

Abstract
Stem cell self-renewal and differentiation must be carefully controlled during development and tissue homeostasis. In the Drosophila optic lobe, neuroepithelial cells first divide symmetrically to expand the stem cell population and then transform into asymmetrically dividing neuroblasts, which generate medulla neurons. The mechanisms underlying this cell fate transition are not well understood. Here, we show a crucial role of some cell cycle regulators in this transition. We find that loss of function in replication protein A (RPA), which consists of three highly conserved protein subunits and functions in DNA replication, leads to disintegration of the optic lobe neuroepithelium and premature differentiation of neuroepithelial cells into medulla neuroblasts. Clonal analyses of RPA loss-of-function alleles indicate that RPA is required to prevent neuroepithelial cells from differentiating into medulla neuroblasts. Inactivation of the core cell cycle regulators, including the G1/S regulators E2F1, Cyclin E, Cdk2, and PCNA, and the G2/M regulators Cyclin A, Cyclin B, and Cdk1, mimic RPA loss-of-function phenotypes, suggesting that cell cycle progression is required for both maintaining neuroepithelial cell identity and suppressing neuroblast formation. We further find that RPA or E2F1 inactivation in the neuroepithelial cells correlates with downregulation of Notch signaling activity, which appears to result from Numb mislocalization. Thus, we have shown that the transition from neuroepithelial cells to neuroblasts is directly regulated by cell cycle regulators and propose a model in which the inhibition of neuroepithelial cell cycle progression downregulates Notch signaling activity through Numb, which leads to the onset of neurogenesis.
Introduction
During mammalian cerebral cortex development, neuroepithelial (NE) cells first undergo symmetric divisions to expand the pool of proliferating progenitor cells. They then transit to become radial glial cells that undergo asymmetric, neurogenic divisions to generate the neurons and glial cells in the brain (Götz and Huttner, 2005). The mechanisms underlying this transition are not well understood. The development of the Drosophila optic lobe shares a similar transition pattern of symmetric to asymmetric division (Doe, 2008; Knoblich, 2008; Brand and Livesey, 2011) and can be used as a simpler genetic model to study the regulatory mechanisms underlying neurogenesis during brain development.
The Drosophila optic lobe is the visual processing center of the brain that consists of the lamina, the medulla, and the lobula complex (Fig. 1A–C; Meinertzhagen and Hanson, 1993). Each optic lobe develops from 30 to 40 embryonic ectoderm cells. After larval hatching, these NE cells start to divide symmetrically and soon separate into an outer proliferation center (OPC) and an inner proliferation center (IPC) (Fig. 1A). Neurogenesis in the medulla begins at late-second instar, when the columnar NE cells located on the medial edge of the OPC undergo a transition to become neuroblasts (NBs), which generate medulla neurons (Nassif et al., 2003; Egger et al., 2007; Hayden et al., 2007; Fig. 1B,C). NE cells transform into NBs in a precisely regulated manner, and the JAK/STAT (Janus kinase/signal transducer and activator of transcription protein) (Yasugi et al., 2008; Wang et al., 2011b) and Notch pathways (Egger et al., 2010; Ngo et al., 2010; Orihara-Ono et al., 2011; Wang et al., 2011a) negatively regulate the differentiation of NE cells into NBs; the Fat-Hippo pathway has been shown to promote neuroepithelial growth (Reddy et al., 2010), while the EGFR (epidermal growth factor receptor) pathway drives the proneural wave from the medial to the lateral NE cells, inducing the transformation of NE cells into NBs (Yasugi et al., 2010).
Figure 1.

RPA3 is expressed in the optic lobe neuroepithelia. A, The larval CNS. B, Lateral view of the optic lobe, which harbors three optic ganglia: the medulla (me), lamina (la), and lobula complex (lo). LF, lamina furrow; ED, eye imaginal disc; os, optic stalk. Anterior is to the right. C, Frontal view of the brain lobe with the laterally located optic lobe (OL) and the medially located central brain (CB). Along the lateral–medial axis, symmetrically dividing NE cells give rise to asymmetrically dividing NBs at the medial edge. LPC, lamina precursor cells. D, A multiple sequence alignment between Drosophila melanogaster CG15220, Homo sapiens RPA3, Macaca mulatta RPA3, Rattus norvegicus RPA3, Mus musculus RPA3, and Danio rerio RPA3. Conserved and similar amino acid residues are shaded in black, red, and blue. E, E', RPA3 protein is expressed in the NE cells, medulla and central brain NBs, GMCs, lamina precursor cells, and immature lamina neurons. RPA3 staining is shown in green. F, RPA3 is not expressed in medulla neurons. G, RPA3 expression was greatly reduced or eliminated in RPA3RNAi flip-out clones. Scale bars: A, 70 μm; B, 40 μm; C, 20 μm (for C, E, E', F, G).
Cell cycle regulation may be an important mechanism underlying the transition from proliferative to differentiative divisions of NE cells (Dehay and Kennedy, 2007). In vertebrates, lengthening of the cell cycle of NE cells in vitro by inhibition of cyclin-dependent kinases (CDKs) induces premature generation of neurons (Calegari and Huttner, 2003), while overexpression of Cdk4 and cyclin D1 together (Lange et al., 2009; Artegiani et al., 2011) leads to the expansion of neural progenitor cells in the mouse brain. In the Drosophila optic lobe, there is also a cell cycle arrest that corresponds to the transition from NE cells to medulla NBs (Reddy et al., 2010; Orihara-Ono et al., 2011). These observations suggest a tight link between the rate of cell cycle progression and the switch of NE cell proliferation to neurogenesis. However, the mechanisms underlying the transition of division modes are still not well understood.
Here, we show that replication protein A (RPA), as well as the core cell cycle regulators, regulates the transition of NE cell division in the optic lobe. Loss of RPA and core cell cycle regulator function causes precocious NE-to-NB transition, during which Notch signaling activity is downregulated and the distribution of the Notch antagonist Numb is disturbed.
Materials and Methods
Fly strains and genetic crosses.
Strain w1118 was used as the wild-type strain. The following transgenic fly lines and mutations were used: UAS-RPA3RNAi (VDRC stock: v15380), UAS-RPA2RNAi (v30570), UAS-RPA1RNAi (v11210), UAS-E2F1RNAi (v108837), UAS-CycERNAi (v110204, Bloomington stock: BL29314), UAS-Cdk2RNAi (v104959), UAS-CycDRNAi (v29023, v29024), UAS-Cdk4RNAi (v40576, v40577), UAS-PCNARNAi (v51253, National Institute of Genetics stock 9193R-1), UAS-CycARNAi (BL29313, v103595), UAS-CycBRNAi (BL34544), UAS-Cdk1RNAi (v41838, BL28368), UAS-pntRNAi (Tsinghua stock: THU1668), UAS-numbRNAi (THU1674), UAS-RPA3 (this study), UAS-E2F1 (gift from B. Edgar), UAS-CycE (BL4781 and gift from B. Edgar), UAS-E2f1 UAS-Dp (BL4770, BL4774), UAS-CycA (BL6633), UAS-Cdk1 (BL6638, BL6642), UAS-string (BL4777, BL4778), hs-Flp122; UAS-CycD UAS-Cdk4; UAS-E2f1 UAS-Dp (gift from K. Irvine), UAS-EGFRDN (BL5364), UAS-NotchFL (gift from G. Struhl), UAS-numb-Myc (gift from C.-Y. Lee), UAS-yki:V5S168A (BL28818), UAS-3HA-stat92EΔNΔC (gift from E. Bach); tubP-GAL80ts, RPA2KG00759, l(1)G0241G0241 (referred to as RPA3G0241, Peter et al., 2002), CycEKG00239, E2F1729, PCNAEY09082, and pnt▵88 [described in Flybase (http://flybase.bio.indiana.edu)]. E(spl)m8-lacZ is a lacZ reporter of the E(spl)m8 gene (Kramatschek and Campos-Ortega, 1994). Gal4 lines used include c855a-Gal4 (Hrdlicka et al., 2002), c768-Gal4 (Manseau et al., 1997), and inscuteable-Gal4 (insc-Gal4) (Betschinger et al., 2006). Drosophila stocks and crosses were kept under standard conditions at 25°C unless otherwise indicated.
Mosaic clones were generated by FLP/FRT-mediated (flippase/flippase recombination target-mediated) somatic recombination (Xu and Rubin, 1993). FRT19A or RPA3G0241 FRT19A/ FM7c (DGRC stock 111825) females were crossed with FL122 Ub-GFP FRT19A/Y males; FRT40A, RPA2KG00759 FRT40A/ CyO (DGRC stock 114619) or CycEKG00239 FRT40A/CyO (DGRC stock 111513) females with y w FL122; Ub-GFP FRT40A/CyO males; FRT42D or FRT42D PCNAEY0908 (DGRC stock 114577) females with y w FL122; FRT42D Ub-GFP/CyO males; FRT82B, FRT82B pntΔ88/TM3, Sb, or FRT82B E2F1729/TM3, Sb females with y w hs Flp1/Y; FRT82B Ub-GFP, Minute/TM3, Sb males. Larval progeny from these crosses were subjected to a 1 h heat shock at 38°C ∼48 or 72 h after larval hatching (ALH) to induce somatic recombination. Late-third instar larvae were dissected for analyses.
For RNAi and overexpression experiments, female flies carrying a UAS-RNAi or UAS-X construct were crossed with c768-Gal4 or c855a-Gal4 males and the progeny cultured at 25 or 31°C (for UAS-E2F1RNAi, UAS-CycERNAi, UAS-Cdk2RNAi and UAS-CycBRNAi, and UAS-3HA-stat92EΔNΔC) until various larval stages before analyses. In some experiments involving RPA3 RNAi knockdown, we also combined c768-Gal4 or c855a-Gal4 with a temperature-sensitive Gal80 repressor construct (Gal80ts) to temporally control RPA3 expression. In this experiment, UAS-RPA3RNAi females were crossed with tub-Gal80ts/Y; c768-Gal4/+ or tub-Gal80ts/Y; c855a-Gal4/+ males. The progeny from these crosses were first cultured at 17°C until early-third instar and then shifted to 31°C to inactivate Gal80ts and initiate RPA3 RNA knockdown. To induce ectopic expression in clones using the flip-out method (Struhl and Basler, 1993), UAS-RPA3RNAi, UAS-RPA2RNAi, UAS-RPA1RNAi, UAS-Cdk1RNAi, or UAS-CycARNAi females were crossed to y w hsFlp1/Y; actin<y+<Gal4, UAS-GFP males and second-instar larvae were heat-shocked at 38°C for 30 min and then cultured at 25°C. Mid-third or late-third instar larvae were used for analyses.
Generation of anti-RPA3 antibody.
A rabbit polyclonal antiserum against Drosophila RPA3 was generated using His-tagged RPA3 fusion protein as an antigen source. To raise the antibody, we amplified the entire coding sequence of RPA3 by PCR using Drosophila genomic DNA as template and the following primer set: forward primer 5′CCGGAATTCATGGATGCCTTTGATCC′ and reverse primer 5′CCGCTCGAGCTAGCCGCTGCGATAG3′. The resulting PCR product of 339 base pairs encoding 112 aa of the RPA3 polypeptide was digested with EcoR I and Xho I and subcloned into the EcoR I/XhoI sites of the plasmid vector pET-28a(+). A His-tagged RPA3 protein of the expected size (13 kDa) was expressed in Escherichia coli BL21 (DE3), purified with Ni-NTA agarose (Qiagen) and used to immunize New Zealand white rabbits (Abmart). The antibody was affinity-purified using protein A/G agarose beads.
Immunohistochemistry and BrdU labeling.
Larval brain staining was performed as previously described (Wang et al., 2011a). The following primary antibodies were used: guinea pig anti-Deadpan (1:1000, gift from J. Skeath), guinea pig anti-Miranda (1:500, gift from C. Doe), rabbit anti-Asense (1:1000, gift from Y. N. Jan), guinea pig anti-Numb (1:1000, gift from J. Skeath), rabbit anti-PntP1 (1:500; gift from J. Skeath), rabbit anti-PatJ (1:1000, gift from H. Bellen), rabbit anti-β-galactosidase (1:1000, Cortex Biochem); mouse anti-Discs large [4F3, 1:50, Developmental Studies Hybridoma Bank (DSHB)], mouse anti-Notch intracellular domain (C17.9C6, 1:100, DSHB), mouse anti-Delta (C594.9B, 1:100, DSHB), mouse anti-Dachshund (mAbdac2-3, 1:100, DSHB), rat anti-Elav (7E8A10, 1:100, DSHB), mouse anti-Prospero (MR1A, 1:100, DSHB), mouse anti-c-Myc (9E10, 1:100, DSHB), mouse anti-β-tubulin (E7, 1:20, DSHB), mouse anti-V5 (Cat. No. R96025, 1:400, Invitrogen), mouse anti-HA (Cat. No. 326700, 1:400, Invitrogen), mouse anti-dpMAP kinase (Cat. No. m8159, 1:1000; Sigma-Aldrich), rabbit anti-DE-cadherin (sc-33743, 1:100, Santa Cruz Biotechnology), rabbit anti-aPKC (sc-216, 1:1000, Santa Cruz Biotechnology), rabbit anti-phospho-histone H3 (Cat. No. 06-570, 1:1000; Millipore), rabbit anti-activated caspase-3 (Cat. No. 9661s, 1:75, Cell Signaling Technology), rat anti-BrdU (ab6326–250, 1:100, Abcam), and rabbit anti-RPA3 (1:100, this study). Secondary antibodies used were as follows: Alexa Fluo-488 goat anti-rabbit (1:200) (Invitrogen); Cy3-conjugated goat anti-mouse (1:200), Cy3-conjugated goat anti-rabbit (1:200), Cy5-conjugated goat anti-mouse (1:200), Cy5-conjugated goat anti-rat (1:200), and Cy5-conjugated donkey anti-guinea pig (1:200) (Jackson ImmunoResearch Laboratories). To stain F-actin, Alexa Fluo-488 phalloidin or Alexa Fluo-568 phalloidin (Invitrogen) was added at 1:200 during secondary antibody incubation.
For BrdU-labeling experiment, brains were dissected from mid-third instar larvae in Schneider S2 medium. The brains were then incubated in the medium containing 200 μg/ml BrdU for 1 h at room temperature, after which the brains were fixed as described above.
Before incubation with anti-BrdU antibody, larval brains were incubated in 2N HCl for 30 min and then neutralized by washing with 0.1 m boric acid two times for 2 min each. Confocal images were captured by an Olympus FV1000 confocal microscope (objective, 60×; numerical aperture, 1.4) and processed using Imaris (Bitplane) and Adobe Photoshop 7.0 software.
Results
RPA3 is expressed in the optic lobe neuroepithelia
To identify genes that regulate larval neuroepithelial stem cell maintenance and NB formation in the Drosophila optic lobe, we used the UAS/GAL4 system (Brand and Perrimon, 1993) to screen a collection of genes for the RNA knockdown phenotypes in optic lobe development. Two different Gal4 lines are used: c768-Gal4 and c855a-Gal4. These are both active in the OPC and IPC neuroepithelia starting from first instar through late-third instar stages (Egger et al., 2007; Wang et al., 2011a). We found that RNA knockdown of CG15220 caused severe defects in optic lobe development. A multiple sequence alignment revealed that the protein encoded by CG15220 shows high evolutionary conservation with RPA3, which is involved in DNA metabolism, such as DNA replication, repair, and recombination (Fig. 1D).
We performed antibody-staining experiments to assay RPA3 protein expression patterns. In late-third instar larval brains, RPA3 is localized in the nucleus and is expressed strongly in the NE cells in the OPC, in the lamina precursor cells, and in developing lamina cortex neurons; medulla NBs also strongly express RPA3 protein, while the ganglion mother cells (GMCs) express the protein more weakly (Fig. 1E,E'); medulla and lamina neurons do not express RPA3 (Fig. 1E',F). NBs in the central brain (Fig. 1E') and ventral nerve cord (VNC) also strongly express RPA3. The IPC NE cells express RPA3 weakly (data not shown). The specificity of this antibody was confirmed by the specific elimination of RPA3 protein in RPA3RNAi flip-out clones (Fig. 1G) or in the brain lobes in which RPA3 expression was knocked down by RNAi using the c768-Gal4 driver (see Fig. 3S).
Figure 3.
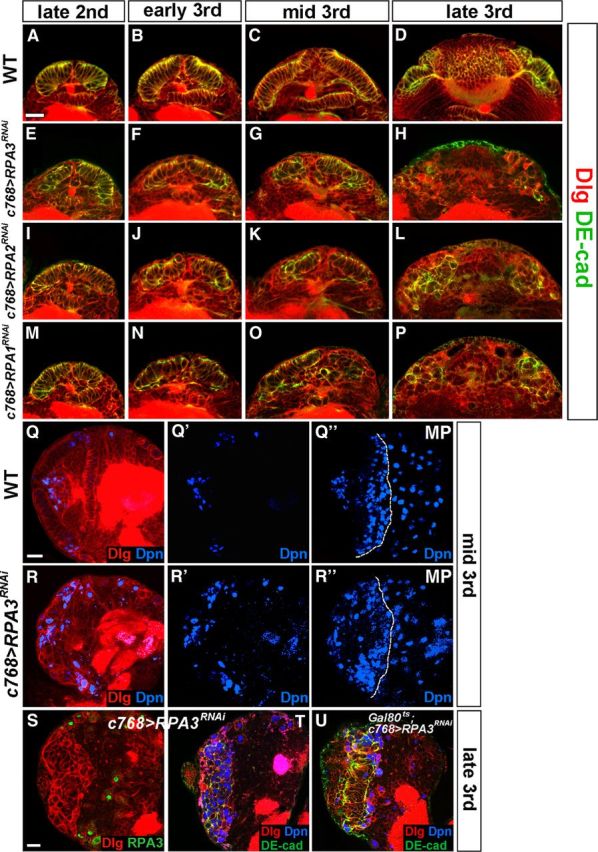
RPA maintains neuroepithelial integrity in the optic lobe. Markers are as indicated. A–P, Time course studies of optic lobe neuroepithelial growth and expansion. A–D, Wild-type brain lobes at late-second (A), early-third (B), mid-third (C), and late-third instar (D). E–P, Brain lobes in which RPA3 (E–H), RPA2 (I–L), or RPA1 (M–P) expression was knocked down by RNAi using the c768-Gal4 driver. Loss of RPA3, RPA2, or RPA1 activity led to the gradual loss of NE cells; the NE cells were partially lost by mid-third instar and completely lost by late-third instar. Lateral is up and medial down. Scale bar: A, 20 μm (for A–P). Q–R”, Loss of RPA3 activity leads to premature differentiation of NE cells into medulla NBs. Mid-third instar brains were analyzed. Q, Q', Q”, Wild-type brain lobe stained with Dpn and Dlg. MP, maximal projection. R, R', R”, c768-Gal4/UAS-RPA3RNAi brain lobe. Dpn+ NBs formed prematurely in RPA3 mutant brains compared with wild-type brains. Lateral is to the left and dorsal up. Scale bar: Q, 20 μm (for Q–R”). S–U, RPA3RNAi brain lobes from late-third instar larvae showing disintegration of the optic lobe neuroepithelium and their differentiation into NBs. S, RPA3 expression was specifically knocked down in the optic lobe NE cells using the c768-Gal4 driver. T, This led to the loss of NE cells and premature differentiation of NE cells into medulla NBs. U, Gal80ts; c768-Gal4/UAS-RPA3RNAi brains retained more NE cells by late-third instar stages compared with c768-Gal4/UAS-RPA3RNAi brain lobes. Lateral is to the left and dorsal up. Scale bar: S, 20 μm (for S–U).
RPA is required for lamina and medulla development in the optic lobe
We next examined the effect of loss of RPA3 activity on optic lobe development. The wild-type optic lobe contains three visual ganglia: the medulla, the lamina, and the lobular complex (Fig. 1B). The crescent-shaped lamina can be visualized with anti-Dachshund (Dac) antibody, the dome-shaped medulla cortex with anti-Elav antibody, and the boundary between the medulla cortex and the central brain with anti-Prospero antibody, which stains central brain neurons strongly but stains medulla GMCs weakly (Fig. 2A,B). We found that the brain did not develop a lamina when RPA3 expression was knocked down in the optic lobe using either c768-Gal4 or c855a-Gal4 driver (Fig. 2C', compare with wild type in Fig. 2A', 100%, n = 39); the medulla cortex was also severely reduced in size (Fig. 2D, compare with wild type in Fig. 2B, 100%, n = 45). These results suggest that neurogenesis in the lamina and medulla is affected in RPA3 mutant brains.
Figure 2.
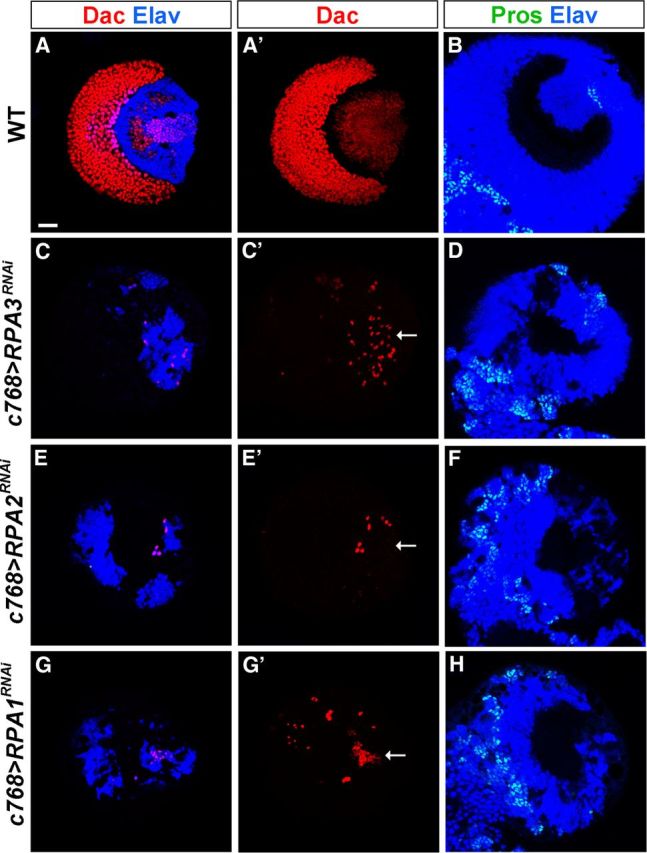
RPA is essential for lamina and medulla development in the optic lobe. Late-third instar larval brains were stained with the antigens indicated. A, A', B, Wild-type brain lobes showing the crescent-shaped lamina (A, A') and the dome-shaped medulla (B). The border between the medulla and the central brain is revealed by strong Prospero (Pros) staining of central brain neurons. C, C', D, c768-Gal4/UAS-RPA3RNAi brain. The lamina is absent (C, C', arrow indicates lobular complex cells) and the medulla is smaller compared with wild type (D, compare with B). E, E', F, c768-Gal4/UAS-RPA2RNAi brains. The lamina is absent (E, E') and the medulla (F) is severely reduced in size. G, G', H, c768-Gal4/UAS-RPA1RNAi brains. The mutant brain also did not develop the lamina (G, G') and had a smaller medulla (H, compare with B). Anterior is to the left and dorsal up. Scale bar, 20 μm.
Mammalian RPA is a single-stranded DNA-binding protein complex composed of 70, 32, and 14 kDa subunits (referred to as RPA1, RPA2, and RPA3, respectively), which are highly conserved in eukaryotes. RPA plays essential roles in many processes of DNA metabolism, including DNA replication, DNA repair, and homologous recombination (for reviews, see Wold, 1997; Iftode et al., 1999; Binz et al., 2004). Because RPA3 is essential for optic lobe development, we expected that RPA2 and RPA1 might also function in the optic lobe. We used RNAi to knock down RPA2 or RPA1 expression in the optic lobe. As with loss of RPA3 function, loss of RPA2 or RPA1 function also caused a loss of the lamina (Fig. 2E',G', 100%, n = 30) and a smaller medulla (Fig. 2F,H, 100%, n = 30). The eye imaginal disc appeared normal (data not shown), indicating that the defects in lamina and medulla development resulted from the optic lobe rather than from the eye. These observations show that the RPA complex is required for lamina and medulla development in the optic lobe.
RPA is essential for NE cell maintenance and expansion in the optic lobe
Since the OPC NE cells are the progenitor cells that generate lamina and medulla neurons, the defects in RPA mutant brains could be caused by a deficiency in the maintenance and/or expansion of NE cells. We thus followed the progression of NE cell proliferation and their differentiation into NBs in mutant brain lobes using several molecular markers. The NE cells have a columnar shape and express apical epithelial proteins PatJ and atypical protein kinase C (aPKC), and adherens junction proteins DE-cadheren (DE-cad) and Armadillo, whereas medulla NBs have a rounded shape, downregulate the expression of apical proteins, and express Deadpan (Dpn), Asense (Ase), and Miranda (Mira) (Egger et al., 2007). In the wild type, the optic lobe NE cells divide symmetrically to expand the progenitor population during early larval stages and reach a maximal number by mid-third instar (Fig. 3A–C). Neurogenesis in the medulla starts at the late-second instar stage, when the NE cells at the medial edge of the OPC lose their columnar shape and transform into rounded medulla NBs. Thus, by late-third instar, the OPC neuroepithelium decreases in size while the number of NBs increases in the medial region of the optic lobe (Fig. 3D); and by early pupal stages, all the NE cells have differentiated into lamina and medulla neurons.
To assess the role of RPA in neuroepithelial maintenance and proliferation, we used RNAi to knock down the expression of RPA components. RPA3 RNA knockdown caused defects in the OPC neuroepithelium at late-second instar, which was manifested by abnormal epithelial cell morphology (Fig. 3E, 83%, n = 18). RPA3RNAi brain lobes gradually lost the NE cells during early-third to mid-third instars (Fig. 3F,G, 100%, n = 51; compare with wild type in Fig. 3B,C); and by late-third instar, there were very few NE cells left in the RPA3RNAi brain lobe (Fig. 3H, 100%, n = 48; compare with wild type in D) and almost all cells in the OPC differentiated into NBs (Fig. 3T). Similar to RPA3 RNA knockdown, RPA2 or RPA1 RNA knockdown also led to disintegration of the OPC neuroepithelium during early-third to mid-third instars (Fig. 3J,K,N,O, 100%, n = 35) and eventually almost complete loss of OPC NE cells by late-third instar (Fig. 3L,P, 100%, n = 39).
Concomitant with a decrease in NE cell number in the OPC during early-third to mid-third instar, an increased number of medulla NBs appeared in comparison with wild type (Fig. 3R,R',R”, 100%, n = 14; compare with Fig. 3Q,Q',Q”), suggesting that the NE cells in the mutant brains had prematurely transformed into medulla NBs. By contrast, during comparable stages in the wild type, Dpn+ NBs were generated only at the medial edge of the OPC neuroepithelium (Fig. 3Q,Q',Q”). Since knockdown of RPA3 by c768-Gal4 or c855a-Gal4 strongly affected neuroepithelial integrity (Fig. 3S,T), we alternatively combined c768-Gal4 or c855a-Gal4 with a Gal80ts construct to temporally control RPA3 expression (see Materials and Methods). By shifting the culture from a low permissive temperature (18°C) to the nonpermissive high temperature (31°C), we can temporally control the stage of RPA3 knockdown. When RPA3 expression was knocked down at early-third instar, we found that there were some NE cells left at late-third instar. These cells were undergoing morphological change and became NBs (Fig. 3U, 100%, n = 24). Together, these observations indicate that RPA is required for the maintenance and expansion of neuroepithelial stem cells in the OPC and that loss of RPA activity leads to early disintegration of the OPC neuroepithelium and premature medulla NB formation.
RPA mutant cells in clones are extruded from the neuroepithelium and undergo premature neurogenesis
To further examine the effect of removing RPA activity on brain development, mosaic clones were induced using the FLP/FRT system (Xu and Rubin, 1993). RPA3 mosaic clones were induced using a strong loss-of-function allele RPA3G0241 at late-second or mid-third instar and the brains were analyzed at late-third instar. We first characterized RPA3 mutant clones located in the OPC neuroepithelium. The RPA3 mutant epithelial clones were small and composed of only a few cells (Fig. 4A,B) and costaining for Dpn and DE-cad or PatJ showed that the mutant cells did not change NE cell identity (PatJ+) or cell fate (Dpn−) (Fig. 4A–B', 100%, n = 41). Some mutant cells in epithelial clones were found to be extruded from the neuroepithelium and basally enter the medulla cortex (Fig. 5A,B). Costaining of RPA3 mutant cells for Dpn and DE-cad or Discs large (Dlg), which outlines all cell cortices, showed that once the mutant cells had been extruded from the neuroepithelium, they began to change cell morphology to rounded or irregular shape; and one to several cells located in the more medial region of the clone began to express Dpn (Fig. 5A,B; 100%, n = 14). In wild-type control clones, the epithelial cells always stayed in the OPC neuroepithelium and were not extruded to the medulla cortex (Fig. 4I,I'), whereas those control clones located entirely in the medulla cortex contained differentiated neurons but no NBs, which should have originated from an NB that generated a lineage of medulla neurons (Fig. 4J,J'). We conclude that loss of RPA3 caused the extrusion of the mutant cells from the neuroepithelium, which prematurely become NBs.
Figure 4.
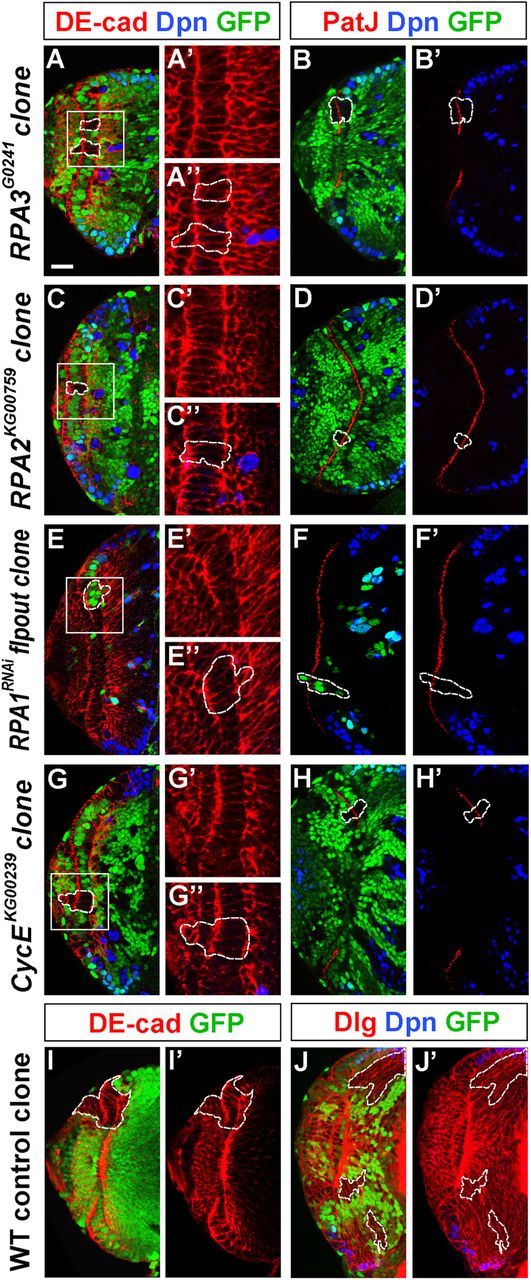
RPA and CycE mutant NE cells in small clones do not disrupt epithelial cell integrity. Brain lobes from late-third instar larvae were stained for Dpn and DE-cad or PatJ. A–F', RPA3 mosaic clones (A–B'), RPA2 mosaic clones (C–D'), and RPA1RNAi flip-out clones (E–F'). RPA mutant cells in epithelial clones retained columnar NE cell morphology, expressed the apical marker PatJ, and did not express the NB marker Dpn. G–H', CycE mosaic clones. CycE mutant cells in small NE clones also retained epithelial morphology and identity, and did not express Dpn. I–J', Wild-type control clones. The epithelial cells in the wild-type clone always stayed in the OPC neuroepithelium (I, I') and those clones entirely localized in the medulla do not ectopically express Dpn (J, J'). Lateral is to the left and medial to the right. Scale bar, 20 μm.
Figure 5.

Loss of RPA activity in epithelial cell clones causes extrusion of mutant epithelial cells and premature NB formation. Brain lobes from late-third instar larvae were stained with the antigens indicated; selected clones are identified by dashed lines. A–I, RPA3 mosaic clones. A–B, RPA3 mutant cells in epithelial clones were segregating from surrounding wild-type neuroepithelium and entering the medulla cortex. Some mutant cells located in the more medial region of the clone became NBs (Dpn+, arrowheads). C–I, Mutant clones entirely localized in the medulla cortex. The ectopic NBs expressed Dpn (C), Mira (D), and Ase (E); had asymmetric aPKC and Mira localization during mitosis (F); and had the ability to divide (G) to generate GMC progeny (Pros+) and neurons (Elav+) (H, I). J–L, RPA3RNAi flip-out clones. The mutant cells in RPA3RNAi flip-out clones located in the medulla expressed NB markers (J). Mitotic cells in the mutant clone underwent asymmetric division as Numb or aPKC was localized in one pole of the mitotic cell perpendicular to the spindle orientation (K, L, arrows). M–R, RPA2 mosaic clones. S–X, RPA1RNAi flip-out clones. Like RPA3 mutant clones, mutant NE cells in RPA2 (M, N) and RPA1 clones (S, T) were extruded from the OPC neuroepithelium and entered the medulla cortex, causing some of the mutant cells located in the more medial region of the clones to express Dpn. In medulla clones (O–R; U–X), ectopic NBs were generated that underwent asymmetric divisions and generated GMCs and neurons. Lateral is to the left and dorsal up. Scale bar, 20 μm.
RPA3 mutant clones were more frequently found to be localized entirely in the medulla cortex, which were presumed to have originated from the neuroepithelium. In medulla clones, typically several to >12 mutant cells ectopically expressed NB-specific markers Dpn (Fig. 5C, 94%, n = 115), Mira (Fig. 5D, 91%, n = 36), and Ase (Fig. 5E, 100%, n = 28). These Dpn+/Mira+/Ase+ cells had diameters of 7–10 μm and behaved like NBs as they went through metaphase with asymmetric aPKC and Mira localization (Fig. 5F) and were dividing [labeled by anti-phospho-histone H3 (PH3) staining] (Fig. 5G), generating GMC progeny (Prospero+) and a limited number of neurons (Elav+) (Fig. 5H,I). Similar results were also found in RPA3RNAi flip-out clones (Fig. 5J–L). In contrast to the very limited number of cells in RPA3 mutant epithelial cell clones, RPA3 mutant clones entirely localized in the medulla cortex contained a much larger number of progeny, ranging between 15 and 50 cells (Fig. 5C,H), although we noticed that the number of differentiated neuronal progeny were more limited (Fig. 5I) compared with the number of neurons in wild-type control NB clones (Fig. 4J,J').
RPA2 mutant clones were induced using the strong loss-of-function allele RPA2KG00759 and RPA1RNAi mutant clones were induced using the flip-out method. The overall results for RPA2 and RPA1 mutants were similar to those for RPA3 mutants. The mutant epithelial clones were small and mutant cells in the clone retained the epithelial cell morphology and cell fate (Fig. 4C–F', 100%, n = 35). Some mutant cells in epithelial clones were found to be extruded from the neuroepithelium and basally enter the medulla cortex (Fig. 5M,N,S,T). These extruded cells changed cell morphology and began to express the NB marker Dpn (Fig. 5M,N,S,T). In clones entirely localized in the medulla cortex, there were always several to >12 ectopic NBs that divided asymmetrically and generated GMC progeny and neurons (Fig. 5O–R,U–X; and data not shown). Again, the sizes of medulla clones were quite large, comparable to wild-type control clones (Fig. 5O,U). Thus, our molecular and morphological data indicate that loss of RPA activity in epithelial clones leads to the extrusion of mutant epithelial cells and their premature differentiation into medulla NBs.
Loss of RPA activity causes NE cells to proliferate more slowly
From the mosaic clone and RNAi flip-out clone data described above, we noticed that the cell number in RPA mutant NE clones was much smaller than that in wild-type control clones (Fig. 4). To determine whether RPA mutant epithelial cells have a proliferative defect, we first determined the proliferation rate using the flip-out system. Wild-type and RPA3RNAi clones were induced at late-second instar (48 h ALH), when the OPC is comprised of an expanding epithelial sheet.
We then counted cell numbers in epithelial cell clones at early-third, mid-third, and late-third instar stages. At early-third instar (60 h ALH), the cell number in an RPA3RNAi clone (3.5 ± 0.7, n = 34) was close to that in a wild-type clone (3.6 ± 0.8, n = 38) (Fig. 6A,D); at mid-third instar (72 h ALH), there were fewer cells found in RPA3RNAi clones (5.3 ± 0.6, n = 39) compared with wild type (8.1 ± 0.8, n = 40) (Fig. 6B,E); by late-third instar (96 h ALH), the cells in a wild-type clone proliferated to 28.9 ± 4.9 (n = 36), while the number of cells in RPA3 mutant clones appeared to have changed little (5.9 ± 1.3, n = 25) (Fig. 6C,F). Quantification of the proliferative rates is shown in Figure 6K. BrdU labeling was used to directly assay the proliferation states of RPA3 mutant cells and we found that many fewer mutant NE cells, compared with wild-type NE cells, were incorporating BrdU (Fig. 6I,J, n = 39; compare with wild type in Fig. 6G,H, n = 37). Cell death was not induced in RPA mutant brains as assayed with anti-activated caspase-3 antibody, which recognizes apoptotic cells (data not shown). Similarly, cell proliferation was also strongly inhibited in RPA2 and RPA1 mutant NE cells (data not shown). Thus, RPA is required for NE cell proliferation in the optic lobe. RPA mutant clones in the central brain contained a similar number of progeny as those in central brain control clones (data not shown). Thus, it appears that RPA is specifically required for the proliferation of NE cells in the optic lobe.
Figure 6.

RPA3 mutant NE cells have a proliferative defect. Dlg outlines all cell cortices in larval brains and GFP marks flip-out clones. Clones were induced at late-second instar and assayed at early-third (A, D), mid-third (B, E), or late-third instar stage (C, F). A–C, Wild-type control clones. The NE cell in a clone proliferated to expand the epithelial cell pool. D–F, RPA3RNAi flip-out clones. Mutant epithelial cells in RPA3RNAi clones proliferated poorly. Arrow indicates NE clone. G–J, BrdU labeling of wild-type (G, H) and RPA3RNAi (I, J) mutant brains at mid-third instar stage. Two sections were shown for each brain lobe. Fewer RPA3 mutant epithelial cells incorporated BrdU compared with wild type. K, Quantification of cell numbers per epithelial cell clone for wild-type (WT; white bars) and RPA3RNAi flip-out brains (RPA3; black bars). n, total number of clones analyzed; the average number of cells per clone is shown between brackets. *p < 0.05, ***p < 0.001; Student's t test. Error bars represent SDs. Lateral is to the left and dorsal up. Scale bar, 20 μm.
Cell cycle control in optic lobe development
The observation that loss of RPA activity caused premature differentiation of NE cells into NBs suggests a correlation between cell cycle length and neurogenesis in the optic lobe.
We hypothesized that lengthening or inhibition of NE cell cycle progression may be causally linked with the transition from symmetric proliferative division of NE cells to asymmetric division of medulla NBs.
To directly test this hypothesis, we analyzed the effects on optic lobe development of a number of core cell cycle regulators, including E2F1, PCNA, Cyclin E, Cyclin D, Cyclin B, Cyclin A, Cdk1, Cdk2, and Cdk4. Studies have shown that E2F1, Cdk2, and its binding partner cyclin E are involved in G1/S transition (Dynlacht et al., 1994; Knoblich et al., 1994; Neufeld et al., 1998), proliferating cell nuclear antigen (PCNA) is important for both DNA synthesis and DNA repair during S phase (Essers et al., 2005), and Cdk4 and its binding partner CycD are involved in cell growth (Datar et al., 2000; Meyer et al., 2000; Emmerich et al., 2004), while Cdk1, as well as cyclin A and cyclin B are required for G2/M transition (Lehner and O'Farrell, 1990; Knoblich and Lehner, 1993; Lindqvist et al., 2009; McCleland et al., 2009). Indeed, we found that loss of many of these regulators except CycD and Cdk4 caused defects in optic lobe development (Fig. 7B–H'). For instance, RNA knockdown of PCNA expression in the optic lobe led to the gradual loss of NE cells during early-third to late-third instars and premature NB formation (Fig. 7E,E', 100%, n = 37). RNA knockdown of E2F1, CycE, CycA, CycB, Cdk1, or Cdk2 expression in the optic lobe similarly led to defects in neuroepithelial maintenance and premature formation of NBs (Fig. 7B–D', F–H'; for E2F1, 100%, n = 37; CycE, 100%, n = 29; CycA, 100%, n = 25; CycB, 100%, n = 34; Cdk1, 100%, n = 26; Cdk2, 100%, n = 23). To verify that the loss of these cell cycle regulators indeed affected cell cycle progression of NE cells, we used BrdU labeling to examine the G1/S transition and anti-PH3 staining to examine the G2/M transition in RNAi brains. We found that in E2F1 or CycE RNAi brains, BrdU labeling in NE cells was reduced compared with wild-type NE cells, consistent with a role of these G1/S regulators in the transition from G1 to S phase (Fig. 7J,J',K,K'; for E2F1, 14.9 BrdU+ cells/section, n = 10; for CycE, 8.4 BrdU+ cells/section, n = 11; compare with wild type in I,I', 23.5 BrdU+ cells/section, n = 12); while in CycB or Cdk1 RNAi brains, there was an increase in mitotic NE cells (PH3+) (Fig. 7N,N′,O,O', for CycB, 5.9 PH3+ cells/section, n = 11; for Cdk1, 9.3 PH3+ cells/section, n = 12; compare with wild type in M,M', 1.4 PH3+ cells/section, n = 14) but a reduction in BrdU-labeled cells compared with wild-type NE cells (Fig. 7L,L'; for Cdk1, 9.5 BrdU+ cells/section, n = 12; compare with wild type in I,I'), consistent with a cell cycle delay or arrest in the G2/M phase in the mutant NE cells.
Figure 7.
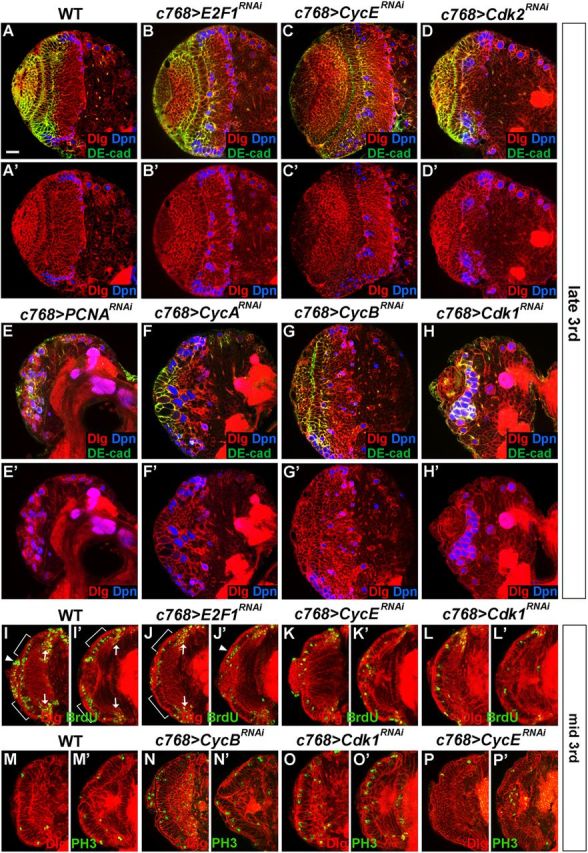
Cell cycle regulators are required for the maintenance and expansion of NE cells in the optic lobe. Brain lobes from late-third instar (A–H') or mid-third (I–P') instar larvae were stained with the antigens indicated. A, A', Wild type. B, B', c768-Gal4/UAS-E2F1RNAi brains. C, C', c768-Gal4/UAS-CycERNAi brains. D, D', c768-Gal4/UAS-Cdk2RNAi brains. E, E', c768-Gal4/UAS-PCNARNAi brains. F, F', c768-Gal4/UAS-CycARNAi brains. G, G', c768-Gal4/UAS-CycBRNAi brains. H, H', c768-Gal4/UAS-Cdk1RNAi brains. In brains mutant for each of these cell cycle genes, some Dpn+ NBs formed prematurely. For PCNA (E, E'), CycA (F, F'), and Cdk1(H, H') mutant brains, the NE cells were largely lost by late-third instar and most mutant cells in the optic lobe differentiated into NBs (Dpn+). I–L', BrdU labeling of NE cells at the mid-third instar stage. I, I', Wild type. J, J', c768-Gal4/UAS-E2F1RNAi brains. K, K', c768-Gal4/UAS-CycERNAi brains. L, L', c768-Gal4/UAS-Cdk1RNAi brains. In the mutant NE cells, compared with wild-type NE cells, BrdU labeling was sharply reduced. Two sections are shown for each brain lobe. The region of NE cells is indicated by brackets. Arrow indicates medulla NBs and GMCs incorporating BrdU. Filled arrowheads indicate BrdU labeling of lamina precursor cells and/or NBs from the IPC. M–P', Anti-phospho-histone H3 labeling of NE cells at the mid-third instar stage. M–M', Wild type. N, N′, c768-Gal4/UAS-CycBRNAi brains. O, O', c768-Gal4/UAS-Cdk1RNAi brains. P, P', c768-Gal4/UAS-CycERNAi brains. Compared with wild-type NE cells (M, M'), PH3 labeling in NE cells was significantly increased in c768-Gal4/UAS-CycBRNAi (N, N′) or c768-Gal4/UAS-Cdk1RNAi (O, O') brains, while the number of PH3-labeled NE cells in c768-Gal4/UAS-CycERNAi (P, P') brains was close to that in wild type. Two sections are shown for each brain lobe. Lateral is to the left and dorsal up. Scale bar, 20 μm.
Mosaic clones using strong loss-of-function mutant alleles were induced to further assess the effect of loss of these cell cycle regulators on optic lobe development. CycE clones were induced by using the null allele CycEKG00239. In small CycE mutant epithelial clones, the mutant cells still expressed the epithelial marker PatJ (Fig. 4H,H'), but not the NB marker Dpn (Fig. 4G–H'). More typically, CycE mutant epithelial clones were found to contain both NE cells and cells that entered the medulla cortex (Fig. 8A–C). In such clones, a number of mutant cells located in the medulla cortex expressed Dpn (Fig. 8A',B',C'). In contrast to RPA mutant epithelial clones, which were usually small (Fig. 5A,B), CycE mutant epithelial clones were quite large, suggesting that CycE mutant NE cells proliferated faster than RPA mutant epithelial cells. In such large clones, the mutant epithelial cells changed cell morphology to rounded or irregular shape (Fig. 8A,A', 100%, n = 16) and had reduced or lost PatJ expression (Fig. 8C,C', 100%, n = 21), suggesting that loss of CycE function affected NE cell integrity. Again, many mutant clones were entirely localized in the medulla cortex (Fig. 8D–F) and in such clones many ectopic NBs were found that expressed Dpn and Mira and had the ability to generate GMCs (Prospero+) and large numbers of neurons (Elav+) (Fig. 8D–F, n = 56).
Figure 8.
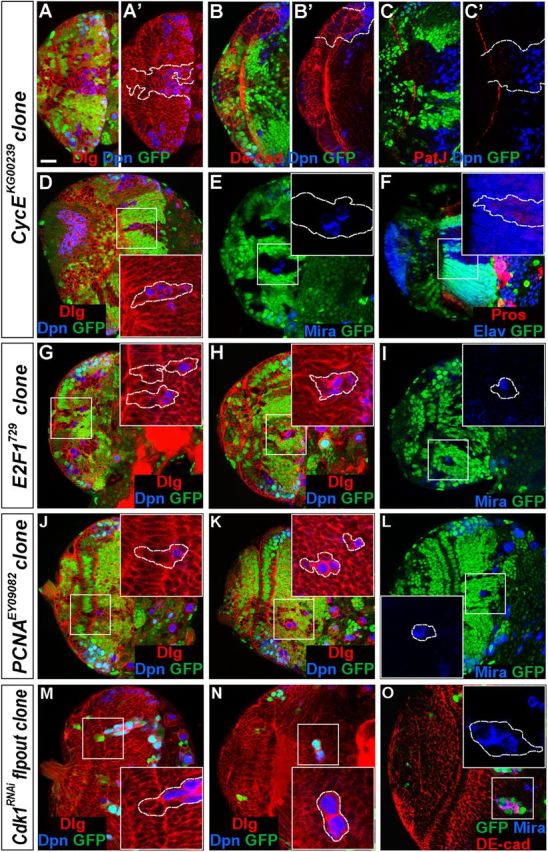
Loss of cell cycle regulator function in mutant clones causes premature differentiation of NE cells into medulla NBs. Brain lobes from late-third instar larvae were stained with the antigens indicated; selected clones are identified by dashed lines. A–F, CycEKG00239 mosaic clones. A–C', Mutant clones that had both NE cells and cells that entered the medulla cortex. The mutant NE cells in the clone expressed PatJ (C'), but not Dpn (A', B'), while a number of the cells that entered the medulla expressed Dpn (A–C). In large mutant clones, the NE cells had reduced or lost PatJ expression (C'). D–F, CycE mutant clones entirely localized in the medulla. Ectopic NBs formed (D, E) that generated medulla neurons (F). G–O, E2F1729 mosaic clones (G–I), PCNAEY09082 mosaic clones (J–L), and Cdk1RNAi flip-out clones (M–O). The mutant epithelial cells in the clones were being extruded from the neuroepithelium (G, J, M) and entered the medulla cortex where ectopic NBs formed prematurely. Lateral is to the left and dorsal up. Scale bar, 20 μm.
We also conducted mosaic clone analyses for E2F1 and PCNA using strong loss-of-function or null alleles and flip-out clone analyses for Cdk1 using RNAi. Clones of cells mutant for these genes exhibited phenotypic similarities to those of RPA mutants in the optic lobe (Fig. 8G–O; and data not shown). E2F1, PCNA, and Cdk1 mutant epithelial cell clones were each very small, similar to RPA3 mutant epithelial cell clones (Fig. 8G,J,M). Mutant epithelial cells were found to be extruded from the NE and enter the medulla cortex where the mutant cells started to express the NB marker Dpn (Fig. 8G,J,M). E2F1, PCNA, or Cdk1 mutant clones in the medulla were also found to contain several ectopic NBs and a quite large number of progeny (Fig. 8H,K,O). Together, our data from these systematic analyses of cell cycle regulators suggest that the slowing down or inhibition of cell cycle progression in either G1/S or G2/M phase can lead to the transition of symmetric NE cell division to asymmetric NB division.
In contrast to the effect on neuroepithelial fate transformation, RNA knockdown of these cell cycle regulators (E2F1, CycE, CycA, Cdk1, RPA3, RPA2, and RPA1) using insc-Gal4, which is active in medulla NBs and in central brain and VNC NBs but not active in the optic lobe NE cells, did not cause obvious defects in medulla development (data not shown). Furthermore, loss of E2F1, Cyclin E, PCNA, RPA3, or RPA2 function in mosaic NB clones in the central brain did not appear to affect NB self renewal (always 1 single NB per clone) and generation of a quite normal number of neuronal progeny, with the exception of Cdk1, whose loss of function in flip-out RNAi clones in the central brain led to a limited number of ectopic NBs but no neuronal progeny (data not shown). We conclude that the loss of cell cycle regulators is specifically linked with the transition of division modes in the optic lobe NE cells.
Notch signaling activity is downregulated in RPA and E2F1 mutant NE cells
Genetic studies indicate that Numb negatively regulates Notch signaling activity (Frise et al., 1996) and the numb loss-of-function phenotype is epistatic to the CycA mutant phenotype in embryonic neurogenesis (Wai et al., 1999), suggesting a potential interaction between cell cycle regulators and Notch signaling. We investigated the impact of loss of RPA and E2F1 function on Notch signaling activity, which has recently been shown to play an important role in controlling the growth and differentiation of NE cells in the optic lobe (Egger et al., 2010; Ngo et al., 2010; Reddy et al., 2010; Yasugi et al., 2010; Orihara-Ono et al., 2011; Wang et al., 2011a; Weng et al., 2012).
To examine whether Notch signaling activity is affected in RPA mutant NE cells, we crossed the E(spl)m8-lacZ line, a reporter that responds to Notch activation, into the RPA mutant background. In wild-type brain lobes, E(spl)m8-lacZ is expressed throughout the OPC neuroepithelium with a peak level of activity near the medial transition zone where medulla NBs form (Fig. 9A,A'; Wang et al., 2011a). We made RPA3 or RPA2 mosaic epithelial clones and found that E(spl)m8-lacZ expression was significantly reduced in RPA3 (Fig. 9B,B', 91%, n = 35) or RPA2 mutant NE cells (Fig. 9C,C', 92%, n = 37) compared with surrounding wild-type NE cells. Knockdown of RPA3 expression in the NE using the c768-Gal4 driver also significantly reduced E(spl)m8-lacZ expression levels (Fig. 9D,D', 100%, n = 12, compare with wild type in A,A'). Similarly, knockdown of E2F1 expression in the NE under c768-Gal4 control significantly reduced E(spl)m8-lacZ expression levels in patches of the mutant NE cells (Fig. 9E,E', 100%, n = 26). Thus, it appears that inhibition of cell cycle progression is associated with downregulation of Notch signaling activity.
Figure 9.

Notch signaling activity is downregulated in RPA and E2F1 mutant NE cells. Brain lobes from late-third instar larvae were stained with the antigens indicated; the clones are marked by the lack of GFP expression and by dashed lines. Arrow indicates the lamina furrow (LF). A, A', E(spl)m8-lacZ expression in wild type. B–C', E(spl)m8-lacZ expression was dramatically downregulated in RPA3 (B, B') or RPA2 (C, C') mosaic NE clones (filled arrowheads). There is also a medulla clone immediately underneath the NE that had lost E(spl)m8-lacZ expression (C', open arrowhead). D–E', E(spl)m8-lacZ expression in RPA3RNAi brains (D, D') and E2F1RNAi brains (E, E'). E(spl)m8-lacZ expression was downregulated in RPA3 or E2F1 mutant brains. F–G', Notch expression was not affected in RPA3 (F, F') or RPA2 (G, G') mutant epithelial clones. H–I', Delta expression was not influenced in RPA3 (H, H') or RPA2 (I, I') mutant epithelial clones. Note that in mutant clones located entirely in the medulla, there was an increase of both Notch and Delta expression (G', I', double arrowheads). J–L', Numb localization in wild type (J, J'; yellow arrows indicate the apical membrane domain of NE cells), RPA3RNAi brains (K, K'), and E2F1RNAi brains (L, L'). Open arrows indicate the basal domain of NE cells. Numb is mislocalized and strongly concentrated along the subapical region and the lateral region of the NE cells in RPA3 and E2F1 mutant brains. M–N′, PatJ expression in wild-type (M, M') and RPA3RNAi brains (N, N′). The apical marker PatJ was reduced or almost eliminated in RPA3RNAi brains (N, N′). Lateral is to the left and dorsal up. Scale bar, 20 μm.
To further explore the relationship between cell cycle progression and Notch pathway activity, we searched for possible defects in the expression of Notch components, but we found that neither Notch nor Delta expression was apparently affected in these mutant epithelial cells (Fig. 9F–I', 100%, n = 30; but note that mutant clones localized entirely in the medulla cortex showed upregulation of both Delta and Notch expression levels, indicated by double arrows in Fig. 9 G',I'). However, we find that the localization of the Notch antagonist Numb was disturbed. Numb protein is expressed in wild-type NE cells with a basolateral localization (Fig. 9J'; small yellow arrow indicates apical domain of the NE cells; large arrow indicates the basal membrane of the NE cells; Wang et al., 2011a). In RPA3 or E2F1 RNAi brains, Numb was mislocalized such that the protein was found to be strongly concentrated in both the subapical and the lateral region of the NE cells (Fig. 9K',L', 100%, n = 35; compare with wild-type Numb localization in J'). The apicobasal polarity of NE cells was disturbed in RPA3 RNAi brains, as revealed by reduced or lost expression of the apical marker PatJ (Fig. 9N,N′, n = 13; compare with wild type in M,M') and aPKC (data not shown). These results provide a plausible model to explain how the slowing down or cell cycle arrest in RPA or E2F1 mutant NE cells might cause the NE-to-NB transition: the prolonged time during the cell cycle arrest might allow diffusion and thus mislocalization of Numb protein, which could downregulate Notch activity at the subapical region, leading to the NE-to-NB transition.
To test the functional significance of Notch inactivation by RPA loss of function, we ectopically expressed a full-length wild-type Notch construct (UAS-NFL) in RPA3 RNAi brains. Remarkably, the NE growth was significantly rescued (Fig. 10A–D'). When RPA3 expression was knocked down at 25°C using the c768-Gal4 driver, there was virtually no NE cells left at the late-third instar stage (Fig. 10B,B'); however, when full-length Notch was coexpressed in these mutant brains, we observed a significant number of NE cells in the optic lobe at the late-third instar stage (Fig. 10D,D', 100%, n = 9). Ectopic expression of full-length Notch alone did not affect NE growth in the optic lobe (Fig. 10C,C'). Using RNAi to knock down both RPA3 and numb expression, we further tested whether Numb might also interact with RPA3. As has been shown before (Wang et al., 2011a), numb RNAi caused neuroepithelial overgrowth (Fig. 10E,E') and tumor development in the optic lobe (data not shown). When numb and RPA3 RNAs were knocked down together, we also observed a significant rescue of NE growth (Fig. 10F,F', 100%, n = 8; compare with B,B'). Together, these results demonstrate that the loss of Notch activity is largely responsible for the loss of NE cells in RPA3 RNAi brains and for the transition from NE to NBs.
Figure 10.

RPA3 interacts with several signaling pathways in the optic lobe. Brain lobes from late-third instar larvae were stained with the antigens indicated, as well as phalloidin, which stains F-actin in the cell cortex; the clones are marked by the lack of GFP expression and by dashed lines. A–B', Compared with wild type (A, A'), there are very few NE cells left in the c768-Gal4/UAS-RPA3RNAi brain lobes (B, B'). C–D', c768-Gal4/UAS-NFL (C, C') and c768-Gal4/UAS-RPA3RNAi,UAS-NFL brains (D, D'). The loss of NE cells in c768-Gal4/UAS-RPA3RNAi brains was significantly rescued by ectopically expressing full-length Notch (NFL) (D, D'). E–F', c768-Gal4/UAS-numbRNAi (E, E') and c768-Gal4/UAS-RPA3RNAi,UAS-numbRNAi brains (F, F'). Loss of numb activity led to neuroepithelial overgrowth (E, E'); when RPA3 and numb RNAs were simultaneously knocked down, the loss of NE cells in RPA3 RNAi brains was significantly rescued (F, F'). G–H', c768-Gal4/UAS-yki:V5s168A (G, G') and c768-Gal4/UAS-RPA3RNAi,UAS-yki:V5s168A brains (H, H'). Ectopic Yorkie activation led to neuroepithelial overgrowth (G, G'), while the loss of NE cells in RPA3 RNAi brains was significantly rescued by coexpression of activated Yorkie (H, H'). I–J', c768-Gal4/UAS-3HA-statACT (I, I') and c768-Gal4/UAS-RPA3RNAi,UAS-3HA-statACT brains (J, J'). Overexpression of activated stat92E (UAS-3HA-statACT [= UAS-3HA-stat92EΔNΔC]) led to neuroepithelial overgrowth (I, I'); however, the loss of NE cells in RPA3 RNAi brains was not rescued by coexpression of activated stat92E (J, J'). K–L', RPA3 mosaic clones. Loss of RPA3 in mosaic clones led to MAP kinase activation, revealed by anti-dpMAP kinase staining (K, K'). Correspondingly, Pointed P1 (Pnt1) expression in RPA3 mosaic clones was upregulated (L, L'). Open arrows indicate ectopic Pnt1 expression in RPA3 mutant cells in the medulla adjacent to the NE cells. Lateral is to the left and dorsal up. Scale bar, 20 μm.
RPA3 interacts with Fat/Hippo and EGFR signaling to control NE-to-NB transition
Several additional pathways are also implicated in neuroepithelial maintenance and proliferation in the optic lobe: namely, the JAK/STAT, Fat/Hippo, and EGFR pathways. We tested for an interaction between the Fat/Hippo pathway and RPA3 by ectopic expression of an activated form of Yorkie (Yki), which functions as an effector in the Fat-Hippo pathway. Overexpression of activated Yorkie alone led to neuroepithelial overgrowth (Fig. 10G,G'; Reddy et al., 2010). Overexpression of activated Yorkie also significantly rescued NE growth in RPA3 RNAi brains (Fig. 10H,H', 100%, n = 18; compare with RPA3 RNAi brains in B,B'). Thus, we found that both the Notch and Yorkie pathways interact with and probably act downstream of RPA3 to control the NE-to-NB transition in the optic lobe.
To test whether the JAK/STAT pathway interacts with RPA3, we overexpressed an activated STAT92E construct (UAS-3HA-stat92EΔNΔC; Ekas et al., 2010) in the optic lobe NE cells. Overexpression of this activated stat92E construct alone led to neuroepithelial overgrowth (Fig. 10I,I, 87%, n = 14), as has been shown for overexpression of activated JAK (UAS-hopTum-l) (Yasugi et al., 2008; Wang et al., 2011b). However, we found that activation of JAK/STAT signaling did not rescue the RPA3 loss-of-function phenotypes (Fig. 10J,J', n = 12), suggesting that JAK/STAT does not function downstream of RPA3.
In addition, we found that inactivation of RPA3 in mosaic clones or flip-out RNAi mutant clones caused activation of EGFR signaling, as revealed by increased levels of dual phospho-mitogen-activated protein (MAP) kinase staining (Fig. 10K,K', 73.9%, n = 23), which is indicative of EGFR pathway activation (Gabay et al., 1997), and, correspondingly, an upregulation of Pointed P1(PntP1) (Fig. 10L,L', 78.6%, n = 42), which is a major target gene of the EGFR pathway. The activation of MAP kinase occurred in RPA3 mutant cells extruded from and adjacent to the NE cells (Fig. 10K,K'), foreshadowing the differentiation of RPA3 mutant cells into ectopic medulla NBs that would emerge a few cells more medially (see the location of ectopic NBs in Fig. 5A,B). The EGFR pathway activation has previously been shown to drive the proneural wave from medial to lateral NE cells and promote the NE-to-NB transition (Yasugi et al., 2010). Here, we found that loss of RPA3 induced activation of the EGFR pathway, thus promoting the NE-to-NB transition.
Discussion
In this study, we have analyzed the role of RPA in cell cycle progression in the Drosophila optic lobe and found that loss of RPA function causes premature differentiation of NE cells into medulla NBs. Furthermore, inactivation of the core cell cycle regulators in NE cells, including E2F1, Cyclin E, PCNA, Cyclin A, Cyclin B, Cdk1, and Cdk2, each also caused the precocious NE-to-NB transition. Thus, our data indicate that inhibition of NE cell cycle progression is causally linked with the transition of symmetric proliferative division to asymmetric neurogenic division in the optic lobe neuroepithelium and this transition is likely achieved through Numb-mediated downregulation of Notch signaling activity.
The RPA complex encodes highly conserved proteins involved in a variety of DNA metabolism, including DNA replication, DNA repair, and recombination processes (Wold, 1997; Iftode et al., 1999; Binz et al., 2004; Bochkarev and Bochkareva, 2004). However, a role of any of these RPA components in animal development has not been reported before. Our study represents the first demonstration that the RPA complex plays a crucial role in developmental processes and in particular in the regulation of neural stem cell division modes. A recent study identified many genes that play a role in mitotic division and maintenance of chromosome integrity in Drosophila S2 cells, among which are RPA3 and RPA1 (Somma et al., 2008). Consistent with a role of RPA in DNA replication, loss of RPA function resulted in severely reduced BrdU labeling and in proliferation defects in mutant NE cells. Also, time course study indicated that cell cycle progression in RPA mutant epithelial cells was severely inhibited or arrested (Fig. 6). Thus, our data suggest a model whereby RPA regulates the transition of neuroepithelial proliferative division to asymmetric NB division through slowing down or arresting the cell cycle.
The cell cycle control has been noted for its close correlation between cell cycle length and the modes of neural progenitor division in vertebrates. For example, it has been shown that the duration of the cell cycle increases as the nervous system development proceeds and lengthening of the cell cycle is associated with the onset of neurogenesis (Takahashi et al., 1995; Dehay et al., 2001; Lukaszewicz et al., 2002; Calegari et al., 2005). Artificially increasing the cell cycle length by specific inhibition of cyclin-dependent kinases can induce premature neurogenesis in vitro (Calegari and Huttner, 2003), while overexpression of the Cdk4/cyclin D complex (Lange et al., 2009; Artegiani et al., 2011) or even cyclin D or cyclin E alone (Pilaz et al., 2009) delays neurogenesis and enhances the generation of basal progenitors. A transient cell cycle arrest has also been observed in the Drosophila optic lobe neuroepithelium that coincides with the transition from NE to NBs (Reddy et al., 2010; Orihara-Ono et al., 2011; Weng et al., 2012). These studies provided strong support for a close association of cell cycle lengthening and the switch of progenitor division mode. Our results extend these previous findings and provide direct in vivo evidence that inhibition of NE cell cycle progression causes the change of cell division modes and the onset of neurogenesis in the Drosophila optic lobe. Since the loss of either G1/S regulators E2F1, Cyclin E, Cdk2, PCNA, and RPA, or G2/M regulators Cyclin A, Cyclin B, and Cdk1 each leads to precocious NE-to-NB transition in the optic lobe, our data suggest that it is the overall rate of cell cycle progression, not only G1 phase lengthening, that is critical for the switch of progenitor division modes. However, it should be noted that the cell cycle regulators show some degrees of specificity regarding their roles in the regulation of cell division mode. Contrary to the enhanced progenitor proliferation induced by CycD and Cdk4 co-overexpression in the mouse brain, loss of CycD or Cdk4 function did not affect neuroepithelial maintenance or differentiation in the optic lobe. These results suggest that CycD and Cdk4 may be nonessential for cell cycle progression in the optic lobe neuroepithelium or that they may be functionally redundant with other cyclins and Cdks.
In contrast to findings from loss-of-function studies, forced expression of RPA3, E2F1, E2F1 and Dp, CycE, CycA, Cdk1, or string (stg) had no effects on the maintenance or differentiation of the neuroepithelial stem cells (data not shown). We reasoned that, although loss of activity of a single cell cycle regulator might slow down cell cycle progression to induce precocious NE-to-NB transition, overexpression of a single cell cycle regulator might not be sufficient to accelerate cell cycle progression. Perhaps coexpression of more cell cycle regulators can accelerate the cell cycle and delay neuroepithelial differentiation. Indeed, the coexpression of CycD, Cdk4, and E2F1 and Dp together did appear to delay neurogenesis to some extent in the optic lobe (Reddy et al., 2010).
Notch signaling plays a conserved role in Drosophila and mammalian brain development in maintaining the progenitor status and inhibiting neurogenesis (de la Pompa et al., 1997; Hitoshi et al., 2002; Grandbarbe et al., 2003; Yoon and Gaiano, 2005). In our present study, we find that Notch signaling activity is downregulated in RPA3 and RPA2 mutant NE cells in the optic lobe (Fig. 9B–D'). This is also the case when the core cell cycle regulator E2F1 is inactivated (Fig. 9E,E'). These data point to a common mechanism underlying Notch inactivation by the cell cycle: the rate of cell cycle progression determines whether Notch is inactivated or not. We find that when the cell cycle progression is delayed or inhibited, Numb is mislocalized and strongly expressed in the subapical region and the lateral region of mutant NE cells (Fig. 9K–L'). Thus, perhaps, the delaying or arrest of the cell cycle would allow the diffusion of Numb protein, which is normally localized in the basolateral domain of NE cells, into the more apical sides of NE cells, where it can interact with the Notch receptor, thereby inhibiting Notch activity. The demonstration that the NE growth defects in RPA3 RNAi brains can be significantly rescued by either increasing Notch expression or decreasing Numb expression provides strong support for the notion that it is Notch inactivation that leads to the transition from the NE to NBs.
Further analyses revealed the involvement of additional signaling pathways in regulating NE growth and differentiation in the optic lobe. Overexpression of activated Yorkie or inactivation of Fat or Warts promotes NE overgrowth and suppresses NE-to-NB transition (Reddy et al., 2010). Interestingly, we found that the epithelial defects in RPA3 RNAi brains also could be significantly rescued by expression of activated Yorkie, suggesting that Yorkie may act downstream of the cell cycle regulator. Yorkie has recently been shown to be activated noncell-autonomously by Notch in wing imaginal discs (Graves et al., 2012). This observation would be consistent with our finding that both Notch and Yorkie expression can lead to a rescue of the neuroepithelial defects in RPA3 RNAi brains.
The EGFR signaling pathway is activated in the medial optic lobe NE cells by Rhomboid, which promotes Pnt1 expression; Pnt1 then induces the expression of the proneural gene lethal of scute (l'sc) thereby driving the transition from NE to NBs (Yasugi et al., 2010). Surprisingly, we found that the loss of RPA3 activity led to EGFR pathway activation and upregulation of Pnt1 expression. This suggests that cell cycle progression itself may suppress EGFR activity, while slowing down or arresting the cell cycle would result in EGFR activation during NE-to-NB transition.
In the optic lobe neuroepithelium at the third-instar stage, there is a transient cell cycle arrest in the transition zone between the lateral NE cells and the medial NBs (Reddy et al., 2010; Orihara-Ono et al., 2011; Weng et al., 2012). Our findings could explain the functional importance of a cell cycle arrest in the transition of symmetrically dividing NE cells to asymmetrically dividing NBs. It seems that the cell cycle arrest does two things at the same time: inactivation of Notch signaling and activation of the EGFR pathway, both of which facilitate the NE-to-NB transition. Questions remain as to what causes the cell cycle arrest in the transition zone. We speculate that the JAK/STAT pathway might act at the top tier in promoting NE growth and suppressing their differentiation into NBs. JAK/STAT activity is graded along the lateral-to-medial direction of the OPC neuroepithelium (Wang et al., 2011b); its decreasing activity in the medial NE cells may be sufficiently low to cause the cell cycle arrest in the transition zone of the OPC neuroepithelium.
In conclusion, our systematic analyses of the RPA complex and core cell cycle regulators show that cell cycle progression is required for the maintenance of NE cell identity and suppression of their transformation into NBs. Our studies provide novel insight into a critical process in neurogenesis: the transition from symmetric proliferative NE cell division to asymmetric neurogenic NB division and may help us understand the control mechanism underlying cerebral cortex development in mammals.
Footnotes
This work was supported by grants from National Science Foundation of China (Grant 30671035) and National Basic Sciences Research Programs (Grant 2007CB947203). We thank Drs. E. Bach, A. Baonza, H. Bellen, C. Doe, N. Dyson, B. Edgar, M. Freeman, K. Irvine, Y.- N. Jan, J. Knoblich, C.-Y. Lee, J. Skeath, G. Struhl, T. Tabata, Z. Wang, and B. Zhou for antibodies and fly stocks; the Bloomington Stock Center, National Institute of Genetics stock center (Japan), Drosophila Genetic Resource Center, Vienna Drosophila RNAi Center, and the Tsinghua stock center (China) for fly stocks; and the Developmental Studies Hybridoma Bank for antibodies. We thank H. Wang, who helped in the initial screen to identify genes that affect optic lobe development.
References
- Artegiani B, Lindemann D, Calegari F. Overexpression of cdk4 and cyclinD1 triggers greater expansion of neural stem cells in the adult mouse brain. J Exp Med. 2011;208:937–948. doi: 10.1084/jem.20102167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124:1241–1253. doi: 10.1016/j.cell.2006.01.038. [DOI] [PubMed] [Google Scholar]
- Binz SK, Sheehan AM, Wold MS. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair. 2004;3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- Bochkarev A, Bochkareva E. From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr Opin Struct Biol. 2004;14:36–42. doi: 10.1016/j.sbi.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Brand AH, Livesey FJ. Neural stem cell biology in vertebrates and invertebrates: more alike than different? Neuron. 2011;70:719–729. doi: 10.1016/j.neuron.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Calegari F, Huttner WB. An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci. 2003;116:4947–4955. doi: 10.1242/jcs.00825. [DOI] [PubMed] [Google Scholar]
- Calegari F, Haubensak W, Haffner C, Huttner WB. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci. 2005;25:6533–6538. doi: 10.1523/JNEUROSCI.0778-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datar SA, Jacobs HW, de la Cruz AF, Lehner CF, Edgar BA. The Drosophila cyclin D-Cdk4 complex promotes cellular growth. EMBO J. 2000;19:4543–4554. doi: 10.1093/emboj/19.17.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Pompa JL, Wakeham A, Correia KM, Samper E, Brown S, Aguilera RJ, Nakano T, Honjo T, Mak TW, Rossant J, Conlon RA. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development. 1997;124:1139–1148. doi: 10.1242/dev.124.6.1139. [DOI] [PubMed] [Google Scholar]
- Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci. 2007;8:438–450. doi: 10.1038/nrn2097. [DOI] [PubMed] [Google Scholar]
- Dehay C, Savatier P, Cortay V, Kennedy H. Cell-cycle kinetics of neocortical precursors are influenced by embryonic thalamic axons. J Neurosci. 2001;21:201–214. doi: 10.1523/JNEUROSCI.21-01-00201.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CQ. Neural stem cells: balancing self-renewal with differentiation. Development. 2008;135:1575–1587. doi: 10.1242/dev.014977. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Brook A, Dembski M, Yenush L, Dyson N. DNA-binding and trans-activation properties of Drosophila E2F and DP proteins. Proc Natl Acad Sci U S A. 1994;91:6359–6363. doi: 10.1073/pnas.91.14.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger B, Boone JQ, Stevens NR, Brand AH, Doe CQ. Regulation of spindle orientation and neural stem cell fate in the Drosophila optic lobe. Neural Dev. 2007;2:1. doi: 10.1186/1749-8104-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger B, Gold KS, Brand AH. Notch regulates the switch from symmetric to asymmetric neural stem cell division in the Drosophila optic lobe. Development. 2010;137:2981–2987. doi: 10.1242/dev.051250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekas LA, Cardozo TJ, Flaherty MS, McMillan EA, Gonsalves FC, Bach EA. Characterization of a dominant-active STAT that promotes tumorigenesis in Drosophila. Dev Biol. 2010;344:621–636. doi: 10.1016/j.ydbio.2010.05.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich J, Meyer CA, de la Cruz AF, Edgar BA, Lehner CF. Cyclin D does not provide essential Cdk4-independent functions in Drosophila. Genetics. 2004;168:867–875. doi: 10.1534/genetics.104.027417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers J, Theil AF, Baldeyron C, van Cappellen WA, Houtsmuller AB, Kanaar R, Vermeulen W. Nuclear dynamics of PCNA in DNA replication and repair. Mol Cell Biol. 2005;25:9350–9359. doi: 10.1128/MCB.25.21.9350-9359.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frise E, Knoblich JA, Younger-Shepherd S, Jan LY, Jan YN. The Drosophila Numb protein inhibits signaling of the Notch receptor during cell–cell interaction in sensory organ lineage. Proc Natl Acad Sci U S A. 1996;93:11925–11932. doi: 10.1073/pnas.93.21.11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay L, Seger R, Shilo BZ. In situ activation pattern of Drosophila EGF receptor pathway during development. Science. 1997;277:1103–1106. doi: 10.1126/science.277.5329.1103. [DOI] [PubMed] [Google Scholar]
- Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Grandbarbe L, Bouissac J, Rand M, Hrabé de Angelis M, Artavanis-Tsakonas S, Mohier E. Delta-Notch signaling controls the generation of neurons/glia from neural stem cells in a stepwise process. Development. 2003;130:1391–1402. doi: 10.1242/dev.00374. [DOI] [PubMed] [Google Scholar]
- Graves HK, Woodfield SE, Yang CC, Halder G, Bergmann A. Notch signaling activates Yorkie non-cell autonomously in Drosophila. PLoS One. 2012;7:e37615. doi: 10.1371/journal.pone.0037615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MA, Akong K, Peifer M. Novel roles for APC family members and Wingless/Wnt signaling during Drosophila brain development. Dev Biol. 2007;305:358–376. doi: 10.1016/j.ydbio.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia AJ, Nye JS, Conlon RA, Mak TW, Bernstein A, van der Kooy D. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002;16:846–858. doi: 10.1101/gad.975202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdlicka L, Gibson M, Kiger A, Micchelli C, Schober M, Schöck F, Perrimon N. Analysis of twenty-four Gal4 lines in Drosophila melanogaster. Genesis. 2002;34:51–57. doi: 10.1002/gene.10125. [DOI] [PubMed] [Google Scholar]
- Iftode C, Daniely Y, Borowiec JA. Replication protein A (RPA): the eukaryotic SSB. Crit Rev Biochem Mol Biol. 1999;34:141–80. doi: 10.1080/10409239991209255. [DOI] [PubMed] [Google Scholar]
- Knoblich JA. Mechanisms of asymmetric stem cell division. Cell. 2008;132:583–597. doi: 10.1016/j.cell.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Knoblich JA, Lehner CF. Synergistic action of Drosophila cyclins A and B during the G2-M transition. EMBO J. 1993;12:65–74. doi: 10.1002/j.1460-2075.1993.tb05632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, Lehner CF. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell. 1994;77:107–120. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- Kramatschek B, Campos-Ortega JA. Neuroectodermal transcription of the Drosophila neurogenic genes E(spl) and HLH-m5 is regulated by proneural genes. Development. 1994;120:815–826. doi: 10.1242/dev.120.4.815. [DOI] [PubMed] [Google Scholar]
- Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell. 2009;5:320–331. doi: 10.1016/j.stem.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Lehner CF, O'Farrell PH. The roles of Drosophila cyclins A and B in mitotic control. Cell. 1990;61:535–547. doi: 10.1016/0092-8674(90)90535-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist A, Rodríguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz A, Savatier P, Cortay V, Kennedy H, Dehay C. Contrasting effects of basic fibroblast growth factor and neurotrophin 3 on cell cycle kinetics of mouse cortical stem cells. J Neurosci. 2002;22:6610–6622. doi: 10.1523/JNEUROSCI.22-15-06610.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manseau L, Baradaran A, Brower D, Budhu A, Elefant F, Phan H, Philip AV, Yang M, Glover D, Kaiser K, Palter K, Selleck S. GAL4 enhancer traps expressed in the embryo, larval brain, imaginal discs, and ovary of Drosophila. Dev Dynamics. 1997;209:310–322. doi: 10.1002/(SICI)1097-0177(199707)209:3<310::AID-AJA6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- McCleland ML, Farrell JA, O'Farrell PH. Influence of cyclin type and dose on mitotic entry and progression in the early Drosophila embryo. J Cell Biol. 2009;184:639–646. doi: 10.1083/jcb.200810012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinertzhagen IA, Hanson TE. The development of the optic lobe. In: Bate M, Martinez-Arias A, editors. The development of Drosophila melanogaster. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 1993. pp. 1363–1491. [Google Scholar]
- Meyer CA, Jacobs HW, Datar SA, Du W, Edgar BA, Lehner CF. Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 2000;19:4533–4542. doi: 10.1093/emboj/19.17.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassif C, Noveen A, Hartenstein V. Early development of the Drosophila brain: III. The pattern of neuropile founder tracts during the larval period. J Comp Neurol. 2003;455:417–434. doi: 10.1002/cne.10482. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Ngo KT, Wang J, Junker M, Kriz S, Vo G, Asem B, Olson JM, Banerjee U, Hartenstein V. Concomitant requirement for Notch and Jak/Stat signaling during neuro-epithelial differentiation in the Drosophila optic lobe. Dev Biol. 2010;346:284–295. doi: 10.1016/j.ydbio.2010.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orihara-Ono M, Toriya M, Nakao K, Okano H. Downregulation of Notch mediates the seamless transition of individual Drosophila neuroepithelial progenitors into optic medullar neuroblasts during prolonged G1. Dev Biol. 2011;351:163–175. doi: 10.1016/j.ydbio.2010.12.044. [DOI] [PubMed] [Google Scholar]
- Peter A, Schöttler P, Werner M, Beinert N, Dowe G, Burkert P, Mourkioti F, Dentzer L, He Y, Deak P, Benos PV, Gatt MK, Murphy L, Harris D, Barrell B, Ferraz C, Vidal S, Brun C, Demaille J, Cadieu E, et al. Mapping and identification of essential gene functions on the X chromosome of Drosophila. Supplementary material. EMBO Rep. 2002;3:34–38. doi: 10.1093/embo-reports/kvf012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilaz LJ, Patti D, Marcy G, Ollier E, Pfister S, Douglas RJ, Betizeau M, Gautier E, Cortay V, Doerflinger N, Kennedy H, Dehay C. Forced G1-phase reduction alters mode of division, neuron number, and laminar phenotype in the cerebral cortex. Proc Natl Acad Sci U S A. 2009;106:21924–21929. doi: 10.1073/pnas.0909894106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy BV, Rauskolb C, Irvine KD. Influence of fat-hippo and notch signaling on the proliferation and differentiation of Drosophila optic neuroepithelia. Development. 2010;137:2397–2408. doi: 10.1242/dev.050013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somma MP, Ceprani F, Bucciarelli E, Naim V, De Arcangelis V, Piergentili R, Palena A, Ciapponi L, Giansanti MG, Pellacani C, Petrucci R, Cenci G, Vernì F, Fasulo B, Goldberg ML, Di Cunto F, Gatti M. Identification of Drosophila mitotic genes by combining co-expression analysis and RNA interference. PLoS Genet. 2008;4:e1000126. doi: 10.1371/journal.pgen.1000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Basler K. Organizing activity of wingless protein in Drosophila. Cell. 1993;72:527–540. doi: 10.1016/0092-8674(93)90072-x. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 1995;15:6046–6057. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai P, Truong B, Bhat KM. Cell division genes promote asymmetric interaction between Numb and Notch in the Drosophila CNS. Development. 1999;126:2759–2770. doi: 10.1242/dev.126.12.2759. [DOI] [PubMed] [Google Scholar]
- Wang W, Liu W, Wang Y, Zhou L, Tang X, Luo H. Notch signaling regulates neuroepithelial stem cell maintenance and neuroblast formation in Drosophila optic lobe development. Dev Biol. 2011a;350:414–428. doi: 10.1016/j.ydbio.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Wang W, Li Y, Zhou L, Yue H, Luo H. Role of JAK/STAT signaling in neuroepithelial stem cell maintenance and proliferation in the Drosophila optic lobe. Biochem Biophys Res Commun. 2011b;410:714–720. doi: 10.1016/j.bbrc.2011.05.119. [DOI] [PubMed] [Google Scholar]
- Weng M, Haenfler JM, Lee CY. Changes in Notch signaling coordinates maintenance and differentiation of the Drosophila larval optic lobe neuroepithelia. Dev Neurobiol. 2012;72:1376–1390. doi: 10.1002/dneu.20995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Yasugi T, Umetsu D, Murakami S, Sato M, Tabata T. Drosophila optic lobe neuroblasts triggered by a wave of proneural gene expression that is negatively regulated by JAK/STAT. Development. 2008;135:1471–1480. doi: 10.1242/dev.019117. [DOI] [PubMed] [Google Scholar]
- Yasugi T, Sugie A, Umetsu D, Tabata T. Coordinated sequential action of EGFR and Notch signaling pathways regulates proneural wave progression in the Drosophila optic lobe. Development. 2010;137:3193–3203. doi: 10.1242/dev.048058. [DOI] [PubMed] [Google Scholar]
- Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci. 2005;8:709–715. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]