

Abstract
Cabotegravir is an investigational integrase inhibitor in development for the treatment and pre‐exposure prophylaxis of HIV‐1 infection. Liver disease is a major cause of morbidity and mortality in HIV‐infected individuals and can impact the pharmacokinetics (PK) of HIV medications. This phase 1 study evaluated the PK of cabotegravir in individuals with moderate hepatic impairment (n = 8) versus healthy controls (n = 8). Participants received a single oral cabotegravir 30‐mg tablet and underwent PK sampling to determine total and unbound plasma cabotegravir concentrations. Calculated geometric least‐squares mean ratios (90% confidence intervals) for individuals with hepatic impairment versus healthy controls were 0.73 (0.50‐1.06) for AUC0‐∞, 0.69 (0.51‐0.93) for Cmax, 1.40 (0.80‐2.46) for unbound concentration (CU) 2 hours postdose, 1.55 (0.82‐2.94) for CU at 24 hours, 2.14 (1.57‐2.90) for unbound fraction (FU) at 2 hours, and 1.90 (1.14‐3.18) for FU at 24 hours. Adverse events (AEs) occurred in 2 individuals with hepatic impairment and 3 healthy controls and were grade 1/2 in severity. No participant discontinued because of AEs. Increased FU resulted in a modest decrease in total plasma exposure not considered clinically relevant. We conclude that cabotegravir may be administered without dose adjustment in patients with mild to moderate hepatic impairment.
Keywords: cabotegravir, hepatic impairment, pharmacokinetics, protein binding
Cabotegravir is a potent integrase strand transfer inhibitor in clinical development for the treatment and pre‐exposure prophylaxis (PrEP) of HIV‐1 infection with both oral tablet and long‐acting injectable formulations.1, 2, 3 The long‐acting injectable formulation is being developed as monthly or bimonthly treatments for the maintenance of viral suppression in patients infected with HIV‐1 and as PrEP in at‐risk individuals.4 Oral cabotegravir is being developed as lead‐in therapy to assess safety and tolerability prior to switching to the long‐acting injectable regimen as well as an oral bridging therapy for instances when monthly or bimonthly injections may be missed. The long‐acting injectable formulation of cabotegravir, supported by oral tablet lead‐in and bridging therapy, may provide an alternative therapeutic option for both HIV treatment and PrEP in populations that may find it challenging to adhere to a daily oral regimen. Both oral and long‐acting formulations of cabotegravir had low rates of adverse event–related discontinuations in clinical trials.1, 3
Oral cabotegravir is rapidly absorbed and has a long apparent plasma half‐life of 38.5 hours following single‐dose administration in healthy individuals.5 Oral cabotegravir 30 mg once daily achieves concentrations at the end of the dosing interval following repeat administration that exceed the protein‐binding‐adjusted 90% viral inhibition constant by 20‐fold.6 Findings from a pharmacokinetic (PK) study of single‐dose, long‐acting cabotegravir showed that once‐monthly or even less frequent dosing was well tolerated and generally safe for both intramuscular and subcutaneous routes of administration.4 The terminal phase half‐life (t1/2) after administration of long‐acting cabotegravir reflects absorption and ranges from approximately 25 to 54 days.4 In vitro, cabotegravir is metabolized in the liver by phase II glucuronidation via the uridine diphosphoglucuronosyl transferase (UGT) pathway primarily by UGT1A1 and minor involvement of UGT1A9.7 The absolute bioavailability of cabotegravir has not been determined, but the low intrinsic clearance values for the glucuronide metabolite (M1) formation by recombinant UGT enzymes 1A1 and 1A9 (4.5 and 2.2 µL/min/mg, respectively) suggest that cabotegravir is a very low extraction‐ratio drug. In humans, oral [14C]cabotegravir (dose, 28.2 mg/kg) is excreted in feces (mean, 58.5% recovered; range, 46.9%‐72.7% recovered) and urine (mean, 26.8% recovered; range, 17.4%‐41.2% recovered). Following a single dose of [14C]cabotegravir, cabotegravir is detected in human feces (mean, 46.8% dose), and M1 is detected in urine (mean, 20.0% dose).
Among individuals infected with HIV‐1, liver disease is a major cause of mortality and morbidity and accounts for 14% to 18% of all HIV‐associated deaths.8, 9 Predisposing factors for hepatic impairment associated with HIV infection include chronic hepatitis B virus (HBV) infection, hepatitis C virus (HCV) infection, or both; drug‐induced liver injury; nonalcoholic fatty liver disease; and alcohol abuse.9 Liver disease can impact liver blood flow and modify synthesis of plasma proteins and drug metabolism, which may influence the PK of antiretroviral therapy.10
Given the primary route of elimination of cabotegravir via UGT metabolism, it is important to characterize the effect of hepatic impairment on cabotegravir PK. Cabotegravir is highly bound to plasma proteins with less than 1% free drug in circulation and is highly bound to albumin.1, 6, 11 Hepatic impairment is commonly associated with reductions in serum albumin concentrations that may impact cabotegravir protein binding, resulting in increases in free drug concentrations, variable distribution, and alterations in cabotegravir elimination.12 For individuals with hepatic impairment, a comorbidity for which free circulating drug may be greatly increased, the usual therapeutic window or level of toxicity of any drug should be determined.13 The purpose of this study was to evaluate the impact of moderate hepatic impairment on the PK parameters and safety of cabotegravir.
Methods
Study Design
A phase 1 open‐label, parallel‐group, single‐dose adaptive study was designed to evaluate the impact of moderate hepatic impairment on cabotegravir PK. The study was conducted at DaVita Clinical Research (Lakewood, Colorado, and Minneapolis, Minnesota) and Orlando Clinical Research Center (Orlando, Florida) in 16 adults: 8 with moderate hepatic impairment categorized by the Child‐Pugh classification system (Child‐Pugh score, 7‐9) and 8 healthy controls with normal hepatic function (Child‐Pugh score < 5) matched by sex, age (±10 years), and body mass index (BMI, ±25%) at 3 centers in the United States from June 22, 2015, to September 16, 2016. A single oral dose of cabotegravir 30 mg was administered in the fasted state to all participants, who underwent serial PK sampling to assess PK parameters. A follow‐up visit occurred 10 to 14 days after cabotegravir was administered.
The primary end point was comparison of cabotegravir area under the concentration‐time curve from time 0 extrapolated to infinity (AUC0‐∞) and the maximum observed concentration (Cmax) among participants with moderate hepatic impairment and matched healthy controls. Secondary end points included assessments of plasma protein binding, unbound concentration of cabotegravir, unbound fraction in plasma of cabotegravir at 2 and 24 hours postdose, AUC from time 0 to the last time of quantifiable concentration (AUC0‐t), the percentage of AUC0‐∞ obtained by extrapolation (%AUCex), concentration at 24 hours postdose (C24), cabotegravir plasma t1/2, apparent clearance following oral dosing, lag time before observation of drug concentrations (tlag), time of occurrence of Cmax (tmax), apparent volume of distribution, and safety parameters, including the monitoring of adverse events (AEs), clinical laboratory tests, vital signs, and findings on electrocardiography (ECG) and physical examinations.
The study was compliant with the principles stated in the Declaration of Helsinki. Integreview Institutional Review Board (Austin, Texas) approved the research protocol, and the study was conducted at 3 US clinical sites: Lakewood, Colorado; Orlando, Florida; and Minneapolis, Minnesota. Written informed consent was obtained from all individuals prior to initiation of the study (Clinicaltrials.gov identifier, NCT02354950; ViiV‐clinicalstudyregister.com identifier, 201479).
Selection of Study Participants
Adults aged ≥18 and ≤70 years with a body weight of ≥50 kg and a BMI between 19.0 and 41.0 kg/m2 were enrolled in the study. Women were eligible for enrollment if they were not pregnant, lactating, or of reproductive potential. Matched control participants were determined by a health‐care professional to be healthy based on findings from a physical examination, medical history, laboratory values, and ECG. Participants with moderate hepatic impairment were included if they were clinically stable for 1 month prior to screening, had a Child‐Pugh score of 7 to 9, and had confirmation on liver biopsy or medical imaging modality of cirrhosis associated with an unambiguous history (eg, evidence of portal hypertension). Key exclusion criteria for all individuals were: cardiac abnormalities, a positive HIV antibody test result, history of regular alcohol consumption ≤ 6 months prior to the study, and a positive result on drug/alcohol screening. Other exclusion criteria included evidence of recent infection (duration < 6 months) with HBV, HCV, or both; history of cholecystectomy and inflammatory bowel disease; presence of hepatopulmonary or hepatorenal syndrome; presence of primary cholestatic liver diseases; and history of liver transplant. Concomitant medication was considered on a case‐by‐case basis and permitted if it did not jeopardize the interpretation of the data or safety of the study participant. Doses of acetaminophen (≤2 g/day) were permitted for use in healthy volunteers only. Unless a concomitant medication was considered by the investigator not to interfere with the study, participants were required to abstain from potential enzyme inhibitors within 7 days, for 14 days if taking potential enzyme inducers, or for 5 half‐lives (whichever was longer) prior to the first dose of study medication until completion of the follow‐up visit. In addition, the following were withheld on the day of study dosing: antacids, vitamins, calcium and iron supplements, and other medications containing polyvalent cations or with the ability to interfere with gastric absorption.
Bioanalytical Methods
Cabotegravir (30 mg) was administered orally under fasted conditions with serial plasma PK samples collected predose and 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, 72, 120, and 168 hours postdose. Two and 24 hours after the dose of cabotegravir, plasma samples were collected to determine bound and unbound plasma cabotegravir concentrations. Cabotegravir plasma concentrations were determined using a validated analytical method previously published.7
Briefly, samples of plasma were extracted by protein precipitation with acetonitrile containing the internal standard, [13C2H2 15N]‐cabotegravir. An aliquot (2‐7 µL) was injected onto a Luna C18(2)‐HST column (Phenomonex, Torrance, California) and eluted with a mobile phase (A: 0.4% formic acid in water; B: 25 mM ammonium formate in acetonitrile:water [80:20, v/v]) using the following gradient: 0.0 to 0.3 minutes, 40% B; 0.3 to 2.0 minutes, 40% to 90% B; and 2.0 to 2.5 minutes, 90% B. A Sciex API‐4000 mass spectrometer (AB Sciex, Framingham, Massachusetts) with an electrospray source that employed positive‐ion mode and multiple‐reaction monitoring detected the analytes (cabotegravir transition mass‐to‐charge ratio, 406.0‐263.0; internal standard transition mass‐to‐charge ratio, 410.1‐263.0). Cabotegravir plasma samples were processed by Covance Laboratories (Madison, Wisconsin). The linear range of detection for cabotegravir from a 25‐µL plasma sample was 25 to 25 000 ng/mL.
Plasma protein binding of cabotegravir was determined in quadruplicate by equilibrium dialysis using rapid equilibrium dialysis (RED) device inserts (membrane with a molecular weight cutoff of 8000 Da) and base plate from Thermo Fisher Scientific (Rockford, Illinois). Fortified plasma (300 µL) was added to each donor side of the RED device inserts, and blank plasma ultrafiltrate (PUF; 500 µL) was added to each receiver side of the RED device inserts. Samples were then incubated at 37°C in a chamber containing 5% CO2 with relative humidity maintained at 95% and rotated at 500 rpm for 5 hours. After incubation, plasma and dialysate from each device insert were transferred to a 96‐well plate. A 20‐µL sample from the donor side of each RED device was diluted with 180 µL of blank PUF (10× dilution) prior to analysis. A 180‐µL sample from the receiver side of each RED device was diluted with 20 µL of blank plasma (1.11× dilution) prior to analysis. All PUF samples were analyzed with a validated analytical method using high‐performance liquid chromatography with tandem‐mass spectrometry. The linear range of detection for cabotegravir from a 50‐µL sample of PUF was 1 to 2500 ng/mL.
Each of the plasma samples for protein binding were dialyzed in quadruplicate and reported as cabotegravir total plasma concentration (4 records) and cabotegravir unbound plasma (4 records). For each participant, mean total plasma concentration and mean unbound concentration were calculated. Total and unbound cabotegravir concentrations at 2 and 24 hours by cohort were summarized.
Experimental acceptance criteria for determining the concentration of plasma cabotegravir as well as bound and unbound plasma cabotegravir concentrations were as follows. Quality‐control samples containing cabotegravir at 3 concentrations were analyzed with each batch of participant samples and compared against separately prepared calibrated standards. For the analysis to be acceptable, no more than one‐third of the quality‐control results could deviate >15% from the nominal concentration, and ≥50% of the results from each quality‐control concentration had to be within 15% of the nominal concentration.
Pharmacokinetic Assessments
Plasma cabotegravir concentration‐time data were analyzed by noncompartmental PK analysis with Phoenix WinNonlin version 6.3 or higher (Certara, Princeton, New Jersey). With sex and treatment group as fixed effects and age and BMI as continuous variables, log‐transformed PK parameters, except %AUCex, tmax, and tlag, were analyzed by analysis of covariance. The point estimates of geometric least‐squares mean ratios of the hepatic impairment group compared with healthy controls and associated 90% confidence intervals (CIs) for comparisons for cabotegravir for each PK parameter were generated by a mixed‐linear model within the SAS/STAT module of SAS (version 9.3 or higher; SAS Institute, Cary, North Carolina). The PK parameters for Tmax, tlag, and %AUCex were summarized descriptively. The unbound fraction (fu) was calculated using the total and unbound plasma concentrations of cabotegravir generated 2 and 24 hours postdose in the following formula,
where C(unbound) and C(total) are the unbound and total concentrations of cabotegravir in plasma, respectively. The PK concentration population included all participants in the study who had evaluable cabotegravir assays following plasma PK sampling.
The relationship between plasma cabotegravir PK primary and secondary end points and liver function measurements that included Child‐Pugh score (overall score and liver synthetic ability [albumin, bilirubin, and prothrombin time]) was assessed by Pearson correlation and linear and/or nonlinear regression methods. For the purpose of Pearson correlation, parameters of hepatic function, including total Child‐Pugh, serum albumin, and serum bilirubin scores and prothrombin time (international normalized ratio), were treated as continuous variables, and participants with normal hepatic function were considered to have a score of 0.
Safety Assessments
Safety assessments included assessments of vital signs, ECG findings, clinical laboratory tests, and monitoring of AEs. Assessments were continued through the 8 days of the trial and the follow‐up period (10‐14 days postdose).
Results
Study Disposition
Sixteen adults were enrolled, including 8 participants with moderate hepatic impairment and 8 healthy matched controls; all completed the study. Most study participants were male (75%) and Caucasian (identifying as white or of European heritage; 75%) with a mean age ± standard deviation (SD) and mean BMI ± SD of 58.6 ± 5.1 years and 29.2 ± 3.9 kg/m2, respectively (Table 1). Demographic and baseline characteristics for both treatment groups were similar. In participants with hepatic impairment, 75% had Child‐Pugh scores of 7 and 8 (n = 3 each), and 25% had Child‐Pugh scores of 9 (n = 2). Because the Child‐Pugh scoring system is based on assessments (ie, clinical and biochemical assessments of encephalopathy, ascites, serum bilirubin and albumin levels, and international normalized ratio) not entirely specific to liver disease, all participants with moderate hepatic impairment also exhibited clinical evidence of chronic liver disease, cirrhosis, or both conditions. Seven participants had a history of ascites, 4 participants had a history of alcohol abuse or alcohol‐induced cirrhosis, 4 participants had a history of chronic HCV infection (duration > 6 months), and 3 participants had a history of hepatic encephalopathy. One participant from each treatment group had 1 protein‐binding sample lost during shipment prior to analysis; therefore, no data were generated for those 2 individuals.
Table 1.
Demographics of Study Participants
| Moderate Hepatic Impairment | Matched Healthy Controlsa | Total | |
|---|---|---|---|
| Demographic | (n = 8) | (n = 8) | (n = 16) |
| Sex, n (%) | |||
| Female | 2 (25) | 2 (25) | 4 (25) |
| Male | 6 (75) | 6 (75) | 12 (75) |
| Age (y), mean (SD) | 60.3 (3.20) | 56.9 (6.17) | 58.6 (5.06) |
| BMI (kg/m2), mean (SD) | 29.3 (3.78) | 29.2 (4.17) | 29.2 (3.85) |
| Race/ethnicity, n (%) | |||
| Black/African American | 2 (25) | 2 (25) | 4 (25) |
| White/Caucasian/European heritage | 6 (75) | 6 (75) | 12 (75) |
| Child‐Pugh total score, n (%) | |||
| 7 | 3 (37.5) | — | — |
| 8 | 3 (37.5) | — | — |
| 9 | 2 (25.0) | — | — |
| Plasma albumin (g/L), mean (SD) | 36.0 (6.5) | 43.3 (5.1) | — |
| AAG (g/L), mean (SD) | 0.73 (0.45) | 0.92 (0.15) | — |
| ALP (IU/L), mean (SD) | 141.5 (73) | 87.9 (16) | — |
| ALT (IU/L), mean (SD) | 45.5 (27) | 21.9 (5.2) | — |
| AST (IU/L), mean (SD) | 61.3 (32) | 24.1 (4.4) | — |
| Direct bilirubin (µM), mean (SD) | 12.4 (6.6) | 3.4 (0.9) | — |
| Bilirubin (µM), mean (SD) | 24.4 (10.6) | 9.4 (2.9) | — |
All laboratory values reported were conducted at screening (30 days prior to first dose).
AAG, alpha‐1 acid glycoprotein; ALT, alanine aminotransferase; ALP, alkaline phosphatase; AST, aspartate aminotransferase; BMI, body mass index; SD, standard deviation.
Healthy controls were matched to participants with hepatic impairment by sex, age (±10 years), and BMI (±25%).
Pharmacokinetics
Maximum plasma concentrations of cabotegravir were achieved 2 hours postdose with a plasma t1/2 of 32 hours in participants with moderate hepatic impairment and 38 hours in healthy controls (Table 2; Figure 1A). Geometric mean oral clearance in participants with moderate hepatic impairment was 0.30 L/h, 38% higher than in matched controls at 0.24 L/h. As a result, the geometric means (95%CIs) of plasma AUC0‐∞ and Cmax were 102 µg·h/mL and 2.70 µg/mL, respectively, in participants with moderate hepatic impairment compared with 127 µg·h/mL and 3.55 µg/mL, respectively, in the matched healthy controls (Table 2).
Table 2.
Summary of Select Cabotegravir Pharmacokinetic Parameters
| Moderate Hepatic Impairment | Matched Healthy Controls | ||||
|---|---|---|---|---|---|
| (n = 8) | (n = 8)a | ||||
| Pharmacokinetic Parameter | Mean (SD) | Geometric Mean (95%CI) [CVb%] | Mean (SD) | Geometric Mean (95%CI) [CVb%] | GLS Mean Ratio (90%CI) |
| AUC0‐∞, µg·h/mL | 108 (38.8) | 102 (75.2‐138) [37.3] | 134 (47.4) | 127 (94.7‐170) [36.2] | 0.73 (0.50‐1.06) |
| AUC0‐t, µg·h/mL | 104 (35.8) | 98.2 (73.3‐132) [36.2] | 128 (44.2) | 121 (91.0‐162) [35.4] | 0.73 (0.51‐1.05) |
| Cmax, µg/mL | 2.88 (1.04) | 2.70 (1.94‐3.76) [41.1] | 3.64 (0.883) | 3.55 (2.90‐4.33) [24.3] | 0.69 (0.51‐0.93) |
| C24, µg/mL | 1.28 (0.377) | 1.23 (0.956‐1.58) [30.8] | 1.59 (0.564) | 1.50 (1.13‐2.01) [35.6] | 0.73 (0.53‐1.02) |
| t1/2, h | 32.1 (9.37) | 30.8 (23.7‐40.1) [32.2] | 37.5 (4.95) | 37.2 (33.4‐41.5) [13.1] | 0.82 (0.65‐1.04) |
| CL/F, L/h | 0.312 (0.110) | 0.30 (0.218‐0.399) [37.3] | 0.249 (0.085) | 0.24 (0.176‐0.317) [36.2] | 1.38 (0.95‐2.01) |
| CU2H, µg/mL | 0.00948 (0.00661) | 0.0074 | 0.00589 (0.00227) | 0.0053 | 1.40 (0.80‐2.46) |
| CU24H, µg/mL | 0.00501 (0.00317) | 0.0041 | 0.00266 (0.00107) | 0.0026 | 1.55 (0.82‐2.94) |
| FU2H, %b | 0.338 (0.172) | 0.31 (0.202‐0.467) [47.6] | 0.164 (0.050) | 0.16 (0.119‐0.207) [30.5] | 2.14 (1.57‐2.90) |
| FU24H, %b | 0.384 (0.286) | 0.32 (0.184‐0.564) [66.6] | 0.170 (0.039) | 0.17 (0.134‐0.207) [23.9] | 1.90 (1.14‐3.18) |
AUC, area under the concentration‐time curve; AUC0‐∞, AUC from time 0 extrapolated to infinity; AUC0‐t, AUC from time 0 to last quantifiable concentration; C24, concentration 24 hours postdose; CI, confidence interval; CL/F, apparent oral clearance; Cmax, maximum observed plasma concentration; CU2H, unbound concentration at 2 hours; CU24H, unbound concentration at 24 hours; CV, coefficient of variation; CVb, between‐participant coefficient of variation; FU2H, unbound fraction at 2 hours; FU24H, unbound fraction at 24 hours; GLS, geometric least squares; SD, standard deviation; t1/2, terminal elimination phase half‐life.
Healthy controls were matched to participants with hepatic impairment by sex, age (±10 years), and body mass index (±25%).
Protein‐binding samples from 1 participant from each treatment group were not received for analysis (n = 7).
Figure 1.
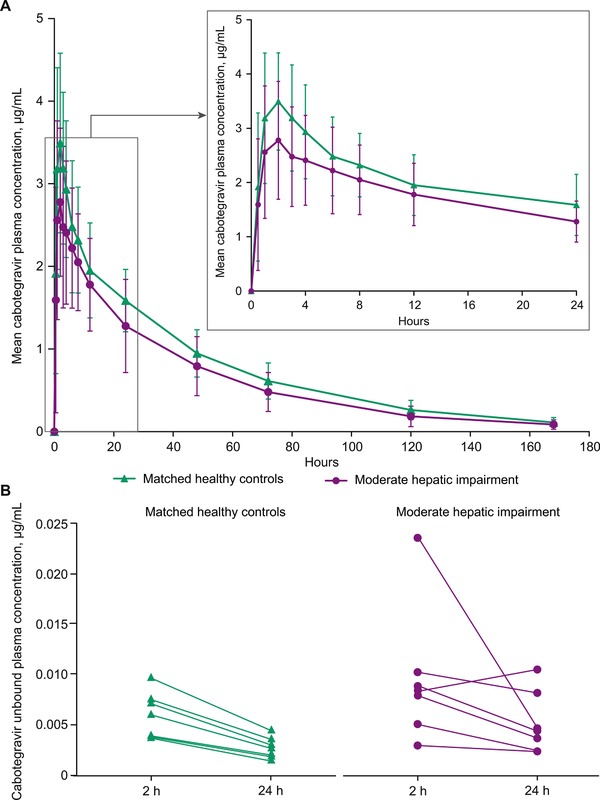
Mean ± standard deviation of (A) total cabotegravir plasma concentrations from 0 to 180 hours after dosing and inset, highlighting 0 to 24 hours, and (B) mean unbound cabotegravir plasma concentrations 2 and 24 hours after dosing.
The geometric mean of unbound plasma cabotegravir concentrations 2 and 24 hours postdose in participants with moderate hepatic impairments were 40% and 55% higher, respectively, than those of the matched healthy controls (Table 2; Figure 1B). Furthermore, the geometric mean free fraction of cabotegravir in participants with moderate hepatic impairment was 0.31% 2 hours postdose and 0.32% 24 hours postdose compared with 0.16% 2 hours postdose and 0.17% 24 hours postdose in the matched healthy controls (Table 2). From 2 and 24 hours postdose, the unbound fraction of cabotegravir remained stable across the plasma cabotegravir peak‐to‐trough concentration range. According to Pearson correlation between cabotegravir PK parameters and hepatic function variables (Child‐Pugh total score; serum albumin and bilirubin values; international normalized ratio; and concentrations of serum albumin, total bilirubin, α‐1 acid glycoprotein, and total protein), unbound cabotegravir fractions 2 and 24 hours postdose were negatively correlated with serum albumin concentrations (P < .001 and P = .016, respectively), indicating that as the lower the albumin concentration decreases, the fraction unbound increases (Figure 2). A correlation was observed between the cabotegravir unbound fraction and total protein 2 hours postdose (P = .009), but no correlation was observed 24 hours postdose (P = .195; Table 3).
Figure 2.
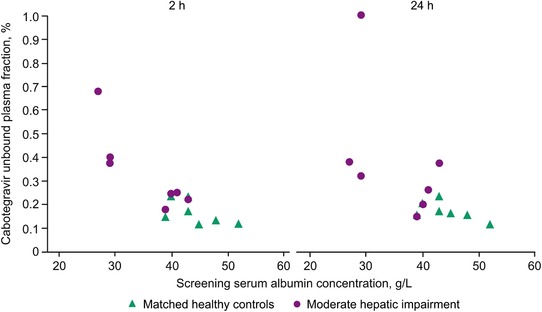
Scatterplot of unbound plasma cabotegravir fraction versus serum albumin concentration at 2 and 24 hours.
Table 3.
Summary of Pearson Correlation Between Plasma Cabotegravir PK Parameters and Hepatic Function
| PK Parameter | n | Hepatic Function Parameter | Pearson Correlation Coefficient | P |
|---|---|---|---|---|
| FU2H, % | 14 | Albumin concentration, g/L | −0.85 | <.001 |
| Total Child‐Pugh score | 0.61 | .021 | ||
| Serum albumin score | 0.91 | <.001 | ||
| Serum bilirubin score | 0.52 | .058 | ||
| PT (INR) | 0.60 | .025 | ||
| Total bilirubin, µmol/L | 0.42 | .132 | ||
| α‐1 Acid glycoprotein, g/L | 0.28 | .337 | ||
| Total protein, g/L | −0.67 | .009 | ||
| FU24H, % | 14 | Albumin concentration, g/L | −0.63 | .016 |
| Total Child‐Pugh score | 0.53 | .049 | ||
| Serum albumin score | 0.60 | .022 | ||
| Serum bilirubin score | 0.33 | .251 | ||
| PT (INR) | 0.49 | .073 | ||
| Total bilirubin, µmol/L | 0.37 | .188 | ||
| α‐1 Acid glycoprotein, g/L | −0.30 | .304 | ||
| Total protein, g/L | −0.37 | .195 |
Hepatic function parameters of total Child‐Pugh score, serum albumin, serum bilirubin, and PT (INR) were treated as continuous variables. Participants with normal hepatic function were considered to have a score of 0. FU2H, unbound fraction at 2 hours; FU24H, unbound fraction at 24 hours; INR, international normalized ratio; PT, prothrombin time.
Safety
Five participants (31%; hepatic impairment, n = 2; healthy controls, n = 3) developed AEs during the study (Table 4). None of the AEs were reported more than once in either treatment group. Two grade 2 AEs, hypertension and upper respiratory tract infection (both from the matched healthy control group), were reported. Three participants had AEs that the investigator considered to be related to the study treatment: 1 participant from the hepatic impairment group reported grade 1 constipation, and 2 participants from the matched healthy control group reported grade 2 upper respiratory infection (n = 1) and grade 1 headache (n = 1). None of the AEs led to withdrawal from the study, and no deaths or serious AEs were reported.
Table 4.
Summary of Adverse Events Among Study Participants
| Moderate Hepatic Impairment | Matched Healthy Controlsa | Overall | |
|---|---|---|---|
| AE, n (%) | (n = 8) | (n = 8) | (n = 16) |
| Participant with any AE | 2 (25) | 3 (38) | 5 (31) |
| Folliculitis | 1 (13) | 0 | 1 (6) |
| Gastroenteritis | 1 (13) | 0 | 1 (6) |
| Upper respiratory tract infectionb | 0 | 1 (13) | 1 (6) |
| Constipationb | 1 (13) | 0 | 1 (6) |
| Hypertensionb | 0 | 1 (13) | 1 (6) |
| Back pain | 0 | 1 (13) | 1 (6) |
| Headacheb | 0 | 1 (13) | 1 (6) |
| Papule | 1 (13) | 0 | 1 (6) |
AE, adverse event.
Healthy controls were matched to participants with hepatic impairment by sex, age (±10 years), and body mass index (±25%).
Considered related to treatment by study investigator.
No clinically significant vital signs or ECG abnormalities were observed in the study. Laboratory abnormalities were observed for 13 participants from the hepatic impairment treatment group (grade 2, n = 9; grade 3, n = 3; grade 4, n = 1) and 1 participant from the control group (grade 3). Grade 3/4 laboratory abnormalities included decreased lymphocyte count (n = 3), increased direct bilirubin (grade 3, n = 1), and increased lipase (grade 3, n = 1). None of these observations were considered clinically significant, and none were reported as AEs.
Discussion
In 2015, an estimated 325 million people were chronically infected with HBV or HCV, and 5% to 25% of persons infected with HIV‐1 were chronically coinfected with HBV, HCV, or both viruses, thereby increasing the likelihood that antiviral therapy could be administered to patients with reduced hepatic function.8, 14 Moreover, nonalcoholic fatty liver disease, an increasingly leading cause of hepatic dysfunction, is becoming an important cause of morbidity in patients with HIV infection as their life expectancy increases.15 Although a paucity of data exists on patients with HIV infection and comorbid nonalcoholic fatty liver disease, this hepatic disease is more prevalent among patients with HIV infection than in the general population, with recent estimates ranging from 13% to 55% in this patient group.15 Other causes of hepatic impairment in people with HIV infection include antiretroviral drug toxicity and alcohol abuse.9 The primary objective of this study was to characterize plasma PK parameters of a single oral dose of cabotegravir in participants with hepatic impairment compared with those observed in healthy controls matched for sex, age, and BMI.
Cabotegravir is metabolized in the liver by phase II glucuronidation via the UGT pathway primarily by UGT1A1 with minor involvement of UGT1A9.7 For individuals with moderate hepatic impairment, the potential for significant changes in cabotegravir exposure is predicted to be low, because cirrhosis does not significantly impact the K m value for glucuronidation of different classes of drugs.16 The results of the present study demonstrate reductions of 27%, 31%, and 27% for total plasma AUC0‐∞, Cmax, and C24, respectively, in participants with hepatic impairment compared with healthy matched controls. The reduction in cabotegravir exposure is equivalent to a one‐third dose reduction to approximately 20 mg (of cabotegravir). When it is coadministered with rilpivirine 25 mg once daily, cabotegravir 10 mg once daily achieves steady‐state trough concentrations 8‐fold above protein‐adjusted 90% inhibitory concentration (PA‐IC90) and demonstrable, durable maintenance of viral suppression in patients with HIV infection without moderate or severe hepatic impairment.1 The PA‐IC90 for cabotegravir when administered with rilpivirine is 0.166 µg/mL. In this study, the observed geometric mean 24‐hour cabotegravir concentration following the administration of a single dose of cabotegravir 30 mg in participants with moderate hepatic impairment was 1.23 µg/mL, 7.4‐fold above the PA‐IC90. Assuming a 2.4‐fold accumulation based on the terminal‐phase t1/2, the steady‐state trough concentration following once‐daily dosing in these individuals is predicted to be 18‐fold above PA‐IC90, consistent with a 20‐mg dose of cabotegravir. Therefore, the exposure differences between groups are not considered clinically relevant, and oral cabotegravir 30 mg can be safely administered to patients with moderate hepatic impairment without dose adjustment.
Cabotegravir is highly bound to serum plasma proteins with less than 1% free drug in circulation.6 Consistent with the pathophysiology of liver disease, reductions in albumin levels were reported in the current study among participants with hepatic impairment.17 Furthermore, positive correlation was observed between higher unbound fractions of cabotegravir and higher albumin scores (lower albumin concentration) and higher Child‐Pugh scores. Although the absolute unbound drug concentrations were 40% to 55% higher in participants with moderate hepatic impairment, the free drug fraction of cabotegravir remained constant over time in participants with moderate hepatic impairment (2 hours, 0.31%; 24 hours, 0.32%) because of a compensatory decrease in total plasma concentrations. This decrease in total plasma concentrations is reflected as an apparent increase in oral clearance and an apparent decrease in AUC0‐∞ in the participants with moderate hepatic impairment and is based on total plasma concentrations.13 Although participants with moderate hepatic impairment had higher exposure to unbound drug, the unbound concentrations remained very low and in the single‐digit ng/mL range. Furthermore, cabotegravir 60 mg once daily, which would be expected to have an approximately 2‐fold higher unbound concentration, was studied in participants with HIV infection for nearly 2 years and had few adverse event‐related discontinuations.1 Thus, the observed increase in exposure to unbound drug in participants with moderate hepatic impairment was not considered clinically relevant.
Moderate hepatic impairment had a similar impact on the free fraction of dolutegravir, a structural analogue of cabotegravir also primarily metabolized by UGT1A1.7, 18, 19, 20 In participants with moderate hepatic impairment, an approximately 2‐fold higher mean unbound fraction (0.44%) was observed 24 hours following a single, 50‐mg oral dose of dolutegravir compared with the unbound fraction (0.23%) detected in healthy individuals. However, unlike cabotegravir, the similar increase in unbound fraction did not result in lower total plasma dolutegravir exposures, which were attributed to decreased intrinsic clearance and impaired metabolizing enzyme capacity.
One study limitation is that there may be differences in the PK characteristics of persons infected with HIV,10 a patient population excluded from this study. In addition, the conclusions of the present study cannot be extrapolated to patients with severe hepatic impairment because the impact of serum albumin concentrations on unbound concentrations of cabotegravir and the resultant plasma exposure are unknown in this population.
In summary, moderate hepatic impairment had modest impact on total plasma cabotegravir PK, even with an approximately 90% to 114% increase of the unbound fraction of cabotegravir. Based on these results, evaluation of cabotegravir PK in mild hepatic impairment is considered unnecessary. As such, cabotegravir may be administered without dose adjustment in patients with mild to moderate hepatic impairment. Although the concentration‐time profile of long‐acting injectable cabotegravir differs from that associated with oral dosing of cabotegravir, systemic clearance is common to both routes of administration.4 Therefore, a similar, modest increase in clearance is expected among participants with hepatic impairment receiving long‐acting cabotegravir. The use of oral or long‐acting cabotegravir in patients with severe hepatic impairment, however, cannot be recommended without further study.
Declaration of Conflicting Interests
J.S.B.S., S.L.F., and K.K.B. are currently employed by GlaxoSmithKline. Y.L. and Z.Z. are employees of PAREXEL International. Z.Z. received grant support from ViiV Healthcare for statistical analysis. A.R.T., C.T., W.R.S., and P.P. are currently employed by ViiV Healthcare and are shareholders in GlaxoSmithKline.
Acknowledgments
All listed authors meet the criteria for authorship set forth by the International Committee of Medical Journal Editors. Editorial assistance was provided under direction of the authors by Leila Strickland and Sherri Damlo, MedThink SciCom, and was funded by ViiV Healthcare.
Funding
This study was funded by ViiV Healthcare.
Author Roles
J.S.B.S. was involved in the analysis and interpretation of the data, drafting the manuscript, critically revising the manuscript for important intellectual content, and approval of the final version of the manuscript for publication. S.L.F. was involved in the design of the study; the acquisition, analysis, and interpretation of the data; critically revising the manuscript for important intellectual content; and approval of the final version of the manuscript for publication. Y.L. was involved in the conception and design of the study, the acquisition and interpretation of the data, critically revising the manuscript for important intellectual content, and approval of the final version of the manuscript for publication. Z.Z. was involved in the analysis of the data. K.K.B. was involved in the acquisition and interpretation of the data, critically revising the manuscript for important intellectual content, and approval of the final version of the manuscript for publication. A.R.T. was involved in the design of the study, the acquisition and interpretation of the data; critically revising the manuscript for important intellectual content, and approval of the final version of the manuscript for publication. C.T. and W.R.S. were involved in the conception and design of the study, interpreting the data, critically revising the manuscript for important intellectual content, and approval of the final version of the manuscript for publication. P.P. was involved in the acquisition and interpretation of the data, drafting the manuscript, critically revising the manuscript for important intellectual content, and approval of the final version of the manuscript for publication.
Fellows of the American College of Clinical Pharmacology: none.
These data were presented in part at IDWeek; October 4–8, 2017; San Diego, CA.
References
- 1. Margolis DA, Brinson CC, Smith GH, et al; LAI116482 study team . Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral‐naive adults with HIV‐1 infection (LATTE): a randomised, phase 2b, dose‐ranging trial. Lancet Infect Dis. 2015;15(10):1145–1155. [DOI] [PubMed] [Google Scholar]
- 2. Margolis DA, Gonzalez‐Garcia J, Stellbrink HJ, et al. Long‐acting intramuscular cabotegravir and rilpivirine in adults with HIV‐1 infection (LATTE‐2): 96‐week results of a randomised, open‐label, phase 2b, non‐inferiority trial. Lancet. 2017;390(10101):1499–1510. [DOI] [PubMed] [Google Scholar]
- 3. Markowitz M, Frank I, Grant RM, et al. Safety and tolerability of long‐acting cabotegravir injections in HIV‐uninfected men (ECLAIR): a multicentre, double‐blind, randomised, placebo‐controlled, phase 2a trial. Lancet HIV. 2017;4(8):e331–e340. [DOI] [PubMed] [Google Scholar]
- 4. Spreen W, Ford SL, Chen S, et al. GSK1265744 pharmacokinetics in plasma and tissue after single‐dose long‐acting injectable administration in healthy subjects. J Acquir Immune Defic Syndr. 2014;67(5):481–486. [DOI] [PubMed] [Google Scholar]
- 5. Ford SL, Sutton K, Lou Y, et al. Effect of rifampin on the single‐dose pharmacokinetics of oral cabotegravir in healthy subjects. Antimicrob Agents Chemother. 2017;61(10):e00487–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spreen W, Min S, Ford SL, et al. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials. 2013;14(5):192–203. [DOI] [PubMed] [Google Scholar]
- 7. Bowers GD, Culp A, Reese MJ, et al. Disposition and metabolism of cabotegravir: a comparison of biotransformation and excretion between different species and routes of administration in humans. Xenobiotica. 2016;46(2):147–162. [DOI] [PubMed] [Google Scholar]
- 8. World Health Organization . Global hepatitis report, 2017. http://apps.who.int/iris/bitstream/10665/255016/1/9789241565455-eng.pdf?ua=1. Accessed August 2, 2018.
- 9. Price JC, Thio CL. Liver disease in the HIV‐infected individual. Clin Gastroenterol Hepatol. 2010;8(12):1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Podany AT, Scarsi KK, Fletcher CV. Comparative clinical pharmacokinetics and pharmacodynamics of HIV‐1 integrase strand transfer inhibitors. Clin Pharmacokinet. 2017;56(1):25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Margolis DA, Boffito M. Long‐acting antiviral agents for HIV treatment. Curr Opin HIV AIDS. 2015;10(4):246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arroyo V, Garcia‐Martinez R, Salvatella X. Human serum albumin, systemic inflammation, and cirrhosis. J Hepatol. 2014;61(2):396–407. [DOI] [PubMed] [Google Scholar]
- 13. Greenblatt DJ, Sellers EM, Koch‐Weser J. Importance of protein binding for the interpretation of serum or plasma drug concentrations. J Clin Pharmacol. 1982;22(5‐6):259–263. [DOI] [PubMed] [Google Scholar]
- 14. Easterbrook P, Johnson C, Figueroa C, Baggaley R. HIV and hepatitis testing: global progress, challenges, and future directions. AIDS Rev. 2016;18(1):3–14. [PubMed] [Google Scholar]
- 15. Tafesh ZH, Verna EC. Managing nonalcoholic fatty liver disease in patients living with HIV. Curr Opin Infect Dis. 2017;30(1):12–20. [DOI] [PubMed] [Google Scholar]
- 16. Furlan V, Demirdjian S, Bourdon O, Magdalou J, Taburet AM. Glucuronidation of drugs by hepatic microsomes derived from healthy and cirrhotic human livers. J Pharmacol Exp Ther. 1999;289(2):1169–1175. [PubMed] [Google Scholar]
- 17. Bernardi M, Maggioli C, Zaccherini G. Human albumin in the management of complications of liver cirrhosis. Crit Care. 2012;16(2):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song IH, Borland J, Savina PM, et al. Pharmacokinetics of single‐dose dolutegravir in HIV‐seronegative subjects with moderate hepatic impairment compared to healthy matched controls. Clin Pharmacol Drug Develop. 2013;2(4):342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Castellino S, Moss L, Wagner D, et al. Metabolism, excretion, and mass balance of the HIV‐1 integrase inhibitor dolutegravir in humans. Antimicrob Agents Chemother. 2013;57(8):3536–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johns BA, Kawasuji T, Weatherhead JG, et al. Carbamoyl pyridone HIV‐1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J Med Chem. 2013;56(14):5901–5916. [DOI] [PubMed] [Google Scholar]