conformed to the Canadian animal care guidelines at Université de Montréal and McGill University. Hippocampal slice cultures were prepared and maintained as described previously (Stoppini et al., 1991; Ran et al., 2009; Bidinosti et al., 2010). Briefly, male and female 4–6-d-old wild-type C57BL/6 (Charles River) and homozygous 4E-BP2+/+ or 4E-BP2−/− mice (Banko et al., 2005) were anesthetized and decapitated. The brains were removed and dissected in HBSS-based medium (Invitrogen). Corticohippocampal slices (400 μm thick) were obtained using a McIlwain tissue chopper (Campden Instruments). Three or 4 slices were placed on Millicell culture plate inserts (Millipore) and incubated for 3 d in OptiMem-based medium (Invitrogen) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Thereafter, the medium was replaced every 2 d until onset of experiments after 8–15 d in culture.
Dissociated cortical neuron cultures
Intact cortices were isolated from brains of E16 mouse embryos from 4E-BP2+/+ mice in dissection buffer [HBSS (Invitrogen ) supplemented with 0.2% HEPES-KOH (Sigma), pH 7.2]. The meninges and blood vessels were carefully removed. Cells were mechanically dissociated with 0.025% trypsin (Wisent) and plated on 150 μg/ml poly-D-lysine (Sigma) coated plates. Growth medium was Neurobasal (Invitrogen ) supplemented with 2% B27 supplement (Invitrogen ) and 2 mm GlutaMAX (Invitrogen ). After 4 d in culture, glial cell growth was inhibited with 1 μm cytosine β-d-arabinofuranoside (Sigma).
Whole-cell recordings
On experiment day, slices were transferred to oxygenated artificial CSF (ACSF) at room temperature containing (in mm) 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 4 MgSO4, 4 CaCl2, 26 NaHCO3, and 10 d- (+) glucose (pH 7.3–7.4; 295–305 mOsmol), and allowed to recuperate for at least 45 min. Subsequently, slices were transferred to a submersion chamber perfused (2 ml/min) with oxygenated ACSF at 31 ± 0.5°C. CA1 pyramidal neurons were identified based on their soma shape and position in stratum pyramidale with an upright microscope (Nikon Eclipse, E600FN) equipped with a long-range water-immersion objective (40×, Nomarski Optics) and an infrared camera (4980 Series, Cohu). Whole-cell recordings were obtained from pyramidal neurons using borosilicate pipettes (3–6 MΩ). The intracellular recording solution contained the following (in mm): 130 CsMeSO3, 5 CsCl, 2 MgCl2, 5 diNa-phosphocreatine, 10 HEPES, 2 ATP-Tris, 0.4 GTP-Tris, and 0.1% biocytin (pH 7.2–7.3; 275–285 mOsmol). Recordings were made in the voltage-clamp mode using a Multiclamp 700A amplifier (Molecular Devices). Unless stated otherwise, the cell holding potential was maintained at −60 mV and series resistance was routinely monitored. Recorded signals were low-pass filtered at 2 kHz, digitized at 20 kHz, and stored on a PC. Data acquisition and off-line analyses were performed using 1322A Digidata acquisition board and pClamp 10 software (Molecular Devices). Data were only included if the holding current was stable or series resistance varied <25% of initial value.
Evoked, spontaneous, and miniature EPSCs and IPSCs
EPSCs mediated by AMPARs were recorded in the presence of DL-2-amino-5-phosphonovaleric acid (AP5; 50 μm; Tocris Biosciences) and gabazine (5 μm; Tocris Biosciences), to block NMDARs and GABAA receptors, respectively. A bipolar θ-glass electrode filled with ACSF and positioned in the stratum radiatum was used to evoke pairs of EPSCs at 0.5 Hz by constant DC current pulses (50 μs duration; 50 ms interpulse interval). Putative single-fiber EPSCs were evoked using minimal stimulation at previously described (Raastad et al., 1992; Perez et al., 2001; Ran et al., 2009). In brief, stimulus strength was adjusted to a value that generated ∼50% successes (range 40–60%). Amplitude of mean EPSC (including failures), failure rate (expressed as percentage of total number of stimulations), and potency (amplitude of successful EPSCs only) were extracted from 5 min bins over a 10–20 min period. To measure rectification ratio of AMPAR-mediated EPSCs, spermine (0.1 mm) was added to the intracellular solution and EPSCs were evoked (2× minimal stimulation strength) at a holding potential of −70, 0, and +40 mV. Rectification was expressed as percentage of the peak EPSC amplitude at +40 mV relative to −70 mV. Miniature EPSCs (mEPSCs) were recorded in the presence of TTX (0.5 μm; Alomone Laboratories) coapplied with AP5 and gabazine. For analysis, mEPSCs were detected on a running template (mean of ∼20 events) with a well-defined baseline, using pClamp-10 software. Detection threshold was set at 3 pA, and 150–200 events were sampled per neuron. Miniature inhibitory postsynaptic currents (mIPSCs) were recorded using a CsCl-based intracellular solution, which contained (in mm): 135 CsCl, 10 HEPES, 2 MgCl2, and 4 MgATP (pH 7.25). In these experiments, 6,7-dinitroquinoxaline-2,3-dione (DNQX; 5 μm; Tocris Bioscience) and AP5 (50 μm) were present to block excitatory transmission via AMPARs and NMDARs. In forskolin-induced L-LTP experiments (see below for induction protocol), we first recorded spontaneous EPSCs at a holding potential of −70 mV for a 10–20 min period and 150–200 events were sampled per neuron. For total charge transfer, the cumulative sum of all detected events was taken for a 10 min period. Subsequently, to measure AMPAR/NMDAR EPSC ratios, we evoked EPSCs, by stimulating Schaffer collateral axons at double the strength of minimal stimulation, at −70, 0, and +40 mV in the presence of gabazine (5 μm). The AMPAR component was taken at the peak EPSC at −70 mV, whereas the NMDAR component was measured at 50 ms after stimulus at +40 mV. To reduce polysynaptic responses and spontaneous bursting activity, an A1 adenosine receptor agonist, 2-Cl-adenosine (6 μm; Tocris Bioscience), was continuously applied throughout the duration of forskolin L-LTP experiments (Otmakhov et al., 2004). In silent synapse experiments, we first stimulated Schaffer collateral axons at a minimal stimulus intensity, which evoked a clear AMPAR-mediated EPSC at a holding potential of −60 mV (n = 30 events) (Isaac et al., 1995). Subsequently, we gradually decreased the stimulus strength to a value yielding 100% of failures. If at the same stimulus intensity and with the membrane potential clamped at +40 mV, a successful NMDAR-mediated EPSC was evoked, the synapse was considered silent (no AMPAR component). In contrast, in cases where the same procedure yielded no EPSCs at −60 or at +40 mV, the synaptic test was considered a failure at this intensity and the synapse recruited at the previous threshold stimulus intensity was deemed active (with AMPAR and NMDAR components). If a silent synapse was not found at a given stimulation site in a neuron, the stimulation electrode was moved to another stimulation site in stratum radiatum and the test procedure was repeated for as long as the recording remained stable.
Protocol for forskolin-induced late LTP
For L-LTP induction, slice cultures were first transferred to filtered normal ACSF for a 15 min period in a sterile culture hood. Subsequently, slices were treated for 30 min in a filtered solution containing forskolin (10 or 50 μm; Calbiochem), the phosphodiesterase inhibitor rolipram (0.1 μm; Sigma-Aldrich), and 0 mm Mg2+ ACSF (Otmakhov et al., 2004; Kopec et al., 2006). After forskolin stimulation, slices were incubated for a 90 min recovery period in normal (4 mm Mg2+) ACSF in a 37°C humidified incubator. For sham treatment, slices were similarly treated for 30 min but with vehicle (DMSO) containing normal ACSF. Slices were then transferred to a submersion chamber for patch-clamp recordings. In some experiments, PP242 (2.5 μm; Sigma-Aldrich) was applied from 30 min before to 30 min after forskolin or sham treatment.
shRNA constructs and transfection
Short hairpin RNA (shRNA)-IRES-EGFP constructs were biolistically transfected in slice cultures (8–13 d in vitro) using a Helios gene gun (Bio-Rad) as previously described (Bourdeau et al., 2007). The nontargeting shRNA (shCtl) and mouse 4E-BP2 shRNA (sh4E-BP2-TRCN TRCN0000075614, Mission, Sigma) were custom cloned in the pLKO.1-IREStGFP vector (Mission, Sigma). For nucleofection experiments, 106 cortical cells in suspension were used, following the AMAXA Nucleofector (Lonza) mouse cortical neuron kit protocol with 5 μg of plasmids encoding the scrambled shRNA or the shRNA targeting 4E-BP2 to assess knockdown. After 10 d in culture, cells were lysed in RIPA buffer.
Western blotting
Western blots were performed on extracts from whole hippocampi or stimulated CA1 mini-slices as described previously (Topolnik et al., 2006; Gobert et al., 2008). All extracts were dissociated in RIPA buffer containing protease inhibitors (inhibitor Mixture Set I from Calbiochem), 2 mm sodium-o-vanadate (Sigma), and 1 mm sodium fluoride (Sigma). Antibodies against indicated proteins were as follows: GluA1 (Alomone Laboratories) and GluA2 (Millipore); GluN1, GluN2A, and GluN2B (Sigma); PSD95, GFAP, 4E-BP2, eIF4E, phospho-rpS6, rpS6, and β-actin (Cell Signaling Technology); Arc/Arg3.1 (Santa Cruz Biotechnology); anti-GFP (BD Biosciences, Living Colors); HA (Sigma); secondary anti-mouse (GE Healthcare) and anti-rabbit (Jackson ImmunoResearch Laboratories) and anti-goat (Santa Cruz Biotechnology). Quantification of immunoblots was performed using ImageQuant (Bio-Rad). Values were normalized to β-actin, rpS6, or another control as specified. For S6 phosphorylation Western blot assays, acute CA1 mini-slices were obtained as previously described (Topolnik et al., 2006; Gobert et al., 2008). Briefly, 19–21-d-old 4E-BP2−/− or wild-type 4E-BP2+/+ mice (Banko et al., 2005) were anesthetized with halothane (Halocarbon Laboratories), and the brains were rapidly removed into ice-cold, oxygenated modified ACSF containing (mm): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1.3 MgSO4, 2.5 CaCl2 and 10 glucose (pH 7.4, 300 mosmol l−1). Acute slices (400 μm thick) were cut using a vibratome (Leica VT1000S, Leica Microsystems) and transferred to a chamber with ice-cold oxygenated modified ACSF where mini-slices consisting of CA1 region were surgically isolated under a dissection microscope. CA1 mini-slices were collected in slice holding chambers in heated (31°C) oxygenated modified ACSF and allowed to recover for 1 h. Afterward slices were treated for 30 min with forskolin in the absence or presence of PP242 (30 min pretreatment), transferred to cold modified ACSF, and collected in a tube on dry ice. Each sample containing five or six slices from two different animals was stored at −80°C until biochemical analysis.
Polysome analysis
Polysome profile analysis was performed as previously described (Costa-Mattioli et al., 2005). Intact hippocampi were washed with ice-cold PBS containing 100 μg/ml cycloheximide and subsequently lysed in a hypotonic lysis buffer (5 mm Tris-HCl (pH 7.5), 2.5 mm MgCl2, 1.5 mm KCl, 100 μg/ml cycloheximide, 2 mm DTT, 0.5% Triton X-100, and 0.5% sodium deoxycholate). Lysate concentration was balanced using a Bradford assay (Bio-Rad) and by measuring total RNA concentration using a NANODROP2000 spectrophotometer (Thermo Scientific). Lysates were loaded onto 10–50% sucrose density gradients (20 mm HEPES-KOH, pH 7.6, 100 mm KCl, 5 mm MgCl2) and centrifuged at 35,000 rpm for 2 h at 4°C. Gradients were fractionated and the optical density at 254 nm was continuously recorded using an ISCO fractionator (Teledyne ISCO). Total RNA from each fraction was isolated using Trizol (Invitrogen) and reverse transcribed using the Superscript III kit (Invitrogen).
Quantitative RT-PCR
Total or polysome reverse transcribed RNA was analyzed using a Bio-Rad iQ SYBR Green Supermix kit as previously described (Costa-Mattioli et al., 2007). The primers used for all experiments are summarized in Table 1. Results are presented in arbitrary units as relative amounts using serial dilutions of cortical or hippocampal RNA as quantitative PCR concentration standards.
Table 1.
Primers used for quantitative RT-PCR
| Forward | Reverse | |
|---|---|---|
| Grin1 | TGATCATTTCCGGGTGGATTGCGA | AAGGATGCCTTCGCGTACTAGCTT |
| Grin2a | ACTGGGTCACAGAGCAACATGCTA | AGTCCATTCGCGAGGAGTTCATGT |
| Grin2b | AGTGCTGTTCTCCCAAGTTCTGGT | TGGAAGTCATCTTTCTCGTGGGCA |
| Dlg4 | ATCGGTGACGACCCATCCATCTTT | TCCCGGACATCCACTTCATTGACA |
| Actb | ATCGATCGGCTAGCCTGTCGAGTCGCGTCCACCC | CGATCGATAAGCTTGGCGAACTGGTGGCGGGTGTG |
| Gria1 | TGTAATTTAGGGACCCATTAGGGACTCCA | GTCTCCAGTTTGGATACATAGAAACCTTGT |
| Gria2 | TCCTGTTGAGTTTCGTACACGCCT | ATGCCCTCGCTTACCTCCTTCATT |
| Arg3.1 | TGCCTACATGGGTTCCAAGACA | TGGCTTGTCTTCACCTTCAGCTC |
Synaptosome preparation
Intact hippocampi were dissected in ice-cold PBS and homogenized in 320 mm sucrose, 1 mm EDTA, 5 mm Tris-HCl, pH 7.4, and 25 μm DTT. Synaptosomes were isolated on a discontinuous Percoll (GE Healthcare) gradient (Dunkley et al., 2008). The fraction between 10% and 23% Percol was isolated and resuspended in 2× SDS-PAGE sample buffer.
Spine analysis
Organotypic slices were first tranfected with a plasmid encoding enhanced yellow fluorescent protein (EYFP; Clontech Laboratories). At 48 h after transfection, slices were fixed in 4% paraformaldehyde overnight at 4°C, washed in phosphate buffer, and mounted on glass slides for confocal imaging with ProLong Gold Antifade (Invitrogen). EYFP-transfected CA1 pyramidal neurons were randomly selected based on fluorescence and characteristic morphology. Confocal images were obtained from secondary branches of apical dendrites using a confocal laser scanning microscope (LSM 510; Carl Zeiss) equipped with a 63× oil-immersion objective (numerical aperture, 1.4; Carl Zeiss). Sequential z-stacks consisted of 10–20 sections (512 × 512 pixels, 50–100 μm dendritic segments) spaced at 0.2–0.4 μm apart. Spine length and shape were analyzed using LSM 510 software. To examine changes in different classes of spines, we categorized spines in three types on the basis of length and shape (Matsuzaki et al., 2001; McKinney, 2005; Lebeau et al., 2008): (1) filopodia, long protrusions (>1 μm) without a spine head; (2) long spines, long protrusions (>1 μm) with a small head at the tip; and (3) regular spines, short protrusions (<1 μm), including stubby and mushroom-type spines.
Statistical analysis
Unless otherwise stated, group comparison was performed using an unpaired t test. For comparisons of more than two groups, an ANOVA was used followed by a post hoc Tukey test. In some cases, a Kolmogorov–Smirnov test was used to compare cumulative distributions of mEPSC parameters between groups (150 events per neuron). Data were expressed as mean ± SEM and a P value < 0.05 was considered significant.
Results
Facilitation of excitatory synaptic transmission by 4E-BP2 knock-down
To examine the mechanisms by which 4E-BP2 translation inhibition regulates excitatory synaptic transmission at the single cell level, we used transgenic 4E-BP2−/− knock-out mice (Banko et al., 2005) and whole-cell recordings from CA1 pyramidal cells in hippocampal slice cultures. First we validated that removal of 4E-BP2 translation inhibition upregulated AMPAR-mediated miniature excitatory synaptic activity (Bidinosti et al., 2010). Spontaneous mEPSCs were augmented in amplitude (57 ± 17% increase) and frequency (95 ± 14% increase) in CA1 pyramidal cells in slice cultures from 4E-BP2−/− mice compared with wild-type 4E-BP2+/+ mice (Fig. 1A). The rise time and decay of mEPSCs were unchanged (data not shown). There was no difference in input resistance between neurons from wild-type and 4E-BP2−/− slices (126 ± 11 MΩ vs 125 ± 15 MΩ; n = 5 each), indicating that the absence of 4E-BP2 does not alter passive membrane properties of CA1 pyramidal neurons.
Figure 1.

Facilitation of AMPAR-mediated unitary excitatory synaptic transmission in 4E-BP2−/− mice. A, Increased spontaneous miniature synaptic activity in 4E-BP2−/− mice. Top: Continuous 1 s recordings (in TTX, AP-5, and gabazine), showing more frequent and larger amplitude mEPSCs in pyramidal cells of 4E-BP2−/− mice. Bottom: Summary bar graphs for all cells, showing a greater mEPSC amplitude and frequency in slices from 4E-BP2−/− mice. B, Representative EPSCs evoked by minimal stimulation in pyramidal neurons illustrating larger responses in slices from 4E-BP2−/− mice. Left: Superimposed 20 successive traces (EPSC successes + failures; gray) and mean response of 100 events (including failures; black). Middle: Mean EPSC pairs evoked by paired-pulse stimulation (50 ms interpulse interval), showing similar paired-pulse facilitation in 4E-BP2−/− mice. The first and second EPSCs are superimposed on the right. Right: Superimposed scaled EPSC pairs showing that paired-pulse ratio is unchanged in slices from 4E-BP2−/− mice. C, Summary bar graphs showing facilitation of EPSC potency (mean EPSC without failures) and similar failure rate and paired-pulse ratio in 4E-BP2−/− mice. Data are mean ± SEM. *p < 0.05. Number of cells provided above bars.
Given that spontaneous mEPSCs reflect unitary synaptic transmission independent of action potentials and originating from many inputs, we next explored the consequences of 4E-BP2 knock-out on AMPAR-mediated unitary EPSCs evoked at Schaffer collateral synapses using minimal stimulation (weak stimulation at ∼50% failure rate; Fig. 1B, C). The potency of EPSCs (mean EPSC excluding failures) was nearly doubled in pyramidal cells from 4E-BP2−/− compared with wild-type mice. EPSCs evoked by paired-pulse stimulation showed facilitation, and paired-pulse facilitation was unchanged in 4E-BP2−/− compared with wild-type (Fig. 1B, C). These results suggest that removal of 4E-BP2 translation inhibition increases AMPAR-mediated transmission at Schaffer collateral synapses via an increase in postsynaptic responsiveness at threshold stimulation. However, possible alterations in presynaptic release probability cannot be ruled out.
Changes in basal synaptic transmission in 4E-BP2−/− mice could be attributed to compensatory mechanisms resulting from the general loss of 4E-BP2 alleles during development (Banko et al., 2005). Thus, to examine such nonspecific compensatory effects, we characterized the consequences of removal of 4E-BP2 translation represssion using shRNA to selectively downregulate 4E-BP2 expression in CA1 pyramidal neurons in mature slice cultures (10–15 DIV) from wild-type mice (Fig. 2). First, the efficacy of sh4E-BP2 in downregulating 4E-BP2 expression was verified using Western blot and dissociated cortical neuron cultures transfected with sh4E-BP2 compared with shCtl controls (reduction by 54.4 ± 12.3%; Fig. 2A). Slice cultures were then biolistically transfected with a bicistronic vector to express GFP and shRNA targeting 4E-BP2 (sh4E-BP2) or a control nontargeting sequence (shCtl; Fig. 2B). After a 48 h recovery period, whole-cell recordings of mEPSCs were obtained from GFP-labeled transfected and untransfected pyramidal neurons in the same slice (Fig. 2). In sh4E-BP2-transfected neurons, mEPSC amplitude and frequency (normalized to untransfected neighboring cells) were greater (31 ± 8% and 26 ± 8% increase, respectively) compared with cells transfected with nontargeting shCtl (Fig. 2C–E). The kinetics of mEPSCs was unaltered in sh4E-BP2 transfected neurons (data not shown). In contrast, there was no difference in mEPSC amplitude or frequency in neurons transfected with a shCtl relative to untransfected cells recorded in the same slice. Coupled to the previous observation that overexpression of 4E-BP2 rescues the changes in mEPSCs in pyramidal cells of 4E-BP2−/− mice (Bidinosti et al., 2010), our results suggest that basal transmission at AMPAR-mediated excitatory synapses is regulated by 4E-BP2 levels in mature hippocampal pyramidal neurons.
Figure 2.

Facilitation of AMPAR-mediated spontaneous miniature synaptic activity by shRNA downregulation of 4E-BP2 in slice cultures. A, Reduction by shRNA of 4E-BP2 levels. Left, Representative Western blots from dissociated cortical neuron cultures confirming efficacy of sh4E-BP2 construct. The characteristic three band distribution of 4E-BP2 in untransfected or cells transfected with nontargeting control shRNA (shCtl-IRES-GFP) almost disappears in sh4E-BP2-IRES-GFP transfected cells. Immunoblots are also shown for phosphorylated S6 (rpS6), eIF4E, actin, and GFP, showing the specificity of the knockdown. Note robust GFP expression in shCtl and sh4E-BP2 transfected cells. Right, Summary bar graphs for all experiments (n = 3 experiments) showing that 4E-BP2 protein amount (normalized to β-actin) was reduced after transfection with sh4E-BP2 compared with shCtl. Data are mean ± SEM. *p < 0.05. B, Diagram of transfection and recording protocol. Hippocampal slice cultures were biolistically transfected with a bicistronic plasmid encoding GFP and an shRNA targeting 4E-BP2 (sh4E-BP2-IRES-GFP) or a control nontargeting sequence (shCtl-IRES-GFP). At 48 h after transfection, whole-cell recordings were obtained from GFP-expressing and untransfected CA1 pyramidal neurons. C, Representative examples of spontaneous mEPSCs recorded at 48 h after transfection. C1, Continuous current traces showing enhancement of mEPSCs in cells with shRNA knockdown of 4E-BP2. mEPSCs were more frequent and larger in amplitude in sh4E-BP2 transfected neurons (right) compared with untransfected cells recorded in the same slice (left). C2, Transfection with a control nontargeting shRNA (shCtl) had no effect on mEPSC amplitude or frequency. D, Scatter plots of mEPSC amplitude and frequency values for sh4E-BP2 and shCtl transfected versus untransfected neurons. Diagonal line indicates unity. Data points deviate substantially from unity only for sh4E-BP2 (●) and not shCtl (○) transfected neurons, indicating a facilitation of mEPSC amplitude and frequency in cells with 4E-BP2 downregulation. E, Summary bar graphs of mEPSC amplitude and frequency for shRNA experiments, showing increased mEPSC amplitude and frequency (expressed as percentage of values from untransfected cells in the same slice) in sh4E-BP2-transfected cells relative to shCtl transfected cells. Data are mean ± SEM. *p < 0.05 (t test). Number of cells are indicated above bar.
Selective modulation of AMPAR function in 4E-BP2 knock-out mice
The efficacy of hippocampal excitatory transmission can be regulated postsynaptically via synthesis and insertion of AMPARs (Nayak et al., 1998; Ju et al., 2004; Smith et al., 2005). However, similar activity-dependent regulation of NMDARs can also affect synaptic function (Bellone and Nicoll, 2007; Chen and Bear, 2007). Therefore, we examined next whether suppression of 4E-BP2 translation inhibition also modifies NMDAR-mediated synaptic transmission. First we determined whether the AMPAR/NMDAR EPSC ratio was altered by recording AMPAR and NMDAR-mediated components of EPSCs at holding potentials of −70 and +40 mV, respectively (Fig. 3A). In pyramidal cells of 4E-BP2−/− mice, AMPAR/NMDAR EPSC ratio was increased by 71 ± 45% compared with wild-type controls (Fig. 3B), indicating an effective modulation of AMPAR over NMDAR-mediated transmission. Next we examined whether the modulation AMPAR transmission was selective, by recording isolated NMDAR component of EPSCs in the presence of the AMPAR blocker DNQX (Fig. 3C, D). The current–voltage relation of NMDAR-mediated EPSCs was unaltered in pyramidal cells from 4E-BP2−/− mice compared with wild-type controls (Fig. 3D). In addition, the amplitude of NMDAR-mediated EPSCs (Fig. 3C) was not different [peak amplitude at +40 mV: 69 ± 14 pA in 4E-BP2−/− group (n = 10) and 70 ± 32 pA in wild-type (n = 7); p > 0.05, unpaired t test], indicating that removal of 4E-BP2 translation repression does not affect NMDAR-mediated synaptic transmission and, thus, selectively regulates AMPAR-mediated synaptic function.
Figure 3.

Selective modulation of AMPAR-mediated transmission in 4E-BP2−/− mice. A, Examples of EPSCs composed of AMPAR and NMDAR-mediated components. Arrows indicate points at which amplitude of AMPAR and NMDAR components was measured at holding potentials of −70 mV and +40 mV, respectively. Note the increase in the AMPAR-mediated component and unchanged NMDAR component in the cell from 4E-BP2−/− mice. Arrowhead indicates time of stimulation. B, Summary bar graph showing the increase in AMPAR/NMDAR EPSC ratio in 4E-BP2−/− mice. Data are mean ± SEM. *p < 0.05. C–D, Representative traces (C) and mean current–voltage relations (D) of NMDAR-mediated EPSCs recorded at different holding potentials in the presence of the AMPAR antagonist DNQX, showing intact amplitude and voltage–current relation of NMDAR-mediated EPSCs in cells of 4E-BP2−/− mice. E, Decrease in the proportion of silent synapses in 4E-BP2−/− mice. Left, Example of EPSCs at AMPAR silent synapse in a cell from wild-type mice (WT). NMDAR-mediated EPSCs were evoked at +40 mV holding potential, and no AMPAR-mediated EPSC was detected at −60 mV. Example of EPSCs from a cell from 4E-BP2−/− mice (4E-BP2−/−) with both NMDAR and AMPAR-mediated components. Right, Summary plot of proportion (expressed as percentage total) of cells with AMPAR silent or functional synapses, showing reduced incidence of silent synapses in 4E-BP2−/− mice. F, No change in current–voltage relation of AMPAR-mediated EPSCs in 4E-BP2−/− mice. Left, Representative traces (average of 30 events) showing AMPAR-mediated EPSCs (stimulation intensity, 2 × threshold) recorded in ACSF containing AP5 (50 μm) and gabazine (5 μm) to block NMDA and GABA receptors, respectively. EPSCs were evoked at different membrane potentials (−70, 0, and +40 mV) in CA1 pyramidal neurons from wild-type (left traces, black) and 4E-BP2−/− mice (right traces, gray). Right, Summary current–voltage plots for all cells showing unchanged linear voltage–current relation in 4E-BP2−/− mice.
Our observation of increased AMPAR/NMDAR ratio and unaltered NMDAR component is reminiscent of the activation of postsynaptically silent synapses reported during LTP (Isaac et al., 1995; Liao et al., 1995). Therefore, we examined whether 4E-BP2 deletion affected the proportion of AMPAR silent synapses (expressing only NMDARs) in pyramidal cells. Schaffer collateral stimulation was applied at weak, threshold intensity, and NMDAR-mediated EPSCs were monitored at a holding potential of +40 mV and AMPAR-mediated EPSCs at −60 mV (Isaac et al., 1995; Liao et al., 1995). In cells from wild-type mice, we found AMPAR silent synapses (EPSCs with an NMDAR component but no AMPAR component) in ∼37% of cases (6 of 16 neurons). However, in all 15 cells from 4E-BP2−/− mice, no such silent synapses were detected, and EPSCs were composed of AMPAR and NMDAR components (Fig. 3E). Thus, a reduced incidence of silent synapses in pyramidal cells of 4E-BP2−/− mice suggests that the facilitation of AMPAR synaptic transmission may involve activation of AMPARs at previously silent synapses.
Facilitation of AMPAR-mediated transmission may occur by postsynaptic insertion of GluA2-lacking Ca2+-permeable AMPARs (CP-AMPARs) with higher conductance at synapses (Liu and Cull-Candy, 2000; Plant et al., 2006). CP-AMPARs show voltage-dependent inward rectification resulting from intracellular polyamine block (Liu and Cull-Candy, 2000; Plant et al., 2006). Therefore, we examined voltage-dependent rectification of EPSCs using a spermine-containing (0.1 mm) intracellular solution. AMPAR-mediated EPSCs in cells of 4E-BP2−/− mice showed a linear current–voltage relation that was similar to cells from wild-type mice (Fig. 3F). The unaltered voltage–current relation and absence of inward rectification of EPSCs suggest that enhancement of AMPAR-mediated synaptic transmission in 4E-BP2−/− mouse does not involve a change in AMPAR subunit composition.
Changes in spine morphology in 4E-BP2 knock-out mice
Protein synthesis-dependent long-term potentiation at hippocampal synapses involves an increase in AMPAR-mediated synaptic currents that is associated with a sustained enlargement of dendritic spines (Matsuzaki et al., 2001, 2004; Nimchinsky et al., 2002; Tanaka et al., 2008). To determine whether facilitation of AMPAR-mediated synaptic transmission by removal of 4E-BP2 translation repression is associated with changes in spine density or morphology, we examined EYFP-transfected CA1 pyramidal neurons in cultured slices of wild-type and 4E-BP2−/− mice (Fig. 4). Three independent experiments were performed, and a total of 2003 spine protrusions were analyzed in cells of wild-type (866 protrusions, 38 neurons) and 4E-BP2−/− (1137 protrusions, 34 neurons) slices. First, we found no obvious alterations in the general dendritic arborization of transfected cells from 4E-BP2−/− mice compared with wild-type (Fig. 4A), but the density of spines was increased by 43 ± 8% in dendrites of neurons from 4E-BP2−/− mice (Fig. 4B). Second, morphology of spines was changed. Spine length was 22 ± 2% shorter in slices from 4E-BP2−/− mice relative to wild-type (Fig. 4C), and this was associated with a 52 ± 7% increase in the proportion of regular spines and a 67 ± 5% decrease in filopodia in 4E-BP2−/− mice (Fig. 4D). Thus, our analysis suggests that suppression of 4E-BP2 translation inhibition results, first, in an increase in spine density and, second, in a decrease in spine length resulting from a shift in spine shape to more regular spines and less filopodia in 4E-BP2−/− mice. These changes in spine density and shape are consistent with the heightened efficacy of AMPAR-mediated synaptic transmission in 4E-BP2−/− mice.
Figure 4.

Increase in spine density and alteration in spine morphology of pyramidal neurons in 4E-BP2−/− mice. A, Confocal images of representative EYFP-transfected CA1 pyramidal neurons (left) and enlarged dendritic segments (middle) in slice cultures from wild-type (top) and 4E-BP2−/− (bottom) mice. Individual spines are shown at a higher magnification (right). Scale bars: left, 25 μm; middle, 5 μm; right, 1 μm. B, Summary bar graph showing an increase in spine density in dendrites of neurons from 4E-BP2−/− mice. C, Cumulative plots of the distribution of spine length for each group. Inset, Summary bar graph of spine length, showing decreased spine length in neurons from 4E-BP2−/− mice. D, Summary bar graph of number of regular, long, and filopodia type of spine in cells of each group, showing an increase in regular spines and decrease in filopodia in neurons of 4E-BP2−/− mice. Data are mean ± SEM. *p < 0.05.
Selective increase in GluA1 and GluA2 translation in 4E-BP2 knock-out mice
Given the selective regulation of AMPAR-mediated synaptic function in 4E-BP2−/− mice, we hypothesized that the mechanisms by which 4E-BP2 translation repression control hippocampal synapses might be through selective translation of specific mRNAs. Schaffer collateral synapses contain AMPARs composed of GluA1 and GluA2 subunits (Arai et al., 1997; Tsuzuki et al., 2000), and the intact EPSC current–voltage relation in cells of 4E-BP2−/− mice is consistent with a GluA1/GluA2 subunit composition at synapses in these mice. Thus, we tested whether 4E-BP2 knockdown regulates the synthesis of AMPA receptor subunits by performing polysome profile analysis of hippocampal lysates from 4E-BP2−/− and wild-type mice (Fig. 5). Although general translation was not significantly altered in 4E-BP2−/− relative to wild-type hippocampi (Fig. 5A), GluA1 (Gria1) and GluA2 (Gria2) mRNA distribution was shifted to heavier polysome fractions (Fig. 5B) in 4E-BP2−/−, compared with wild-type mice, indicating enhanced translation. As expected, protein amounts of GluA1 and GluA2 were significantly increased, especially in synaptosomal fractions (Fig. 5C, E). Notably, there was no change in the polysome profile distribution or protein amounts of the scaffolding protein PSD95 (Dlg4), the NMDAR subunits GluN1 (Grin1), GluN2A (Grin2a), and GluN2B (Grin2b), or the immediate early gene Arc (Arg3.1) (Fig. 5B, C, E). Interestingly, there was no change in the total mRNA amounts for any of the genes examined (Fig. 5D), suggesting that transcription is not involved. Thus, by directly controlling the synthesis of a specific subset of AMPAR subunits within synaptic compartments, 4E-BP2 translation repression can regulate fast excitatory synaptic function and spine shape at hippocampal synapses.
Figure 5.
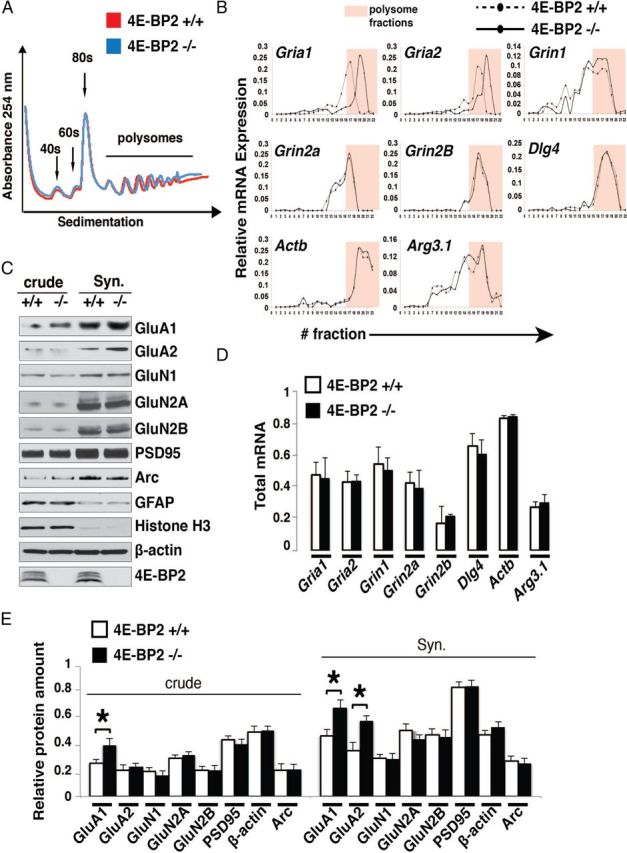
Selective increase in translation of GluA1 and GluA2 subunits in 4E-BP2−/− mice. A, Polysome profile analysis of hippocampi from 4E-BP2−/− and wild-type (4E-BP2+/+) mice. No change is observed in the general translation profile as measured by the 254 nm absorbance of different fractions. The 40, 60, 80 s, and polysome fractions are shown. B, Quantitative RT-PCR of polysome fractions from 4E-BP2−/− and wild-type (4E-BP2+/+) hippocampi. The relative abundance of GluA1, GluA2, GluN1, GluN2A/B, PSD95 (Dlg4), and Arc mRNAs is measured with quantitative RT-PCR across the different fractions of the polysome profiles from 4E-BP2−/− and wild-type mouse hippocampi (n = 4), showing selective shift of GluA1 and GluA2 mRNA to heavier polysome fractions. Polysome fractions are highlighted in pink. C, Representative Western blot analysis (n = 3) of total (crude) and synaptosomal (Syn.) fraction extracts from 4E-BP2−/− (−/−) and wild-type (+/+) mice. Lysates are probed for GluA1, GluA2, GluN1, GluN2A, GluN2B, PSD95, and Arc. GFAP and histone H3 are markers for the purity of the synaptosomes, whereas actin is used as loading control. Note selective increase in expression of GluA1 and GluA2 in 4E-BP2−/− relative to wild-type mice. D, Total mRNA amounts of GluA1 (Gria1), GluA2 (Gria2), GluN1 (Grin1), GluN2A (Grin2a), GluN2B (Grin2b), Arc (Arg3.1), and PSD95 (Dlg4) in hippocampal lysates from 4E-BP2−/− and wild-type (4E-BP2+/+) mice. No change is observed in the total amounts (n = 4). E, Quantification of Western blot analysis from 4E-BP2−/− and wild-type (4E-BP2+/+) mouse hippocampi. Relative protein amounts of GluA1 (increase crude: 34.3 ± 9.2%; Syn.: 31.4 ± 7.1%), GluA2 (increase Syn.: 38.7 ± 5.3%), GluN1, GluN2A, GluN2B, Arc, and PSD95 normalized to the actin signal are shown. *p < 0.02 (one-way ANOVA) (n = 3).
Alteration in long-term synaptic plasticity in 4E-BP2 knock-out mice
4E-BP2 is implicated in translational control during long-term synaptic plasticity, including L-LTP (Banko et al., 2005) and metabotropic glutamate receptor-mediated long-term depression (Banko et al., 2006). However, how these effects are related to the alterations in AMPAR-mediated synaptic function remains unknown. Thus, we explored the role of removal of 4E-BP2 translation repression in L-LTP, taking into account facilitation of AMPAR function, using whole-cell recordings from pyramidal cells in cultured slices of 4E-BP2−/− mice. We used a chemical induction protocol consisting of bath application of the adenylyl cyclase activator forskolin to induce cL-LTP, which shares essential properties of electrically induced L-LTP, including sensitivity to inhibitors of protein synthesis (anisomycin) and mTOR (rapamycin), and is elicited at a large proportion of pyramidal neuron synapses (Frey et al., 1993; Otmakhov et al., 2004; Kopec et al., 2006; Gobert et al., 2008). We used a 30 min bath application of forskolin (50 μm) in the presence of the phosphodiesterase inhibitor rolipram (0.1 μm) and in 0 mm Mg2+ to relieve voltage-dependent blockade of NMDARs (Fig. 6A). After a 90–210 min recovery period, whole-cell recordings of spontaneous AMPAR-mediated EPSCs (sEPSCs) were obtained from pyramidal neurons in forskolin- or sham-(DMSO) treated slices from 4E-BP2−/− or wild-type mice (Fig. 6B). As expected in sham-treated slices, sEPSC amplitude and frequency were greater in cells from 4E-BP2−/− compared with wild-type mice (Fig. 6C). Moreover, application of forskolin at 50 μm (FSK) produced a long-term potentiation of the amplitude (79 ± 19% increase) and frequency (96 ± 35% increase) of sEPSCs, relative to sham treatment in wild-type slices (Fig. 6B, C). Total charge transfer of sEPSCs (cumulative sum of individual sEPSC charge over a 10 min acquisition period), a measure reflecting both amplitude and frequency of sEPSCs, was increased by 184 ± 17% (Fig. 6C). The effects were dose-dependent, and applications of a lower concentration of forskolin (10 μm) did not elicit long-term changes in sEPSCs in wild-type slices (Fig. 6B, C). In contrast, in cells of 4E-BP2−/− slices, forskolin (50 μm) failed to induce changes in sEPSC frequency and only increased amplitude by 18 ± 8%, resulting in no change in sEPSC total charge transfer (Fig. 6C) and indicating an impairment of cL-LTP in pyramidal cells of 4E-BP2−/− mice. Interestingly, in cells of 4E-BP2−/− slices, a low concentration of forskolin (10 μm) was effective in potentiating sEPSC amplitude (26 ± 5%), frequency (99 ± 32%), and total charge transfer (143 ± 11%) relative to sham treatment. Thus, our sEPSC recordings in single neurons show both a lowering of the induction threshold for cL-LTP and an impairment of cL-LTP using normal induction stimulation in 4E-BP2−/− mice, indicating a critical role of suppression of 4E-BP2 translation inhibition in L-LTP induction at single synapses.
Figure 6.

Alterations in cL-LTP at single synapses in 4E-BP2−/− mice. A, Diagram of experimental protocol for cL-LTP induction. On experimental day, cultured slices from wild-type and 4E-BP2−/− mice were first placed in normal ACSF solution for a 15 min period and subsequently treated with forskolin (10 or 50 μm, 30 min duration; black bar) in the presence of rolipram (0.1 μm, gray bar) and 0 Mg2+-containing ACSF, or sham treatment (DMSO in ACSF). After a 90–210 min recovery period in normal ACSF, whole-cell recordings were obtained from CA1 pyramidal neurons. B, Representative continuous recordings of spontaneous synaptic activity (sEPSCs) in cells of wild-type (top) and 4E-BP2−/− (bottom) slices, illustrating that a long-term increase in sEPSC activity was induced by 50 μm forskolin treatment (right) in wild-type slices and by 10 μm forskolin treatment (middle) in 4E-BP2−/− slices, relative to sham-treatment (left). C, Summary bar graphs for all cells showing increased basal transmission in 4E-BP2−/− slices and cL-LTP of sEPSCs in different conditions. In sham-treated (S) 4E-BP2−/− slices (filled bars), sEPSC amplitude, frequency, and total charge were larger than in wild-type slices (wt, open bars). Data are mean ± SEM. #p < 0.05 (unpaired t tests). In wild-type slices, 50 μm forskolin treatment (F) induced long-term increases in sEPSC amplitude, frequency, and total charge relative to sham (S), but 10 μm forskolin (LF) did not. In 4E-BP2−/− slices, 10 μm forskolin (LF) induced long-term increases in sEPSC amplitude, frequency, and total charge relative to sham, but 50 μm forskolin (F) only increased amplitude. *p < 0.05 (one-way ANOVA). Number of cells in each group is indicated above bars.
To further explore the synaptic mechanisms implicated during cL-LTP, we examined next the AMPAR/NMDAR ratio of evoked EPSCs at 90–210 min after forskolin treatment in cells of 4E-BP2−/− and wild-type slices. In cells of wild-type slices, application of 50 μm forskolin resulted in a long-term increase of 108 ± 58% in the AMPAR-mediated component of EPSCs but not in the NMDAR-mediated component, resulting in an increase of 61 ± 24% in AMPAR/NMDAR EPSC ratio (Fig. 7). No long-term effects were found with a lower concentration of forskolin (10 μm). In cells of 4E-BP2−/− slices, first, as expected, the AMPAR component of EPSCs was increased by 62 ± 6% relative to wild-type slices after sham-treatment, but not the NMDAR component. In addition, in cells of 4E-BP2−/− slices, 50 μm forskolin failed to alter AMPAR or NMDAR-mediated components of EPSCs, resulting in no long-term change in the AMPA/NMDA EPSC ratio and indicating an impairment of cL-LTP using normal induction conditions after 4E-BP2 deletion. However, a low dose of forskolin (10 μm) induced a long-term increase of 75 ± 22% of the AMPAR component of EPSCs and resulted in a facilitation of AMPAR/NMDAR EPSC ratio by 69 ± 26% (Fig. 7), giving another indication of a facilitation of the threshold of cL-LTP induction after 4E-BP2 knockdown. Thus, despite a selective upregulation of AMPAR-mediated synaptic function by removal of 4E-BP2 translation inhibition, cL-LTP can be induced at a lower threshold and involves a further selective long-term increase in AMPAR-mediated synaptic function in cells of 4E-BP2−/− slices.
Figure 7.

Facilitation of AMPAR/NMDAR ratio of evoked EPSCs during cL-LTP in 4E-BP2−/− mice. A, Top, Representative recordings of evoked AMPAR and NMDAR-mediated components of EPSCs (2 × minimal stimulation threshold) in cells of wild-type slices at 90–210 min after treatment, with AMPAR component measured at holding potential of −70 mV and NMDAR component measured at + 40 mV, at points indicated with arrows (sham treatment, left). Arrowhead indicates time of stimulation. The ratio of AMPAR/NMDAR-mediated components of EPSCs was increased after application of forskolin (50 μm). Bottom: Example of evoked AMPAR and NMDAR-mediated components of EPSCs in 4E-BP2−/− slices, showing facilitation of AMPAR/NMDAR EPSC ratio only by low forskolin concentration (10 μm). B, Summary bar graphs for all cells showing effects of sham (S), 10 μm (LF), and 50 μm (F) forskolin treatment on EPSCAMPA, EPSCNMDA, and EPSCAMPA/EPSCNMDA ratio in wild-type (open bars) and 4E-BP2−/− (filled bars) slices. Note selective long-term increase in EPSCAMPA and EPSCAMPA/EPSCNMDA ratio. Data are mean ± SEM. *p < 0.05 (one-way ANOVA). Number of cells in each group is indicated above bars.
Relief from mTOR dependence of long-term synaptic plasticity in 4E-BP2 knock-out mice
Activation of mTOR is critical for the induction of L-LTP, and 4E-BP2 has been proposed to be a principal downstream effector of mTOR in regulating cap-dependent translation in long-term plasticity (Tang et al., 2002; Banko et al., 2005). In this scenario, genetic removal of 4E-BP2 should bypass activity-dependent mTOR activation and result in mTOR-independent cL-LTP induction because of the absence of 4E-BP2 repression of translation. Therefore, we examined the extent to which mTOR signaling contributes to long-term synaptic plasticity in wild-type and 4E-BP2−/− mice using the potent and selective active-site mTOR inhibitor, PP242 (Apsel et al., 2008; Choo and Blenis, 2009; Feldman et al., 2009; Dowling et al., 2010; Janes et al., 2010) (Fig. 8A). First, we confirmed the effective inhibition of mTOR signaling by PP242 by examining its effects on ribosomal S6 phosphorylation, a read-out of mTOR kinase activity, using Western blot analysis of hippocampal slices from wild-type and 4E-BP2−/− mice after forskolin and sham treatment (Fig. 8A). Applications of 50 μm forskolin in wild-type slices and 10 μm forskolin in 4E-BP2−/− slices increased S6 phosphorylation (139 ± 24% and 110 ± 5%, respectively; Fig. 8A). Forskolin-induced S6 phosphorylation was inhibited by PP242 in both wild-type and 4E-BP2−/− slices, confirming effective inhibitory actions on mTOR signaling in both conditions (Fig. 8A).
Figure 8.

Loss of mTOR dependence of cL-LTP in 4E-BP2−/− slices. A, Inhibition by PP242 of forskolin-induced ribosomal S6 phosphorylation. Left, Representative Western blot from hippocampal CA1 mini-slices showing increases in S6 phosphorylation by 50 μm forskolin application (F) in wild-type slices and by 10 μm forskolin (LF) in 4E-BP2−/− slices that were prevented by PP242 (F+PP; LF+PP). Right, Summary bar graphs for all experiments (n = 4 experiments) showing block by PP242 of forskolin-induced S6 phosphorylation (normalized to S6 level) in both wild-type (open bars) and 4E-BP2−/− (filled bars) slices. B, Diagram of induction and recording protocol. Cultured hippocampal slices from 4E-BP2−/− or wild-type mice were treated first with PP242 (2.5 μm; dark gray bar), or anisomycin (40 μm), in 4 mm Mg2+ ACSF for 30 min. Subsequently, 10 or 50 μm forskolin (black bar) was coapplied with rolipram (0.1 μm, light gray bar) and PP242 (2.5 μm) or anisomycin (40 μm) in 0 mm Mg2+ ACSF for a 30 min period. Slices were then exposed for 30 min to PP242 or anisomycin in 4 mm Mg2+ ACSF and for an additional 90–210 min recovery period in 4 mm Mg2+ ACSF. For controls, slices were treated with vehicle (DMSO) as sham treatment, or only PP242 or anisomycin. C, Recordings from representative cells in each group, showing long-term increase in sEPSC activity induced by forskolin (middle), which is blocked by PP242 (left) in wild-type slices (top) but not in 4E-BP2−/− slices (bottom). D, Summary bar graphs for all cells showing the block of forskolin-induced long-term changes in sEPSCs by PP242 in wild-type (open bars) but not 4E-BP2−/− (filled bars) slices. In wild-type slices, the increases in sEPSC amplitude, frequency, and total charge induced by 50 μm forskolin (F) relative to PP242 alone (PP), were prevented by coapplication of forskolin and PP242 (F+PP242). In 4E-BP2−/− slices, PP242 was ineffective in blocking the potentiation of sEPSC amplitude, frequency, and total charge induced by 10 μm forskolin (LF vs LF+PP), whereas the translation inhibitor anisomycin (40 μm; ANI vs LF+ANI; gray bars) was effective. Dotted lines in bar graphs correspond to respective sham-treated groups. Data are mean ± SEM. *p < 0.05. Number of cells in each group is indicated above bars.
We next examined the effects of PP242 on cL-LTP induction. In cells of wild-type slices, 2.5 μm PP242, a dose that effectively suppresses all mTOR activity and 4E-BP phosphorylation (Feldman et al., 2009; Dowling et al., 2010), prevented cL-LTP of AMPAR-mediated sEPSCs. The long-term increases in sEPSC amplitude (43 ± 3%), frequency (136 ± 68%), and total charge (230 ± 20%) induced by 50 μm forskolin were virtually abolished in the presence of PP242 (Fig. 8C, D), indicating that forskolin-induced cL-LTP was mTOR-dependent in wild-type slices, as previously observed using extracellular field recordings (Gobert et al., 2008). By itself, PP242 application did not alter sEPSC amplitude, frequency, and total charge compared with sham treatment (Fig. 8C, D), ruling out nonspecific effects on AMPAR-mediated synaptic transmission. In contrast, in cells of 4E-BP2−/− slices, cL-LTP induced by low-dose forskolin was not affected by PP242. The long-term increases in sEPSC amplitude (27 ± 5%), frequency (53 ± 16%), and total charge (142 ± 13%) induced by 10 μm forskolin were unchanged by PP242 application (Fig. 8C, D), suggesting that suppression of 4E-BP2 translation inhibition removes the requirement for mTOR activation in cL-LTP induction and, thus, that 4E-BP2 is the critical downstream effector of mTOR activation in cL-LTP induction. In addition, these data suggest that mTOR-mediated S6 phosphorylation does not play a critical role in cL-LTP in 4E-BP2−/− slices. Finally, to confirm that cL-LTP induced by low-dose forskolin in 4E-BP2−/− slices was translation-dependent, we examined its sensitivity to the protein synthesis inhibitor, anisomycin (40 μm). Effectively, application of low-dose forskolin in the presence of anisomycin produced no change in sEPSC amplitude, frequency, or total charge, compared with a control group treated with anisomycin alone (Fig. 8D).
Overall, our results indicate that mTOR and 4E-BP2-mediated translational control mechanisms contribute to the induction of cL-LTP by selectively regulating AMPAR function and are consistent with 4E-BP2 being the predominant downstream target of mTOR-dependent signaling pathways.
Discussion
Using transgenic and shRNA knockdown approaches, we demonstrate that removal of 4E-BP2 translational repression selectively upregulates AMPAR function assayed at single hippocampal synapses, without affecting NMDAR-mediated transmission. 4E-BP2 deletion also affected translation-dependent chemically induced L-LTP assayed at single synapses, lowering induction-threshold and removing mTOR signaling requirement while impairing induction by normal stimulation. Pyramidal cell dendritic spines showed alterations in density and shape, consistent with increased excitatory synaptic function. Finally, polysome profiles indicated that 4E-BP2 knock-out selectively increased translation of GluA1 and GluA2 mRNAs, without affecting translation of other synaptic mRNAs (GluN1, GluN2A/B, PSD95, Arc) or general translation. Thus, relief of 4E-BP2 translational repression selectively activates GluA subunit synthesis, upregulates AMPAR-mediated synaptic function, and alters long-term plasticity of hippocampal synapses.
4E-BP2 regulation of excitatory synapse function
We uncover mechanisms by which 4E-BP2-dependent translational control regulates excitatory synaptic function. Our finding that removal of 4E-BP2 translational repression augments basal synaptic transmission differs from previous observations using extracellular field recordings (Banko et al., 2005), probably resulting from high-resolution recording techniques used here to isolate unitary excitatory synaptic currents (Gkogkas et al., 2013). Furthermore, we extend previous findings of upregulation of basal transmission at hippocampal synapses after removal of 4E-BP2 translation inhibition (Bidinosti et al., 2010) and establish that AMPAR function is selectively upregulated. The mechanisms implicated appear largely postsynaptic with increased potency of AMPAR-mediated EPSCs, reduced proportion of silent synapses, and increased amplitude of AMPAR-mediated mEPSCs. Although AMPAR-mediated mEPSCs increased in frequency, the paired-pulse ratio of evoked EPSCs was unaltered, suggesting that release probability was unaffected. Contributions of presynaptic mechanisms cannot be ruled out, and recruitment of functional release sites with similar release probability may occur, as during translation-dependent L-LTP induced by cAMP (Bolshakov et al., 1997).
Removal of 4E-BP2 translational repression resulted in alterations in pyramidal cell dendritic spines in line with enhanced AMPAR synaptic function. The increase in spine density concurred with enhanced mEPSC frequency. Moreover, because shorter regular spines are associated with more synaptic AMPARs than long spines and because filopodia contain fewer synaptic AMPARs (Matsuzaki et al., 2001), the decrease in spine length, increase in proportion of regular spines, and reduction in filopodia agree with fewer silent synapses. We did not use deconvolution methods required for estimating spine head area or volume (Richards et al., 2005); however, such an approach might reveal additional insights on alterations of the spine fine structure by 4E-BP2 regulation. Thus, our spine analysis suggests that suppression of 4E-BP2 translational inhibition increases incorporation of AMPARs at synapses with associated changes in spine density and shape.
Our findings indicate that 4E-BP2 levels may be critical for regulating the strength of AMPAR synaptic function. First, effects on minimal stimulation-evoked EPSCs and mEPSCs gave two independent measures of enhanced unitary synaptic transmission in 4E-BP2−/− slices. Second, shRNA knockdown of 4E-BP2 in mature neurons of wild-type slices revealed similar effects on AMPAR mEPSCs. Finally, overexpression of wild-type 4E-BP2 in pyramidal cells rescues the augmented mEPSC transmission in 4E-BP2−/− slices (Bidinosti et al., 2010). Although it remains to be determined whether overexpression of 4E-BP2 in wild-type mice negatively regulates AMPAR function, the evidence so far suggests that 4E-BP2 expression level may inversely set the strength of AMPAR synapses. We found that, with shRNA knockdown of 4E-BP2, the effects on mEPSC amplitude and frequency were less than in 4E-BP2−/− mice. shRNA knockdown reduced 4E-BP2 levels in dissociated neuron cultures by ∼55%, suggesting that the discrepancy may be the result of incomplete 4E-BP2 knockdown with shRNA. However, possible developmental effects in 4E-BP2−/− mice cannot be ruled out in addition.
Our results also indicate that 4E-BP-dependent translational control of AMPAR function is cell-specific. Hippocampal inhibitory interneurons express both 4E-BP1 and 4E-BP2, the deletion of which does not alter basal AMPAR synaptic function. Minimal stimulation-evoked EPSCs and mEPSCs are intact in interneurons of 4E-BP1/2−/− knock-out mice (Ran et al., 2009). Thus, excitatory synaptic inputs onto pyramidal cells and interneurons may be differentially regulated in hippocampal synaptic networks by 4E-BP-dependent translational control.
4E-BP2 regulation of long-term plasticity of excitatory synapses
The effects of removal of 4E-BP2 translational repression on chemically induced long-term synaptic plasticity (Bolshakov et al., 1997; Otmakhov et al., 2004) extend a previous report of altered synaptic plasticity using field recordings in 4E-BP2−/− slices (Banko et al., 2005). We show that, at the single-cell/synapse levels, and taking into account increased basal transmission, 4E-BP2 deletion produces two effects on chemically induced L-LTP at AMPAR synapses: facilitation of L-LTP threshold and impairment of L-LTP induced by normal stimulation. Suppression of 4E-BP2 translation inhibition was insufficient to induce L-LTP because L-LTP was induced at a lower threshold even though basal transmission was enhanced, demonstrating an absence of L-LTP occlusion. Moreover, L-LTP facilitation was not the result of increased NMDAR function because NMDAR-mediated synaptic transmission was intact in slices of 4E-BP2−/− mice. However, consistent with a necessary role of 4E-BP2 translational control in L-LTP, cL-LTP of sEPSCs was blocked by the mTOR active site inhibitor PP242 in wild-type but not 4E-BP2−/− mice. Thus, elimination of 4E-BP2 translation repression removes the mTOR signaling requirement in cL-LTP. Interestingly, cL-LTP in 4E-BP2−/− mice was prevented by the translation inhibitor anisomycin (Fig. 8D) indicating that translation of plasticity-related mRNAs is derepressed in these slices. Similarly, translation-dependent mGluR LTD is mTOR-independent in 4E-BP2−/− mice (Banko et al., 2006). Translational control via mTORC1 implicates additional 4E-BP2-independent downstream pathways, including S6K and 5′ TOP-mRNAs (Hay and Sonenberg, 2004; Gobert et al., 2008; Laplante and Sabatini, 2012). Because L-LTP facilitation in 4E-BP2−/− mice is translation-dependent (Banko et al., 2005) and because PP242 did not impair L-LTP induced by weak stimulation while blocking S6 phosphorylation in 4E-BP2−/− slices, our findings suggest that L-LTP induction normally requires removal of 4E-BP2 translation repression via mTOR, independent of downstream translational control via S6K.
The impairment of cL-LTP induced with normal stimulation in 4E-BP2−/− mice appears a predominant effect on synapse function because hippocampal-dependent learning and memory are impaired in these mice (Banko et al., 2005). Impairment of cL-LTP induced by normal stimulation in 4E-BP2−/− slices was not the result of occlusion by enhanced basal synaptic transmission because cL-LTP could be induced by weak stimulation. Thus, excessive eIF4F complex formation and translation initiation by normal L-LTP-inducing stimuli in 4E-BP2−/− mice may trigger nonspecific translation of proteins detrimental to L-LTP maintenance (Banko et al., 2005), perhaps as a homeostatic mechanism to protect from excessive translation in pyramidal cells. Interestingly, L-LTP threshold is also lowered in hippocampal interneurons from 4E-BP1/2−/− mice but without impairment in plasticity induced by normal stimulation (Ran et al., 2009), further highlighting the cell-specific nature of 4E-BP translational control mechanisms in synaptic plasticity.
Selective increase in translation of GluA1/GluA2 subunits
4E-BP2 appears to regulate excitatory synaptic function by controlling recruitment of specific mRNAs in translating polysomes. Consistent with the notion that AMPARs at mature Schaffer collateral synapses are predominantly composed of GluA1/GluA2 heteromers (Traynelis et al., 2010), 4E-BP2 deletion triggered translation of GluA1 and GluA2 subunits, without affecting synthesis of GluN1 and GluN2A/B subunits of NMDARs, synaptic scaffolding protein PSD95, or Arc. Suppression of 4E-BP2 translation inhibition recruits GluA1 and GluA2 mRNAs to translating ribosomes, without changing transcription of these mRNAs or general translation. These effects are also consistent with the unchanged NMDAR function. Thus, 4E-BP2 translational control of synaptic function regulates specific mRNAs while preserving receptor subunit composition. A microarray screen of mRNAs whose translation is stimulated by BDNF via mTOR in cultured neurons similarly found increased translation of GluA1, but also augmented translation of GluN1, GluN3, and the scaffolding protein Homer2 (Schratt et al., 2004). These differences suggest that mTOR may differentially regulate translation of specific mRNAs via its distinct effector pathways 4E-BP, S6K, and 5′ TOP-mRNAs (Hay and Sonenberg, 2004; Gobert et al., 2008; Laplante and Sabatini, 2012; Thoreen et al., 2012), with translation of AMPAR mRNAs controlled via 4E-BP2, and NMDAR mRNAs via S6K or 5′ TOP mRNAs. Recent observations that 4E-BP deletion augments the translation of 5′ TOP mRNAs in fibroblasts (Thoreen et al., 2012) raise the possibility that 4E-BP knock-out may cause a general increase in protein synthesis through enhanced translation of 5′ TOP mRNAs. However, we have recently shown that no changes in general translation were detected in 4E-BP2−/− mice, as evidenced by [35S]Met/Cys-metabolic labeling of acute hippocampal slices and unaltered polysome/monosome ratio (Gkogkas et al., 2013).
mTOR is implicated in regulating dendrite growth (Jaworski et al., 2005; Kumar et al., 2005), spine formation (Kumar et al., 2005; Tavazoie et al., 2005; Chen and Napoli, 2008), and synaptic function (Tavazoie et al., 2005; Lee et al., 2011). mTOR regulation of dendrite morphogenesis implicates both 4E-BP and S6K effector pathways (Jaworski et al., 2005), whereas the downstream effector mechanisms of mTOR regulation of spine and synaptic function are not identified. Our results indicate that removal of 4E-BP2 translational repression by mTOR mediates the upregulation of AMPAR synaptic function and spine formation (Kumar et al., 2005; Chen and Napoli, 2008; Lee et al., 2011). Interestingly, downregulation of the upstream negative mTOR regulator TSC1/2 activates mTOR, yielding similar increases in AMPAR function without affecting release probability, but with different morphological alterations (decreased spine density and increased length) (Tavazoie et al., 2005). The different morphological effects may be attributed to TSC/mTOR regulation of S6K or 5′ TOP mRNAs effector pathways, or to mTOR-independent interactions of TSC with Rho GTPase and actin depolymerization factor cofilin (Tavazoie et al., 2005).
Finally, our findings demonstrate a correlation between increased GluA1 and GluA2 subunit translation and AMPAR synaptic function. The augmented GluA synthesis is probably not sufficient alone to increase synaptic activity (Hayashi et al., 2000). Indeed, additional polysome profile analysis and Western blot analysis revealed that translation of neuroligins is upregulated in 4E-BP2−/− mice, whereas siRNA downregulation of neuroligin 1 in 4E-BP2−/− mice rescued the increased excitatory synaptic activity and autism-like behavioral phenotype (Gkogkas et al., 2013). Thus, 4E-BP2 regulates excitatory synaptic function by controlling the specific recruitment of AMPAR subunit and synaptic adhesion molecule mRNAs to translating polysomes, representing a selective mechanism for regulation of AMPAR-mediated synaptic function during long-term plasticity and hippocampal-dependent memory processes. However, its dysregulation may lead to pathological conditions, such as autism spectrum disorders.
Footnotes
This work was supported by the Canadian Institutes of Health Research Operating Grant MOP-10848 to J.-C.L. and the Fonds de la Recherche en Santé du Québec (Groupe de Recherche sur le Système Nerveux Central). J.-C.L. is the recipient of the Canada Research Chair in Cellular and Molecular Neurophysiology. I.R. received a Fellowship from the Savoy Foundation. We thank Dr. Wayne Sossin for critical comments on earlier versions of the manuscript.
The authors declare no competing financial interests.
References
- Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y, Mizuguchi M, Takashima S. Developmental changes of glutamate receptors in the rat cerebral cortex and hippocampus. Anat Embryol. 1997;195:65–70. doi: 10.1007/s004290050025. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Merhav M, Stern E, Sonenberg N, Rosenblum K, Klann E. Behavioral alterations in mice lacking the translation repressor 4E-BP2. Neurobiol Learn Mem. 2007;87:248–256. doi: 10.1016/j.nlm.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Bellone C, Nicoll RA. Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 2007;55:779–785. doi: 10.1016/j.neuron.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Bidinosti M, Ran I, Sanchez-Carbente MR, Martineau Y, Gingras AC, Gkogkas C, Raught B, Bramham CR, Sossin WS, Costa-Mattioli M, DesGroseillers L, Lacaille JC, Sonenberg N. Postnatal deamidation of 4E-BP2 in brain enhances its association with raptor and alters kinetics of excitatory synaptic transmission. Mol Cell. 2010;37:797–808. doi: 10.1016/j.molcel.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Bourdeau ML, Morin F, Laurent CE, Azzi M, Lacaille JC. Kv4.3-mediated A-type K+ currents underlie rhythmic activity in hippocampal interneurons. J Neurosci. 2007;27:1942–1953. doi: 10.1523/JNEUROSCI.3208-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Napoli JL. All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARα. FASEB J. 2008;22:236–245. doi: 10.1096/fj.07-8739com. [DOI] [PubMed] [Google Scholar]
- Chen WS, Bear MF. Activity-dependent regulation of NR2B translation contributes to metaplasticity in mouse visual cortex. Neuropharmacology. 2007;52:200–214. doi: 10.1016/j.neuropharm.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, Imataka H, Cuello AC, Seidah N, Sossin W, Lacaille JC, Ron D, Nader K, Sonenberg N. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjevic K, Lacaille JC, Nader K, Sonenberg N. eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007;129:195–206. doi: 10.1016/j.cell.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, Kozma SC, Thomas G, Sonenberg N. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–1176. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille JC, Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert D, Topolnik L, Azzi M, Huang L, Badeaux F, Desgroseillers L, Sossin WS, Lacaille JC. Forskolin induction of late-LTP and up-regulation of 5′ TOP mRNAs translation via mTOR, ERK, and PI3K in hippocampal pyramidal cells. J Neurochem. 2008;106:1160–1174. doi: 10.1111/j.1471-4159.2008.05470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau G, Maher-Laporte M, Topolnik L, Laurent CE, Sossin W, Desgroseillers L, Lacaille JC. Staufen1 regulation of protein synthesis-dependent long-term potentiation and synaptic function in hippocampal pyramidal cells. Mol Cell Biol. 2008;28:2896–2907. doi: 10.1128/MCB.01844-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Huang CC, Hsu KS. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology. 2011;61:867–879. doi: 10.1016/j.neuropharm.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney RA. Physiological roles of spine motility: development, plasticity and disorders. Biochem Soc Trans. 2005;33:1299–1302. doi: 10.1042/BST0331299. [DOI] [PubMed] [Google Scholar]
- Nayak A, Zastrow DJ, Lickteig R, Zahniser NR, Browning MD. Maintenance of late-phase LTP is accompanied by PKA-dependent increase in AMPA receptor synthesis. Nature. 1998;394:680–683. doi: 10.1038/29305. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, Lisman J. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol. 2004;91:1955–1962. doi: 10.1152/jn.00941.2003. [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, Lacaille JC. A hebbian form of long-term potentiation dependent on mGluR1a in hippocampal inhibitory interneurons. Proc Natl Acad Sci U S A. 2001;98:9401–9406. doi: 10.1073/pnas.161493498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Raastad M, Storm JF, Andersen P. Putative single quantum and single fibre excitatory postsynaptic currents show similar amplitude range and variability in rat hippocampal slices. Eur J Neurosci. 1992;4:113–117. doi: 10.1111/j.1460-9568.1992.tb00114.x. [DOI] [PubMed] [Google Scholar]
- Ran I, Laplante I, Bourgeois C, Pépin J, Lacaille P, Costa-Mattioli M, Pelletier J, Sonenberg N, Lacaille JC. Persistent transcription- and translation-dependent long-term potentiation induced by mGluR1 in hippocampal interneurons. J Neurosci. 2009;29:5605–5615. doi: 10.1523/JNEUROSCI.5355-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA, Mateos JM, Hugel S, de Paola V, Caroni P, Gähwiler BH, McKinney RA. Glutamate induces the rapid formation of spine head protrusions in hippocampal slice cultures. Proc Natl Acad Sci U S A. 2005;102:6166–6171. doi: 10.1073/pnas.0501881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24:7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WB, Starck SR, Roberts RW, Schuman EM. Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron. 2005;45:765–779. doi: 10.1016/j.neuron.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topolnik L, Azzi M, Morin F, Kougioumoutzakis A, Lacaille JC. mGluR1/5 subtype-specific calcium signalling and induction of long-term potentiation in rat hippocampal oriens/alveus interneurones. J Physiol. 2006;575:115–131. doi: 10.1113/jphysiol.2006.112896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki K, Isa T, Ozawa S. Subunit composition of AMPA receptors expressed by single hippocampal neurons. Neuroreport. 2000;11:3583–3587. doi: 10.1097/00001756-200011090-00036. [DOI] [PubMed] [Google Scholar]