

Abstract
Cortical spreading depression (CSD) is associated with release of arachidonic acid, impaired neurovascular coupling, and reduced cerebral blood flow (CBF), caused by cortical vasoconstriction. We tested the hypothesis that the released arachidonic acid is metabolized by the cytochrome P450 enzyme to produce the vasoconstrictor 20-hydroxyeicosatetraenoic acid (20-HETE), and that this mechanism explains cortical vasoconstriction and vascular dysfunction after CSD. CSD was induced in the frontal cortex of rats and the cortical electrical activity and local field potentials recorded by glass microelectrodes, CBF by laser Doppler flowmetry, and tissue oxygen tension (tpO2) using polarographic microelectrodes. 20-HETE synthesis was measured in parallel experiments in cortical brain slices exposed to CSD. We used the specific inhibitor HET0016 (N-hydroxy-N′-(4-n-butyl-2-methylphenyl)formamidine) to block 20-HETE synthesis. CSD increased 20-HETE synthesis in brain slices for 120 min, and the time course of the increase in 20-HETE paralleled the reduction in CBF after CSD in vivo. HET0016 blocked the CSD-induced increase in 20-HETE synthesis and ameliorated the persistent reduction in CBF, but not the impaired neurovascular coupling after CSD. These findings suggest that CSD-induced increments in 20-HETE cause the reduction in CBF after CSD and that the attenuation of stimulation-induced CBF responses after CSD has a different mechanism. We suggest that blockade of 20-HETE synthesis may be clinically relevant to ameliorate reduced CBF in patients with migraine and acute brain cortex injuries.
Introduction
Cortical spreading depression (CSD) is a depolarization wave that propagates over the cerebral cortex at a speed of 3–5 mm/min (Leao, 1986). CSD is the mechanism underlying migraine with aura (Lauritzen, 1994) and is involved in progressive neural injury in stroke and acute brain trauma (Strong et al., 2002), intracerebral and subarachnoid hemorrhage (Dreier et al., 2006; Fabricius et al., 2006), and malignant stroke (Dohmen et al., 2008; Lauritzen et al., 2011). CSD induces a persistent reduction in cerebral blood flow (CBF), a persistent increase in the metabolic rate of oxygen (CMRO2), causing tissue hypoxia, and impaired neurovascular coupling (Piilgaard and Lauritzen, 2009; Lauritzen et al., 2011), which is moderately expressed in the otherwise normally perfused brain and severe in the acutely injured animal or human brain cortex (Dreier et al., 2009; Bosche et al., 2010; Lauritzen et al., 2011). Therefore, the identification of vascular mechanisms that improve O2 delivery and energy metabolism after CSD is of great interest.
Pretreatment with l-arginine, the substrate for nitric oxide synthase, ameliorates the reduction in CBF and the reduced vascular reactivity after CSD (Fabricius et al., 1995). This beneficial effect may relate to the interaction of nitric oxide (NO) with vascular smooth muscle-soluble guanylate cyclase leading to increased levels of guanosine 3′,5′-cyclic monophosphate (cGMP), but NO may also hinder vasoconstriction by inhibiting cytochrome P (CYP)-450 ω-hydroxylase activity, which catalyzes the conversion of arachidonic acid (AA) to 20-hydroxyeicosatetraenoic acid (20-HETE) (Sun et al., 2000; Roman, 2002; Mulligan and MacVicar, 2004). This mechanism is relevant because CSD causes a 10-fold increase in tissue AA levels (Lauritzen et al., 1990). In patients, the level of 20-HETE in the CSF is associated with delayed cerebral ischemia and poor outcomes after aneurysmal subarachnoid hemorrhage (Crago et al., 2011), and the beneficial effects of reducing 20-HETE synthesis may relate to increased collateral blood flow to the ischemic penumbra during ischemia or alterations in cerebral hemodynamic function after reperfusion.
Alternatively, one might consider the possibility that inhibitors of 20-HETE synthesis interfere with vascular effects produced by CSD in brain tissue. Here, we report that single episodes of CSD in the rat cerebral cortex induce time-dependent increases in cortical levels of 20-HETE with persistent reduction in CBF after CSD via direct assessment of 20-HETE synthesis in rat cortical brain slices under conditions that mimic in vivo experiments and in vivo data of colocalized changes in electrocorticography (ECoG) activity, evoked local field potential (LFP) responses, tissue oxygen tension (tpO2), and CBF. Inhibition of 20-HETE synthesis by HET0016 prevented both the CSD-induced increase in 20-HETE levels and the persistent reduction in basal CBF after CSD, whereas neurovascular coupling (i.e., the vascular response to increments in nerve cell activity) remained impaired. The data suggest that 20-HETE synthesis is directly involved in CSD-induced vasoconstriction.
Materials and Methods
Brain slice preparation.
Cortical slices (400 μm thick) were prepared from juvenile (postnatal age 18–22 d) Sprague Dawley rats (total n = 95 slices from 16 rats). Slices were recovered at 32°C for 30 min, followed by recovery at room temperature for 30 min as per standard protocols (Brenowitz et al., 2006). CSD was induced by transferring slices to a chamber containing high K+ (20 mm) solution at 32°C for 10 min. We confirmed in some experiments that CSD was consistently evoked with this protocol as we previously reported (Basarsky et al., 1998; Zhou et al., 2010), by imaging the intrinsic optical signal of the spreading wave. Slices were then collected 10, 20, 60, and 120 min after the beginning of CSD induction. Pairs of slices were homogenized in 100 μl of triphenylphosphine (0.01 mg/4 ml, Sigma), and 20-HETE was quantified using the 20-HETE ELISA kit (Detroit R&D Inc.). Aliquots (50 μl) of homogenate from each sample were freeze-dried using a lyophilizer, and 20-HETE ELISA results were normalized to the dry weight. To assess the effects of 2-amino-5-phospho-valeric acid (APV), a glutamate NMDA receptor antagonist, and HET0016 on 20-HETE levels, slices were prepared and recovered for 30 min at 32°C and 30 min at room temperature. Slices were then incubated with APV (100 μm) or HET0016 (100 nm) for 30 min at room temperature before CSD induction (Fig. 1).
Figure 1.
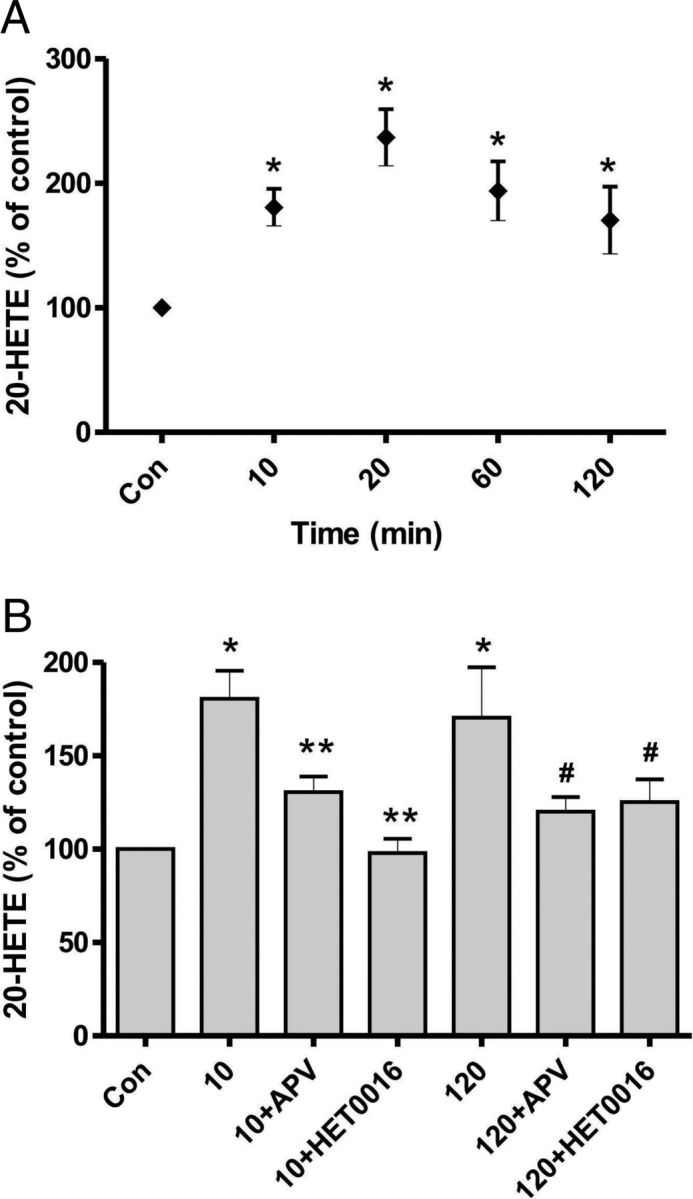
CSD triggers a prolonged increase in 20-HETE synthesis in cortical brain slices. A, After a single episode of CSD triggered by a short application of high external [K+], ELISA showed that 20-HETE synthesis peaked within 20 min and remained elevated up to 120 min. *p < 0.05 compared with control. The n values range from 5 to 9 samples per group. B, Preventing CSD by inhibiting NMDA receptors with APV blocked any increase in 20-HETE induced by high external [K+]. The increase in 20-HETE observed after CSD was blocked by HET0016, a CYP-450 enzyme inhibitor. The n values range from 6 to 18 samples per group. *p < 0.05 compared with control. **p < 0.05 compared with 10 min following CSD induction. #p < 0.05 compared with 120 min after CSD.
Animal anesthesia and preparation.
All experiments were conducted in compliance with the guidelines of the European Community on the Care and Use of Laboratory Animals and approved by the Danish National Ethics Committee. Eighteen adult male Wistar rats (250 ± 50 g; Charles River) were anesthetized with isoflurane (4% at induction and 2% during surgery; Vapor) in 50% O2 and 50% air. Catheters were placed in the left femoral artery and vein and continuously perfused with physiological saline. Continuous monitoring of arterial blood pressure and regularly collected blood samples for the determination of arterial pH, pO2, and pCO2 assured the maintenance of basic physiological parameters. Body temperature was maintained at 36°C to 37°C using a temperature-controlled heating pad (Harvard Apparatus). The trachea was cannulated for mechanical ventilation with a respirator to maintain arterial pH at 7.34 ± 0.01, PaCO2 at 33.7 ± 1.2 mmHg, and PaO2 at 115 ± 2 mmHg. The head was fixed in a stereotaxic frame. Two craniotomies were performed over homologous regions of the somatosensory cortex. The craniotomies were superfused with artificial CSF (composition in mm): 120 NaCl, 2.8 KCl, 22 NaHCO3, 1.45 CaCl, 1.0 Na2HPO4, and 0.876 MgCl2, pH 7.43, heated to 37°C and bubbled with 5% CO2, 21% O2, 74% atmospheric air. A small craniotomy was performed 2 mm rostral and 2 mm lateral to the right of bregma. After surgery, the anesthesia was changed to intravenous α-chloralose HBC complex dissolved in saline (0.5 g/ml) (bolus: 1.6 ml/kg intravenously; continuous infusion: 1.1 ml/kg per hour). The level of anesthesia was checked by observing arterial blood pressure during stimulation and by tail pinch. The experiments were performed after a postoperative recovery period, allowing a stable level of anesthesia, stable laser Doppler baseline, and an arterial blood pressure of 90–120 mmHg. Arterial blood pressure remained stable during electrical stimulation. Thus, CBF increases were unrelated to fluctuations in blood pressure. The software program Spike 2 Version 7.2a with a power 1401 interface (CED) was used for data sampling and offline calculations of LFP amplitudes. At the end of the experiment, the heart was stopped by an intravenous bolus injection of air and the zero level of recording parameters observed. Three of 10 animals in the control group and one of eight rats in the intervention group were excluded for analysis because of cerebral damage or bleedings.
Transcallosal fiber stimulation.
Recording electrodes and laser Doppler probes were placed as close as possible to each other in a region devoid of large vessels (>100 μm) at coordinates 3 mm posterior to bregma and 6 mm lateral to the right side of the midline, corresponding to the barrel cortex. A coated bipolar stainless steel electrode (SNEX 200, contact separation: 0.25 mm; RMI) was lowered 0.5 mm into the left sensory cortex, 3.0 mm posterior to bregma and 6.0 mm lateral to the midline, using stereotactic instruments. We used direct current stimulation (ISO-flex; AMPI) given as square wave pulses (0.2 ms, 2–6 mA) at 4 Hz in trains lasting for 15 s and at 2, 4, 6, 10, 20, and 30 Hz in trains lasting for 4 s. Each of the 15 s trains was run three times and each of the 4 s trains once with interstimulus intervals of 2 min, allowing both the EEG and basal CBF measurements to reestablish stable baseline values between stimulations (Piilgaard and Lauritzen, 2009). Cortical stimulation elicited episodes of CSD on the stimulated side. To circumvent this problem, we applied the NMDA receptor antagonist MK801 (1 mm) to the stimulated (left) cortex (Fig. 2A), which prevented the occurrence of CSD (Hoffmeyer et al., 2007). This procedure ensured reproducible tpO2, LFP, and CBF responses in the activated (right) cortex. Synaptic and vascular responses were stable for hours using these stimulus variables (Piilgaard and Lauritzen, 2009).
Figure 2.
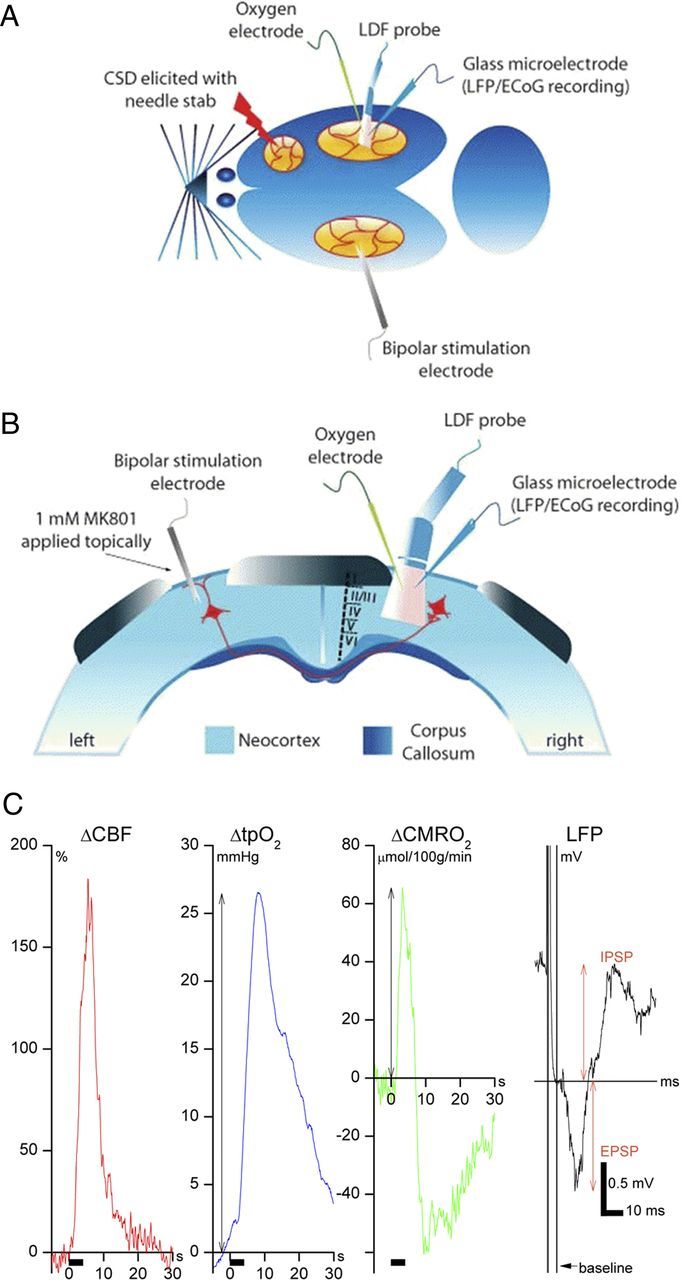
Experimental design. A, Rat brain viewed from above. CSD was elicited in the right frontal cortex with a brief needle stab and propagated into the right somatosensory cortex, monitored by a glass microelectrode recording ECoG activity. Using the same microelectrode, we recorded LFPs evoked by a bipolar stimulation electrode placed in the left somatosensory cortex. Baseline and evoked changes in CBF were recorded by LDF, whereas tpO2 was recorded using a Clark-type polarographic glass microelectrode. B, Rat brain in coronal section. The cortical surface of the stimulation side was superfused with 1 mm MK801 (NMDA receptor blocker) to avoid generation of seizure activity and CSD in that hemisphere, which was produced by placement of the stimulation electrode and transcallosal stimulation (Hoffmeyer et al., 2007). C, Evoked CBF responses were defined as the area under the curve, whereas tpO2 and CMRO2 responses were defined as peak amplitudes from stimulation onset to 20 s later. LFP recordings were EPSPs, defined as a negative peak amplitude, and IPSPs, defined as the positive peak amplitude. All evoked responses were normalized to the pre-CSD value for 20 or 30 Hz stimulation within each animal.
CSD elicitation.
CSD was elicited with the stab of a fine needle coated with potassium acetate (Amemori and Bures, 1988) in the right frontal cortex 2 mm rostral to bregma and 2 mm lateral to bregma, avoiding cortical bleeding. The CSD propagated into the right somatosensory cortex, monitored by a glass microelectrode recording ECoG activity at a distance of ∼6 mm from the site of elicitation.
Drug protocol.
Three groups of animals were used for in vivo experiments: control recordings taken immediately before and after CSD (n = 6), recordings taken during a control period after 1 h of incubation with topically applied CSF and after CSD (n = 4, sham), and recordings taken during a control period after 1 h of incubation with HET0016 to block 20-HETE synthesis and after CSD (n = 8). HET0016 is a selective inhibitor of ω-hydroxylase, which converts AA to 20-HETE, with a IC50 < 15 nm in renal microsomes, which is 100 times lower than any other inhibitor of this pathway (Miyata et al., 2001; Kehl et al., 2002). HET0016 was dissolved in ethanol and diluted in artificial CSF to a concentration of 1 μm and 0.1% ethanol (Liu et al., 2008). This concentration inhibits the formation of 20-HETE in renal microsomes by 90% without inhibiting epoxyeicosatrienoic acid formation (Kehl et al., 2002). Recordings were taken in all groups in 7 min periods up to 20, 50, 80, and 110 min after CSD.
Laser Doppler flowmetry.
CBF was continuously recorded using laser Doppler flowmetry (LDF). The LDF probe was at a fixed position 0.3 mm above the pial surface in a region devoid of large blood vessels (wavelength, 780 nm; fiber separation, 250 mm; Periflux 4001 Master, Perimed). With this wavelength and fiber separation, changes in CBF were recorded to a cortical depth of 1 mm (Fabricius et al., 1997). After stable baseline recordings were obtained, the probe was left for the duration of the experiment. The signal was A/D converted and recorded using the CED power 1401 interface and CED Spike 2 Version 7.2a software (100 Hz digital sampling rate). The LDF monitor displays blood flow readings in arbitrary units that do not allow for measurement of CBF in terms of absolute values, but the method is valid in determining relative changes of CBF during moderate flow increases (Fabricius and Lauritzen, 1996).
Measurement of tissue partial pressure of oxygen.
We used a modified Clark-type polarographic oxygen microelectrode (OX-10; Unisense) with a guard cathode for tpO2 measurements. The advantage of this electrode type is its small tip size (10 μm) and its built-in guard cathode, which removes all oxygen from the electrolyte reservoir enabling us to measure tpO2 over time and among different treatment conditions with excellent long-term stability (signal drift, 0–0.5% per hour). The field of sensitivity is a sphere two times the tip diameter. The electrodes used in this study were constructed so that 90% of the response time was <1 s and the stirring sensitivity nearly negligible at <0.8%. Oxygen microelectrodes responded linearly to changes in oxygen concentration. Each electrode was calibrated before each experiment in both air-saturated Milli-Q water and oxygen-free solution consisting of 0.1 m sodium l-ascorbate and 0.1 m NaOH dissolved in saline. Mean tpO2 ± SEM was 18.3 ± 1.7 mmHg (n = 14). The oxygen electrode was vertically inserted stepwise into the somatosensory cortex and positioned at the same cortical depth as the glass microelectrode (300–600 μm). The distance between the two electrodes was <200 μm. The electrodes and LDF probe were kept in the same position throughout the experiment and recorded signals from the same column of tissue with spatial overlap, even if the sampling volumes of the techniques differed. No linear drift in baseline occurred during the experiments. The oxygen electrodes were connected to a high-impedance Pico-amperemeter (PA 2000; Unisense) that sensed the currents of the oxygen electrodes. Signals were A/D converted and recorded at 100 Hz (Power 1401 and Spike 2 Version 7.2a; CED).
Electrophysiology.
We used single-barreled glass microelectrodes filled with isotonic saline (impedance, 2–3 MΩ; tip, 2 μm). Extracellular LFPs were recorded with a single electrode at a depth of 300–600 μm in the right somatosensory cortex. An Ag/AgCl ground electrode was placed in the well. The preamplified (×10) signal was A/D converted, amplified (×100), bandpass filtered at 0.1–5000 Hz, and digitized using the power 1401 hardware (CED) connected to a PC running the Spike 2 Version 7.2a software. The digital sampling rates for LFP and ECoG were 20 and 1 kHz, respectively.
Calculation of CMRO2.
To evaluate the effect of stimulation on oxygen metabolism, we calculated CMRO2 from simultaneous recordings of tpO2 and CBF. Baseline values of tpO2 and CBF were taken as the mean of a 20 s period obtained before the onset of stimulation. These values were then combined with values for CBF and CMRO2 reported in the literature using α-chloralose anesthesia (53 ml/100 g of per minutes and 219 mmol/100 g per minutes, respectively) (Zhu et al., 2002) to calculate the corresponding value of L, the effective diffusion coefficient of oxygen in brain tissue. CBF and tpO2 recordings were used to calculate CMRO2 (Gjedde et al., 2005). The relationship between the three variables is as follows:
 |
where P50 is the half-saturation tension of the oxygen–hemoglobin dissociation curve, h is the Hill coefficient of the same dissociation curve, and Ca is the arterial oxygen concentration. Standard values used in the calculations were as follows: P50 = 36 mmHg, h = 2.7, and Ca = 8 mmol/ml. We calculated an L value of 4.18 mmol/100 g per minutes per mmHg.
Data and statistical analysis.
For all analyzed parameters, we found no difference between values before and after incubation with CSF (n = 4) or in the control dataset for the group in which CSD was elicited just after control recordings (n = 6). Thus, we were able to merge the two groups and use the data in both groups just before CSD as our CSF control group (n = 10). Because of changes in the strength of the stimulation between the different animals, all evoked responses were normalized to the value from the 20 or 30 Hz stimulation during the control period for each animal. Simultaneously recorded LFP, CBF, and tpO2 signals were used for analysis. The LFP amplitudes were calculated for each stimulation period as the difference between the baseline and first negative peak (EPSP, EPSP), and between the baseline and first positive peak (IPSP, IPSP) (Creutzfeldt, 1995). Peak values were summed for each stimulation train. Stimulation-induced CBF responses (ΔCBF) were converted from arbitrary units to percentage change from baseline (20 s before stimulation). We used average values from stimulation onset to 20 s after stimulation onset. The tpO2 values were converted from pA to mmHg. For tpO2 and CMRO2 responses, a running average was applied and evoked responses defined as the peak value within the first 20 s of stimulation (Fig. 2C). Baseline values for CBF, tpO2, and CMRO2 were calculated as the average of 20 s before stimulation onset. A running average was applied to the resting baseline values for CBF, tpO2, and CMRO2 for periods after CSD and calculated as the average (CBF), minimum value (tpO2), or peak value (CMRO2) of 7 min periods and compared with the average value at 2 min before CSD onset. This procedure was repeated for almost every time period, except for a few instances for which we used 3 min periods because of noise problems. Group values were obtained by summing individual animal values and dividing by the number of animals. Custom-made analysis programs based on Matlab 7.3 (MathWorks) were used to calculate CMRO2 as already described. R Version 2.12.1 (R development core team) was used for statistical analyses on log-transformed data. Linear regression was used to examine the frequency dependence of neurovascular and neurometabolic coupling. Mixed models for means and unpaired t tests were used for statistical analysis when comparing between groups; ANOVA and paired t tests were used when comparing within groups. Values are expressed as mean ± SEM. Values were considered significant at p < 0.05.
Results
20-HETE synthesis in brain slices
We measured the 20-HETE levels in rat brain slices after CSD induction using a 20-HETE ELISA kit. The 20-HETE level in the control condition (Fig. 1A) was 3.9 ± 0.7 ng/ml. Imaging the intrinsic optical signal revealed that application of 20 mm K+ for 10 min consistently induced a single episode of CSD in slices (data not shown) as previously shown (Zhou et al., 2010). The 20-HETE levels were significantly increased after CSD was triggered by 20 mm K+. The increase was immediate (at 10 min), peaked at 20 min after CSD, and remained elevated up to 120 min (Fig. 1A). The percentage increase at each time point (compared with control, n = 5) measured after CSD was 180.66 ± 14.90% at 10 min (n = 9, p < 0.05), 236.82 ± 22.74% at 20 min (n = 8, p < 0.05), 193.87 ± 23.64% at 60 min (n = 6, p < 0.05), and 170.35 ± 27.05% at 120 min (n = 6, p < 0.05). In contrast, slices incubated for 120 min without CSD induction showed no 20-HETE elevation compared with control (93.04 ± 9.16% (n = 3, p > 0.05), data not shown). We also measured 20-HETE 10 and 120 min after 20 mm K+ perfusion in the presence of the NMDA receptor antagonist APV (100 μm); the increase in 20-HETE was significantly blocked by APV (Fig. 1B; 10 min + APV: 130.53 ± 8.42%, n = 12, p < 0.05; 120 min + APV: 120.00 ± 7.99%, n = 11, p < 0.05). Application of APV did not affect 20-HETE levels in control slices (116.16 ± 8.64%, n = 8, p > 0.05, data not shown).
Finally, we examined the effect of a specific inhibitor of 20-HETE synthesis, HET0016 (100 nm), on the CSD-induced production of 20-HETE. The increase in 20-HETE was significantly inhibited in the presence of HET0016 (Fig. 1B; 10 min + HET0016: 97.86 ± 7.65%, n = 10, p < 0.05; 120 min + HET0016: 125.33 ± 12.14%, n = 12, p < 0.05). In contrast, HET0016 had no effect on 20-HETE levels in control slices (93.37 ± 14.47%, n = 4, p > 0.05, data not shown). These data indicate that 20-HETE is generated by a single episode of CSD and remains elevated for 120 min in rat brain slices.
Baseline and evoked responses after incubation with HET0016 under control conditions
In agreement with previous studies, we found no effect of HET0016 on resting CBF at baseline (Liu et al., 2008), and HET0016 also had no effect on baseline CMRO2. In addition, we observed no effects on any of the evoked responses after 1 h of incubation with HET0016 in animals pretreated with HET0016 (n = 8). This observation suggests that HET0016 does not interfere with the basic control mechanisms of baseline CBF and CMRO2 or with evoked synaptic, vascular, or metabolic responses.
Acute changes in CBF, CMRO2, and tpO2
A small but significant decrease in baseline CBF after the needle stab preceded the onset of CSD as described previously (Fabricius et al., 1995; Chuquet et al., 2007). CBF decreased to 67.2 ± 2.8% of baseline values (p = 7.3 × 10−6) for the control group (n = 10), and no difference was observed between control and intervention animals in which CBF decreased to 69.0 ± 1.8% of baseline values (n = 8) (Fig. 3B,D). CSD onset was defined by a loss of spontaneous ECoG activity and large DC shift, indicating that the depolarization wave reached the somatosensory cortex where recordings were taken (Fig. 3A), which happened 1–2 min after the needle stab. At the same time as the DC shift, we observed a large increase in CBF that peaked at 352.8 ± 37.2% of baseline values for the control group (p = 1.4 × 10−6). The peak value of this hyperemia lasted for a couple of minutes and was not different in the intervention group (358.7 ± 40.0% of baseline; Fig. 3D). Similar to our previous study, we found both a tpO2 peak from a pre-CSD value of 16.5 ± 3.1 mmHg to 31.6 ± 3.9 mmHg and a tpO2 decrease to 2.9 ± 1.5 mmHg in the control group (Piilgaard and Lauritzen, 2009). No differences were found in the amplitudes of these changes between the two groups, and we did not find a difference between the groups when we looked at the significant increase in CMRO2 associated with CSD (Fig. 3D).
Figure 3.
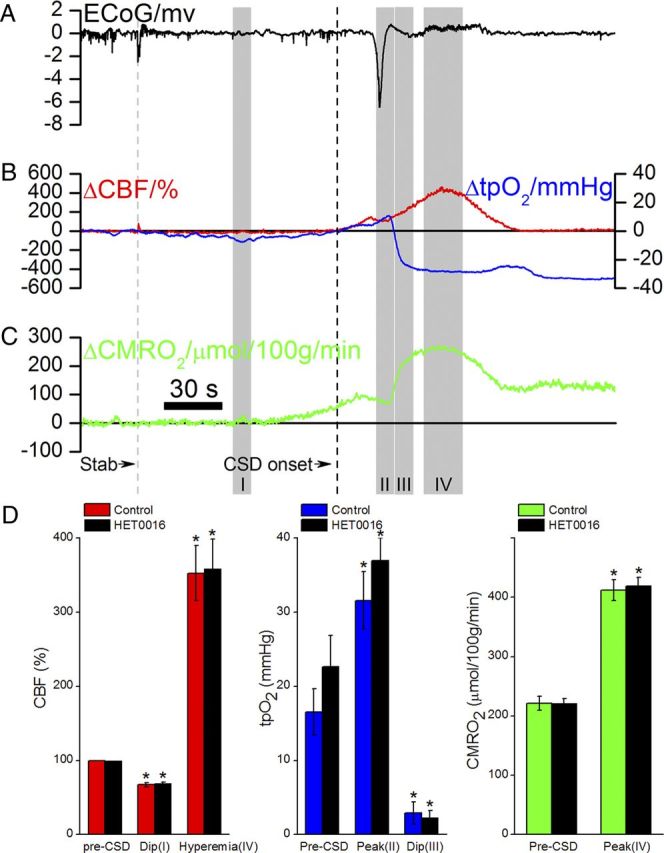
Vascular and metabolic changes during a single episode of CSD. A–C, Data are from one animal. Average data from all animals are displayed in D. A, ECoG showing extinction of spontaneous electrocorticographic activity during CSD and a large DC shift, both returning to normal after a few minutes. B, CSD induced a small decrease followed by increased CBF, whereas tpO2 showed a biphasic response with an initial increase, followed by a persistent decrease below pre-CSD values. C, CMRO2 increased markedly during CSD for 2 min. D, Group values for maximum changes in CBF, tpO2, and CMRO2 accompanying CSD. Time indications on the abscissa correspond to time periods marked in gray in A–C. *p < 0.05, compared with pre-CSD values in same group. No significant difference was found between the control and HET0016 groups. Data are mean ± SEM.
Persistent changes in baseline CBF, tpO2, and CMRO2 1–2 h after CSD
We measured changes for periods of 7 min at 20, 50, 80, and 110 min after CSD and compared them with the average baseline values before CSD (Fig. 4A). In the time interval 13–20 min after CSD, CBF was significantly decreased (−17.9 ± 3.7% of baseline values; p = 0.001) for the control group (n = 10). For rats that received HET0016, the baseline CBF remained constant for the entire 2 h period after CSD (p = 0.615, n = 8), and this was significantly different from untreated rats exposed to CSD (p = 0.005). We observed a significant decrease in resting baseline tpO2 (−9.8 ± 4.8 mmHg, p = 0.014), but only a tendency for increased CMRO2 (10.5 μmol/100 g/min ± 14.5 μmol/100 g/min, p = 0.063), after CSD. HET0016 had no effect on resting baseline tpO2, which remained low after CSD, despite constant CBF and nonsignificant increase in CMRO2 after CSD (Fig. 4B,C).
Figure 4.
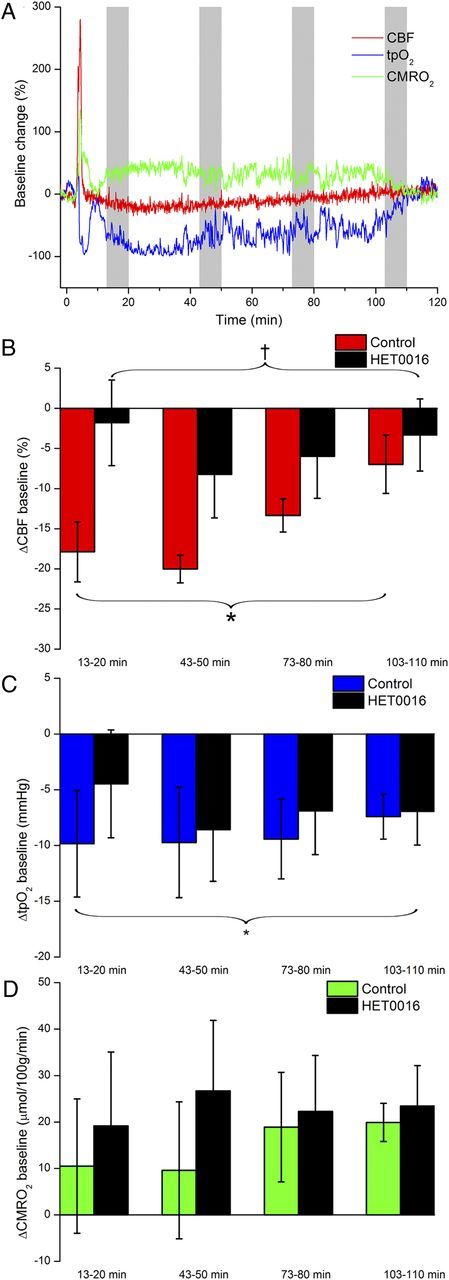
Time course of long-term baseline changes in CBF, tpO2, and CMRO2 after a single episode of CSD. A, CBF and CMRO2 increased in the first minutes after onset of CSD, followed by a persistent decrease in CBF and TpO2 and increase in CMRO2, shown here as original data for one control animal. B, After a single episode of CSD, CBF decreased with a peak at 13–20 min and returned toward pre-CSD levels within the following 2 h. Pretreatment with HET0016 prevented this reduction in CBF. C, The CBF reduction was accompanied by reduced tpO2, which was unaffected by HET0016, despite amelioration of the post-CSD reduction in CBF. D, CMRO2 showed a tendency to increase in the post-CSD period, but this did not reach significance. No difference was observed between control and HET0016-treated rats. Calculation of the group baseline changes after CSD was based on average values taken from time periods indicted by gray columns in A. Abscissa shows time after CSD in minutes. *p < 0.05 compared with pre-CSD values in the same group. †p < 0.05 compared with the control group. Data are mean ± SEM.
Evoked CBF responses before and after CSD
All evoked responses were normalized to the pre-CSD values in the same animal. We tested two stimulation paradigms: stimulations at 4 Hz for 15 s (Fig. 5) and stimulation trains of five different frequencies for 4 s each (Fig. 6). The 2 paradigms examined stimulation-induced responses under steady-state conditions (15 s) and during a transient condition (4 s). For both paradigms, we saw a significant drop in evoked CBF and tpO2 responses after CSD. Evoked responses to 4 Hz stimulations for 15 s decreased from 46.2 ± 5.3% to 16.5 ± 2.1% for CBF (p = 4.7 × 10−9) and from 52.8 ± 11.1% to 17.2 ± 7.3% for tpO2 (p = 0.023). No difference was found in the magnitude of the reduction of the evoked responses between the control and intervention groups. Evoked CBF responses gradually returned toward pre-CSD levels >2 h, whereas evoked tpO2 responses remained attenuated throughout the experiment. Stimulations at 4 Hz for 15 s led to an increase in evoked CMRO2 responses (55.4 ± 6.1% to 67.3 ± 17.0%) after CSD in control animals (p = 0.040), whereas the intervention group did not show significant changes after CSD. By using stimulation trains of different frequencies, we observed increases in evoked CMRO2 responses in control animals (p = 0.033) and significantly reduced responses in intervention animals (p = 1.2 × 10−8). The two groups were significantly different (p = 0.023) and remained significant throughout the experiment.
Figure 5.

Changes in evoked CBF, tpO2, and CMRO2 responses after CSD using stimulation at 4 Hz for 15 s. HET0016 treatment had no effect on any of the evoked parameters. A, Evoked CBF responses decreased after CSD with no difference between control and intervention groups (control = 10 rats, HET0016 = 8 rats). Responses returned toward control values over the next 2 h. B, Evoked tpO2 responses also decreased after CSD but showed no recovery over the next 2 h and no differences between the control and intervention groups. C, Evoked CMRO2 responses were larger after CSD in the control group compared with the group treated with HET0016. *p < 0.05 compared with pre-CSD values in the same group. Abscissa shows the time after CSD in minutes. Data are mean ± SEM.
Figure 6.
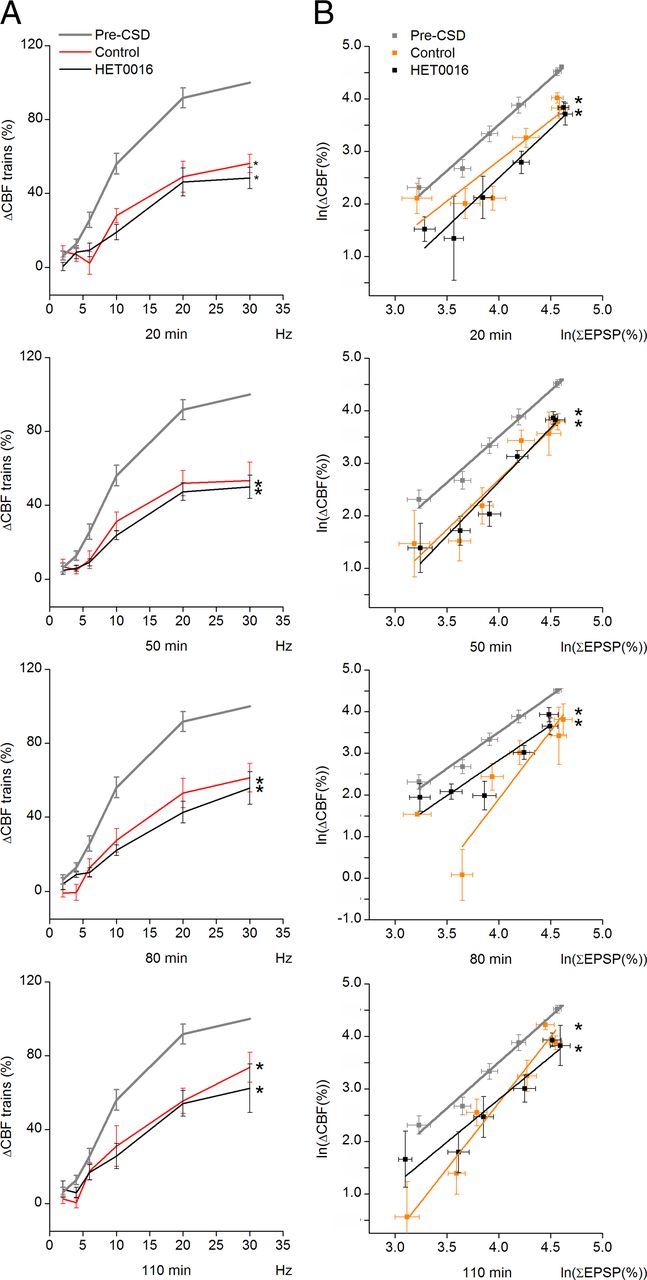
Changes in neurovascular coupling after CSD using different stimulation frequencies. Panels show findings for different times after CSD; the periods are marked in gray in Figure 4A. A, CSD decreased the CBF responses at all times up to 110 min and all frequencies (2–30 Hz). The reduction was unaffected by HET0016 treatment. No difference was found in the time course of recovery toward pre-CSD values between the two groups, and CBF responses did not return to pre-CSD values by 110 min. *p < 0.05 compared with pre-CSD values in same group. Abscissa shows frequencies in Hz (control = 10 rats, HET0016 = 8 rats). B, Neurovascular coupling (i.e., the relationship between synaptic activity as indicated by the summed EPSP responses [∑EPSP] and CBF) was altered after CSD, and this was unaffected by HET0016. Each panel shows the linear regression of log-transformed data for synaptic activity (∑EPSP) versus ΔCBF. *p < 0.05 compared with pre-CSD values in the same group. Data are mean ± SEM.
Frequency dependence of evoked synaptic responses before and after CSD
Synaptic transmission in the transcallosal pathway is glutamatergic and results in activation of postsynaptic glutamate and GABA receptors (Kawaguchi, 1992). Transcallosal stimulation in trains of 4 s duration evoked reproducible, frequency-dependent LFP responses in the contralateral homologous somatosensory cortex. The recorded LFP responses (Fig. 2C) were biphasic, consisting of an initial negative potential indicating synaptic excitation (i.e., EPSP) and a longer-lasting positive potential indicating synaptic inhibition (i.e., IPSP). The sum of the EPSP and IPSP amplitudes increased as a function of the stimulation frequency, whereas the IPSP became undetectable at stimulation frequencies >20 Hz. After CSD, the sum of the EPSP and IPSP amplitudes increased as a function of the stimulation frequency. For the EPSP, we found no effect of time on the size of the responses after CSD or a difference between the control and intervention groups. The IPSP responses decreased in size after CSD and continued to decrease throughout the 2 h of recording (p = 0.0001) in the control group. This decrease in the IPSP after CSD was not observed in the intervention group, which significantly differed from the control group (p = 0.0004, n = 8).
Neurovascular and neurometabolic coupling before and after CSD
Figure 6A compares the stimulus- and frequency-dependent changes in CBF after CSD in control and intervention animals. Mixed-model analysis of the evoked CBF responses versus frequency revealed a significant difference between pre- and post-CSD (p = 0.0001), with gradual return toward pre-CSD values. Our results suggest alteration of evoked vascular responses after CSD. We then assessed neurovascular coupling after CSD, defined as the correlation between excitatory synaptic activity and CBF. We calculated the summed EPSP amplitudes (ΣEPSP) for each stimulus and experimental condition as an indicator of the total excitatory synaptic activity during the stimulation period and plotted this variable versus the observed changes in CBF. A significant change in neurovascular coupling (ΔCBF vs ΣEPSP) was found in response to CSD (p = 0.042) with no difference between groups. Neurometabolic coupling showed no significant changes after CSD for CMRO2 versus ΣEPSP in the control group, whereas the intervention group exhibited impaired neurometabolic coupling (p = 0.018). These findings suggest that neurovascular, but not neurometabolic, coupling is affected after an episode of CSD under normal circumstances, and that blocking the synthesis of 20-HETE reduces neurometabolic coupling responses after CSD.
Discussion
In this study, we showed that CSD triggers a persistent increase in the cortical levels of 20-HETE that is almost congruent with the time course of the reduction in baseline CBF, and that both the increase in 20-HETE levels and the reduction in CBF were blocked by HET0016, a specific blocker of 20-HETE synthesis (Miyata et al., 2001; Kehl et al., 2002). The reduced neurovascular coupling after CSD was unaffected by HET0016, suggesting that different mechanisms control baseline CBF and neurovascular coupling after CSD. CSD triggered an increase in 20-HETE synthesis in brain slices with a distinct time course, showing maximal elevations at 10–20 min after CSD onset. The elevations were specific for the induction of CSD because the 20-HETE levels remained constant when the NMDA receptor antagonist APV, which blocks CSD (Lauritzen and Hansen, 1992), was added to the medium. Furthermore, the increase in 20-HETE synthesis was explained by increased activity of the CYP-450 enzyme, as the specific inhibitor HET0016 (Miyata et al., 2001) blocked the CSD-induced increase in 20-HETE levels. The increments in total tissue 20-HETE levels represented a doubling of control values, which is comparable to, but smaller than, the 10-fold increase in 20-HETE in the CSF induced by subarachnoid hemorrhage in rats (Kehl et al., 2002) and the sixfold increase in rat cerebral arteries during increases in systemic blood pressure (Gebremedhin et al., 2000).
In an attempt to translate from brain slice to in vivo data, we measured 20-HETE concentrations in brain samples taken during in vivo experiments using a protocol that previously was used for sampling brain tissue for measurements of AA release (Lauritzen et al., 1990) However, a CSD in vivo did not evoke sufficient accumulation of 20-HETE for it to be detectable with a high enough signal-to-noise ratio in the sample, indicating a higher variability of 20-HETE for the in vivo than for the slice preparation. In vivo, a temperature gradient during the freezing process may exist in the tissue within the first seconds that may allow for undesired breakdown of 20-HETE. Other sources of errors may include swelling of frozen brain, sample degradation during storage and shipment, and washout of 20-HETE with the blood in the post-CSD period.
In agreement with previous studies, our data showed that HET0016 had no effect on basal or evoked CBF responses (Gebremedhin et al., 2000; Kehl et al., 2002; Liu et al., 2008). In addition, we report that blockade of 20-HETE synthesis had no effect on basal or evoked synaptic activity or CMRO2. These findings are consistent with the in vivo level of 20-HETE in the rat cerebral cortex, being below the minimum limits for detection during rest (Renic et al., 2009) and with very low levels of AA, the substrate for CYP-450, under control conditions (Lauritzen et al., 1990).
We hypothesize that the mechanism for CSD-induced AA release relates to glutamate receptor-induced increases in cytosolic Ca2+ levels in neurons and astrocytes that stimulates phospholipases A and C, which catalyze the breakdown of phospholipids and maximize AA concentration increases during the first 4–5 min of CSD (Lauritzen et al., 1990). Blockade of phospholipase A2 may ameliorate the initial arteriole constriction, preceding the DC shift in young rats (Chuquet et al., 2007). This effect could be consistent with blockade of AA formation and subsequent 20-HETE synthesis, but our data indicate that the initial hypoperfusion was unaffected by blockade of 20-HETE synthesis, suggesting that different substances mediate the initial vasoconstriction in very young and mature rats. In rats, CSD causes a 2–3 min increase in CBF after the initial vasoconstriction (Busija et al., 2008), whereas CSD triggers profound vasoconstriction in mice (Ayata et al., 2004). The increase in CBF in rats may change if multiple CSD episodes are elicited (Fabricius et al., 1995) and with the vascular supply of the tissue, specifically the perfusion pressure (Sukhotinsky et al., 2010). In disease states, such as subarachnoid hemorrhage, the polarity of the vascular reaction during CSD may change to strong vasoconstriction reminiscent of the strong vasoconstriction in mice (Ayata et al., 2004). This produces localized damage in the cerebral cortex in rats and patients, without evidence of a vasodilator phase (Dreier et al., 1998). We found that the increase in CBF (and CMRO2) during CSD was unaffected by blockade of 20-HETE synthesis. This finding suggests that 20-HETE synthesis does not play a major role in the net vascular reaction during the first minutes of CSD, which may relate to the involvement of multiple signaling pathways in the acute phase of CSD (Busija et al., 2008).
The time course of changes in the concentration of 20-HETE in cortical slices correlated with the persistent decrease in CBF 1–2 h after CSD. Despite the amelioration of the post-CSD reduction in CBF by HET0016, we found that blocking the synthesis of 20-HETE did not change the reduction of evoked vascular responses after CSD. CBF responses to transcallosal fiber stimulation depends on NO and cyclooxygenase products (Hoffmeyer et al., 2007) generated by cortical pyramidal cells that contain COX-2 and inhibitory interneurons that contain nitric oxide synthase (Enager et al., 2009). In addition, astrocytic mechanisms control CBF (Mulligan and MacVicar, 2004; Gordon et al., 2007; Enager et al., 2009; Lecrux et al., 2011). The differential effect of 20-HETE blockade after CSD on basal CBF and neurovascular coupling responses is consistent with 20-HETE synthesis being implicated in arteriolar constriction after CSD and not in neurovascular coupling. In comparison, l-arginine pretreatment ameliorates the reduction of CBF after CSD and increases the rate of recovery for cerebrovascular responsiveness to hypercapnia. Thus, a decreased supply of NO contributes to the reduced CBF after CSD (Fabricius et al., 1995), which may be consistent with the observation that NO inhibits CYP-450 and in consequence 20-HETE synthesis (Attwell et al., 2010). Finally, topical application of NO donors or the cell-permeable cGMP analog 8-Br-cGMP reestablishes resting CBF to pre-CSD values and reverses CSD-induced attenuation of the cerebrovascular response to hypercapnia when given after CSD (Scheckenbach et al., 2006). We hypothesize that decreased NO availability and increased 20-HETE production account for the CBF reduction and impaired neurovascular coupling after CSD.
After CSD, the EPSP amplitude rapidly returned to normal, whereas the IPSP did not, which is consistent with impaired function of inhibitory neurons after CSD (Kruger et al., 1996; Piilgaard and Lauritzen, 2009). This was previously observed to correlate with the change in neurovascular coupling after CSD (Piilgaard and Lauritzen, 2009), but in the present study, we observed that pretreatment with HET0016 prevented the reduction of the IPSP amplitude after CSD without affecting neurovascular coupling. Therefore, this study did not support the hypothesis that impaired interneuronal function explains reduced vascular reactivity after CSD. In addition, we observed stimulation-induced increases in CMRO2 after CSD in the control group, which were attenuated in the HET0016 group. No data are available in the literature to explain this effect of 20-HETE on neuronal function and CMRO2, and these two issues will need to be addressed in further studies.
In conclusion, increased 20-HETE synthesis during CSD leads to cortical vasoconstriction with slow recovery of function after outwash or breakdown of the vasoconstrictor. Increased synthesis of 20-HETE may explain a great proportion of the prolonged CBF reduction after CSD. Thus, 20-HETE synthesis by CYP-450 contributes mechanistically to reduced CBF accompanying CSD. We suggest that drugs targeting CYP-450 or effector molecules downstream from 20-HETE synthesis (e.g., PKC or Ca2+-gated K+ channels) may be useful for treating the acute decrease in CBF in patients with migraine or in acutely injured cerebral cortex (Lauritzen et al., 2011).
Footnotes
This work was supported by the Danish Council for Independent Research: Medical Sciences, the Lundbeck Foundation via the Lundbeck Foundation Centre for Neurovascular Signalling, the NOVO Nordisk Foundation, NORDEA Foundation for Center for Healthy Aging, the Canadian Institutes for Health Research, the Canadian Stroke Network, and Fondation Leducq. We thank Oscar Borries, MS, for Matlab programming and Associate Professor Lars Jørn Jensen (Section for Biochemistry, Physiology & Nutrition, Department of Basic Animal and Veterinary Science, University of Copenhagen, Frederiksberg C, Denmark) for supplying HET0016.
The authors declare no competing financial interests.
References
- Amemori T, Bures J. Functional ablation of the olfactory bulb by spreading depression: unit activity changes and transient anosmia. Brain Res Bull. 1988;20:421–427. doi: 10.1016/0361-9230(88)90072-x. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Salomone S, Ozdemir-Gursoy Y, Boas DA, Dunn AK, Moskowitz MA. Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab. 2004;24:1172–1182. doi: 10.1097/01.WCB.0000137057.92786.F3. [DOI] [PubMed] [Google Scholar]
- Basarsky TA, Duffy SN, Andrew RD, MacVicar BA. Imaging spreading depression and associated intracellular calcium waves in brain slices. J Neurosci. 1998;18:7189–7199. doi: 10.1523/JNEUROSCI.18-18-07189.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosche B, Graf R, Ernestus RI, Dohmen C, Reithmeier T, Brinker G, Strong AJ, Dreier JP, Woitzik J, Woitzik J. Recurrent spreading depolarizations after subarachnoid hemorrhage decreases oxygen availability in human cerebral cortex. Ann Neurol. 2010;67:607–617. doi: 10.1002/ana.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Best AR, Regehr WG. Sustained elevation of dendritic calcium evokes widespread endocannabinoid release and suppression of synapses onto cerebellar Purkinje cells. J Neurosci. 2006;26:6841–6850. doi: 10.1523/JNEUROSCI.1280-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busija DW, Bari F, Domoki F, Horiguchi T, Shimizu K. Mechanisms involved in the cerebrovascular dilator effects of cortical spreading depression. Prog Neurobiol. 2008;86:417–433. doi: 10.1016/j.pneurobio.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci. 2007;27:4036–4044. doi: 10.1523/JNEUROSCI.0721-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crago EA, Thampatty BP, Sherwood PR, Kuo CW, Bender C, Balzer J, Horowitz M, Poloyac SM. Cerebrospinal fluid 20-HETE is associated with delayed cerebral ischemia and poor outcomes after aneurysmal subarachnoid hemorrhage. Stroke. 2011;42:1872–1877. doi: 10.1161/STROKEAHA.110.605816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creutzfeldt O. Cortex cerebri. Oxford: Oxford Science; 1995. General neurophysiology of the cortex; pp. 131–163. [Google Scholar]
- Dohmen C, Sakowitz OW, Fabricius M, Bosche B, Reithmeier T, Ernestus RI, Brinker G, Dreier JP, Woitzik J, Strong AJ, Graf R, Graf R. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63:720–728. doi: 10.1002/ana.21390. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Körner K, Ebert N, Görner A, Rubin I, Back T, Lindauer U, Wolf T, Villringer A, Einhäupl KM, Lauritzen M, Dirnagl U. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-l-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab. 1998;18:978–990. doi: 10.1097/00004647-199809000-00007. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, Lehmann TN, Sarrafzadeh A, Willumsen L, Hartings JA, Sakowitz OW, Seemann JH, Thieme A, Lauritzen M, Strong AJ. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129:3224–3237. doi: 10.1093/brain/awl297. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, Tolias C, Oliveira-Ferreira AI, Fabricius M, Hartings JA, Vajkoczy P, Lauritzen M, Dirnagl U, Bohner G, Strong AJ, Strong AJ. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132:1866–1881. doi: 10.1093/brain/awp102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enager P, Piilgaard H, Offenhauser N, Kocharyan A, Fernandes P, Hamel E, Lauritzen M. Pathway-specific variations in neurovascular and neurometabolic coupling in rat primary somatosensory cortex. J Cereb Blood Flow Metab. 2009;29:976–986. doi: 10.1038/jcbfm.2009.23. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Lauritzen M. Laser-Doppler evaluation of rat brain microcirculation: comparison with the [14C]-iodoantipyrine method suggests discordance during cerebral blood flow increases. J Cereb Blood Flow Metab. 1996;16:156–161. doi: 10.1097/00004647-199601000-00018. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Akgoren N, Lauritzen M. Arginine-nitric oxide pathway and cerebrovascular regulation in cortical spreading depression. Am J Physiol. 1995;269:H23–H29. doi: 10.1152/ajpheart.1995.269.1.H23. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Akgören N, Dirnagl U, Lauritzen M. Laminar analysis of cerebral blood flow in cortex of rats by laser-Doppler flowmetry: a pilot study. J Cereb Blood Flow Metab. 1997;17:1326–1336. doi: 10.1097/00004647-199712000-00008. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, Lauritzen M. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129:778–790. doi: 10.1093/brain/awh716. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Lowry TF, Taheri MR, Birks EK, Hudetz AG, Narayanan J, Falck JR, Okamoto H, Roman RJ, Nithipatikom K, Campbell WB, Harder DR. Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ Res. 2000;87:60–65. doi: 10.1161/01.res.87.1.60. [DOI] [PubMed] [Google Scholar]
- Gjedde A, Johannsen P, Cold GE, Ostergaard L. Cerebral metabolic response to low blood flow: possible role of cytochrome oxidase inhibition. J Cereb Blood Flow Metab. 2005;25:1183–1196. doi: 10.1038/sj.jcbfm.9600113. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Mulligan SJ, MacVicar BA. Astrocyte control of the cerebrovasculature. Glia. 2007;55:1214–1221. doi: 10.1002/glia.20543. [DOI] [PubMed] [Google Scholar]
- Hoffmeyer HW, Enager P, Thomsen KJ, Lauritzen MJ. Nonlinear neurovascular coupling in rat sensory cortex by activation of transcallosal fibers. J Cereb Blood Flow Metab. 2007;27:575–587. doi: 10.1038/sj.jcbfm.9600372. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Receptor subtypes involved in callosally-induced postsynaptic potentials in rat frontal agranular cortex in vitro. Exp Brain Res. 1992;88:33–40. doi: 10.1007/BF02259126. [DOI] [PubMed] [Google Scholar]
- Kehl F, Cambj-Sapunar L, Maier KG, Miyata N, Kametani S, Okamoto H, Hudetz AG, Schulte ML, Zagorac D, Harder DR, Roman RJ. 20-HETE contributes to the acute fall in cerebral blood flow after subarachnoid hemorrhage in the rat. Am J Physiol Heart Circ Physiol. 2002;282:H1556–H1565. doi: 10.1152/ajpheart.00924.2001. [DOI] [PubMed] [Google Scholar]
- Krüger H, Luhmann HJ, Heinemann U. Repetitive spreading depression causes selective suppression of GABAergic function. Neuroreport. 1996;7:2733–2736. doi: 10.1097/00001756-199611040-00065. [DOI] [PubMed] [Google Scholar]
- Lauritzen M. Pathophysiology of the migraine aura: the spreading depression theory. Brain. 1994;117:199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab. 1992;12:223–229. doi: 10.1038/jcbfm.1992.32. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ, Kronborg D, Wieloch T. Cortical spreading depression is associated with arachidonic acid accumulation and preservation of energy charge. J Cereb Blood Flow Metab. 1990;10:115–122. doi: 10.1038/jcbfm.1990.14. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:17–35. doi: 10.1038/jcbfm.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão AA. Spreading depression. Funct Neurol. 1986;1:363–366. [PubMed] [Google Scholar]
- Lecrux C, Toussay X, Kocharyan A, Fernandes P, Neupane S, Lévesque M, Plaisier F, Shmuel A, Cauli B, Hamel E. Pyramidal neurons are “neurogenic hubs” in the neurovascular coupling response to whisker stimulation. J Neurosci. 2011;31:9836–9847. doi: 10.1523/JNEUROSCI.4943-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li C, Falck JR, Roman RJ, Harder DR, Koehler RC. Interaction of nitric oxide, 20-HETE, and EETs during functional hyperemia in whisker barrel cortex. Am J Physiol Heart Circ Physiol. 2008;295:H619–H631. doi: 10.1152/ajpheart.01211.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata N, Taniguchi K, Seki T, Ishimoto T, Sato-Watanabe M, Yasuda Y, Doi M, Kametani S, Tomishima Y, Ueki T, Sato M, Kameo K. HET0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. Br J Pharmacol. 2001;133:325–329. doi: 10.1038/sj.bjp.0704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab. 2009;29:1517–1527. doi: 10.1038/jcbfm.2009.73. [DOI] [PubMed] [Google Scholar]
- Renic M, Klaus JA, Omura T, Kawashima N, Onishi M, Miyata N, Koehler RC, Harder DR, Roman RJ. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2009;29:629–639. doi: 10.1038/jcbfm.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Scheckenbach KE, Dreier JP, Dirnagl U, Lindauer U. Impaired cerebrovascular reactivity after cortical spreading depression in rats: restoration by nitric oxide or cGMP. Exp Neurol. 2006;202:449–455. doi: 10.1016/j.expneurol.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Fabricius M, Boutelle MG, Hibbins SJ, Hopwood SE, Jones R, Parkin MC, Lauritzen M. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738–2743. doi: 10.1161/01.str.0000043073.69602.09. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Yaseen MA, Sakadzić S, Ruvinskaya S, Sims JR, Boas DA, Moskowitz MA, Ayata C. Perfusion pressure-dependent recovery of cortical spreading depression is independent of tissue oxygenation over a wide physiologic range. J Cereb Blood Flow Metab. 2010;30:1168–1177. doi: 10.1038/jcbfm.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CW, Falck JR, Okamoto H, Harder DR, Roman RJ. Role of cGMP versus 20-HETE in the vasodilator response to nitric oxide in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2000;279:H339–H350. doi: 10.1152/ajpheart.2000.279.1.H339. [DOI] [PubMed] [Google Scholar]
- Zhou N, Gordon GR, Feighan D, MacVicar BA. Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb Cortex. 2010;20:2614–2624. doi: 10.1093/cercor/bhq018. [DOI] [PubMed] [Google Scholar]
- Zhu XH, Zhang Y, Tian RX, Lei H, Zhang N, Zhang X, Merkle H, Ugurbil K, Chen W. Development of (17)O NMR approach for fast imaging of cerebral metabolic rate of oxygen in rat brain at high field. Proc Natl Acad Sci U S A. 2002;99:13194–13199. doi: 10.1073/pnas.202471399. [DOI] [PMC free article] [PubMed] [Google Scholar]