

Abstract
Objective
To ensure accurate and appropriate reporting of non‐invasive prenatal testing (NIPT) results, the standard of testing should be measured and monitored by participation in external quality assessment (EQA) schemes. The findings from international pilot EQAs for NIPT for the common trisomies are presented.
Methods
In the first pilot, three EQA providers used artificially manufactured reference materials to deliver an EQA for NIPT. The second pilot used clinically collected maternal plasma samples. The testing and reporting for aneuploidy status was performed by participating laboratories using routine procedures. Reports were assessed against peer ratified criteria and EQA scores were returned to participants.
Results
Forty laboratories participated in the first. Genotyping accuracy was high; four laboratories reported a critical genotyping error (10%) and two reported partial results. Eighty seven laboratories participated in the second pilot using maternal plasma, two reporting a critical genotyping error (2.3%). For both rounds, report content was variable with key information frequently omitted or difficult to identify within the report.
Conclusions
We have successfully delivered an international pilot EQA for NIPT. When compared with currently available manufactured materials, EQA for NIPT was best performed using clinically collected maternal plasma. Work is required to define and improve the standard of reporting.
Short abstract
What is already known about this topic?
To ensure laboratories deliver accurate, appropriate, and effective reporting of tests performed in their laboratories participation in external quality assessment (EQA) schemes is required by accrediting bodies.
There is no international EQA scheme available for NIPT for aneuploidy.
What does this study add?
EQA for the common trisomies can be successfully delivered across multiple laboratories worldwide.
The EQA is best delivered using clinically collected maternal plasma samples which can be transported at room temperature.
Reporting standards are very variable with key information often omitted.
Further work is required to develop internationally acceptable EQA for NIPT aneuploidy including reporting standards.
The high level of participation in these pilots suggests the need for continued delivery of EQA.
1. INTRODUCTION
Non‐invasive prenatal testing (NIPT) based on analysis of cell free DNA (cfDNA) in maternal plasma is now recognised as being a highly sensitive screening test for trisomies 13,18, and 21. It has been implemented rapidly across the globe.1 NIPT can be delivered using a variety of methodologies but is generally reported to have high sensitivities and specificities regardless of the platform and methodologies used.2, 3 There are however many factors that might influence results including relatively low fetal fraction with the majority of circulating cfDNA being maternal in origin,4 although fetal levels increase with gestation and vary with maternal weight,5, 6, 7, 8 placental size, some pregnancy complications, and multiple pregnancies. In addition, as the majority of cfDNA in maternal plasma is maternal in origin and the fetal component derives from the placenta,8 maternal abnormalities and abnormalities confined to the placenta may be reflected in the results.9 Testing is largely delivered by commercial providers, either through their own laboratories or though transferring their technology to both public and private sector laboratories, although there has been some local development of testing.10, 11 There has, however, been very little by way of standardisation. Indeed, a recent review that included international opinions demonstrated considerable variation in reporting NIPT results and recommended development and participation in quality assurance schemes.12
To ensure high standards of testing and reporting, it is generally accepted that it should be delivered by laboratories that are “accredited.” Internationally, the standard required is defined by ISO15189.13 External quality assessment (EQA), or proficiency testing (PT), provides a mechanism by which the clinical, analytical, and interpretation performance of genetic testing centres can be externally assessed against international standards and compared with other laboratories. To achieve and maintain accreditation for testing, participation in EQA(s) for all diagnostic services offered is required, and EQA performance is utilised by national accreditation bodies as a tangible measure of the quality of a laboratory's performance. Participation in EQA provides evidence of the standard of testing and reporting (including accuracy, reliability, and appropriateness), benchmarks the service against other laboratories providing comparable testing and is usually undertaken at least annually for every aspect of the diagnostic service. If a centre fails to meet the required standard (based on best practice guidelines) to pass the individual EQA distribution, the performance is designated poor, enabling the laboratory to investigate and perform a root cause analysis to rectify the problem.
EQAs are designed to check and sometimes challenge laboratory screening and/or testing for a particular genetic disease/disorder or gene/target combination. Ideally, EQA will cover all aspects of the diagnostic process through the distribution of the relevant clinical samples and associated clinical scenarios, ie, the pre‐analytical, analytical, and post analytical phases (including the clinical interpretation of results). EQA in genetics is generally qualitative (detecting the presence or absence of a variant) and the EQAs discussed here are an example of where such a qualitative approach is used. EQA should be autonomous from professional and national bodies so independent external verification of the quality of service is provided as well as giving confidence to the laboratory, host institution, and users of the service that the laboratory's performance is satisfactory.13 There are two main genetics EQA providers in Europe offering an international service: genomic quality assessment (GenQA) (https://www.genqa.org/), and the european molecular quality network (EMQN) (https://www.emqn.org/). GenQA offers a wide repertoire of cytogenomic, genomic and clinical genetics EQAs, and EMQN, which is independent of GenQA, offers a wide range of genomic EQAs.
There are a number of unique challenges in delivering an EQA for NIPT, mainly providing EQA material for both (a) a variety of assay platforms and (b) a potentially large number of participants. The major challenge is to provide sufficient material that is of a suitable quality and has the relevant genetic aberration. The ideal EQA programme would provide every EQA participant with a batch of material that is identical to the sample type they normally receive for routine testing (ie, the same material type provided in the same quantity, collected, stored, and transported in the optimal manner). To allow inter‐laboratory comparison, the material provided should ideally come from a single or small number of batches, the genotype of which has been independently validated by at least two different laboratories, prior to provision of the EQA. The EQA provider also needs to ensure that the samples have not deteriorated in the interval between validation and testing by the participant laboratories and to also consider the cost of sourcing the materials and transportation requirements. Whilst real clinical samples are ideally used for EQA, in some circumstances, artificial material may be appropriate to meet these requirements.
The main advantage of artificial reference materials is that large quantities can be manufactured and validated prior to distribution. Secondly, these samples are stable at ambient temperature, and consequently, handling and shipping problems/costs can be reduced. Thirdly, a single batch of material reduces problems associated with sample heterogeneity, resulting in more reliable inter‐laboratory comparisons. Finally, the use of artificial materials allows for the engineering of specific characteristics of interest into the sample. For example, in a Chinese NIPT EQA, the effect of low fetal fraction was investigated by limiting the amount of sheared fetal DNA in the serum.14 However, there are some disadvantages when using artificial materials as they are not identical to real samples and consequently their performance in an assay might differ, potentially leading to a suboptimal assessment. For example, with NIPT, mechanical shearing of DNA produces fragments which differ in size distribution from those found in maternal serum, with less small DNA fragments (less than 100 bp) present.14 The fragments produced by mechanical shearing may also be problematic for paired‐end next generation sequencing (NGS) approaches. In addition, all commercially available reference materials are not currently matched for the parent/child relationship and therefore NIPT platforms using single nucleotide polymorphism (SNP) analysis is not possible using these materials.
Real clinical materials have known performance characteristics and can be tested using all clinically relevant methodologies therefore represent the gold standard. However, many laboratories require up to 4 ml of plasma to complete NIPT testing thus it is not possible to obtain sufficient amounts of donated plasma from a single pregnant patient required for a large EQA programme. Any EQA for NIPT will therefore need to use multiple samples/sources for the sample distribution, which limits the reliability of inter‐laboratory comparison. Additionally, any validation requirements reduce the volume of sample available for participants to test. In addition, as handling, transit time/conditions, and freeze/thawing are all factors that affect the relative proportions of maternal and fetal DNA in plasma,15 careful consideration is required to ensure the EQA samples arrive in an optimal condition. In order to guarantee stability of real samples, it may be necessary to consider low temperature distribution on dry ice thereby increasing the costs and logistical difficulties of exporting the EQA materials. Finally, the restrictive availability of material from pregnancies with the rarer trisomies, eg, Edwards and Patau syndromes, are an additional limiting factor which makes offering a comprehensive range of different NIPT EQA samples a challenge.
To inform the development of an international EQA scheme, we describe two pilot EQA rounds for NIPT in singleton pregnancies delivered by the two main genetics EQA providers, GenQA and EMQN. The first uses artificial materials and the second real clinical materials.
What is already known about this topic?
To ensure laboratories deliver accurate, appropriate, and effective reporting of tests performed in their laboratories participation in external quality assessment (EQA) schemes is required by accrediting bodies.
There is no international EQA scheme available for NIPT for aneuploidy.
What does this study add?
EQA for the common trisomies can be successfully delivered across multiple laboratories worldwide.
The EQA is best delivered using clinically collected maternal plasma samples which can be transported at room temperature.
Reporting standards are very variable with key information often omitted.
Further work is required to develop internationally acceptable EQA for NIPT aneuploidy including reporting standards.
The high level of participation in these pilots suggests the need for continued delivery of EQA.
2. METHODS
2.1. EQA pilot 1
A commercial reference material provider, SeraCare, was commissioned to manufacture bespoke artificial materials for use in the first EQA pilot. Specific details of manufacture are held by the manufacturer who work to the relevant ISO standards.13 All samples were independently validated by the EQA providers before distribution. One sample contained cfDNA with trisomy 21 at a fetal fraction of 4%, the second sample contained cfDNA with trisomy 18 at a fetal fraction of 10%, and the final sample was low risk of trisomy 13, 18, and 21, with a cell free fetal fraction of 12%. DNA at a concentration of 20 ng/mL, sheared to approximately 170 base pairs in size, was supplied in 1 mL aliquots of human plasma. Participating laboratories were requested to assume that the plasma has been double spun prior to receipt. If laboratories required more than 1 mL of plasma for processing, then they were instructed to dilute the sample with buffer to the required volume for testing.
The expected results were confirmed prior to EQA distribution by independently validating the samples in four laboratories using different testing protocols. The known limitation of the EQA material was that the maternal and placentally‐derived cfDNA was not matched and thus laboratories using methods reliant on comparison between maternal and fetal SNPs or pair‐ended NGS were not eligible to participate. The validating laboratories reported the expected genotypes but commented that the “samples did not perform as well as routine clinical samples.” In order to meet the overwhelming demand for external quality assessment of NIPT, it was decided to proceed with the pilot EQA despite these limitations but without scoring of results or applying performance monitoring.
Each EQA sample was assigned an “EQA patient” name, sample identifier, reason for referral, and a clinical case scenario (see Table 1). Participants were informed that all samples supplied were from singleton pregnancies and NIPT for trisomy 13, 18, and 21 was requested. Laboratories were asked to analyse samples and report results using their local standard operating procedures. These reports were submitted electronically to the EQA provider websites and assessed for genotyping accuracy anonymously by a panel of NIPT experts. Assessor feedback on the genotyping and interpretation of the results, as well as the clerical accuracy of reports was provided, measured against peer ratified criteria when required (Table 2), and an individual laboratory report returned to each laboratory. An incorrect genotype was classified as a critical error.
Table 1.
Clinical cases and expected fetal results
| EQA Round 1 | |||
|---|---|---|---|
| Case details | Clinical case | Sample details | Expected fetal result |
| Alexia LOPEZ(dob 14/05/1981) | Referred for NIPT by a consultant obstetrician for NIPT testing for aneuploidies. | Artificial plasma. | Low risk of trisomy 13, 18, and 21. |
| Gestational age at time of sampling (confirmed by scan) 13 + 1 weeks. | |||
| Fetal fraction of 12%. | |||
| Margaret MURPHY(dob 20/02/1987) | Referred for NIPT by a consultant clinical geneticist. She has a son who is affected with down syndrome. | Artificial plasma. | High risk of trisomy 21. |
| Gestational age at time of sampling (confirmed by scan) 13 + 2 weeks. | |||
| Low risk of trisomy 13 and 18. | |||
| Fetal fraction 4%. | |||
| Wendy MARTINFIELD(dob 05/10/1983) | Referred by a genetic nurse for NIPT for aneuploidies following routine antenatal screening counselling. | Artificial plasma. | High risk of trisomy 18. |
| Gestational age at time of sampling (confirmed by scan) 12 + 5 weeks. | |||
| Low risk of trisomy 13 and 21. | |||
| Fetal fraction 10%. | |||
| EQA Round 2 | |||
|---|---|---|---|
| Anna‐Maria DeFLOREY(dob 02/10/1981)Height 162.6 cmWeight 89.8 kg | Referred by a genetic nurse for NIPT for aneuploidies following counselling for routine antenatal screening. | Maternal plasma. | Low risk for trisomy 13, 18, and 21. |
| Gestational age at time of sampling (confirmed by scan) 12 + 2 weeks. | |||
| Tabitha HAMILTON(dob 31/12/1988)Height 165.1 cmWeight 64.4 kg | Has previously sent a sample through a web‐based company and received a “high risk” result for trisomy 21. She has been referred for a repeat NIPT by your laboratory by your local consultant clinical geneticist. | Maternal plasma. | Low risk for trisomy 13 and 18. |
| Gestational age at time of sampling (confirmed by scan) 11 + 3 weeks. | |||
| High risk for trisomy 21. | |||
Note. dob: date of birth; EQA: external quality assessment; NIPT: noninvasive prenatal testing. All patient details are mock and created for the purposes of the EQAs.
Table 2.
Marking criteria applied to reports submitted in these EQAs
| EQA Round 1 | Category | Criteria | Marks* |
|---|---|---|---|
| Case | |||
| 1 | Genotyping | No evidence of trisomy 13, 18, and 21 | 2.0 marks |
| 2 | Genotyping | Evidence of trisomy 21. No evidence of trisomy 13 and 18. | 2.0 marks |
| 3 | Genotyping | Evidence of trisomy 18. No evidence of trisomy 13 and 21. | 2.0 marks |
| EQA Round 2 | |||
|---|---|---|---|
| 1 | Genotyping | No evidence of trisomy 13, 18, or 21* | 2.0 marks |
| Interpretation | Low risk for trisomy 13, 18, and 21 | 2.0 marks | |
| 2 | Genotyping | Evidence of trisomy 21. No evidence of trisomy 13 or 18* | 2.0 marks |
| Interpretation | High risk for trisomy 21. Low risk for trisomy 13 and 18. | 2.0 marks | |
| The high‐risk result must be confirmed by invasive prenatal diagnosis |
| Essential elements within report | |||
|---|---|---|---|
| Interpretation—deduction of 0.5 marks if omitted applied in Round 2: | Clerical accuracy—deduction of 0.5 marks if omitted applied in Round 2: | ||
| • Clear clinical interpretation provided | • Unique patient identifiers should be given (mother's name, mother's date of birth) | ||
| • The testing methodology should be given | |||
| • Full limitations of the test should be given | • Gestational age at time of sampling | ||
| • Date of sample collection | |||
| • Date of receipt of sample | |||
| • Correct sample type | |||
| • Reason for referral | |||
| • Report date | |||
| Comments | |||
|---|---|---|---|
| • The use of “positive/negative” or “abnormal/normal” is not recommended as can be misinterpreted |
The maximum score for each category is 2.0 marks. Deductions are made for missing essential elements. If an incorrect genotype is reported then this is classed as a critical genotyping error and 2 marks were deducted.
2.2. EQA pilot 2
Each participating laboratory received two real patient samples (one low‐risk and one high‐risk for trisomy 21), supplied as 4 mL samples obtained from approximately 4000 aliquots derived from 86 real maternal plasma samples, with confirmed pregnancy outcomes (low/high‐risk for common aneuploidy) obtained from the UK National Institute for Health Research Reliable Accurate Prenatal non‐Invasive Diagnosis (RAPID) programme sample biobank.16 These samples were derived from maternal blood collected at the time of invasive testing for clinical indications. Blood was either collected into Streck tubes, before double‐spinning and plasma separation, or into EDTA tubes and spun within 6 hours of blood draw. Plasma was aliquoted into 2 mL tubes and stored at −80°C. The frozen samples were transferred to GenQA and then aliquoted and distributed at ambient temperature. The samples were not pooled and no prior validation was performed because outcome was confirmed for all cases. There was sufficient plasma available for all cases except one to distribute to at least two laboratories. Clinical case scenarios were supplied as outlined for round 1 (Table 1), laboratories were asked to report findings using their standard format and the submitted reports were assessed as described above for round 1 except that a score (out of 2.00) was attributed for each marking category, ie, genotyping, interpretation, and clerical accuracy (Table 2).
3. RESULTS
3.1. EQA pilot 1
Forty five laboratories from 19 different countries in five continents registered to participate, and 40 submitted EQA returns within the allotted reporting period. Four participants failed to submit reports but did not provide a reason and there was one duplicate registration (between two EQA providers), which was subsequently reassigned.
Overall, the standard of genotyping accuracy was high. Four laboratories (10%) reported critical genotyping errors; two laboratories reported the low risk for trisomy 13, 18, and 21, sample to be trisomy 21; and two different laboratories did not report the correct result for the high‐risk trisomy 21 sample (one reported the case as normal and the second reported a borderline trisomy 13 result, as well as trisomy 21). Two laboratories reported partial results for one case each, ie, no conclusive result for one of the chromosomes tested. The EQA providers did not assign scores for that case to that laboratory, as reason for the failure to obtain a reportable result could not be determined. The overall sample failure rate was 18%.
The fetal fraction was reported by 51% of participants, whilst a further 40% did not state on the report either the fraction or whether it had been determined. The remaining 9% participants either reported the fetal fraction only for the “no abnormality detected” case, or only commented when it was not sufficient to meet their internal quality control standards.
3.2. EQA pilot 2
Ninety six laboratories from 30 different countries registered to participate and 86 (89.6%) submitted EQA returns for all cases within the allotted reporting period. Nine participants failed to submit reports for either case but did not provide a reason. One laboratory submitted results for Case 1 only, and three laboratories reported test failures for one or both cases.
The standard of genotyping was higher than in the first EQA, with only two laboratories (2.3%) making a critical genotyping error: one laboratory incorrectly reported the low‐risk case (Case 1) as high‐risk for trisomy 13, and another one incorrectly reported Case 2 as high‐risk for trisomy 18 and low‐risk for 13 and 21. As multiple samples were distributed for each case, then no assessment of fetal fraction was performed. Figure 1 summarises the mean scores obtained by all participating laboratories and Figure 2 summarises the range of scores obtained. There was a wide range of ways to describe the methods used (Figure 3), and several reports contained insufficient information on the limitations of the test performed. Assessors based their evaluation of reports on the international consensus reached previously,12 which is in line with many national recommendations (Table 2).
Figure 1.
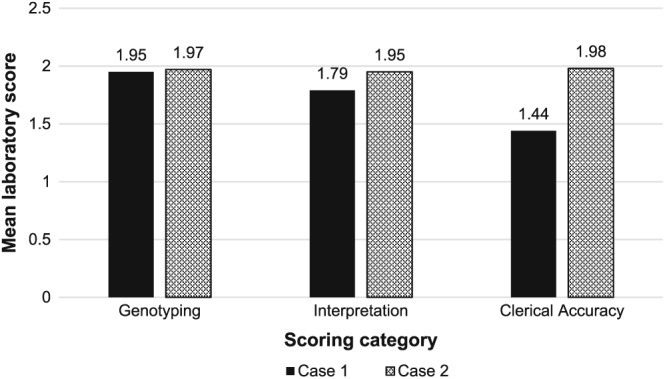
Summary of the mean scores obtained by the participants in the second pilot for Case 1 and Case 2. Maximum mean score obtainable is 2.0
Figure 2.
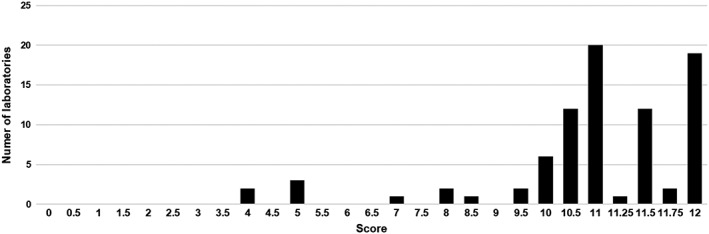
Summary of the range of total scores obtained by the participants in the second pilot. Maximum score obtainable is 12
Figure 3.
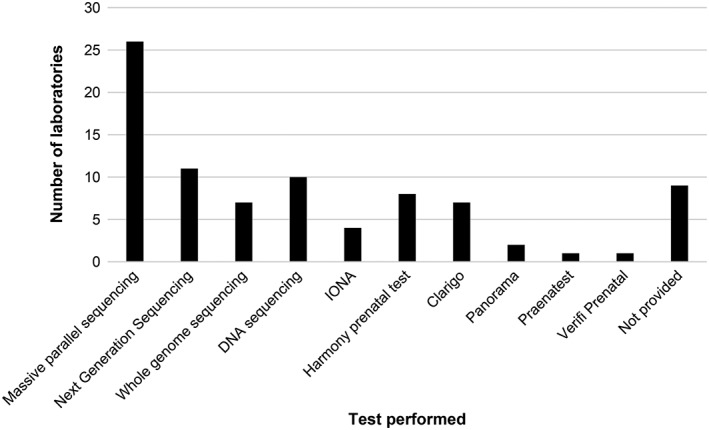
Description of the methodologies tested in pilot 2 as described by participants in their reports showing the wide variation in terminology used, some generic and some using brand names
Risk figures were provided in 27% and 34% of reports for Case 1 (low‐risk case) and case 2 (low‐risk trisomy 13 and 18, high‐risk trisomy 21 case), respectively (Figure 4). Laboratories which provided a risk figure reported the results using a range of different terminologies (Figure 5).
Figure 4.
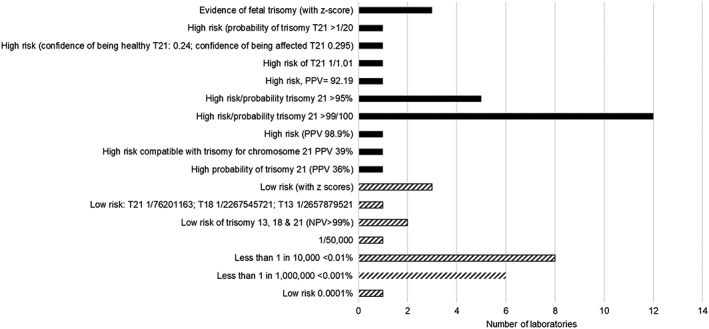
Summary of risk figures provided by laboratories in the second pilot; low‐risk case (hatched) and high‐risk case (block)
Figure 5.
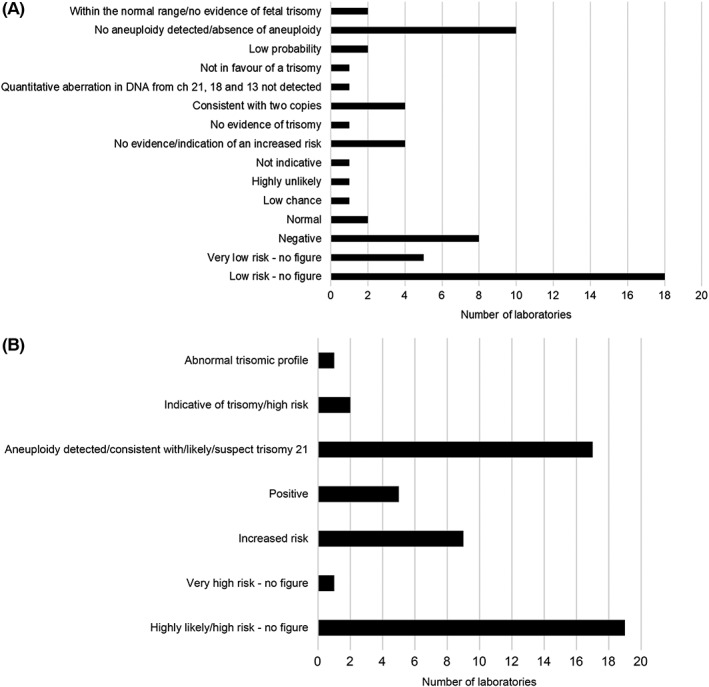
Summary of the terminology used to report the results in the second pilot for (A) the low risk case and (B) the case that was low risk for trisomy 13 and 18, high risk trisomy 21
Three laboratories were unable to obtain a result from the material provided; one did not obtain results for either cases, a second did not obtain a reportable result for Case 1 and a third did not report results for Case 2. All remaining laboratories submitted a result for both cases and the overall sample failure rate in this EQA pilot was 2.3%.
3.3. Report format for both pilot EQA rounds
The format of the reports was very variable for both pilot EQA rounds with essential information missing from the main body of the text, making it difficult to find. Many reports embedded essential information (eg, the requirement to confirm the high‐risk result by invasive testing) either in the footer, a rider, or just generally in a nonprominent position on the report. In some reports submitted for Case 2, only the high‐risk result was stated and the results for other chromosomes tested was omitted. It is generally accepted that clinical reports should include all results.17 Conversely, other laboratories reported the results of “extra extended testing,” which had not been requested. One laboratory reported results for other conditions/abnormalities and nine laboratories reported the fetal sex. Several laboratories used terms such as “positive/negative” or “abnormal/normal” in their reports, the meaning of which could be easily misinterpreted (Figure 5). In Case 2, where the results indicated high risk for trisomy 21, reports often contained a generic comment about additional testing without specifying that invasive prenatal testing is required for confirmation of the result. A number of laboratories appeared to outsource their NIPT to another provider, but none of their submitted reports stated this, although this is a mandatory requirement of ISO 15189.13
Although the majority of laboratories provided details of the test performed, there was considerable variation in the type of information provided (Table 2). Information on the bioinformatic work‐up was not given in the majority of reports, and laboratories that were using a commercial test did not describe the methodology of the assay or give a reference to inform the reader where the information could be accessed.
4. DISCUSSION
We have successfully demonstrated that EQA can be delivered to laboratories performing NIPT for the three common trisomies based on analysis of cfDNA in maternal plasma. The first used artificially created materials which were not applicable to all laboratories and had a higher failure rate than the second, which used real maternal plasma samples applicable to all providers and with a low critical error rate (2.3%). As these are pilot exercises, no official performance monitoring was applied although the EQA providers notified the laboratories of any suboptimal performance and offered help and support to identify and correct the root cause. As the plasma was sent at ambient temperature to 30 different countries in Asia, Australasia, Europe, and North America and the failures were not correlated with an extended time in transit, we have shown the stability of the maternal plasma, allowing future EQA rounds to send the samples globally without risk of significant degradation.
The EQA has also shown considerable variation in the content of the reports, with many containing insufficient information such that the clinician reading them could misinterpret the results or be unaware of the nature and/or limitations of the NIPT screening test. This highlights the need for more standardisation and EQA in this field. For these EQAs, we based the standard on the consensus on report content that has recently been published to evaluate results,12 but these should be reviewed and endorsed EQA providers internationally before further evaluation.
In addition to essential elements required in an NIPT report such as method(s) used, whether targeted or whole genome sequencing, to indicate the extent of testing fetal fraction is another important parameter that may be included in a report. However, there is considerable variation in the way the testing methodology was described, and some providers using a defined fetal fraction as a cut‐off to fail results; others using it as a quality measure and others not commenting at all. There is also variability in the accuracy of fetal fraction measurement and debate as to whether or not this should be included in the report. American College of Medical Genetics and Genomics (ACMG) guidelines18 state that fetal fraction measurement is essential for accurate interpretation of the results and that it should be displayed on the report, whilst an International Society for Peritoneal Dialysis (ISPD) workshop publication reporting consensus opinion for recommended practice showed lack of consensus as to whether or not fetal fraction should be measured or reported.12 This is clearly an area for further discussion as the EQA parameters are refined.
The results should state whether it is a high risk or low risk with results given for all chromosomes tested. As this is a screening test, it is essential that the reader of the report is aware there can be false‐negative and false‐positive results, and the report should state clearly that all high‐risk results should be confirmed by invasive diagnostic testing. The most frequent omission was not stating the limitations of the test performed, which included (a) the sensitivity and specificity of the test, (b) a statement that the NIPT aneuploidy does not cover other genetic abnormalities, and (c) stating the risk of false‐positive or false‐negative results. Discrepant results can be caused by either technical or biological factors such as very low fetal fraction, confined placental mosaicism (CPM), maternal copy number variation, or a vanishing twin.8, 19
It is important that there is no ambiguity in the language when describing the results. A third of laboratories stated the result was either high or low risk whilst two‐thirds provided some estimation of this risk. Some centres gave a precise risk calculation but this figure was not always consistent with the sensitivity of the assay. Overall, there was considerable variation in the risk calculation provided by laboratories regardless of whether it was a high or low risk result. Given the disparity seen and recent debate from lay support groups, further discussion on the terminology used is required, but terms “high‐risk” and “low risk” or “high chance” or “low chance” rather than “normal/abnormal” or “positive/negative” are likely to be more acceptable. The adoption of these terms by laboratories will need to be assessed in future EQA distributions.
Since delivering these pilot EQAs, an improved artificial reference sample has been developed that allows pairing of the maternal and fetal genotypes. This means that it is potentially usable on all platforms currently available. As sourcing clinical samples for large NIPT EQAs is going to be challenging, access to suitable manufactured materials has significant potential advantages. We therefore plan a further pilot EQAs which will distribute both real plasma samples and artificial samples to allow comparison and inclusion of all testing laboratories.
In conclusion, two pilot EQAs for NIPT for aneuploidy in singleton pregnancies have been successfully run and shown that using maternal plasma samples shipped at ambient temperature is superior and more widely applicable. However, there are practical issues that need to be addressed before initiating a formal EQA over a wider range of laboratories, including how to source sufficient plasma samples in the future and terminology to be used in reporting. The significant variation in reporting standards will need to be addressed in future EQAs and further discussion is required to harmonise the approach across the globe, but the data we report here has highlighted the issues that need to be addressed in future EQAs. Finally, the study reported here describes piloting EQA in singleton pregnancies, clearly there are other situations that will require evaluation including multiple pregnancies and those conceived following assisted conception.
CONFLICTS OF INTERESTS
L.S.C. has had collaborations with Roche, Illumina, Oxford Gene Technology, Multiplicom, and Nonacus for the development of NIPT and NIPD. She also received funding from the National Institute for Health Research, Great Ormond Street Children's Charity and Action Medical Research to support research in this area. No other authors have any conflicts of interest to declare.
FUNDING
This work was funded by Genomic Quality Assessment and the European Molecular Quality Network.
ACKNOWLEDGEMENTS
The maternal plasma samples used in the second EQA were obtained from the sample bank collected as part of the NIHR funded “RAPID” Programme Grant for Applied Research (Reliable Accurate Prenatal Non‐Invasive Diagnosis, RP‐PG‐0707‐10107). L.S.C. is partially funded by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health. Thanks to the validating laboratories who supported the delivery of these EQAs.
Deans ZC, Allen S, Jenkins L, et al. Ensuring high standards for the delivery of NIPT world‐wide: Development of an international external quality assessment scheme. Prenatal Diagnosis. 2019;39:379–387. 10.1002/pd.5438
REFERENCES
- 1. Minear MA, Lewis C, Pradhan S, Chandrasekharan S. Global perspectives on clinical adoption of NIPT. Prenat Diagn. 2015;35(10):959‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. Analysis of cell‐free DNA in maternal blood in screening for fetal aneuploidies: updated meta‐analysis. Ultrasound Obstet Gynecol. 2015;45(3):249‐266. [DOI] [PubMed] [Google Scholar]
- 3. Taylor‐Phillips S, Freeman K, Geppert J, et al. Accuracy of non‐invasive prenatal testing using cell‐free DNA for detection of Down, Edwards and Patau syndrome: a systematic review and meta‐analysis. BMJ Open. 2016;18;6(1):e010002. 10.1136/bmjopen-2015-010002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lo YM, Tein MS, Lau TK, et al. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet. 1998;62(4):768‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang E, Batey A, Struble C, Musci T, Song K, Oliphant A. Gestational age and maternal weight effects on fetal cell‐free DNA in maternal plasma. Prenat Diagn. 2013;33(7):662‐666. [DOI] [PubMed] [Google Scholar]
- 6. Wang JC, Sahoo T, Schonberg S, et al. Discordant noninvasive prenatal testing and cytogenetic results: a study of 109 consecutive cases. Genet Med. 2014a;17:234‐236. [DOI] [PubMed] [Google Scholar]
- 7. Wang Y, Chen Y, Tian F, et al. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clin Chem. 2014b;60(1):251‐259. [DOI] [PubMed] [Google Scholar]
- 8. Ashoor G, Syngelaki A, Poon LC, Rezende JC, Nicolaides KH. Fetal fraction in maternal plasma cell‐free DNA at 11‐13 weeks' gestation: relation to maternal and fetal characteristics. Ultrasound Obstet Gynecol. 2013;41(1):26‐32. [DOI] [PubMed] [Google Scholar]
- 9. Alberry M, Maddocks D, Jones M, et al. Free fetal DNA in maternal plasma in anembryonic pregnancies: confirmation that the origin is the trophoblast. Prenat Diagn. 2007;27(5):415‐418. [DOI] [PubMed] [Google Scholar]
- 10. Chitty LS, Wright D, Hill M, et al. Uptake, outcomes, and costs of implementing non‐invasive prenatal testing for Down's syndrome into NHS maternity care: prospective cohort study in eight diverse maternity units. BMJ. 2016;354:i3426:x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oepkes D, Page‐Christiaens GC, Bax CJ, et al. Trial by Dutch laboratories for evaluation of non‐invasive prenatal testing. Part I‐clinical impact. Prenat Diagn. 2016;36(12):1083‐1090. 10.1002/pd.4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deans ZC, Allen S, Jenkins F, et al. Recommended practice for laboratory reporting of non‐invasive prenatal testing (NIPT) of trisomies 13, 18 and 21: a consensus opinion. Prenat Diagn. 2017;37(7):699‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. ISO 15189 :2012. Medical laboratories—Requirements for quality and competence and analysis. https://www.iso.org/standard/56115.html
- 14. Zhang R, Zhang H, Li Y et al. External quality assessment for detection of fetal trisomy 21, 18, and 13 by massively parallel sequencing in clinical laboratories. 2016;18:244–252 [DOI] [PubMed] [Google Scholar]
- 15. Barrett AN, Zimmermann BG, Wang D, Holloway A, Chitty LS. Implementing prenatal diagnosis based on cell‐free fetal DNA. Accurate identification of factors affecting fetal DNA yield. PLOS One. 2011;6:1‐8. 10.1371/journal.pone.0025202.t001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. UK Reliable accurate prenatal non‐invasive diagnosis (RAPID) project sample biobank http://www.rapid.nhs.uk Accessed July 29, 2018.
- 17. Claustres M, Kožich V, Dequeker E, et al. Recommendations for reporting results of diagnostic genetic testing (biochemical, cytogenetic and molecular genetic). Eur J Hum Genet. 2014;22(2):160‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gregg AR, Skotko BG, Benkendorf JL, et al. Noninvasive prenatal screening for fetal aneuploidy, 2016 update: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2016;18(10):1056‐1065. [DOI] [PubMed] [Google Scholar]
- 19. Hartwig TS, Ambye L, Sørensen S, Jørgensen FS. Discordant non‐invasive prenatal testing (NIPT) ‐ a systematic review. Prenat Diagn. 2017;37(6):527‐539. [DOI] [PubMed] [Google Scholar]