

Abstract
The biolipid sphingosine-1-phosphate (S1P) is an essential modulator of innate immunity, cell migration, and wound healing. It is released locally upon acute tissue injury from endothelial cells and activated thrombocytes and, therefore, may give rise to acute post-traumatic pain sensation via a yet elusive molecular mechanism. We have used an interdisciplinary approach to address this question, and we find that intradermal injection of S1P induced significant licking and flinching behavior in wild-type mice and a dose-dependent flare reaction in human skin as a sign of acute activation of nociceptive nerve terminals. Notably, S1P evoked a small excitatory ionic current that resulted in nociceptor depolarization and action potential firing. This ionic current was preserved in “cation-free” solution and blocked by the nonspecific Cl− channel inhibitor niflumic acid and by preincubation with the G-protein inhibitor GDP-β-S. Notably, S1P3 receptor was detected in virtually all neurons in human and mouse DRG. In line with this finding, S1P-induced neuronal responses and spontaneous pain behavior in vivo were substantially reduced in S1P3−/− mice, whereas in control S1P1 floxed (S1P1fl/fl) mice and mice with a nociceptor-specific deletion of S1P1−/− receptor (SNS-S1P1−/−), neither the S1P-induced responses in vitro nor the S1P-evoked pain-like behavior was altered. Therefore, these findings indicate that S1P evokes significant nociception via G-protein-dependent activation of an excitatory Cl− conductance that is largely mediated by S1P3 receptors present in nociceptors, and point to these receptors as valuable therapeutic targets for post-traumatic pain.
Introduction
Ongoing pain regularly results as a consequence of surgical interventions or trauma. Although this represents a major clinical problem and severely affects the patients' recovery, pain relief strategies so far are restricted to the classical opioids or nonopioid analgesics. To develop better strategies based on mechanistic insight, the sequelae of mediators and interactions of injured tissue with the nociceptive system leading to postoperative pain need to be elucidated. Upon tissue injury and damage to blood vessels, vascular leakage, accumulation, and activation of thrombocytes and plasma extravasation occur. Injection of plasma and experimentally activated thrombocytes excite nociceptors and induce pain (Armstrong et al., 1957; Ringkamp et al., 1994). Human platelets contain high concentrations of sphingomyelin-derived sphingosine-1-phosphate (S1P), which can be released upon activation and shape change (Dahm et al., 2006; Ulrych et al., 2011). In blood plasma, S1P levels may reach up to micromolar concentrations, most of which is bound to plasma proteins (Murata et al., 2000; Schmidt et al., 2006; Ohkawa et al., 2008). S1P acts as a lipid growth factor and induces robust endothelial cell activation, resulting in cellular locomotion, vascular maturation, and angiogenesis (Daum et al., 2009). Furthermore, S1P has a general role in postoperative wound healing via either intracellular mode of action or specific targeting of metabotropic membrane receptors (Vogler et al., 2003; Watterson et al., 2007). In general, S1P is the natural ligand of five different G-protein-coupled receptors (GPCRs S1P1-S1P5) that are expressed in various tissues, including neurons and nociceptors (Spiegel and Milstien, 2003; Herr and Chun, 2007; Chi and Nicol, 2010; Mair et al., 2011). Exogenous S1P enhances the excitability of DRG neurons, potentiates heat-sensitive transducer channels and modifies nociceptor heat sensitivity via neuronally expressed S1P1 receptor (Mair et al., 2011). In addition, this receptor has been associated with opioid-induced hyperalgesia (Muscoli et al., 2010; Mair et al., 2011). Because local increases in S1P levels arise from acute tissue injury, damage to blood vessels and activation of platelets after trauma or surgical procedures, we hypothesized that S1P may have a significant role in the mechanism causing post-traumatic pain. In this paper, we address this issue with an interdisciplinary approach and investigate S1P-induced nociceptor excitation in a cellular model. In addition, the relevance of this finding is addressed in vivo in mice and humans.
Materials and Methods
Ethics statement.
All animal experiments have been performed with permission of the Austrian Bundesministerium für Wissenschaft und Forschung (BMWF) ministry (BMWF-66.011/0051-II/10b2008; BMWF-66.011/0113-II/3b/2010; GZ 66.011/85-C/GT/2007) and according to ethical guidelines of the IASP (International Association for the Study of Pain) (Zimmermann, 1986).
Flinching and licking measurement.
All behavioral measurements were done in age-matched awake, unrestrained, male C57BL/6J mice (> 8 weeks old) by individuals who were blinded to the genotype of the mice being analyzed. Spontaneous pain-like behavior was monitored after intracutaneous injection of S1P (15 μl, 500 μm) or vehicle (15 μl, 10% methanol in PBS) into the left hindpaw of wt, S1P3−/−, S1P1fl/fl, or SNS-S1P1−/− mice, which selectively lack the S1P1 receptor subtype in Nav1.8 channel-expressing nociceptive neurons (Mair et al., 2011). Mice were placed in the plastic chamber and allowed to habituate for at least 1 h before the experiment. Flinching and licking behavioral responses were scored over a 30 min period after injection. When mice rapidly shook their hindpaw, it was scored as a positive flinching response and then the number of flinches was assessed manually. Licking behavior was assessed by counting manually the number of licks and the overall duration of all scored licks.
Mouse model of incisional plantar pain.
All mice were anesthetized with ketamine (2 mg/kg) and xylasol (0.2 mg/kg) (both from Ogris Pharma). The plantar aspect of the hindpaw was disinfected with polyvidone-iodine solution (Wundesin, Gebro Pharma) and a 1-cm-longitudinal incision was made with a sterile surgical blade (no. 10), through skin and fascia of the plantar aspect of the paw, starting 0.5 cm from the proximal edge of the heel and extending toward the toes. The plantaris muscle was also incised longitudinally. The skin was sutured with two thread of 5-0 polypropylene on a TF needle (Prolene, Ethicon, Johnson & Johnson Medical Products) and the wound covered with Wundesin. The sutures were removed under anesthesia on postoperative day 2, and typically wounds healed within 5–6 d.
Heat sensitivity.
Heat sensitivity was assessed using the Hargreaves test (Hargreaves et al., 1988). A radiant heat source delivering an increasing heat stimulus was focused under the plantar surface of the hindpaw. The time from initiation of the radiant heat until paw withdrawal (paw withdrawal latency [PWL] in seconds) was measured automatically with an algesiometer (Ugo Basile). Each hindpaw was tested at least three times, and the mean withdrawal latency was calculated. The time interval between two trials on the same hindpaw was at least 1 min.
Mechanical sensitivity.
At least 1 h before testing, mice were placed in a Plexiglas chamber (10.5 × 10.5 × 14 cm) with a metal grid floor. Mechanical sensitivity was tested manually before and after surgery by using calibrated von Frey monofilaments with defined bending forces (1.4, 4, 8, 16, 22.6, 32, and 45.3 mN). Two baseline measurements were taken at 2 and 1 d before surgery. Each filament was applied five times with at least 60 s between applications. Data are expressed as the percentage of paw withdrawals in response to each filament (mechanical response frequency). Positive responses were counted when the paw was sharply withdrawn and flinching of the paw was observed immediately after removal of the filament. All other movements of the paw were considered as unclear responses, and the stimulus application was repeated.
Transgenic mice.
Global S1P3 receptor null mutant mice (S1P3−/−) and mice homozygous for the floxed exon 2 allele of the S1P1 receptor gene (S1P1fl/fl), which encodes for the entire receptor protein, have been described previously (Allende et al., 2003; Kono et al., 2004). S1P1fl/fl mice were cross-bred with SNS-Cre-mice (Agarwal et al., 2004) to obtain homozygous SNS-Cre:S1P1fl/fl (hereinafter referred to as SNS-S1P1−/−) and S1P1fl/fl mice (control littermates; the Cre-recombinase is homozygous or heterozygous) as previously published (Mair et al., 2011).
Flare-reaction measurement.
Five healthy subjects (1 woman and 4 men; mean age 32 years; range, 23 years) participated in this randomized, double-blinded study. Each subject was informed and gave written consent to take part in the study; the experimental protocol was approved by the Ethics Committee of the Medical Faculty of Mannheim of the University Heidelberg. Vehicle solution (saline, 15% cyclodextrin) and S1P (100 μl of 0.125, 0.25, 0.5, and 1 mm) were injected in the central volar forearm at distances of 4 cm between the injection sites. Superficial blood flow around the injection site was assessed before the injection and immediately thereafter repeatedly in 4 min intervals using a laser-Doppler imager (Moor Instruments). An area of 9 × 34.5 cm around the injection sites was scanned. The area of the axon-reflex erythema was determined as the number of cm2 increasing >2-fold compared with the baseline scan by dedicated software (Moorldi Version 5.0, Moor Instruments). Pain scores were assessed for the injections of S1P and vehicle (saline, 15% cyclodextrin); however, the pain upon injection did not differ significantly between vehicle and S1P and there was no lasting pain sensation after retraction of the cannula (data not shown).
Primary sensory neuron culture.
Lumbar (L1-L6) DRG with the cell bodies of primary afferents that project into the hindpaw were harvested from adult C57BL/6J or transgenic mice and dissociated as previously published (Mair et al., 2011). Briefly, ganglia were treated enzymatically with collagenase (Liberase, Roche) and trypsin-EDTA (Invitrogen), and dissociated mechanically with a fire-polished Pasteur pipette. The resulting cell suspension was washed, plated on glass coverslips coated with poly-L-lysine/laminin (Sigma) and cultivated in synthetic, serum-free medium (supplemented TNB, Biochrom) supplemented with nerve growth factor (NGF; 100 ng/ml) at 37°C in 5% CO2 in a humidified incubator for 18–32 h.
Electrophysiology.
Using the whole-cell voltage-clamp configuration of the patch-clamp technique, ionic currents were recorded from isolated neurons at a holding potential of −80 mV as previously published (Obreja et al., 2002, 2005). The external control solution (ECS-low Ca2+) contained the following (in mm): 150 NaCl, 5 KCl, 0.1 CaCl2, 1 MgCl2 (all Sigma), 10 glucose and 10 HEPES (Merck), at pH 7.3 adjusted with NaOH (Merck). Borosilicate glass micropipettes (Science Products) pulled with a horizontal puller (Sutter Instruments) were filled with internal solution (ICS, in mm): 148 KCl, 2 MgCl2, 2 Mg-ATP, 0.2 Na-GTP, 0.1 CaCl2, 1 EGTA (all Sigma) and 10 HEPES (Merck), at pH 7.3 adjusted with KOH (Merck). After filling, electrode resistance was 2.5–3.5 MΩ. Currents were filtered at 2.9 kHz, sampled at 100 Hz, and recorded using an EPC-9 (HEKA) and the Pulse Version 8.74 software (HEKA). The “cation free” extracellular solution contained the following (in mm): 163 NMDG, 5 EGTA, 10 HEPES, 10 d-glucose, 1 MgCl2 at pH 7.3 adjusted with HCl (Merck). The membrane potential was recorded using current-clamp configuration (Ihold = 0 pA) using an ECS containing the following (in mm): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2 (all Sigma), 10 d-glucose and 10 HEPES (Merck), at pH 7.3 adjusted with NaOH (Merck). The pipette solution was composed (in mm) of 45 KCl, 98 K-gluconate, 0.5 CaCl2, 5 EGTA, 10 HEPES, 2 MgATP, 0.2 NaGTP, pH 7.3 adjusted with KOH (Merck). The membrane potenial was sampled at 4 kHz. Experiments were performed at room temperature (∼22°C), and only a single neuron was tested per coverslip. An automated seven-barrel system with common outlet at 100 μm distance of the recorded cell was used for fast S1P administration (Dittert et al., 1998).
Microfluorimetric calcium measurements.
Cells were plated on glass-bottom dishes coated with poly-L-lysine hydrobromide (10 μg/ml) and laminin (10 μg/ml) (both from Sigma). One day cultured cells were incubated with 3 μm of the calcium (Ca2+) sensitive dye Fura-2-acetoxymethyl ester (Invitrogen) in ECS consisting of the following (in mm): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 d-glucose (all from Sigma) and 10 HEPES (Merck), at pH 7.3 adjusted with NaOH (Merck) and were incubated for 25 min at 37°C in 5% CO2, in a humidified incubator. Then cells were washed twice with PBS (PAA) and finally kept in ECS for experiments. For the Ca2+-free solution, CaCl2 was replaced by 5 mm EGTA. Ca2+ recordings were performed using an Olympus IX71 microscope (Olympus) with a 20×/0.85 N.A. oil-immersion objective (Olympus). Fura-2 was excited consecutively at 340 and 380 nm (excitation time: 25 ms) with a polychrome IV monochromator (TILL Photonics), and fluorescence intensities were filtered by a 510 nm LP filter and recorded with a CCD camera (TILL Photonics). The changes of intracellular Ca2+ concentration [Ca2+]i are depicted as ratio of fluorescence intensities collected at 340 nm and 380 nm (ΔF340/380) at a time interval of 1 s (Linhart et al., 2003). For data acquisition, MetaFluor4.6r8 (Universal Imaging) was used and off-line analysis was performed with OriginPro7.SR2 (Origin Lab). The threshold for S1P-positive cells was set to fourfold the SD of the Ca2+ signal evoked by 0.1% methanol (ΔF340/380 = 0.04). All chemicals were purchased from Sigma.
Immunohistochemistry.
Murine DRGs were dissected, fixed in Zamboni's fixative for 24 h, dehydrated through a graded series of ethanol and DMSO, and subsequently embedded in polyethylene glycol (1450 MW: Sigma-Aldrich) (Murphy et al., 1998). DRGs were sectioned at 12 μm and stored in PBS containing 0.01% sodium azide at 4°C before multiple-labeling immunohistochemistry. Free-floating sections were blocked using 10% normal donkey serum and incubated for 48 h with the S1P3 antiserum (a generous gift from S. Mandala). After washing in PBS, the sections were incubated for 2 h with secondary antiserum (Cy3-labeled donkey anti-rabbit IgG, Jackson ImmunoResearch Laboratories), washed in PBS, and coverslipped in buffered glycerol. Likewise, indirect immunofluorescence was performed in cultured neurons fixed in Zamboni's fixative for 2 h and stored in PBS.
Human DRGs (N = 2) were obtained from the South Australian and Victorian Brain Bank from motor neuron disease patients 12–16 h postmortem. DRGs were immersion fixed in Zamboni's fixative for 24–72 h. The use of human material was approved by the Flinders Clinical Research Ethics Committee. Human DRGs were embedded in polyethylene glycol as described previously (Murphy et al., 1998), cut at a thickness of 10 μm, and the free floating sections were incubated for 48 h with S1P3 antiserum. After washing in PBS, the sections were incubated overnight with secondary antiserum, washed in PBS, and coverslipped in buffered glycerol. Images were taken using a confocal laser scanning microscope (Leica SP5).
Immunocytochemistry.
DRG neurons were cultured on coverslips, fixed for 2 h in Zamboni's fixative, washed in PBS, and incubated with primary antisera overnight, followed by washing and 1 h incubation with secondary antisera. Finally, the coverslips were washed in PBS and coverslipped in buffered glycerol (Mair et al., 2011). Preabsorption of the S1P3 antiserum with the corresponding antigen led to the absence of staining (Table 1).
Table 1.
Antigen characteristics
| Antigen | Host | Dilution | Source |
|---|---|---|---|
| S1P2 (directed against C-terminus) | Rabbit | 1:100 | S. Mandala |
| S1P3 (directed against N-terminus) | Rabbit | 1:100 | S. Mandala |
| NF200 | Mouse, clone N52 | 1:1000 | Sigma |
| I-B4 | Bandeira simplifolicia | 1:1000 | Sigma |
| Secondary antisera | |||
| Cy3 anti-rabbit Ig | Donkey | 1:100 | Jackson |
| Cy5 anti-mouse Ig | Donkey | 1:50 | Jackson |
mRNA in situ hybridization.
The protocol used was modified from an earlier method used by Obernosterer et al. (2007). Cryostat sections of mice lumbar DRGs were fixed with Zamboni's fixative for 24 h, washed in PBS, digested with proteinase K (2 mg/ml), fixed with 4% PFA, acetylated (triethanolamine/HCl/acetic anhydride), and incubated with prehybridization buffer. Target mRNAs were hybridized with 0.34 pmol of specific antisense double digoxigenin-labeled mRNA detection probes (S1P3, 5DigN/ACTGATGAGGAAGGCGATGTAT/3Dig_N) or scrambled probes (Exiqon) or hybridization buffer as negative controls (Obernosterer et al., 2007) overnight at 55°C. Washing with sodium–saline citrate was followed by blocking and overnight incubation with alkaline phosphatase-coupled anti-digoxigenin antiserum. Incubation with secondary antisera for 1 h was followed by washing in PBS. Slides were coverslipped in buffered glycerol and images taken using an epifluorescence microscope (BX50, Olympus). Subsequently coverslips were removed, and the sections incubated with BCIP/NBT (Roche) to visualize S1P3 receptor mRNA.
Data analysis and statistics.
Statistical analysis was performed per animal (N) and per number of cells (n) for the patch-clamp and calcium imaging data. For detailed statistical analysis, the Sigma Stat 3 and GraphPad Prism 5 software were used, and the data are presented as mean ± SEM. Statistical tests were used depending on sample size, distribution, and number of variables: two-way repeated-measures (RM) ANOVA with post Tukey test, the nonparametric Mann–Whitney U test, or the parametric unpaired two-sided t test for comparison between groups; for intraindividual comparisons, the Wilcoxon matched-pairs signed-rank test or the parametric Student's paired t test was used. Fisher's test was used where stated. Differences were considered statistically significant at p < 0.05.
Results
S1P evoked pain-like behavior in mice and flare-reaction in humans
To determine whether S1P was involved in the spontaneous pain induction in vivo, we examined flinching and licking responses in wt mice after S1P intradermal injection. We measured the number of flinches for 10 min after S1P (500 μm, 15 μl) or vehicle (10% methanol in PBS, 15 μl) injection into the left hindpaw (ipsilateral) of wt mice. Interestingly, we found that upon S1P injection mice developed a significant increase in the number of flinches (4.9 ± 2.02 compared with vehicle, p = 0.0007, Mann–Whitney U test, N = 10 mice per condition, Fig. 1A). This was accompanied by an increase in S1P-induced number of licks (5.31 ± 1.53 S1P-injected vs vehicle-injected mice, p = 0.0011, Mann–Whitney U test, N = 7 mice per condition, Fig. 1B) and licking time (26.14 ± 8.27 s compared with vehicle, p = 0.001, Mann–Whitney U test, N = 7 mice per condition, Fig. 1C). No effect on flinching and licking behavior was observed in vehicle-injected control mice (Fig. 1A–C) or at lower S1P doses (10 and 100 μm; data not shown). Furthermore, in control experiments, methanol did not produce any depolarization, Ca2+ rise, or pain-like behavior up to concentrations of 10%. These data indicate that S1P can evoke acute spontaneous pain-like behavior in vivo.
Figure 1.
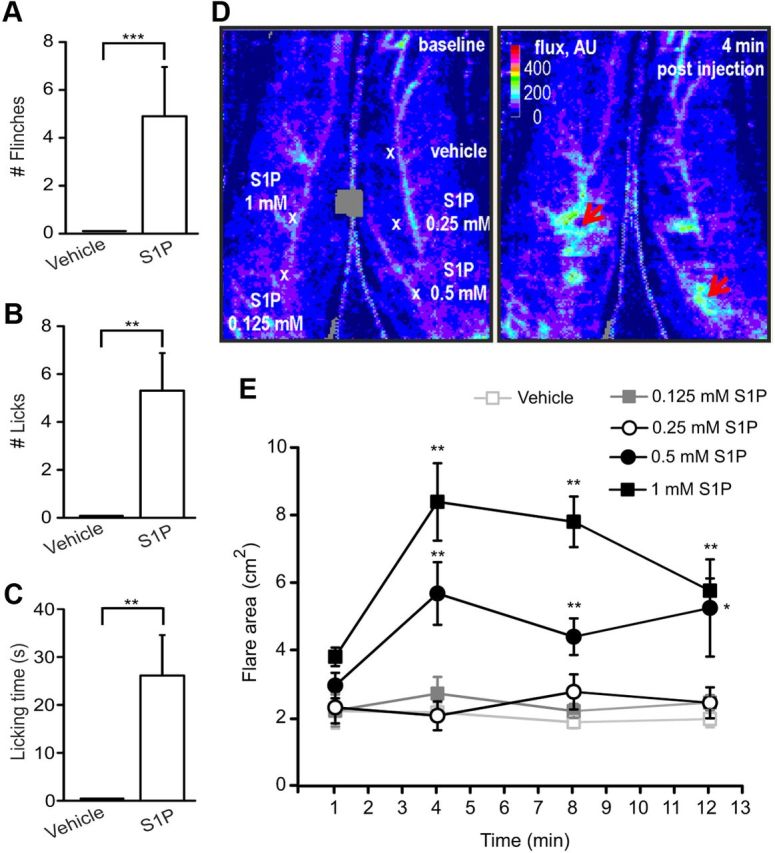
Contribution of S1P to pain-like behavior in mice and axon reflex in humans. Intracutaneous injection of S1P into the ipsilateral hindpaw of wt mice significantly enhanced the number of flinches (A), licks (B), and increased the time of licking response during 10 min upon administration (C) (N = 7–10 mice). D, Specimen (before and 4 min after S1P injections) of the flare reaction recorded by laser Doppler imaging indicating an increased blood flow by lighter colors (flux in arbitrary units [AU], and in a pseudo-color scale) after intradermal injection of increasing concentrations of S1P (100 μl of 0.125, 0.25, 0.5, 1 mm) and vehicle (100 μl, saline, 15% cyclodextrin). Larger flare areas were developed around the injection sites of the two highest concentrations (red arrows). E, Quantitative analysis of the flare reaction area produced by S1P in human skin by repeated laser-Doppler images of the flare profile. The graph represents the time course of the axon reflex flare development (flare area in cm2) around the injection sites of vehicle and S1P (injection after minute 1) at 4 min intervals. *p < 0.05. **p < 0.01. ***p < 0.001.
To investigate whether these findings bear relevance to pain-related behavior in humans, increasing doses of S1P (0.125, 0.25, 0.5, and 1 mm) were injected intradermally. As objective assessment of skin nociceptor excitation the increase of skin blood flow (in pixels) and the corresponding size of the axon-reflex flare area (in cm2) around the different injected sites were monitored with laser Doppler imaging. Cutaneous S1P challenges produced a dose-dependent flare response by increasing the blood flow in the skin immediately after injection (increased intensity in arbitrary units is indicated by red arrows in Fig. 1D, right). Pain scores were assessed for the injections of S1P and vehicle (saline, 15% cyclodextrin); however, the pain upon injection did not differ significantly between vehicle and S1P, and there was no longer lasting pain sensation after retraction of the cannula (data not shown). S1P provoked a massive increase of the axon reflex-mediated blood flow showing the most pronounced effect at 0.5 and 1 mm concentration at 4 min after injection (flare area = 5.82 ± 0.95 cm2 and 8.58 ± 1.16 cm2, respectively) that was significantly higher compared with the vehicle-injected site (flare area = 2.23 ± 0.28 cm2) (p = 0.0000113 compared with 0.5 and 1 mm S1P-injented subjects within the time, two-way RM ANOVA with post Tukey test, N = 5 subjects; p = 0.0079 for 0.5 mm S1P vs vehicle at 4 min after injection, p = 0.0079 for 1 mm S1P vs vehicle at 4 min after injection, Mann–Whitney U test, Fig. 1E). The S1P-evoked flare response was detectable until 12 min after injection, indicating a long-lasting effect of S1P on skin vasodilatation (Fig. 1E). These results support the hypothesis that S1P has a significant role in spontaneous pain behavior indicated by pain like-behavior in mice and skin flare reaction as a sign of nociceptor activation in humans.
S1P-induced membrane depolarization and generation of action potentials in neurons
In the whole-cell current-clamp configuration of the patch-clamp technique, exposure of single dissociated mouse DRG neurons to S1P (1 μm, 10 s) induced a significant membrane depolarization from a resting membrane potential of −54.39 ± 1.02 mV (Ctrl) to −35.68 ± 1.99 mV (S1P) in n = 34 neurons tested (p = 0.0001, t(33) = 10.297, paired t test, N ≥ 3 mice each condition, Fig. 2A,B). Depolarization was sufficient to induce firing of action potentials (APs) upon S1P stimulation in 22 of 34 neurons [64.7%] (fAP = 1.67 ± 0.04 s−1, p = 0.0000004768, T+ = 253, Wilcoxon matched-pairs signed-rank test, Fig. 2C,D). Both depolarization and AP discharge recovered to prestimulation values within 10 min of washout (recovery Em = −49.94 ± 1.23 mV vs S1P application, p = 0.0000511, t(32) = 8.357 and recovery fAP = 0.076 ± 0.068 s−1 vs S1P application, p = 0.00002384, T+ = 8, respectively). These results suggest a direct excitatory effect of S1P on primary afferent neurons.
Figure 2.

Effect of S1P on the membrane potential in murine DRG neurons. A, B, Short application of S1P (1 μm, 10 s) depolarized DRG neurons by changing the membrane potential (Em) from −54.4 ± 1.0 mV in control (Ctrl) condition to −36 ± 2 mV in the presence of S1P. The Em recovery value was measured 10 min after S1P application (−50 ± 1 mV). C, D, Most neurons (64.7%) generated APs in response to S1P (1 μm, 10 s) with a mean firing rate of 1.7 ± 0.4 AP s−1. ***p < 0.001.
S1P-induced slowly activating and slowly inactivating excitatory ionic conductance
Using the whole-cell voltage-clamp configuration of the patch-clamp technique, we observed that S1P (1 μm, 10 s) generated currents with an amplitude of 0.538 ± 0.036 nA (n = 34 cells, Fig. 3A) in most sensory neurons. S1P-evoked currents reached a maximum value after 100.4 ± 4.14 s (n = 57 cells) and were slowly deactivating (τ = 177.90 ± 15.16 s). S1P-activated currents increased in a dose-dependent manner with an EC50 of 0.160 μm (Fig. 3B).
Figure 3.

Ionic nature of the excitatory currents induced by S1P. A, Typical recording of a responsive neuron after a short pulse of S1P (1 μm, 10 s). B, The graph shows the dose–response relationship of the S1P-induced conductance. The EC50 was 0.160 μm S1P. C, Top, Representative Ca2+ imaging measurement of the S1P-evoked Ca2+ transient in DRG neurons in ECS (containing 2 mm Ca2+). Bottom, The Ca2+ rise induced by S1P was completely abolished in Ca2+ free condition (5 mm EGTA, ∼3.5 min exposure) but lightly recovered after washout (2 min). D, E, A small change in [Ca2+]i induced by short application of S1P (1 μm, 60 s) was observed in most DRG neurons. In presence of TG (1 μm, 5 min), the percentage of S1P-responding neurons and the Ca2+ increase magnitude was not significantly different from control conditions. For each Ca2+ measurement, 25–30 neurons (N ≥ 3 mice) were randomly selected in the field. F, Top, Whole-cell recording from S1P-activated current in “cation free” condition (0 Ca2+, 0 K+, 0 Na+, 1 min). Bottom, Quantification of the S1P-induced current density for recordings in F that remained unaltered after neuron exposure to “cation free” solution (3 min after S1P application). n.s., Not significant (p > 0.05). ***p < 0.001.
Because S1P reportedly increases intracellular calcium concentrations ([Ca2+]i) (Hopson et al., 2011), we hypothesized that excitatory currents were at least partially carried by Ca2+ (and Na+) ions. We performed Fura-2-based Ca2+ imaging on cultured neurons and found that S1P-induced [Ca2+]i rises in 182 of 308 [59.1%] neurons (Fig. 3C top, D). Neurons usually responded to S1P within 15 s, and mean ratio amplitudes of evoked Ca2+ responses were ΔF340/380 = 0.14 ± 0.01 (N = 6 mice, n = 308 cells, Fig. 3E). To determine the Ca2+ source of S1P-induced [Ca2+]i elevation, Ca2+ measurements were performed in Ca2+ free solution (5 mm EGTA), and we found S1P-induced Ca2+ transients fully abrogated (ΔF340/380 = 0.0043 ± 0.0005; p = 0.00002 vs S1P, t(126) = 8.791, unpaired t test, N = 3 mice each, n = 127 cells, Fig. 3C bottom, D,E). These results were further corroborated by recordings from isolated DRG neurons after depletion of intracellular Ca2+ stores with thapsigargin (TG), an inhibitor of the endoplasmic reticulum Ca2+-ATPase SERCA (Rogers et al., 1995). Cultured neurons exposed to TG (1 μm, 5 min) did not show any Ca2+ alteration mediated by high potassium-induced depolarization (data not shown). As expected, the percentage of S1P reacting neurons was unchanged (79 of 136 responders [58.1%]; p = 0.917, Fisher's test, Fig. 3D) and neuronal responses to S1P after TG treatment (ΔF340/380 = 0.12 ± 0.01) were similar to untreated controls (p = 0.264, Fisher's test, N = 4 mice, n = 136 cells, Fig. 3E). Together, these data suggest that S1P-evoked Ca2+ transients are the result of a Ca2+ influx rather than a release from intracellular stores.
S1P-activated chloride conductance in primary sensory neurons
To determine the relative contribution of cations to the overall current induced by S1P, whole-cell voltage-clamp recordings were performed using “cation-free” external solution (0 Na+, 0 K+, and 0 Ca2+). Surprisingly, we observed that the S1P-evoked current was fully preserved in “cation-free” ECS (ΔIS1P: 6.18 ± 1.08 pA/pF control vs 5.85 ± 1.38 pA/pF after 1 min “cation free”; p = 0.138, t(15) = 1.568, paired t test N = 3 mice, n = 18 cells, Fig. 3F). This result indicated that excitatory currents were not carried by cations and that Ca2+ influx was likely the result of voltage-gated Ca2+ channel activation secondary to the Ca2+-independent membrane depolarization. Because chloride (Cl−) channels have been associated with the activation of ionic currents by S1P in neuroblastoma cells (Postma et al., 1996), electrophysiological and microfluorometric Ca2+ measurements were performed in the presence of the nonspecific Cl− channel blocker niflumic acid (NFA). We found significant inhibition of IS1P by NFA by reducing the deactivation time constant (τ) from 177.90 ± 15.16 s in control to 44.46 ± 5.38 s with NFA (100 μm) (p = 0.000100839, T = 36, Mann–Whitney U test, N = 3 mice each, n = 18 cells, Fig. 4A,B). Moreover, incubation with NFA (100 μm, 9 min) almost fully prevented S1P-evoked Ca2+ transients (ΔF340/380 = 0.023 ± 0.003, p = 0.0000021, t(434) = 7.581, unpaired t test, N = 4 mice each, n = 128 cells, Fig. 4C,E) and only 18 of 128 (14%) neurons still responded to S1P (p = 0.0000025, Fisher's test, Fig. 4D). Both results support the idea that Cl− is the main ion carrying S1P-induced currents in sensory neurons.
Figure 4.
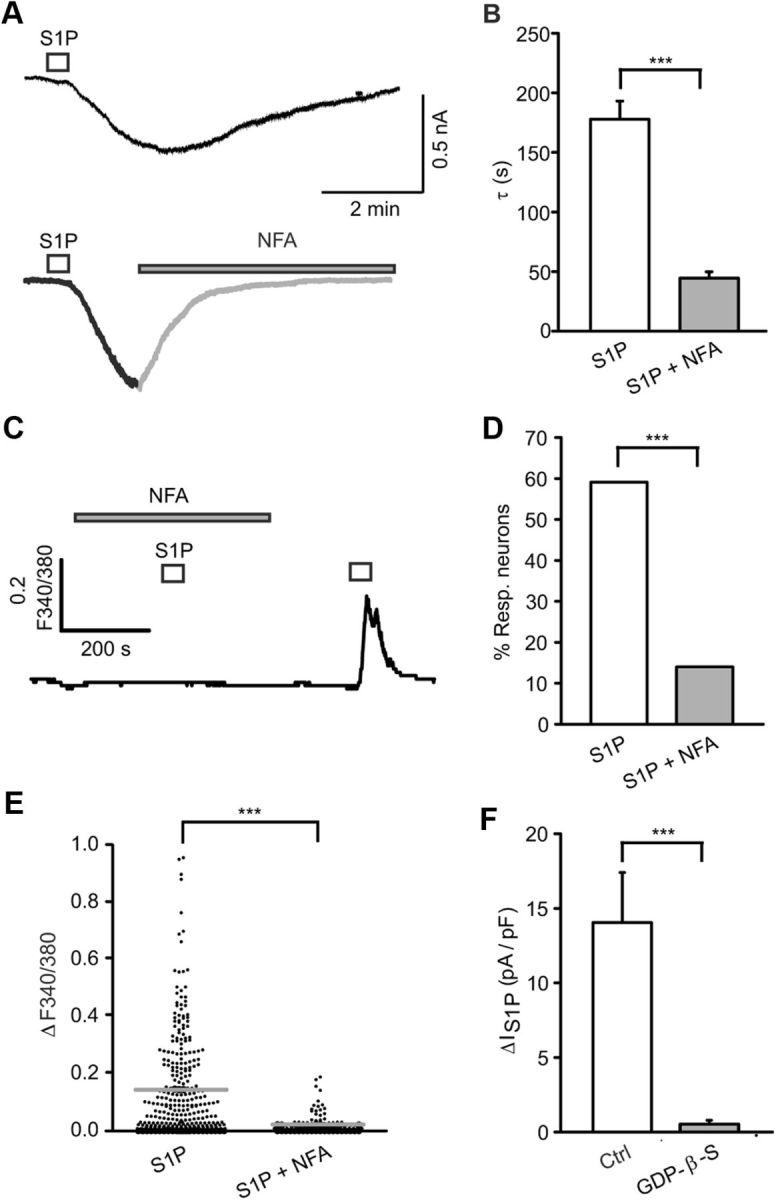
Contribution of Cl− to the ionic conductance of S1P-activated currents. A, B, Application of the nonspecific Cl− channel blocker NFA (100 μm, 5 min) drastically diminished the time constant (τ) of S1P-evoked currents. C, Application of NFA (100 μm, 9 min) prevented the S1P-induced Ca2+ influx. D, Only 14% of neurons [18 of 128] responded to S1P in the presence of NFA. E, S1P-induced Ca2+ response was fully inhibited by NFA. For each Ca2+ measurement, 25–30 neurons from N ≥ 3 mice were selected at random in the field. F, Incubation of DRG neurons with the general G-protein inhibitor GDP-β-S (3 mm, ∼9 min) fully blocked S1P-induced current density. ***p < 0.001.
S1P-induced conductance and excitation is reduced in S1P3 receptor-deficient neurons
Two possible mechanisms of activation may account for S1P-induced current activation: (1) S1P may act as a direct ligand at a channel protein, or (2) channel gating may occur indirectly and requires G-protein coupled S1P receptors. To discriminate between both possibilities, neurons were internally perfused with GDP-β-S (a general G-protein inhibitor; 3 mm, ∼9 min). This treatment fully abolished IS1P in DRG neurons, suggesting a G-protein-dependent mechanism (p = 0.0000015, T = 11, Mann–Whitney U test, N = 3 mice, n = 13 neurons, Fig. 4F).
At the moment, five G-protein-coupled S1P receptors are known: S1P1 receptors were found predominantly expressed in small peptidergic and nonpeptidergic (isolectin B4-positive [I-B4+]) sensory neurons, which are important in regulating behavioral sensitivity during inflammation (Chi and Nicol, 2010; Mair et al., 2011). In addition, S1P2 and S1P3 mRNAs are also detected in DRG neurons (Mair et al., 2011). To address expression patterns for S1P receptors in certain neuronal populations, indirect immunofluorescence was performed with specific antisera for S1P receptors, which were a generous gift from S. Mandala, and colabeling with a marker for large nonnociceptive neurons (Neurofilament 200 [NF200]). S1P2 receptor immunoreactivity was exclusively detected in large (NF200+) peptidergic neurons (Fig. 5A, top). More importantly, S1P3 was the most abundant S1P receptor expressed in virtually all small and large mouse DRG neurons (Fig. 5A, bottom).
Figure 5.
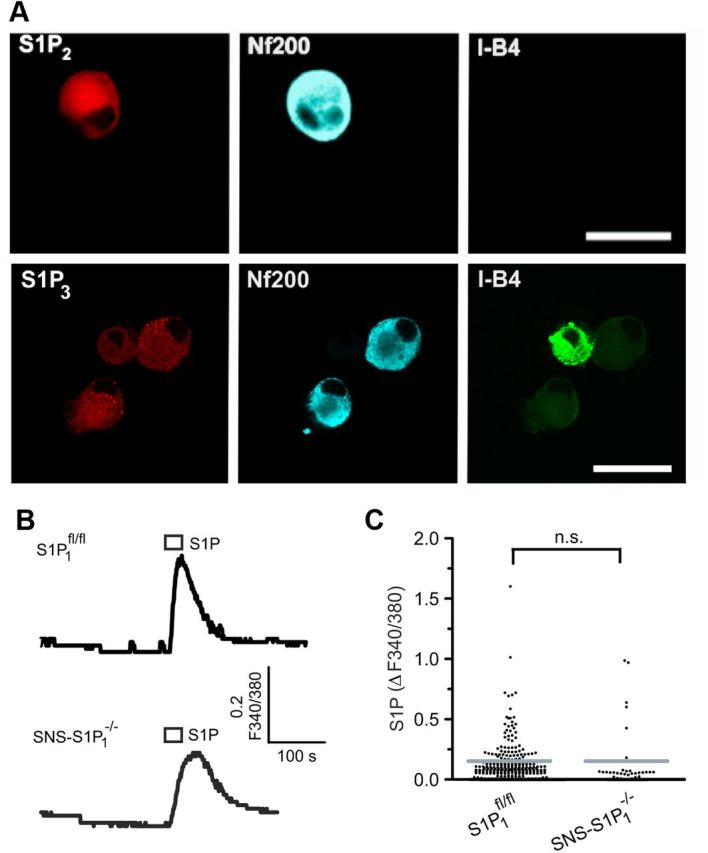
Expression of S1P2 and S1P3 receptors in different populations of mice DRG neurons. A, Top, Representative confocal images of cultured sensory neurons show immunoreactivity for S1P2 receptor subtype present in large myelinated, NF200+ (blue; Cy5 anti-mouse Ig) but not in peptidergic or IB-4 binding sensory neurons. A, Bottom, S1P3 immunoreactivity in cultured DRG neurons colocalized with peptidergic, IB-4 binding (IB-4+), and large diameter (NF200+) sensory neurons. Scale bar, 20 μm. B, C, Characterization of S1P-mediated Ca2+ transients in SNS-S1P1−/− sensory neurons. Typical ratiometric Ca2+ recordings from S1P1fl/fl and SNS-S1P1−/− sensory neurons revealed no difference in the magnitude of the S1P-induced Ca2+ responses (1 μm S1P, 10 s). n.s., Not significant (p > 0.05).
To first address the functional significance of the S1P1 receptor in neuronal responses to S1P, we performed microfluorimetric Ca2+ measurements in DRG neurons from transgenic mice with conditional deletion of S1P1 in nociceptive neurons (SNS-S1P1−/−). S1P-evoked Ca2+ transients in neurons from SNS-S1P1−/− (1 μm, 60 s) were similar to those from S1P1fl/fl mice (p = 0.9688, t(278) = 0.03921, unpaired t test, N = 3 mice per genotype, Fig. 5B,C). This suggests that S1P1 receptor activation may not be quantitatively relevant in S1P-induced pain-like behavior. This is further supported by the small population of S1P1-expressing neurons (Mair et al., 2011), which was in obvious contrast to the finding that IS1P could be evoked in virtually all neurons. Nonetheless, S1P1 may contribute to neuronal responses to S1P in the S1P1-expressing subpopulation of sensory neurons.
In contrast to several reports supporting a role for S1P1 in nociceptor sensitization and hyperexcitability, the significance of S1P3 receptors for nociception has so far not been addressed. To first characterize S1P3 expression in sensory neurons, we performed mRNA in situ hybridization using riboprobes recognizing S1P3 receptor on mouse DRG sections. Distinct, specific signals over the soma of virtually all large- and small-diameter wt DRG neurons were obtained (black arrows in Fig. 6A, left top). Control scramble probes used in wt and S1P3−/− DRG sections (Fig. 6A, right top and bottom) and the prehybridization buffer (data not shown) did not yield any appreciable signals. No S1P3 labeling was detectable in neurons from S1P3−/− (Fig. 6A, left bottom). Importantly, S1P3 receptor immunoreactivity was expressed in virtually all small and large sensory neurons in human and mouse DRG sections (Fig. 6B). When we assessed S1P-induced changes of membrane potential (Em) in S1P3−/− neurons, we observed that S1P-evoked depolarization was drastically decreased compared with wt neurons (ΔEmS1P in S1P3−/− = 4.84 ± 1.1 mV vs wt = 18.71 ± 2.1 mV; p = 0.00000379306, T = 351, Mann–Whitney U test, N = 5 mice, n = 28 neurons, Fig. 6C). In ratiometric Ca2+ measurements, the number of neurons responding to S1P was decreased to ∼20% in S1P3−/− mice (p = 0.0002 vs wt, Fisher's test, N = 6 mice, n = 228 neurons, Fig. 6D) and S1P-evoked Ca2+ transients were significantly diminished (S1P3−/− ΔF340/380 = 0.069 ± 0.006 vs wt = 0.14 ± 0.01; p = 0.00000013, t(622) = 5.327, unpaired t test, Fig. 6E). These data introduce S1P3 receptor as a significant component of S1P-induced nociceptor excitation.
Figure 6.
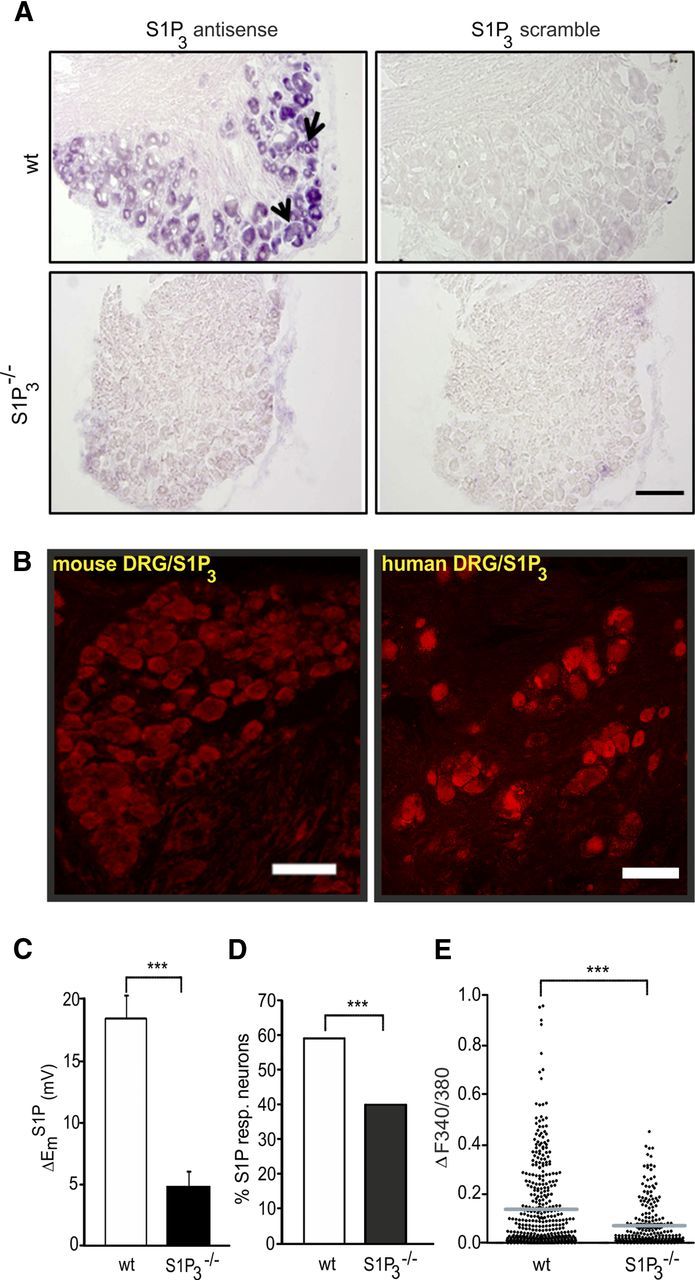
Role of S1P3 receptor in S1P-mediated neuronal excitability. A, mRNA in situ hybridization demonstrates the expression of S1P3 receptor mRNA in populations of small- and large-diameter neurons in mouse DRG sections. A, Bottom left, No signal of labeling in DRG sections from S1P3−/− mice. Right, S1P3 scramble probes (negative control) did not show any noticeable signal in both genotypes. Scale bar, 100 μm. B, Immunohistochemistry revealed the presence of S1P3 receptor protein in small and large neurons in murine and human DRG. Scale bar, 100 μm. C, The depolarizing effect of S1P was significantly decreased in S1P3−/− sensory neurons. D, Analysis of the percentage of neurons that showed a Ca2+ rise evoked by S1P in wt and S1P3−/− sensory neurons. The number of S1P-responsive neurons in S1P3-deficient mice was significantly reduced. E, The magnitude of the S1P-mediated Ca2+ transients was severely diminished in S1P3−/− mice DRG neurons. ***p < 0.001.
Spontaneous S1P-induced pain-like behavior is attenuated in S1P3−/− mice
Finally, we addressed the role of S1P receptors in the generation of spontaneous pain in vivo. After intradermal injection of S1P, licking and flinching behavior responses were similar between SNS-S1P1−/− and S1P1fl/fl mice (p = 0.937, Mann–Whitney U test, N = 7 mice per genotype, Fig. 7A–C). In contrast, the number of flinches was substantially reduced in S1P3−/− mice compared with wt littermates (0.40 ± 0.31 vs 4.9 ± 2.02; p = 0.0075, Mann–Whitney U test, N = 10 mice per genotype, Fig. 7D). In the same line, licking behavior after S1P injection was almost fully prevented in S1P3−/− mice (number of licks S1P3−/− = 1.35 ± 1.10 vs wt = 5.31 ± 1.53; p = 0.0132, Mann–Whitney U test, N = 8 mice, Fig. 7E; and licking time for S1P3−/− mice = 3.13 ± 2.22 s vs wt = 26.14 ± 8.27 s; p = 0.0066, N = 10 mice per genotype, Fig. 7F). The absence of S1P-evoked pain-like behavior in S1P3−/− mice suggests that S1P-induced pain generation is largely mediated by S1P3 receptors in vivo.
Figure 7.

Spontaneous pain behavior in SNS-S1P1−/− and S1P3−/− mice. The number of flinches (A) and licking response (B,C) after S1P injection (500 μm, 15 μl) into the ipsilateral hindpaw of SNS-S1P1−/− mice was not significantly different from control S1P1fl/fl mice. N = 8–10 mice per genotype. D, A significant reduced flinching response was observed in S1P3−/− mice compared with wt mice. E, F, Licking behavior was almost absent in S1P3−/− mice. N = 10 mice per genotype. n.s., Not significant (p > 0.05). *p < 0.05. **p < 0.01.
Role of S1P3 receptors in postoperative pain
Because S1P can be released as a consequence of tissue injury, the incision model of postoperative pain with assessment of responsiveness to mechanical and thermal stimuli was used to address the role of S1P in a more clinically relevant preclinical model (Pogatzki and Raja, 2003). Plantar incision decreased the PWL in wt mice from 8.88 ± 0.37 s (24 h before incision as baseline) to 4.32 ± 0.42 s on postoperative day 1 (Fig. 8A). S1P3−/− mice showed markedly reduced thermal hypersensitivity 24 h after the incision surgery (PWL = 8.14 ± 0.36 s) compared with wt littermates (PWL = 4.32 ± 0.42 s, p = 0.000012, two-way RM ANOVA with post Tukey test, N = 8 mice per genotype, Fig. 8A). Likewise, mechanical hypersensitivity was significantly diminished in S1P3−/− compared with wt mice (p = 0.032 for 4 mN, p = 0.000035 for 8 and 16 mN, two-way RM ANOVA with post Tukey test, N = 8 mice per genotype, Fig. 8B). At later time points, both mouse strains recovered similarly to baseline. These results suggest that S1P via S1P3 receptor has a critical role in postoperative pain.
Figure 8.

Heat and mechanical sensitivity of S1P3−/− and wt littermates after plantar incision. A, In wt mice, plantar incision induced a significant decrease of withdrawal latency (PWL) in response to a noxious heat stimulus at the incision site. The decrease was significantly reduced in the S1P3−/− mice 24 h after plantar incision. B, Likewise, mechanical response frequency was significantly lower in S1P3−/− versus wt mice on postoperative day 1. *p < 0.05. ***p < 0.001.
Together with our reported results in a cellular model of nociception and the findings obtained in humans, this study provides data supporting the hypothesis that S1P-induced activation of S1P3 receptor and a GPCR-dependent Cl− conductance in mice may notably contribute to acute post-traumatic pain.
Discussion
The sphingolipid S1P is a multifaceted immune modulator that can act extracellularly in an autocrine or paracrine manner or intracellularly as a second messenger molecule (Mitra et al., 2006; Alvarez et al., 2007; Takabe et al., 2008). Extracellular and intracellular levels of S1P are tightly regulated by sphingosine kinases (SphKs) and S1P degrading enzymes (Spiegel and Milstien, 2003; van Echten-Deckert and Herget, 2006). Activation of SphK1 is the key event in elevating S1P levels (Maceyka et al., 2002; Taha et al., 2006; Alemany et al., 2007). S1P is deactivated by S1P-phosphatases or degraded by S1P-lyase (Pyne and Pyne, 2000; Le Stunff et al., 2004; Bandhuvula and Saba, 2007). Mice deficient in both SphK1 and SphK2 are not viable, indicating an essential cellular requirement for S1P (Mizugishi et al., 2005).
Chemoattractants, such as TNFα or NGF, can stimulate S1P production in immune cells, epithelia, and even neurons (Zhang et al., 2006b; Alemany et al., 2007), which can release S1P presumably via the specific multidrug resistance-associated protein ABCC1 (Mitra et al., 2006). Although S1P in blood plasma may reach up to micromolar concentrations, it is largely bound to plasma proteins, and free active S1P concentrations are probably very low (Murata et al., 2000; Schmidt et al., 2006; Ohkawa et al., 2008). However, high concentrations of free S1P can, however, arise locally at inflammation sites and presumably at sites of thrombocyte activation (Mitra et al., 2006; Hammad et al., 2008; Ulrych et al., 2011). Together with a previous work of our group on nociceptor excitation by activated platelets that can release S1P upon thromboxane stimulation as it occurs during acute injury of blood vessels (Ringkamp et al., 1994; Ulrych et al., 2011), these findings encouraged us to investigate the significance of S1P in nociception. S1P locally induces nociceptor sensitization and thermal hypersensitivity, which is largely mediated by S1P1 receptor expressed in nociceptors (Mair et al., 2011). In the CNS, the spinal cord analgesic effects have been reported (Coste et al., 2008). However, a number of reports support a proalgesic action of S1P when acting at peripheral nerve terminals via S1P1 (Zhang et al., 2006a,b; Chi and Nicol, 2010; Doyle et al., 2011; Mair et al., 2011), and even an involvement in opioid-induced hyperalgesia at spinal cord level (Muscoli et al., 2010). In addition to the S1P1-mediated proalgesic action, we observed activation of a slowly activating and deactivating inward current in the majority of neurons that were exposed to S1P, and this issue was addressed in the present study. S1P induced a membrane depolarization, which in approximately two thirds of sensory neurons was sufficient to evoke action potentials in vitro, and spontaneous pain-like behavior in vivo in mice. In contrast to the S1P1-mediated thermal sensitization (Mair et al., 2011), the excitatory effect did not require TRPV1 (data not shown) but still was mediated by a GPCR. In general, five metabotropic S1P receptors are known up to date (Spiegel and Milstien, 2003). Several reports support expression of S1P1, S1P2, and S1P3 GPCRs in DRG neurons, whereas expression of S1P4 and S1P5 is controversially discussed (Chi and Nicol, 2010; Mair et al., 2011). Primary afferent neurons in murine DRG are divided into at least three distinct subclasses: sensory neurons expressing NF200 giving rise to myelinated neurons for touch sensation and those being nociceptors. The nociceptor population can be subdivided in neurons expressing neuropeptides, especially calcitonin gene-related peptide-positive, and nonpeptidergic neurons, which express binding sites for I-B4 (isolectin B4, I-B4+ neurons). In a recently published work by our group, we only found evidence of expression of S1P1, S1P2, and S1P3 receptors in mice DRG neurons (Mair et al., 2011). S1P1 receptors were mainly expressed by I-B4+ nociceptors but were also present in a subpopulation of calcitonin gene-related peptide-positive neurons (Mair et al., 2011). In contrast, S1P2 receptor immunoreactivity is dominant in neurons expressing the large nonnociceptive neuron marker NF200. Last, with a combination of in situ hybridization and indirect immune fluorescence, our current results demonstrated that S1P3 receptors were most widely distributed and present in virtually all neurons of all neuron types. The S1P mRNA and protein distribution pattern closely matched the presence of IS1P (S1P current) in virtually all recorded neurons. Furthermore, IS1P required activation of a GPCR and this supported the possibility that S1P-induced excitatory effects may be mediated by S1P3. As expected, the number of S1P responsive neurons was strongly reduced, although in mice with a global null mutation for S1P3, ∼20% of neurons responded to S1P with, however, a small Ca2+ transient. This can be explained by a contribution of S1P1 receptor, which is expressed in ∼20% of sensory neurons (Mair et al., 2011). In vivo, the S1P-induced spontaneous pain behavior was to a large extent abolished in mice lacking S1P3 suggesting that S1P3 largely mediates nociceptor excitation by S1P. Moreover, S1P3-deficient mice were largely protected from thermal and mechanical hyperalgesia induced after plantar incision, revealing a prominent role of S1P3 receptor in postoperative pain.
Regarding the nature of S1P-induced currents, several ion channels may be involved: in particular, lipid sensing is an important aspect of transient receptor potential canonical (TRPC) channel biology enabling integration with other cellular signaling systems (Beech, 2012). There are seven mammalian genes encoding for TRPC channels, of which six are expressed in DRG neurons (Zhu et al., 1996; Montell et al., 2002; Kress et al., 2008; Talavera et al., 2008). TRPC1 and TRPC5 sense the presence of S1P and contribute to S1P effects on Ca2+ homeostasis in a number of cells (Formigli et al., 2009; Beech, 2012). TRPC1 together with C3 and C6 are the most abundant in DRG cells (Kress et al., 2008), and S1P-induced activation of TRPC1 but also TRPC5 could contribute to Ca2+ transients after neuron exposure to S1P in the current study. In contrast to, for example, endothelial cells where S1P promotes endothelial junctional integrity by activating the release of endoplasmic reticulum-Ca2+ (Mehta et al., 2005), Ca2+ transients in the current study fully depended on the presence of extracellular Ca2+, suggesting the Ca2+ influx as a main source of intracellular Ca2+ increase possibly via TRPC1 or TRPC5. However, S1P-induced currents were unaltered in voltage-clamp recordings using nominally “cation free” solution, and this, together with reversal potential shifts observed with variation of extracellular Cl− concentrations (data not shown), strongly supports the idea that IS1P in DRG neurons is probably a Cl− conductance. Like lysophosphatidic acid, S1P activates Cl− currents in keratinocytes and fibroblasts (Postma et al., 1996; Wang et al., 2002). Mature sensory neurons express cation-Cl− cotransporters and maintain high intracellular Cl− concentrations (Mao et al., 2012). Inflammatory mediators and kinases regulate these cotransporters under inflammatory conditions, and activation of Cl− conductances results in neuron excitation rather than inhibition as generally observed in mature neurons in the brain (Funk et al., 2008; Delpire and Austin, 2010). Because S1P induces increases in [Ca2+]i, members of the TMEM16 ion channel subfamily may contribute to S1P-activated Cl− conductance (Yang et al., 2008). Although activation of TMEM16/ANO1 amplifies depolarization and thereby may increase Ca2+ influx (Cho et al., 2012), our observation that S1P-induced Ca2+ transients were fully inhibited with the Cl− channel blocker NFA does not support this possibility: In our experiments, Ca2+ transients in response to S1P were probably evoked by S1P-induced activation of a Cl− conductance and membrane depolarization and consecutive activation of voltage-gated Ca2+ currents as a source of Ca2+ influx.
Direct activation of Cl− channels by GPCRs has not been reported so far; however, first evidence for S1P activation of Gα13 links the RhoA GTPase pathway with activation of intracellular Cl− channels (Postma et al., 2001; Ponsioen et al., 2009). These reports are consistent with the substantial inhibition of S1P-induced currents and Ca2+ influxes in the presence of the general Cl− channel blocker NFA in the present study and support the idea that a depolarizing Cl− conductance is activated by S1P in sensory neurons. The clinical relevance of a Cl− current for activation of nociceptive primary afferents and the ensuing axon reflex flare response has been shown in human skin by blocking the NaK2Cl cotransporter, which reduced histamine-evoked itch-and-flare responses (Willis et al., 2004).
In conclusion, we show, for the first time to our knowledge, that the S1P-induced activation of an excitatory Cl− conductance downstream of S1P3 receptor is a significant inducer of peripheral nociception in mice and humans. Together with recent results, the concept that modulation of S1P receptors can be used to direct neuroprotective and regenerative actions in the CNS is supported, as well as its anti-inflammatory effects (Soliven et al., 2011). Thus, our study supports the idea to extend the pharmacological targeting of S1P receptors into pain therapy.
Footnotes
The authors thank K. Braun, T. Martha, and M. Doblander for expert technical assistance. This work was supported by la Generalitat Valenciana and the Ministerio de Economia y Competitividad (A.V.F.M.), the Australian National Health and Medical Research Council Project Grant 535055 to R.V.H., the Intramural Research Programs of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases to R.L.P., and the Austrian Research Funding Agency FWF Project Grants P20562, P25345, and SPIN to M.K.
The authors declare no competing financial interests.
References
- Agarwal N, Offermanns S, Kuner R. Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis. 2004;38:122–129. doi: 10.1002/gene.20010. [DOI] [PubMed] [Google Scholar]
- Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer Zu Heringdorf D. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:413–428. doi: 10.1007/s00210-007-0132-3. [DOI] [PubMed] [Google Scholar]
- Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- Alvarez SE, Milstien S, Spiegel S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab. 2007;18:300–307. doi: 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Armstrong D, Jepson JB, Keele CA, Stewart JW. Pain-producing substance in human inflammatory exudates and plasma. J Physiol. 1957;135:350–370. doi: 10.1113/jphysiol.1957.sp005715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandhuvula P, Saba JD. Sphingosine-1-phosphate lyase in immunity and cancer: silencing the siren. Trends Mol Med. 2007;13:210–217. doi: 10.1016/j.molmed.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Beech DJ. Integration of transient receptor potential canonical channels with lipids. Acta Physiol (Oxf) 2012;204:227–237. doi: 10.1111/j.1748-1716.2011.02311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi XX, Nicol GD. The sphingosine 1-phosphate receptor, S1PR(1), plays a prominent but not exclusive role in enhancing the excitability of sensory neurons. J Neurophysiol. 2010;104:2741–2748. doi: 10.1152/jn.00709.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Yang YD, Lee J, Lee B, Kim T, Jang Y, Back SK, Na HS, Harfe BD, Wang F, Raouf R, Wood JN, Oh U. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15:1015–1021. doi: 10.1038/nn.3111. [DOI] [PubMed] [Google Scholar]
- Coste O, Brenneis C, Linke B, Pierre S, Maeurer C, Becker W, Schmidt H, Gao W, Geisslinger G, Scholich K. Sphingosine 1-phosphate modulates spinal nociceptive processing. J Biol Chem. 2008;283:32442–32451. doi: 10.1074/jbc.M806410200. [DOI] [PubMed] [Google Scholar]
- Dahm F, Nocito A, Bielawska A, Lang KS, Georgiev P, Asmis LM, Bielawski J, Madon J, Hannun YA, Clavien PA. Distribution and dynamic changes of sphingolipids in blood in response to platelet activation. J Thromb Haemost. 2006;4:2704–2709. doi: 10.1111/j.1538-7836.2006.02241.x. [DOI] [PubMed] [Google Scholar]
- Daum G, Grabski A, Reidy MA. Sphingosine 1-phosphate: a regulator of arterial lesions. Arterioscler Thromb Vasc Biol. 2009;29:1439–1443. doi: 10.1161/ATVBAHA.108.175240. [DOI] [PubMed] [Google Scholar]
- Delpire E, Austin TM. Kinase regulation of Na+-K+-2 Cl− cotransport in primary afferent neurons. J Physiol. 2010;588:3365–3373. doi: 10.1113/jphysiol.2010.190769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittert I, Vlachová V, Knotková H, Vitasková Z, Vyklicky L, Kress M, Reeh PW. A technique for fast application of heated solutions of different composition to cultured neurones. J Neurosci Methods. 1998;82:195–201. doi: 10.1016/s0165-0270(98)00051-x. [DOI] [PubMed] [Google Scholar]
- Doyle T, Finley A, Chen Z, Salvemini D. Role for peroxynitrite in sphingosine-1-phosphate-induced hyperalgesia in rats. Pain. 2011;152:643–648. doi: 10.1016/j.pain.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formigli L, Sassoli C, Squecco R, Bini F, Martinesi M, Chellini F, Luciani G, Sbrana F, Zecchi-Orlandini S, Francini F, Meacci E. Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J Cell Sci. 2009;122:1322–1333. doi: 10.1242/jcs.035402. [DOI] [PubMed] [Google Scholar]
- Funk K, Woitecki A, Franjic-Würtz C, Gensch T, Möhrlen F, Frings S. Modulation of chloride homeostasis by inflammatory mediators in dorsal root ganglion neurons. Mol Pain. 2008;4:32. doi: 10.1186/1744-8069-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammad SM, Crellin HG, Wu BX, Melton J, Anelli V, Obeid LM. Dual and distinct roles for sphingosine kinase 1 and sphingosine 1 phosphate in the response to inflammatory stimuli in RAW macrophages. Prostaglandins Other Lipid Mediat. 2008;85:107–114. doi: 10.1016/j.prostaglandins.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Herr DR, Chun J. Effects of LPA and S1P on the nervous system and implications for their involvement in disease. Curr Drug Targets. 2007;8:155–167. doi: 10.2174/138945007779315669. [DOI] [PubMed] [Google Scholar]
- Hopson KP, Truelove J, Chun J, Wang Y, Waeber C. S1P activates store-operated calcium entry via receptor- and non-receptor-mediated pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2011;300:C919–C926. doi: 10.1152/ajpcell.00350.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu YP, Yamashita T, Proia RL. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279:29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- Kress M, Karasek J, Ferrer-Montiel AV, Scherbakov N, Haberberger RV. TRPC channels and diacylglycerol-dependent calcium signaling in rat sensory neurons. Histochem Cell Biol. 2008;130:655–667. doi: 10.1007/s00418-008-0477-9. [DOI] [PubMed] [Google Scholar]
- Le Stunff H, Milstien S, Spiegel S. Generation and metabolism of bioactive sphingosine-1-phosphate. J Cell Biochem. 2004;92:882–899. doi: 10.1002/jcb.20097. [DOI] [PubMed] [Google Scholar]
- Linhart O, Obreja O, Kress M. The inflammatory mediators serotonin, prostaglandin E2 and bradykinin evoke calcium influx in rat sensory neurons. Neuroscience. 2003;118:69–74. doi: 10.1016/s0306-4522(02)00960-0. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim Biophys Acta. 2002;1585:193–201. doi: 10.1016/S1388-1981(02)00341-4. [DOI] [PubMed] [Google Scholar]
- Mair N, Benetti C, Andratsch M, Leitner MG, Constantin CE, Camprubí-Robles M, Quarta S, Biasio W, Kuner R, Gibbins IL, Kress M, Haberberger RV. Genetic evidence for involvement of neuronally expressed S1P(1) receptor in nociceptor sensitization and inflammatory pain. PLoS One. 2011;6:e17268. doi: 10.1371/journal.pone.0017268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao S, Garzon-Muvdi T, Di Fulvio M, Chen Y, Delpire E, Alvarez FJ, Alvarez-Leefmans FJ. Molecular and functional expression of cation-chloride-cotransporters in dorsal root ganglion neurons during postnatal maturation. J Neurophysiol. 2012;108:834–852. doi: 10.1152/jn.00970.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. 2005;280:17320–17328. doi: 10.1074/jbc.M411674200. [DOI] [PubMed] [Google Scholar]
- Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci U S. 2006;A103:16394–16399. doi: 10.1073/pnas.0603734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25:11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C, Birnbaumer L, Flockerzi V, Bindels RJ, Bruford EA, Caterina MJ, Clapham DE, Harteneck C, Heller S, Julius D, Kojima I, Mori Y, Penner R, Prawitt D, Scharenberg AM, Schultz G, Shimizu N, Zhu MX. A unified nomenclature for the superfamily of TRP cation channels. Mol Cell. 2002;9:229–231. doi: 10.1016/s1097-2765(02)00448-3. [DOI] [PubMed] [Google Scholar]
- Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A, Ui M, Okajima F. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J. 2000;352:809–815. [PMC free article] [PubMed] [Google Scholar]
- Murphy SM, Pilowsky PM, Llewellyn-Smith IJ. Pre-embedding staining for GAD67 versus postembedding staining for GABA as markers for central GABAergic terminals. J Histochem Cytochem. 1998;46:1261–1268. doi: 10.1177/002215549804601106. [DOI] [PubMed] [Google Scholar]
- Muscoli C, Doyle T, Dagostino C, Bryant L, Chen Z, Watkins LR, Ryerse J, Bieberich E, Neumman W, Salvemini D. Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J Neurosci. 2010;30:15400–15408. doi: 10.1523/JNEUROSCI.2391-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat Protoc. 2007;2:1508–1514. doi: 10.1038/nprot.2007.153. [DOI] [PubMed] [Google Scholar]
- Obreja O, Rathee PK, Lips KS, Distler C, Kress M. IL-1 β potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB J. 2002;16:1497–1503. doi: 10.1096/fj.02-0101com. [DOI] [PubMed] [Google Scholar]
- Obreja O, Biasio W, Andratsch M, Lips KS, Rathee PK, Ludwig A, Rose-John S, Kress M. Fast modulation of heat-activated ionic current by proinflammatory interleukin 6 in rat sensory neurons. Brain. 2005;128:1634–1641. doi: 10.1093/brain/awh490. [DOI] [PubMed] [Google Scholar]
- Ohkawa R, Nakamura K, Okubo S, Hosogaya S, Ozaki Y, Tozuka M, Osima N, Yokota H, Ikeda H, Yatomi Y. Plasma sphingosine-1-phosphate measurement in healthy subjects: close correlation with red blood cell parameters. Ann Clin Biochem. 2008;45:356–363. doi: 10.1258/acb.2007.007189. [DOI] [PubMed] [Google Scholar]
- Pogatzki EM, Raja SN. A mouse model of incisional pain. Anesthesiology. 2003;99:1023–1027. doi: 10.1097/00000542-200310000-00041. [DOI] [PubMed] [Google Scholar]
- Ponsioen B, van Zeijl L, Langeslag M, Berryman M, Littler D, Jalink K, Moolenaar WH. Spatiotemporal regulation of chloride intracellular channel protein CLIC4 by RhoA. Mol Biol Cell. 2009;20:4664–4672. doi: 10.1091/mbc.E09-06-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma FR, Jalink K, Hengeveld T, Bot AG, Alblas J, de Jonge HR, Moolenaar WH. Serum-induced membrane depolarization in quiescent fibroblasts: activation of a chloride conductance through the G-protein-coupled LPA receptor. EMBO J. 1996;15:63–72. [PMC free article] [PubMed] [Google Scholar]
- Postma FR, Jalink K, Hengeveld T, Offermanns S, Moolenaar WH. Galpha(13) mediates activation of a depolarizing chloride current that accompanies RhoA activation in both neuronal and nonneuronal cells. Curr Biol. 2001;11:121–124. doi: 10.1016/s0960-9822(01)00030-6. [DOI] [PubMed] [Google Scholar]
- Pyne S, Pyne NJ. Sphingosine 1-phosphate signalling in mammalian cells. Biochem J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringkamp M, Schmelz M, Kress M, Allwang M, Ogilvie A, Reeh PW. Activated human platelets in plasma excite nociceptors in rat skin, in vitro. Neurosci Lett. 1994;170:103–106. doi: 10.1016/0304-3940(94)90249-6. [DOI] [PubMed] [Google Scholar]
- Rogers TB, Inesi G, Wade R, Lederer WJ. Use of thapsigargin to study Ca2+ homeostasis in cardiac cells. Biosci Rep. 1995;15:341–349. doi: 10.1007/BF01788366. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Schmidt R, Geisslinger G. LC-MS/MS-analysis of sphingosine-1-phosphate and related compounds in plasma samples. Prostaglandins Other Lipid Mediat. 2006;81:162–170. doi: 10.1016/j.prostaglandins.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Soliven B, Miron V, Chun J. The neurobiology of sphingosine 1-phosphate signaling and sphingosine 1-phosphate receptor modulators. Neurology. 2011;76:S9–S14. doi: 10.1212/WNL.0b013e31820d9507. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Taha TA, Hannun YA, Obeid LM. Sphingosine kinase: biochemical and cellular regulation and role in disease. J Biochem Mol Biol. 2006;39:113–131. doi: 10.5483/BMBRep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera K, Nilius B, Voets T. Neuronal TRP channels: thermometers, pathfinders and life-savers. Trends Neurosci. 2008;31:287–295. doi: 10.1016/j.tins.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Ulrych T, Böhm A, Polzin A, Daum G, Nüsing RM, Geisslinger G, Hohlfeld T, Schrör K, Rauch BH. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J Thromb Haemost. 2011;9:790–798. doi: 10.1111/j.1538-7836.2011.04194.x. [DOI] [PubMed] [Google Scholar]
- van Echten-Deckert G, Herget T. Sphingolipid metabolism in neural cells. Biochim Biophys Acta. 2006;1758:1978–1994. doi: 10.1016/j.bbamem.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Vogler R, Sauer B, Kim DS, Schäfer-Korting M, Kleuser B. Sphingosine-1-phosphate and its potentially paradoxical effects on critical parameters of cutaneous wound healing. J Invest Dermatol. 2003;120:693–700. doi: 10.1046/j.1523-1747.2003.12096.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Carbone LD, Watsky MA. Receptor-mediated activation of a Cl(−) current by LPA and S1P in cultured corneal keratocytes. Invest Ophthalmol Vis Sci. 2002;43:3202–3208. [PubMed] [Google Scholar]
- Watterson KR, Lanning DA, Diegelmann RF, Spiegel S. Regulation of fibroblast functions by lysophospholipid mediators: potential roles in wound healing. Wound Repair Regen. 2007;15:607–616. doi: 10.1111/j.1524-475X.2007.00292.x. [DOI] [PubMed] [Google Scholar]
- Willis EF, Clough GF, Church MK. Investigation into the mechanisms by which nedocromil sodium, frusemide and bumetanide inhibit the histamine-induced itch-and-flare response in human skin in vivo. Clin Exp Allergy. 2004;34:450–455. doi: 10.1111/j.1365-2222.2004.01898.x. [DOI] [PubMed] [Google Scholar]
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Fehrenbacher JC, Vasko MR, Nicol GD. Sphingosine-1-phosphate via activation of a G-protein-coupled receptor(s) enhances the excitability of rat sensory neurons. J Neurophysiol. 2006a;96:1042–1052. doi: 10.1152/jn.00120.2006. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Intracellular sphingosine 1-phosphate mediates the increased excitability produced by nerve growth factor in rat sensory neurons. J Physiol. 2006b;575:101–113. doi: 10.1113/jphysiol.2006.111575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical considerations in relation to pain in animal experimentation. Acta Physiol Scand. 1986;128:221–233. [PubMed] [Google Scholar]