

Abstract
The postsynaptic scaffolding A-kinase anchoring protein 79/150 (AKAP79/150) signaling complex regulates excitatory synaptic transmission and strength through tethering protein kinase A (PKA), PKC, and calcineurin (CaN) to the postsynaptic densities of neurons (Sanderson and Dell'Acqua, 2011), but its role in inhibitory synaptic transmission and plasticity is unknown. Using immunofluorescence and whole-cell patch-clamp recording in rat midbrain slices, we show that activation of postsynaptic D2-like family of dopamine (DA) receptor in the ventral tegmental area (VTA) induces long-term depression (LTD) of GABAergic synapses on DA neurons through an inositol triphosphate receptor-mediated local rise in postsynaptic Ca2+ and CaN activation accompanied by PKA inhibition, which requires AKAP150 as a bridging signaling molecule. Our data also illuminate a requirement for a clathrin-mediated internalization of GABAA receptors in expression of LTDGABA. Moreover, disruption of AKAP–PKA anchoring does not affect glutamatergic synapses onto DA neurons, suggesting that the PKA–AKAP–CaN complex is uniquely situated at GABAA receptor synapses in VTA DA neurons to regulate plasticity associated with GABAA receptors. Drug-induced modulation of GABAergic plasticity in the VTA through such novel signaling mechanisms has the potential to persistently alter the output of individual DA neurons and of the VTA, which may contribute to the reinforcing or addictive properties of drugs of abuse.
Introduction
GABAA receptor (GABAAR)-mediated GABAergic transmission provides synaptic inhibition at the vast majority of inhibitory synapses in the brain in which it regulates the excitability and function of the individual neurons as well as neural networks. Mounting evidence suggests that synaptic plasticity at GABAergic synapses plays an important role in normal brain functions, such as experience-dependent learning, but also in pathological conditions, including drug addiction (Dacher and Nugent, 2011a). As with plasticity at excitatory synapses, both long-term potentiation (LTP) and long-term depression (LTD) occur at GABAergic synapses in different areas of the brain, including the ventral tegmental area (VTA) (Nugent and Kauer, 2008). Dopamine (DA) release from VTA DA neurons in limbic forebrain areas, such as the nucleus accumbens (NAc), controls reward-motivated learning. Indeed, direct optogenetic activation of VTA DA neurons is reinforcing (Adamantidis et al., 2011), and all major drugs of abuse mediate their reinforcing effects by increasing DA release from the VTA (Di Chiara and Imperato, 1988). The strength of both glutamatergic and GABAergic synapses on VTA DA neurons are shown to be altered in response to drugs of abuse, and this drug-induced plasticity is thought to underlie long-lasting behavioral changes associated with drug experience (Lüscher and Malenka, 2011), yet the precise molecular mechanisms by which addictive drugs alter synaptic plasticity remain unclear.
Recently, we described a novel form of activity-induced GABAergic plasticity in VTA DA neurons (LTDGABA) that was absent in slices from rats that received a single in vivo exposure to morphine (Dacher and Nugent, 2011b). Similarly, LTP at the same GABAergic synapses in VTA DA neurons (LTPGABA) was blocked after a single in vivo morphine treatment (Nugent et al., 2007). These data suggest that GABAergic plasticity in the VTA is an important target for opiates and maybe other addictive drugs in modulating the excitability of DA neurons. To address the question of how drugs of abuse can trigger or modulate GABAergic plasticity at synapses, it is essential to identify the underlying mechanisms that mediate this plasticity. In the present study, we demonstrate a novel role for A-kinase anchoring protein 150 (AKAP150)-dependent signaling in mediating LTDGABA. The AKAP family of scaffold proteins plays an important role in localization of specific kinases [e.g., protein kinases A (PKA) and C (PKC)] and phosphatases [e.g., calcineurin (CaN)] to the synapses that control NMDA, AMPA, and GABAA receptor trafficking and function (Klauck et al., 1996; Luscher et al., 2011; Sanderson and Dell'Acqua, 2011). Moreover, disruption of AKAP150-dependent signaling in the NAc has been found to impair the reinstatement of cocaine-seeking behavior through modulation of AMPA receptor (AMPAR) trafficking, suggesting the critical role of AKAP proteins in susceptibility to relapse (Reissner et al., 2011), although the role of AKAP signaling in synaptic transmission and plasticity in the VTA is unexplored. Here, we provide the first evidence for the selective regulation of GABAAR trafficking-mediated GABAergic plasticity by endogenous DA and D2 receptors (for simplicity, the D2-like family of DA receptors will be identified as D2Rs throughout) that requires an inhibitory G-protein–PKA–inositol triphosphate receptor (IP3R)–Ca2+–CaN–AKAP signaling mechanism.
Materials and Methods
Slice preparation for electrophysiology.
For electrophysiological studies, we used 14- to 22-d-old Sprague Dawley (male and female) rats. Housing and care were the same as described previously (Dacher and Nugent, 2011b). Midbrain horizontal slices (250 μm) were cut and incubated at least 1 h at 34°C before recordings in artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 21.4 NaHCO3, 2.5 KCl, 1.2 NaH2PO4, 2.4 CaCl2, 1.0 MgSO4, 11.1 glucose, and 0.4 ascorbic acid (saturated with 95% O2/5% CO2). Slices were then transferred to a recording chamber and submerged in warm (28°C) ascorbic acid-free ACSF.
Electrophysiology.
Whole-cell recordings were performed using a patch amplifier (Multiclamp 700B) under infrared differential interference contrast microscopy. Data acquisition and analysis were performed using DigiData 1440A and pClamp 10 (Molecular Devices). GABAAR-mediated IPSCs were isolated and recorded as described previously (Dacher and Nugent, 2011b) in ACSF containing 6,7-dinitroquinoxaline-2,3-dione (10 μm), and strychnine (1 μm). In some experiments, AMPAR-mediated EPSCs were isolated and recorded using ACSF containing picrotoxin (100 μm). The patch pipettes were filled with the following (in mm): 125 KCl, 2.8 NaCl, 2 MgCl2, 2 ATP-Na+, 0.3 GTP-Na+, 0.6 EGTA, and 10 HEPES, pH adjusted to 7.28 with KOH (osmolarity adjusted to 275–280 mOsm). A subset of experiments with quinpirole used an internal solution in which 125 mm CsCl was substituted for KCl, and CsOH was used to adjust the pH. Cells were voltage clamped at −70/−80 mV, except during the LTD protocol. The cell input resistance and series resistance were monitored through the experiment, and, if these values changed by >10%, data were not included. Paired GABAA IPSCs or AMPAR-mediated EPSCs were stimulated using a bipolar stainless steel stimulating electrode placed 200–500 mm rostral to the recording site in the VTA at 0.1 Hz (100 μs). The stimulation intensity was adjusted so that the amplitude of synaptic responses ranged from −200 to −800 pA. LTD was induced using low-frequency stimulation (LFS) of 1 Hz for 6 min while cells were voltage clamped at −40 mV (for simplicity, in all figures, “pairing” represents the protocol used for LTD induction). The appearance of an hyperpolarization-activated current (Ih) (≥50 pA) in response to stepping cells from −50 to −100 mV was used to identify VTA DA neurons.
Drug treatment.
Drugs were present in ACSF perfusing slices in the recording chamber at the indicated concentrations for at least 15 min before the LTD protocol. Interleaved control experiments were performed with experiments in which drugs were bath applied. For BAPTA experiments, the intra-pipette solution contained 80–90 mm KCl and 20–30 mm BAPTA, with no added EGTA. Slices were preincubated in 1 μm cyclosporin A for ≥2 h before being transferred into the recording chamber. The drug was also present throughout the experiments. In a subset of experiments, cyclosporine A (1 μm) was used in the intracellular recording pipette. FK506 [(3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy-3-[(1E)-2-[(1R,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propen-1-yl)-15,19-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)tetrone] was dissolved in DMSO and diluted to the final concentration in the intracellular recording pipette. All the drugs were purchased from Sigma, Tocris Bioscience, or Calbiochem. Ht31 and Ht31p (control peptide) were obtained from Promega. Pitstop2 and its control peptide were purchased from Abcam.
Data analyses.
Values are presented as means ± SEM. Statistical significance was determined using repeated-measures ANOVA with significance level of p < 0.05. Levels of LTD are reported as averaged IPSC amplitudes for 5 min just before LTD induction compared with averaged IPSC amplitudes during the 5 min period from 20 to 25 min after the protocol. Levels of quinpirole-/PKI(6–22)-/Ht31-induced depression are reported as averaged IPSC amplitudes for 5 min just before the emergence of drug-induced synaptic depression compared with averaged IPSC amplitudes during the last 5 min period of the peak response. The average time point of drug-induced rundown represents averaged durations after the initiation of the whole-cell configuration and the emergence of drug-induced depression, calculated in each experiment. Paired-pulse ratios (PPRs) (50 ms interstimulus interval) were measured over 5 min epochs of 30 IPSCs as described previously (Nugent et al., 2007).
Immunohistochemistry and image analyses.
Male adult Sprague Dawley rats weighing 300 g (Taconic Farms) were housed individually in a plastic cage and kept on a 12 h light/dark cycle. Food and water were provided ad libitum. Rats were anesthetized with an intraperitoneal injection containing ketamine (85 mg/kg) and xylazine (10 mg/kg) and perfused through the aorta with 300 ml of heparinized 1× PBS, followed by 250 ml of 4% paraformaldehyde (PFA) (USB). The brains were dissected and placed in 4% PFA for 24 h and then cryoprotected by submersion in 20% sucrose for 3 d, frozen on dry ice, and stored at −70°C until sectioned. Sections of the VTA were cut using a cryostat (Leica CM1900) and mounted on slides. Serial coronal sections (20 μm) of the midbrain containing the VTA [from −4.92 to −6.72 mm caudal to bregma (Paxinos and Watson, 2007)]) were fixed in 4% PFA for 5 min, washed in 1× PBS, and then blocked in 10% normal horse serum (NHS) containing 0.3% Triton X-100 in 1× PBS for 1 h. Sections were incubated in rabbit anti-tyrosine hydroxylase (TH) (1:1000; Calbiochem) and goat anti-AKAP150 (1:500; Santa Cruz Biotechnology) in carrier solution (0.5% NHS in 0.1% Triton X-100 in 1× PBS) overnight at room temperature. After rinsing in 1× PBS, sections were incubated for 2 h in Alexa Flour 488-labeled chicken anti-goat IgG and Alexa Flour 568-labeled donkey anti-rabbit IgG (both diluted 1:200). Finally, sections were rinsed in 1× PBS, dried, and coverslipped with Prolong mounting medium containing DAPI to permit visualization of nuclei. Background staining was assessed by omission of primary antibody in the immunolabeling procedure (negative control). VTA tissue sections of rats with previously established presence of TH/AKAP150 immunoreactive neurons were processed as positive control tissue. Images were captured using a Carl Zeiss Pascal Confocal Inverted Microscope System with 100×/1.4 numerical aperture oil-immersion objective.
Results
Synaptic LFS with modest depolarization (a pairing LTD protocol) induced LTD of the GABAAR-mediated evoked IPSCs onto VTA DA neurons as we reported previously (Dacher and Nugent, 2011b). However, in response to the same pairing stimulation paradigm, GABAergic synapses onto VTA GABAergic neurons (lacking Ih current) did not exhibit the IPSC LTDGABA (Fig. 1a–c), suggesting that Ih(−) neurons (presumably GABAergic neurons) differed from Ih(+) neurons because none of the former exhibited LTDGABA. Because of the increasing recognition of heterogeneity among DA neurons in regard to their intrinsic electrophysiological properties, responses to external stimuli, and projection target (Ford et al., 2006; Margolis et al., 2006b; Lammel et al., 2008, 2011), in the present study, we consistently recorded from a region of the VTA (in the dorsal and caudal VTA) that is shown to contain mostly NAc-projecting DA neurons with Ih positivity (Margolis et al., 2006a,b). Therefore, the expression of LTDGABA might be one uniform physiological property of Ih-expressing neurons in the caudal VTA and may be specific to NAc-projecting DA neurons.
Figure 1.
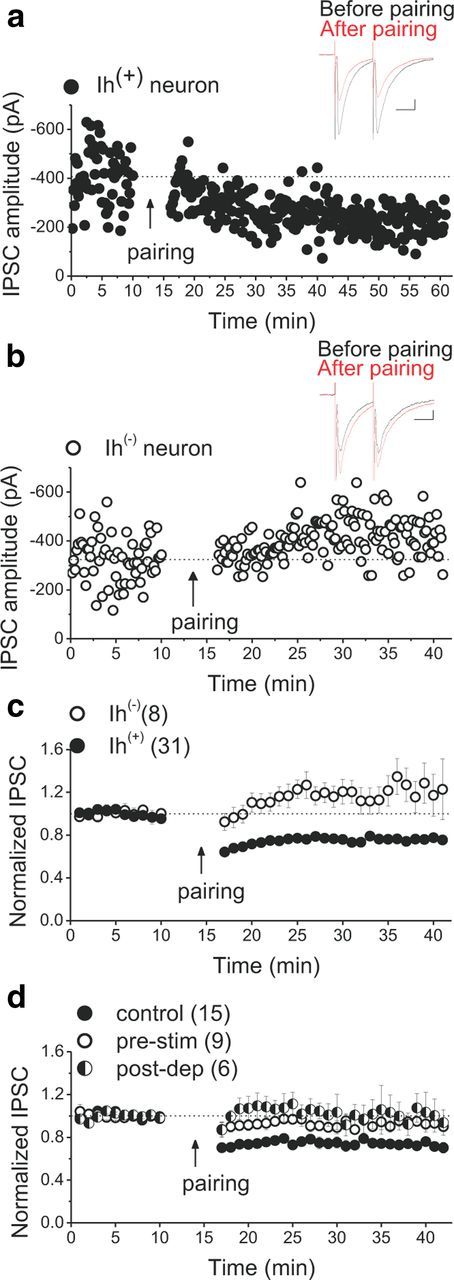
LTDGABA is selectively expressed in DA neurons. a, b, Single experiments showing induction of LTDGABA recorded in Ih(+) (presumably DA) and Ih(−) (presumably GABAergic) neurons, respectively (Ih(+) neurons, 75.9 ± 1.2% of pre-pairing values, repeated-measures ANOVA, F(3,85) = 31.668, p < 0.0001; Ih(−)/GABAergic neurons, 122 ± 5% of pre-pairing values, F(3.6,14.5) = 3.132, p = 0.051). At the arrow, LTD was induced using LFS while cells were depolarized at −40 mV. In this and all figures, “pairing” represents the LTD induction protocol. Insets, Averaged IPSCs before (black) and 25 min after (red) pairing. In this and all figures, 10 consecutive traces from each condition were averaged for illustration as inset. Calibration: 50 pA, 25 ms. c, Average experiments from Ih(+) (filled symbols) and Ih(−) (open symbols) neurons. Ih(+) but not Ih(−) VTA neurons express LTDGABA. All Ih(+) cells recorded as controls throughout the present work (except DMSO control cells) are included for the average from Ih(+) neurons in this graph. d, Average experiments illustrating the absence of LTDGABA in response to low-frequency presynaptic stimulation in which cells were voltage clamped at −70 to −90 mV while receiving LFS (pre-stim, open symbols) or postsynaptic depolarization in which cells were voltage clamped at −40 mV without receiving LFS (post-dep, half-filled symbols). Control experiments (filled symbols) were conducted in response to the LTD pairing protocol (cells were voltage clamped at −40 mV during the delivery of LFS) (control LTD, 74 ± 3% of pre-pairing values, F(4,48.37) = 18.055, p < 0.0001; pre-stim cells, 91 ± 1% of pre-LFS values, F(6,42) = 1.149, p = 0.352; post-dep cells, 102 ± 7% of pre-depolarization values, F(2,6.15) = 0.501, p = 0.633). Pairing of presynaptic stimulation and postsynaptic depolarization is necessary for successful induction of LTDGABA. Values shown throughout figure are the mean ± SEM.
Postsynaptic depolarization or presynaptic stimulation is necessary but not sufficient to induce LTDGABA
The LTD induction protocol is a pairing paradigm so it is unknown whether either low-frequency presynaptic stimulation or postsynaptic depolarization alone is sufficient to induce LTDGABA. To test these individual components of the pairing protocol, we first attempted to induce LTDGABA while cells were voltage clamped at −70 to −95 mV during the induction protocol to minimize postsynaptic depolarization. Under these conditions, low-frequency presynaptic stimulation failed to induce LTDGABA, suggesting that at least some voltage-dependent processes may be critical for the induction of LTDGABA (Fig. 1d, pre-stim experiments). Given that L-type voltage-gated Ca2+ channels (VGCCs) can provide the postsynaptic Ca2+ entry necessary for synaptic plasticity (Morishita and Sastry, 1996; Kreitzer and Malenka, 2005; Adermark and Lovinger, 2007) and the fact that LTDGABA is NMDA receptor (NMDAR)-independent, we tested whether L-type VGCCs were activated by postsynaptic depolarization during the induction protocol to trigger LTDGABA. We attempted to induce LTDGABA in the presence of the L-type VGCC blocker nifedipine (50 μm). Nifedipine did not block LTDGABA (control LTD, 70 ± 0.8% of pre-pairing values, F(10.3,41.2) = 7.721, p < 0.0001, n = 6; nifedipine cells, 69 ± 1% of pre-pairing values, F(3,9.03) = 4.989, p = 0.026, n = 4), suggesting that postsynaptic depolarization engages voltage-dependent signaling molecules other than NMDAR or L-type VGCCs to facilitate LTD induction. Interestingly, postsynaptic depolarization per se was also unable to induce plasticity when the cells were voltage clamped at −40 mV for 6 min without receiving low-frequency presynaptic stimulation (Fig. 1d, post-dep experiments). Altogether, our data suggest that the pairing of presynaptic and postsynaptic events is critical for the successful induction of LTDGABA. Given that electrical stimulation during LTD induction protocol is not specific to GABAergic terminals, it could also trigger somatodendritic release of DA and other neurotransmitters that may contribute to LTD.
Disruption of postsynaptic G-protein signaling blocks LTDGABA
LTDGABA appears to be expressed postsynaptically (through a reduction in the number or conductance of postsynaptic GABAARs), because we showed previously that the PPR and coefficient of variation, the two main indicators of presynaptic plasticity, were unaffected after LTD induction (Dacher and Nugent, 2011b). Additionally, we found that, in contrast to the presynaptic cocaine-induced inhibitory LTD (I-LTD) in the VTA (Pan et al., 2008a), cannabinoid CB1 receptors are not required for induction of LTDGABA, indicating that these two forms of I-LTD in the VTA are triggered by distinct mechanisms. However, both I-LTD and LTDGABA require activation of D2Rs, although it was proposed that the D2Rs involved in I-LTD are located presynaptically on GABAergic terminals (Pan et al., 2008b; Dacher and Nugent, 2011b). To provide additional evidence for the postsynaptic nature of LTDGABA and the involvement of postsynaptic D2Rs in LTDGABA, we blocked the activation of G-protein-coupled receptors (GPCRs) in the postsynaptic cell by including guanosine 5′-O-(β-thiodiphosphate (GDPβS, 200 μm) in the recording pipette. Intra-pipette GDPβS completely blocked LTDGABA, confirming that LTDGABA is postsynaptic and its induction is dependent on activation of postsynaptic GPCRs, such as D2 autoreceptors (Fig. 2a). GPCRs coupling through Gi/Go- or Gq/11-proteins, such as GABABR and group I metabotropic glutamate receptors (mGluRs), respectively, are also located postsynaptically on VTA DA neurons (Johnson and North, 1992; Cameron and Williams, 1994; Fiorillo and Williams, 1998) and are shown to play an important role in the induction of LTD (Bellone and Lüscher, 2005; Kamikubo et al., 2007; Lüscher and Huber, 2010; Tadavarty et al., 2011). Therefore, we further investigated the possible contribution of GABABRs and group I mGluRs in the induction of LTDGABA. We attempted to induce LTDGABA in the presence of a potent GABABR antagonist CGP 54626 ([S-(R*,R*)]-[3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-hydroxypropyl](cyclohexylmethyl)phosphinicacid) (500 nm to 1 μm). CGP 54626 did not block LTD, suggesting that GABABRs are not critical for the induction of LTDGABA (control LTD, 70 ± 0.8% of pre-pairing values, F(0.3,41.2) = 7.721, p < 0.0001, n = 6; CGP54626 cells, 73.5 ± 2% of pre-pairing values, F(6.2,18.8) = 3.951, p = 0.009, n = 5). Using two different group I mGluR blockers, 500 mm (S)-MCPG [(±)-amino-4-carboxy-methyl-phenylacetic acid] and 10 μm A841720 [(4-bromo-3-fluorophenyl)hydrazine hydrochloride], we also tested whether group I mGluR activation is necessary for the induction of LTDGABA. LTDGABA was successfully induced in response to pairing protocol in the presence of either (S)-MCPG or A841720, suggesting that activation of group I mGluRs is also not required for the induction of LTDGABA (control LTD, 70 ± 0.8% of pre-pairing values, F(10.3,41.2) = 7.721, p < 0.0001, n = 6; group I mGluR antagonist cells, 74 ± 2% of pre-pairing values, F(4.7,23.54) = 2.829, p = 0.04, n = 6).
Figure 2.
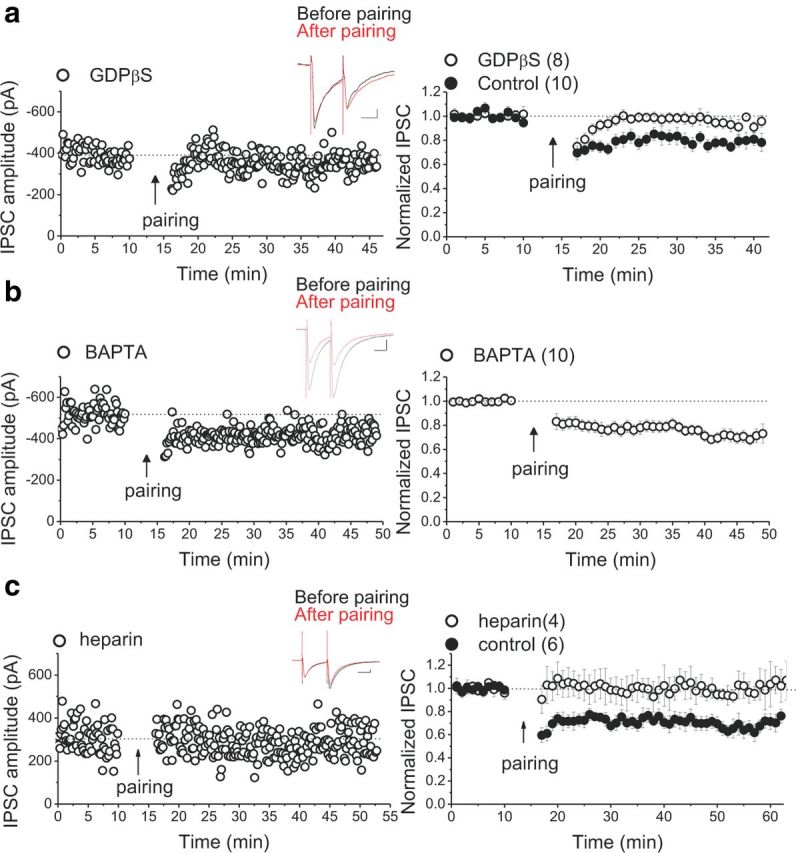
The induction of LTDGABA requires postsynaptic G-protein signaling and IP3R-mediated Ca2+ release, but chelating postsynaptic Ca2+ does not block LTDGABA. a, Single experiment illustrating the block of LTDGABA by intra-pipette GDPβS and averaged experiments with (open symbols) and without (filled symbols) 200 μm GDPβS in the pipette solution (control LTD, 78 ± 1% of pre-pairing values, F(2.28,20.55) = 8.635, p = 0.0014; GDPβS cells, 94 ± 2% of pre-pairing values, F(2.2,11.2) = 0.198, p = 0.846). Inset, Averaged IPSCs before (black) or 25 min after (red) pairing. Calibration: 50 pA, 25 ms. Disruption of postsynaptic G-protein signaling prevents LTDGABA. b, Single experiment illustrating the induction of LTDGABA with intra-pipette BAPTA and averaged experiment with 20–30 mm BAPTA in the pipette solution (open symbols, BAPTA cells, 73 ± 2% of pre-pairing values, F(3,18.39) = 18.37, p < 0.0001). Inset, Averaged IPSCs before (black) or 25 min after (red) pairing. Calibration: 50 pA, 25 ms. Intra-pipette BAPTA does not block LTDGABA. c, Single experiment illustrating the block of LTDGABA by intra-pipette heparin and averaged experiments with (open symbols) and without (filled symbols) 2 mg/ml heparin in the pipette solution (control LTD, 70 ± 0.8% of pre-pairing values, F(10.3,41.2) = 7.721, p < 0.0001; heparin cells, 1.002 ± 3.5% of pre-pairing values, F(6.17,12.35) = 0.634, p = 0.705). Inset, Averaged IPSCs before (black) or 25 min after (red) pairing. Calibration: 50 pA, 25 ms. IP3R blockade abolishes LTDGABA.
IP3R blockade abolishes LTDGABA, whereas chelation of intracellular calcium does not block LTDGABA
Similar to the majority of synaptic plasticity at excitatory synapses, most LTP/LTD of GABAergic synapses is dependent on a rise in postsynaptic Ca2+ and NMDAR activation (Castillo et al., 2011). To test whether postsynaptic Ca2+ signaling is also necessary for LTDGABA, we performed experiments using 20–30 mm BAPTA in the recording pipette to buffer the fast rises in intracellular Ca2+ during LTD induction. We tested LTD at both glutamatergic and GABAergic synapses onto DA neurons using the LTD pairing paradigm. As shown previously (Jones et al., 2000), we confirmed that glutamatergic LTD in VTA DA neurons is calcium dependent and blocked by 30 mm BAPTA (data not shown; control LTD, 78 ± 0.3% of pre-pairing values, F(2.85,11.39) = 3.587, p < 0.05, n = 5; BAPTA cells, 97 ± 2% of pre-pairing values, F(39.66,79.33) = 0.618, p = 0.912, n = 3). Surprisingly, we found that intra-pipette BAPTA was unable to block LTDGABA (Fig. 2b). Consistent with this result, the blockade of VGCCs with nifedipine did not affect LTDGABA, raising the possibility that LTDGABA might be Ca2+ independent. However, it was also possible that a discrete local rise in intracellular Ca2+ as a result of D2R-mediated mobilization of intracellular Ca2+ stores could trigger LTD. In fact, compelling evidence suggests that D2Rs can facilitate Ca2+ signaling through an IP3R-mediated signaling pathway in the striatum and NAc (Hernandez-Lopez et al., 2000; Hu et al., 2005), which could underlie LTDGABA in the VTA. To test this possibility, we attempted to induce LTDGABA in response to the pairing protocol in the presence of the IP3R antagonist heparin (intra-pipette heparin, 2 mg/ml) (Komatsu, 1996). Postsynaptic intra-pipette application of heparin completely blocked LTDGABA, suggesting that a postsynaptic IP3R-induced Ca2+ release occurs during the induction of LTDGABA (Fig. 2c).
Inhibition of CaN activity blocks LTDGABA
Expression of a postsynaptic LTD of GABAergic synapses could be achieved through activation of protein phosphatase (PPs), which dephosphorylate GABAARs or their associated regulatory subunit (Morishita and Sastry, 1996). Several PPs, including PPI, PP2A, and Ca2+/calmodulin-dependent PP (PP2B/also known as CaN), have been shown to participate in postsynaptic as well as presynaptic expression of LTD of excitatory and inhibitory synapses (Collingridge et al., 2010; Castillo et al., 2011). Among them, CaN is of particular interest because it has been shown that stimulation of striatal D2Rs can modulate the striatal excitability through an IP3R–CaN signaling cascade (Hernandez-Lopez et al., 2000; Hu et al., 2005), and D2R activation can cause dephosphorylation of intracellular signaling molecules in a CaN-dependent manner (Nishi et al., 1997; Greengard, 2001). Furthermore, in the context of addiction, the reinforcing effects of chronic amphetamine or morphine are impaired in a mouse strain overexpressing CaN, suggesting a critical role for CaN in the learning mechanisms involved in drug addiction (Biala et al., 2005). Given that LTDGABA is also IP3R and D2R dependent, we next asked whether CaN is an absolute requisite for expression of LTDGABA. LTD induction was attempted in midbrain slices in the presence of two different CaN inhibitors: intra-pipette FK506 (0.6–1 μm) or bath application of cyclosporin A (1 μm) with a previous 2 h incubation of slices in the drug. We found that both CaN inhibitors blocked LTDGABA (Fig. 3a,b). Our data suggest that D2R activation triggers LTDGABA through an IP3–CaN-dependent signaling mechanism.
Figure 3.

LTDGABA requires the activity of postsynaptic CaN. a, Single experiment illustrating the block of LTDGABA by preincubation and bath application of cyclosporin A and averaged experiments with (open symbols) and without (filled symbols) 1 μm cyclosporin (control LTD, 75 ± 2% of pre-pairing values, F(3.9,11.9) = 4.667, p = 0.017; cyclosporine A cells, 105 ± 3% of pre-pairing values, F(2.6,15.6) = 0.716, p = 0.616). Inset, Averaged IPSCs before (black) or 25 min after (red) pairing. Calibration: 50 pA, 25 ms. Cyclosporin A treatment prevents LTDGABA. b, Single experiment illustrating the block of LTDGABA by intra-pipette FK506 and averaged experiments with (open symbols) and without (filled symbols) 0.6–1 μm FK506 in the pipette solution. LTD induction was attempted at 20–30 min or later after the initiation of the whole-cell recording with intra-pipette FK506 (control LTD with intra-pipette DMSO, 79 ± 4% of pre-pairing values, F(2.4,24.72) = 3.466, p = 0.03; FK506 cells, 111 ± 4% of pre-pairing values, F(1.7,6.9) = 0.477, p = 0.614). Inset, Averaged IPSCs before (black) or 25 min after (red) pairing. Calibration: 50 pA, 25 ms. Intra-pipette FK506 prevents LTDGABA.
D2R stimulation mimics and occludes LTDGABA through postsynaptic PKA inhibition with a CaN-dependent mechanism
Inhibition of LTDGABA with CaN blockers suggest that dephosphorylation of GABAARs by CaN could be a mechanism for expression of LTDGABA. D2R activation is also a part of signaling pathway that triggers LTDGABA (Dacher and Nugent, 2011b), and activation of CaN downstream to D2R has been shown to be IP3R–Ca2+ dependent (Hernandez-Lopez et al., 2000; Hu et al., 2005) and facilitated via a Gi–Go–adenylyl cyclase–cAMP–PKA pathway (Hu et al., 2005). Thus, we next examined whether D2R stimulation through its inhibitory effects on postsynaptic PKA is enough to depress GABAergic transmission in a CaN-dependent manner. A continuous or a brief 10 min bath application of 20 μm quinpirole (the D2R agonist) reliably reduced the amplitude of GABAA IPSCs (Fig. 4a,b), without affecting the PPR (IPSC2/IPSC1; PPR before quinpirole, 0.99 ± 0.034; PPR of the peak response to quinpirole, 0.89 ± 0.19; n = 11; F(2.42,16.45) = 1.445, p = 0.265), and this quinpirole-induced depression was CaN dependent because postsynaptic infusion of cyclosporine A (1 μm) significantly prevented this chemical depression (Fig. 4b). Moreover, once the depression with quinpirole reached a stable baseline, the LTD induction protocol using the pairing paradigm (1 Hz LFS at −40 mV) did not induce additional LTDGABA (occlusion) (Fig. 4a,c). Altogether, the present results confirm our previous finding (Dacher and Nugent, 2011b) that transient activation of postsynaptic D2R is sufficient to induce LTDGABA.
Figure 4.
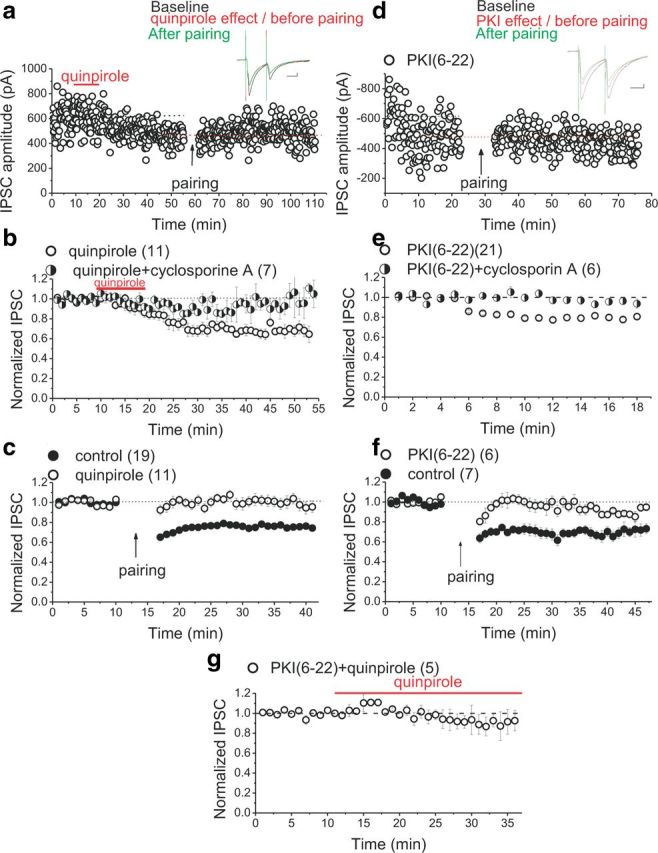
Postsynaptic D2R stimulation and inhibition of PKA mimic and occlude LTDGABA in a CaN-dependent manner. a, Single experiment illustrating the effect of a 10 min (see bar) bath application of 20 μm quinpirole on basal synaptic transmission and the induction of pairing-induced LTDGABA (only 5 min of the baseline before the emergence of the quinpirole-induced depression is shown in this sample experiment). Inset, Averaged IPSCs before (black) or the peak response of quinpirole (red) or 25 min after pairing (green). Calibration: 50 pA, 25 ms. b, Averaged experiments with bath application of 20 μm quinpirole (open symbols) or 20 μm quinpirole plus intra-pipette cyclosporin A (1 μm, half-filled symbols). A continuous or a brief 10 min exposure of midbrain slices to quinpirole induces a rapid rundown of IPSCs that is blocked by cyclosporin A treatment (quinpirole cells, 67 ± 1.6% of the first 5 min baseline values, F(9.6,74.9) = 9.707, p < 0.0001; quinpirole plus cyclosporine A, 95 ± 4% of the pre-quinpirole values, F(2.8,5.64) = 1.106, p = 0.417). c, Averaged LTD induction experiments with (open symbols) or without (filled symbols) quinpirole. Quinpirole-induced depression occludes pairing-induced LTDGABA (control LTD, 75 ± 0.8% of pre-pairing values, F(11,177.36) = 19.121, p < 0.0001; quinpirole, 98 ± 3.7% of pre-pairing values, F(9.4,84.7) = 0.973, p = 0.470). d, Single experiment illustrating the effect of intra-pipette PKI(6–22) on basal synaptic transmission and the induction of pairing-induced LTDGABA in the PKI(6–22)-loaded cell (only 5 min of the baseline before the emergence of the PKI(6–22)-induced depression is shown in this sample experiment). Inset, Averaged IPSCs before (black) or the peak response of PKI(6–22) (red) or 25 min after pairing (green). Calibration: 50 pA, 25 ms. e, Averaged experiments with intra-pipette 10 μm PKI(6–22) (open symbols) or intra-pipette 10 μm PKI(6–22) plus 1 μm cyclosporin A (half-filled symbols). PKI(6–22) induces a rapid rundown of IPSCs that is blocked by cyclosporin A treatment (PKI cells, 75 ± 3% of the first 5 min baseline values, F(2.03,25.5) = 70.525, p < 0.0001; PKI(6–22) plus cyclosporine A, 95 ± 4% of the first 5 min baseline values, F(3,15) = 1.634, p = 0.224). f, Averaged LTD induction experiments with (open symbols) or without (filled symbols) intra-pipette PKI(6–22). LTD induction was attempted at 30 min or later after the initiation of the whole cell recording with intra-pipette PKI. PKI(6–22)-induced depression occludes pairing-induced LTDGABA (control LTD, 71 ± 1% of pre-pairing values, F(5.27,25.35) = 7.167, p < 0.0001; PKI(6–22) cells, 90 ± 4.2% of pre-pairing values, F(1.85,7.4) = 1.899, p = 0.216). g, Averaged experiments with intra-pipette 10 μm PKI(6–22) plus quipirole (open symbols). Intra-pipette PKI(6–22) occludes quinpirole-induced depression of IPSCs (quinpirole plus PKI cells, 90 ± 3% of the pre-quinpirole values, F(3.7,7.434) = 1.127, p = 0.409).
To further demonstrate that quinpirole acts via postsynaptic D2Rs to inhibit the PKA pathway and induce LTDGABA, a membrane-impermeant PKA inhibitor, PKI(6–22), was used in the intracellular pipette. Similar to excitatory synapses in the CA1 region of hippocampus (Kameyama et al., 1998), intracellular inclusion of PKI(6–22) induced a significant reduction in IPSC amplitude within 12 ± 1.2 min after initiation of the whole-cell recording with PKI(6–22)-filled pipettes (Fig. 4d,e), and this PKI(6–22)-induced depression of IPSCs occluded pairing-induced LTDGABA (Fig. 4d–f). Remarkably intra-pipette PKI(6–22)-induced rundown of GABAergic transmission was postsynaptic (PPR values did not change after PKI(6–22) treatment; baseline PPR, 1.02 ± 0.015; PPR of the peak response to PKI(6–22), 1.027 ± 0.3; n = 21; F(1.9,28.69) = 1.448, p = 0.251) and was also prevented by preincubation and bath application of cyclosporine A (1 μm), suggesting that, similar to quinpirole-induced depression and pairing-induced LTDGABA, LTD of PKI(6–22)-loaded neurons is also CaN dependent (Fig. 4e). Moreover, once PKI-induced depression plateaued, we bath applied quinpirole. The PKI-induced depression occluded additional quinpirole-induced synaptic depression, suggesting that D2R activation acts via PKA inhibition to induce LTDGABA (Fig. 4g). It should be noted that, in all experiments performed with quinpirole, CsCl internal solution was used in the intracellular pipette instead of KCl to exclude the possibility that the observed effects of quinpirole were attributable to D2R activation of the G-protein-coupled inwardly-rectifying potassium channels and not through inhibition of PKA. Collectively, these experiments indicate that D2R stimulation through postsynaptic PKA inhibition induces the CaN-dependent LTDGABA.
Disruption of PKA–AKAP150 association at GABAergic but not glutamatergic synapses induces a CaN-dependent LTD
Phosphorylation of GABAARs by PKA seems to mostly stabilize GABAARs in the synapse. Conversely, dephosphorylation of kinase substrates by phosphatases may initiate endocytosis of GABAARs (Luscher et al., 2011; Vithlani et al., 2011). AKAP150 specifically is an attractive candidate as mediator of this phosphorylation–dephosphorylation balance between PKA and CaN for LTD because it is well situated in the synapse to mediate both the effects of PKA and CaN on the regulation of the strength of glutamatergic and GABAergic synapses (Brandon et al., 2003; Snyder et al., 2005; Horne and Dell'Acqua, 2007; Bhattacharyya et al., 2009; Jurado et al., 2010). AKAP150 immunoreactivity has been observed in the midbrain (Glantz et al., 1992); however, the localization of AKAP150 in VTA DA neurons has not been reported previously. Therefore, we first used a double-immunofluorescence staining technique using antibodies against TH (marker for DA neurons) and AKAP150 to visualize DA neurons expressing AKAP150. Colabeled neurons expressing both AKAP150 and TH in the VTA of four rats at two AP levels within the VTA [−4.9 and −5.1 mm caudal to bregma (Paxinos and Watson, 2007)] were scanned and quantified (the total number of cells counted was 447). All TH-positive cells in the studied area of the VTA also expressed AKAP-150 immunoreactivity, whereas AKAP 150-positive cells that did not express TH immunoreactivity were also present (Fig. 5a–c). In the TH-positive cells, the AKAP150-immunoreactivity appeared as granular inclusions throughout the cytoplasm. Our experiments demonstrated for the first time that essentially all dopaminergic neurons in the VTA indeed express AKAP150, although a number of non-DA cells are also AKAP positive (Fig. 5a–c).
Figure 5.
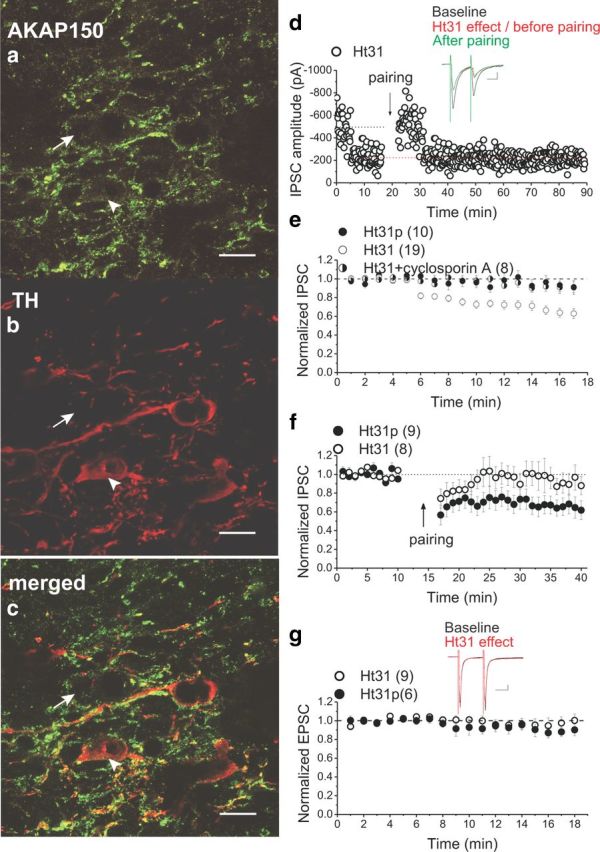
DA neurons of the VTA express endogenous AKAP150, and the disruption of PKA–AKAP150 selectively induces the CaN-dependent LTDGABA. An example of one brain section stained with antibodies to AKAP150 (green) (a), TH (red) (b), and merged from a and b (c), which shows the expression of AKAP150 in TH+ neurons in the VTA. Scale bar, 10 μm. The arrowhead shows a VTA DA neuron with AKAP150 immunoreactivity, and the arrow shows a VTA non-DA neuron with AKAP150 immunoreactivity. d, Single experiment illustrating the effect of intra-pipette Ht31 on basal synaptic transmission and the induction of pairing-induced LTDGABA in the Ht31-loaded cell (only the last 5 min of the baseline responses before the emergence of the Ht31-induced depression is shown in this sample experiment). Inset, Averaged IPSCs before (black) or the peak response of Ht31 (red) or 25 min after pairing (green). Calibration: 50 pA, 25 ms. e, Averaged experiments with intra-pipette 1 μm Ht31 (open symbols) or intra-pipette of 1 μm Ht31p (filled symbols) or intra-pipette 1 μm Ht31 plus 1 μm cyclosporine A (half-filled symbols). Ht31 induces a rapid rundown of IPSCs that is blocked by cyclosporin A treatment (Ht31p cells, 97 ± 8% of the first 5 min baseline values, F(3.0,21.0) = 1.187, p = 0.339; Ht31 cells, 58 ± 6% of the first 5 min baseline values, F(2.2,36.6) = 37.871, p < 0.0001; Ht31 plus cyclosporine A, 92 ± 2% of the first 5 min baseline values, F(2.1,10.4) = 2.044, p = 0.178). f, Averaged LTD induction experiments with control Ht31p peptide (filled symbols) or intra-pipette Ht31 (open symbols). Ht31-induced depression prevents pairing-induced LTDGABA (Ht31p cells, 65 ± 2% of pre-pairing values, F(3.4,26.9) = 8.334, p < 0.0001; Ht31 cells, 90 ± 4% of pre-pairing values, F(3.6,21.4) = 1.112, p = 0.373). g, Averaged experiments with intra-pipette 1 μm Ht31 (open symbols) or intra-pipette 1 μm Ht31p (filled symbols) on AMPAR-mediated EPSCs of Ih(+) neurons. Ht31 does not affect basal glutamatergic transmission mediated by AMPARs (Ht31p, 90 ± 3% of the first 5 min baseline values, F(1.9,9.6) = 3.391, p = 0.078; Ht31 cells, 74 ± 14% of the first 5 min baseline values, F(1.8,12.6) = 0.548, p = 0.574).
Next we tested whether disruption of PKA anchoring to AKAP150 would mimic LTDGABA. We used the anchoring inhibitor peptide Ht31, which binds to the PKA-binding site of a human thyroid AKAP and acts as a competitive antagonist of PKA anchoring to AKAPs but has no effect on PKA activity. In interleaved control experiments, we used Ht31p, the control peptide, as the negative control peptide (Snyder et al., 2005). Intra-pipette Ht31 (1 μm) caused a remarkable rapid rundown of synaptic transmission within 8.45 ± 0.53 min after initiation of the whole-cell recording with Ht31-filled electrodes, whereas intra-pipette Ht31p control peptide (1 μm) did not (Fig. 5d,e). Similar to quinpirole- and PKI(6–22)-induced LTD, intra-pipette Ht31-induced rundown was postsynaptic (PPR values did not change after Ht31 treatment; baseline PPR, 1.056 ± 0.02; PPR of the peak response to Ht31, 1.019 ± 0.2; n = 19; F(1.47,26.54) = 1.334, p = 0.275) and CaN dependent because this rundown was also prevented by preincubation and bath application of 1 μm cyclosporine A (Fig. 5e). To further confirm that Ht31-induced depression of IPSCs and LTDGABA share a similar expression mechanism, we attempted to induce LTD using the LTD induction protocol in Ht31- and Ht31p-loaded cells. Once the depression by Ht31 had plateaued, LFS paired with depolarization failed to induce additional LTD in Ht31-filled neurons, whereas Ht31p-filled neurons still expressed LTDGABA (Fig. 5d,f). The occlusion of LTDGABA with Ht31 further demonstrated that the displacement of PKA from AKAP150 is sufficient to allow for basal CaN activity. Given the established role of AKAP150 in glutamatergic transmission and plasticity, we next examined whether the PKA–AKAP150 association was necessary for AMPAR-mediated synaptic transmission in VTA DA neurons (only Ih(+) neurons were recorded in these experiments). Remarkably, EPSCs were unaffected in Ih(+) cells loaded with Ht31 or Ht31p, suggesting that the PKA–AKAP–CaN complex may be uniquely situated at GABAAR synapses to regulate the trafficking and plasticity associated with GABAARs in VTA DA neurons (Fig. 5g).
Clathrin-mediated endocytosis of GABAARs underlies the AKAP–CaN-dependent LTDGABA
The common mechanism underlying the expression of many forms of postsynaptic glutamatergic LTD seems to involve a clathrin-mediated AMPAR endocytosis (Collingridge et al., 2010). Endocytosis of GABAARs is also regulated through clathrin- and dynamin-dependent mechanisms involving scaffolding protein-mediated interaction of kinases and phosphatases with the GABAAR subunits (Luscher et al., 2011). To test whether clathrin-mediated internalization of GABAARs underlie the expression of LTDGABA in VTA DA neurons, we postsynaptically applied a recently synthesized compound called Pitstop2 that selectively interferes with clathrin-mediated endocytosis of receptors by targeting the terminal domain of clathrin (von Kleist et al., 2011). Postsynaptic loading of cells with 50 μm Pitstop2 prevented LTDGABA, whereas cells loaded with 50 μm Pitstop-control peptide still exhibited LTDGABA in response to the synaptic stimulation (Fig. 6).
Figure 6.
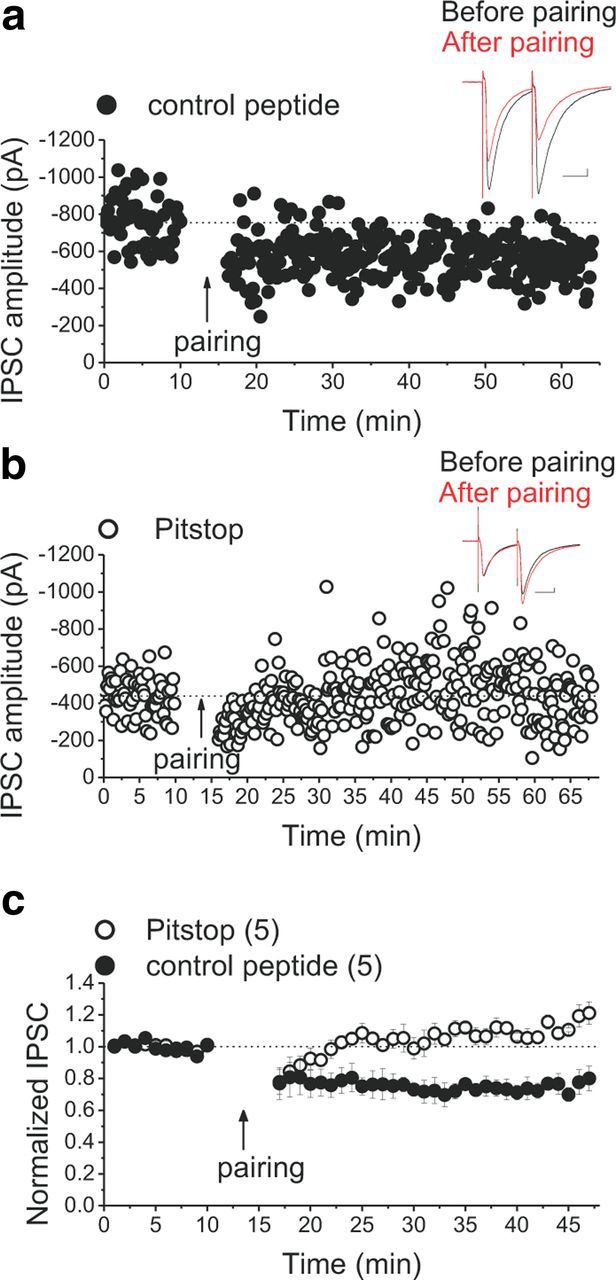
Clathrin-mediated internalization of GABAARs underlies the expression of LTDGABA. a, b, Single experiments showing induction of LTDGABA in cells loaded with Pitstop2 control peptide (filled symbols) or Pitstop2 (open symbols). Insets, Averaged IPSCs before (black) and 25 min after (red) pairing. Calibration: 50 pA, 25 ms. c, Averaged LTD experiments with Pitstop2 control peptide (filled symbols) or Pitstop2 (open symbols). Pitstop2 blocks pairing-induced LTDGABA, whereas control peptide did not affect the induction of LTDGABA (control peptide cells, 76 ± 3% of pre-pairing values, F(5.9,17.88) = 10.149, p < 0.0001; Pitstop cells, 115 ± 5% of pre-pairing values, F(1.39,4.17) = 5.129, p = 0.080).
In summary, we have demonstrated that the induction of LTDGABA results from D2R activation inhibiting PKA, coupled with depolarization- and IP3R-induced Ca2+ release from internal stores, leading to AKAP-mediated activation of CaN and CaN-induced internalization of GABAARs in VTA DA neurons through a clathrin-mediated mechanism.
Discussion
We have identified a novel signaling cascade involving PKA, IP3R, Ca2+, AKAP, and CaN downstream to D2R stimulation that selectively regulates the strength of GABAAR synapses and GABAAR trafficking in VTA DA neurons and provides one expression mechanism for LTDGABA.
Our findings are consistent with the hypothesis that postsynaptic mechanisms are involved in induction and expression of LTDGABA. Postsynaptic disruption of G-protein signaling prevented LTDGABA, confirming that D2 autoreceptors are essential in the induction of LTDGABA. We further elucidated that blockade of group I mGluRs and GABABRs did not impair LTDGABA, excluding the possible roles of these GPCRs in LTDGABA. These findings do not rule out a role for other GPCRs that may be involved in the induction of LTDGABA and also converge on the AKAP–CaN complex to induce LTDGABA with/without involving the PKA signaling pathway.
Because the intracellular inclusion of BAPTA, a fast Ca2+ chelator, did not block LTDGABA, a global increase in intracellular Ca2+ concentration is not necessary, but insensitivity to intra-pipette BAPTA does not rule out local increases in intracellular Ca2+ concentrations close to the point of Ca2+ entry/release. Therefore, a local postsynaptic Ca2+ signaling near the Ca2+ entry site might be sufficient to involve the signaling complex for triggering LTDGABA. In fact, it has been argued that the dissociation constant of BAPTA for Ca2+ makes it an inefficient Ca2+ chelator at relatively low intracellular Ca+2 levels. Therefore, it is likely that CaN is active at low levels of Ca2+ under resting basal conditions (Wang and Kelly, 1997). Moreover, it has been shown that, in the presence of BAPTA, Ca2+ signals might still occur near the Ca2+ source (Issa and Hudspeth, 1996; Nieves-Cintrón et al., 2008). A similar scenario has been reported in arterial smooth muscle cells in which the AKAP150/CaN /NFATc3 signaling is activated by a local PKC-dependent Ca2+ signaling in cells loaded with BAPTA (Nieves-Cintrón et al., 2008). Postsynaptic depolarization is also needed to trigger LTDGABA, suggesting that some voltage-dependent processes facilitate the induction of LTDGABA. We observed that the blockade of L-type VGCCs did not affect LTDGABA, suggesting that the influx of extracellular Ca2+ via these voltage-dependent channels is not needed for LTDGABA. Another source of Ca2+ entry to cytosol is from internal stores, which release Ca2+ through channels such as IP3Rs located on the endoplasmic reticulum (ER) membranes. We also found that the IP3R-mediated Ca2+ release underlie the induction of LTDGABA because intra-pipette application of heparin, an IP3R blocker, completely abolished LTD. Given that D2Rs have direct effects on various voltage-gated ion channels (Na+, K+, and Ca2+ channels) or through inhibition of the cAMP–PKA pathway (Neve et al., 2004), it is likely that D2R activation may involve some of voltage-dependent ion channels located on ER to facilitate the IP3R-mediated Ca2+ release. In fact, it has been suggested that neurons may possess such voltage-dependent mechanisms for intracellular Ca2+ release that could act through G-protein, phospholipase C (PLC), and IP3R activation (Ryglewski et al., 2007). Consistent with our findings, it has been shown that D2R activation can increase the cytosolic levels of Ca2+ via PLC/PKA–IP3R–CaN signaling cascades (Hernandez-Lopez et al., 2000; Hu et al., 2005). Moreover, phosphorylation of IP3Rs by PKA seems to decrease its ability to release Ca2+ (Supattapone et al., 1988; Quinton and Dean, 1992). Whether the D2R–IP3R-mediated local increase in intracellular Ca2+ underlying LTDGABA involves only PKA or also engages PLC remains to be elucidated.
Our next experiments confirmed that the expression of LTDGABA is dependent on CaN; two different CaN inhibitors prevented LTDGABA without affecting the basal synaptic transmission. Similarly, postsynaptic CaN plays an important role in LTD of GABAergic synapses in neurons of deep cerebellar nuclei and in CA1 neurons of the hippocampus (Morishita and Sastry, 1996; Lu et al., 2000; Wang et al., 2003). In the hippocampus, CaN-mediated dephosphorylation of the γ2 subunit of GABAARs increases GABAAR diffusion away from synapses, thereby resulting in less confinement of postsynaptic receptors at GABAergic synapses and LTD expression (Wang et al., 2003). Our experiments with Pitstop2 now clearly show that clathrin-mediated endocytosis of GABAARs underlies the CaN-dependent LTDGABA. However, our results do not exclude a role for other PPs in GABAAR trafficking and LTD because dephosphorylation of β subunits by PP1α and PP2A has also been shown to trigger clathrin-mediated GABAAR internalization (Luscher et al., 2011).
Postsynaptic inhibition of the activity of PKA is sufficient to suppress glutamatergic transmission and occlude an excitatory LTD that is CaN dependent (Kameyama et al., 1998; Tavalin et al., 2002; Snyder et al., 2005). Moreover, the regulation of GABAARs by PKA activity has been widely observed (Brandon et al., 2002). It was very likely that D2R stimulation leads to inhibition of the G-protein-coupled PKA intracellular pathway to allow CaN activity. Consistent with this hypothesis, we found that application of a D2R agonist (quinpirole) or postsynaptic inclusion of a cell-impermeable PKA inhibitor (PKI(6–22)) both induced a chemical form of CaN-dependent LTD that shares a common expression mechanism with LTDGABA. Moreover, PKI-induced depression occluded any additional quinpirole-induced depression, confirming that postsynaptic D2R activation acts via PKA inhibition to trigger LTD. These findings suggest that the basal PKA activity is sufficient to maintain basal GABAergic transmission, and the basal effect of CaN on GABAergic transmission becomes unmasked only in the absence of PKA activity (in this case, D2R stimulation).
How could D2R stimulation and PKA inhibition favor activation of CaN? Several recent studies have shown that LTD and endocytosis of AMPARs at excitatory synapses of the hippocampus requires the binding of endogenous AKAP to CaN and PKA (Kameyama et al., 1998; Tavalin et al., 2002; Snyder et al., 2005; Bhattacharyya et al., 2009; Jurado et al., 2010). In fact, AKAPs can interact directly with CaN and PKC in addition to PKA at these synapses (Sanderson and Dell'Acqua, 2011). The interaction of AKAP150 with GABAARs is not well understood. Using cultured neurons, Brandon et al. (2003) have reported a direct interaction between AKAP150 and the GABAAR β1 and β3 subunits. The PKA phosphorylation of the GABAAR β3 subunits was also reported to be AKAP150-dependent. Another group has argued that the interaction between AKAP150 and the GABAAR β2/3 subunits in the CA1 hippocampal neurons occurs predominantly in the Golgi apparatus rather than at inhibitory synapses in which GABAAR-associated protein, a GABAAR-interacting protein important for GABAAR trafficking, is also present (Lilly et al., 2005). Our immunohistochemical studies strongly suggest that endogenous AKAP is expressed in VTA DA neurons. However, it is still unknown whether a direct interaction between GABAAR subunits and AKAP150 take place at these synapses and whether the targeting of CaN to GABAARs is AKAP150 dependent or also requires other postsynaptic scaffold proteins (Vithlani et al., 2011). If the basal effect of an anchored PKA to AKAP150 is the main determinant of the strength of GABAergic transmission, then the counteracting effect of CaN on GABAergic transmission will only be favored once the PKA–AKAP association is disrupted. Using the AKAP inhibitor peptide Ht31, we demonstrated that, similar to D2R stimulation and postsynaptic inhibition of PKA, postsynaptic disruption of PKA binding from AKAP induces a form of chemical LTD that occludes stimulus-induced LTDGABA. Because of the specific targeting of AKAP150 by Ht31, Ht31 treatment seemed to have a more robust depressing effect on GABAergic transmission than treatments acting more distally to AKAP, presenting the possibility of a floor effect in the Ht31 occlusion experiments. However, similar to quinpirole- and PKI-induced rundown and LTDGABA, the Ht31 rundown was postsynaptic and also CaN dependent. In fact, the magnitudes of rundown induced by PKI, quinpirole, Ht31, and LTDGABA were not significantly different, and the concentrations of drugs used in this study were selected to give a level of receptor/target occupancy that should be in the submaximal range. Remarkably, the effect of Ht31 was selective to GABAAR synapses because AMPAR-mediated glutamatergic transmission was unaffected by Ht31 treatment. Consistent with this finding, postsynaptic stimulation rather than inhibition of PKA activity is suggested to be necessary for induction of glutamatergic LTD in VTA DA neurons (Gutlerner et al., 2002).
We conclude that the PKA-anchored AKAP tonically regulates the strength of GABAAR-mediated transmission and counteracts the basal effects of CaN on GABAA synapses in VTA DA neurons. Once LTD is induced by D2R activation, the basal PKA–AKAP activity is inhibited, allowing CaN to become dissociated from the AKAP complex and becomes active to dephosphorylate GABAARs and promote GABAAR internalization that will ultimately result in LTDGABA.
DA signaling enables reward-related learning through its role in shaping synaptic plasticity and DA cell excitability(Wise, 2008). Therefore, induction of a transient LTDGABA by local release of DA and D2R activation of DA neurons in response to natural rewarding stimuli may serve as a physiological mechanism to decrease the GABAergic inhibitory tone on DA neurons. The resulting increase in DA cell excitability might contribute to the formation of reward-related associative memory. Additionally, addictive drugs increase somatodendritic release of DA (Bradberry and Roth, 1989; Klitenick et al., 1992; Campbell et al., 1996); therefore, it is likely that part of the increased DA cell excitability and DA release in VTA target areas induced by addictive drugs is attributable to an induction of a permanent LTDGABA through D2R stimulation, leading to reduced PKA regulation of AKAP150-anchored CaN activity in the VTA DA neurons. It will be of particular interest to determine whether selective manipulation of GABAergic plasticity through AKAP-dependent signaling in the VTA modulates natural and drug-induced reward. The input specificity of AKAP signaling in VTA DA neurons may provide an important means by which GABAergic inputs onto DA neurons could be selectively manipulated with minimal effects on other inputs. Therefore, pharmacological or genetic targeting of AKAP150 in the VTA DA signaling may present novel and more selective therapeutic interventions to overcome drug addiction.
Footnotes
This work was supported by a Whitehall Foundation grant-in-aid (F.S.N.) and a Department of Defense intramural grant from the Uniformed Services University. We are grateful to Drs. David Lovinger, Stefano Vicini, and Thomas Cote for their critical discussions of previous versions of this manuscript.
The authors declare no competing financial interests.
References
- Adamantidis AR, Tsai HC, Boutrel B, Zhang F, Stuber GD, Budygin EA, Touriño C, Bonci A, Deisseroth K, de Lecea L. Optogenetic interrogation of dopaminergic modulation of the multiple phases of reward-seeking behavior. J Neurosci. 2011;31:10829–10835. doi: 10.1523/JNEUROSCI.2246-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adermark L, Lovinger DM. Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. J Neurosci. 2007;27:6781–6787. doi: 10.1523/JNEUROSCI.0280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. Eur J Neurosci. 2005;21:1280–1288. doi: 10.1111/j.1460-9568.2005.03979.x. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biala G, Betancur C, Mansuy IM, Giros B. The reinforcing effects of chronic d-amphetamine and morphine are impaired in a line of memory-deficient mice overexpressing calcineurin. Eur J Neurosci. 2005;21:3089–3096. doi: 10.1111/j.1460-9568.2005.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradberry CW, Roth RH. Cocaine increases extracellular dopamine in rat nucleus accumbens and ventral tegmental area as shown by in vivo microdialysis. Neurosci Lett. 1989;103:97–102. doi: 10.1016/0304-3940(89)90492-8. [DOI] [PubMed] [Google Scholar]
- Brandon N, Jovanovic J, Moss S. Multiple roles of protein kinases in the modulation of gamma-aminobutyric acid(A) receptor function and cell surface expression. Pharmacol Ther. 2002;94:113–122. doi: 10.1016/s0163-7258(02)00175-4. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Colledge M, Kittler JT, Brandon JM, Scott JD, Moss SJ. A-kinase anchoring protein 79/150 facilitates the phosphorylation of GABA(A) receptors by cAMP-dependent protein kinase via selective interaction with receptor beta subunits. Mol Cell Neurosci. 2003;22:87–97. doi: 10.1016/s1044-7431(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Cocaine inhibits GABA release in the VTA through endogenous 5-HT. J Neurosci. 1994;14:6763–6767. doi: 10.1523/JNEUROSCI.14-11-06763.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AD, Kohl RR, McBride WJ. Serotonin-3 receptor and ethanol-stimulated somatodendritic dopamine release. Alcohol. 1996;13:569–574. doi: 10.1016/s0741-8329(96)00069-9. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Chiu CQ, Carroll RC. Long-term plasticity at inhibitory synapses. Curr Opin Neurobiol. 2011;21:328–338. doi: 10.1016/j.conb.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Dacher M, Nugent FS. Opiates and plasticity. Neuropharmacology. 2011a;61:1088–1096. doi: 10.1016/j.neuropharm.2011.01.028. [DOI] [PubMed] [Google Scholar]
- Dacher M, Nugent FS. Morphine-induced modulation of LTD at GABAergic synapses in the ventral tegmental area. Neuropharmacology. 2011b;61:1166–1171. doi: 10.1016/j.neuropharm.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature. 1998;394:78–82. doi: 10.1038/27919. [DOI] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz SB, Amat JA, Rubin CS. cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol Biol Cell. 1992;3:1215–1228. doi: 10.1091/mbc.3.11.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of dopamine signaling. Biosci Rep. 2001;21:247–269. doi: 10.1023/a:1013205230142. [DOI] [PubMed] [Google Scholar]
- Gutlerner JL, Penick EC, Snyder EM, Kauer JA. Novel protein kinase A-dependent long-term depression of excitatory synapses. Neuron. 2002;36:921–931. doi: 10.1016/s0896-6273(02)01051-6. [DOI] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLCβ1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne EA, Dell'Acqua ML. Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J Neurosci. 2007;27:3523–3534. doi: 10.1523/JNEUROSCI.4340-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XT, Dong Y, Zhang XF, White FJ. Dopamine D2 receptor-activated Ca2+ signaling modulates voltage-sensitive sodium currents in rat nucleus accumbens neurons. J Neurophysiol. 2005;93:1406–1417. doi: 10.1152/jn.00771.2004. [DOI] [PubMed] [Google Scholar]
- Issa NP, Hudspeth AJ. The entry and clearance of Ca2+ at individual presynaptic active zones of hair cells from the bullfrog's sacculus. Proc Natl Acad Sci U S A. 1996;93:9527–9532. doi: 10.1073/pnas.93.18.9527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Kornblum JL, Kauer JA. Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J Neurosci. 2000;20:5575–5580. doi: 10.1523/JNEUROSCI.20-15-05575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC. A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci. 2010;13:1053–1055. doi: 10.1038/nn.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- Kamikubo Y, Tabata T, Kakizawa S, Kawakami D, Watanabe M, Ogura A, Iino M, Kano M. Postsynaptic GABAB receptor signalling enhances LTD in mouse cerebellar Purkinje cells. J Physiol. 2007;585:549–563. doi: 10.1113/jphysiol.2007.141010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- Klitenick MA, DeWitte P, Kalivas PW. Regulation of somatodendritic dopamine release in the ventral tegmental area by opioids and GABA: an in vivo microdialysis study. J Neurosci. 1992;12:2623–2632. doi: 10.1523/JNEUROSCI.12-07-02623.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu Y. GABAB receptors, monoamine receptors, and postsynaptic inositol trisphosphate-induced Ca2+ release are involved in the induction of long-term potentiation at visual cortical inhibitory synapses. J Neurosci. 1996;16:6342–6352. doi: 10.1523/JNEUROSCI.16-20-06342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–773. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70:855–862. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly SM, Alvarez FJ, Tietz EI. Synaptic and subcellular localization of A-kinase anchoring protein 150 in rat hippocampal CA1 pyramidal cells: co-localization with excitatory synaptic markers. Neuroscience. 2005;134:155–163. doi: 10.1016/j.neuroscience.2005.03.039. [DOI] [PubMed] [Google Scholar]
- Lu YM, Mansuy IM, Kandel ER, Roder J. Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron. 2000;26:197–205. doi: 10.1016/s0896-6273(00)81150-2. [DOI] [PubMed] [Google Scholar]
- Luscher B, Fuchs T, Kilpatrick CL. GABA(A) receptor trafficking-mediated plasticity of inhibitory synapses. Neuron. 2011;70:385–409. doi: 10.1016/j.neuron.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006a;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A. 2006b;103:2938–2942. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Sastry BR. Postsynaptic mechanisms underlying long-term depression of GABAergic transmission in neurons of the deep cerebellar nuclei. J Neurophysiol. 1996;76:59–68. doi: 10.1152/jn.1996.76.1.59. [DOI] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Nieves-Cintrón M, Amberg GC, Navedo MF, Molkentin JD, Santana LF. The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc Natl Acad Sci U S A. 2008;105:15623–15628. doi: 10.1073/pnas.0808759105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Kauer JA. LTP of GABAergic synapses in the ventral tegmental area and beyond. J Physiol. 2008;586:1487–1493. doi: 10.1113/jphysiol.2007.148098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008a;28:1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J Neurosci. 2008b;28:14018–14030. doi: 10.1523/JNEUROSCI.4035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Elsevier Academic; 2007. [Google Scholar]
- Quinton TM, Dean WL. Cyclic AMP-dependent phosphorylation of the inositol-1,4,5-trisphosphate receptor inhibits Ca2+ release from platelet membranes. Biochem Biophys Res Commun. 1992;184:893–899. doi: 10.1016/0006-291x(92)90675-b. [DOI] [PubMed] [Google Scholar]
- Reissner KJ, Uys JD, Schwacke JH, Comte-Walters S, Rutherford-Bethard JL, Dunn TE, Blumer JB, Schey KL, Kalivas PW. AKAP signaling in reinstated cocaine seeking revealed by iTRAQ proteomic analysis. J Neurosci. 2011;31:5648–5658. doi: 10.1523/JNEUROSCI.3452-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryglewski S, Pflueger HJ, Duch C. Expanding the neuron's calcium signaling repertoire: intracellular calcium release via voltage-induced PLC and IP3R activation. PLoS Biol. 2007;5:e66. doi: 10.1371/journal.pbio.0050066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Dell'Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF. Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem. 2005;280:16962–16968. doi: 10.1074/jbc.M409693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supattapone S, Danoff SK, Theibert A, Joseph SK, Steiner J, Snyder SH. Cyclic AMP-dependent phosphorylation of a brain inositol trisphosphate receptor decreases its release of calcium. Proc Natl Acad Sci U S A. 1988;85:8747–8750. doi: 10.1073/pnas.85.22.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadavarty R, Rajput PS, Wong JM, Kumar U, Sastry BR. Sleep-deprivation induces changes in GABA(B) and mGlu receptor expression and has consequences for synaptic long-term depression. PLoS One. 2011;6:e24933. doi: 10.1371/journal.pone.0024933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithlani M, Terunuma M, Moss SJ. The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol Rev. 2011;91:1009–1022. doi: 10.1152/physrev.00015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomlin N, Pechstein A, Chau N, Chircop M, Sakoff J, von Kries JP, Saenger W, Kräusslich HG, Shupliakov O, Robinson PJ, McCluskey A, Haucke V. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. Postsynaptic calcineurin activity downregulates synaptic transmission by weakening intracellular Ca2+ signaling mechanisms in hippocampal CA1 neurons. J Neurosci. 1997;17:4600–4611. doi: 10.1523/JNEUROSCI.17-12-04600.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu S, Haditsch U, Tu W, Cochrane K, Ahmadian G, Tran L, Paw J, Wang Y, Mansuy I, Salter MM, Lu YM. Interaction of calcineurin and type-A GABA receptor γ2 subunits produces long-term depression at CA1 inhibitory synapses. J Neurosci. 2003;23:826–836. doi: 10.1523/JNEUROSCI.23-03-00826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Dopamine and reward: the anhedonia hypothesis 30 years on. Neurotox Res. 2008;14:169–183. doi: 10.1007/BF03033808. [DOI] [PMC free article] [PubMed] [Google Scholar]