

Abstract
Objective
The natural history of nonradiographic axial spondyloarthritis (SpA) is incompletely characterized, and there are concerns that nonsteroidal antiinflammatory drugs provide inadequate disease control in patients with active disease. This study was undertaken to investigate the effects of certolizumab pegol (CZP), an anti–tumor necrosis factor treatment, in patients with nonradiographic axial SpA with objective signs of inflammation.
Methods
In this ongoing parallel‐group double‐blind study, adults with active disease were recruited from 80 centers in Australia, Europe, North America, and Taiwan, and were randomized 1:1 to receive placebo or CZP (400 mg at weeks 0, 2, and 4, followed by 200 mg every 2 weeks) in addition to nonbiologic background medication (NBBM). Switching to open‐label CZP (or other biologic) or making background medication changes was permitted at any point during the trial, although changes before week 12 were discouraged. The primary end point was the proportion of patients achieving major improvement (MI) (i.e., a ≥2.0‐point decrease in the score from baseline or achievement of the lowest possible score [0.6]) in the Ankylosing Spondylitis Disease Activity Score (ASDAS) at week 52.
Results
A total of 317 patients were randomized to receive placebo plus NBBM (n = 158) or CZP plus NBBM (n = 159). ASDAS‐MI at week 52 was achieved in 47.2% (75 of 159) of CZP plus NBBM patients, which was significantly greater (P < 0.0001) than the 7.0% (11 of 158) of placebo plus NBBM patients in whom ASDAS‐MI was achieved. Of the placebo plus NBBM patients, 60.8% (96 of 158) switched to open‐label treatment before week 52 compared to 12.6% (20 of 159) of the CZP plus NBBM patients.
Conclusion
Adding CZP to background medication is superior to adding placebo in patients with active nonradiographic axial SpA. These results indicate that remission in nonradiographic axial SpA treated without biologics occurs infrequently, demonstrating the need for treatment beyond nonbiologic therapy.
Introduction
Axial spondyloarthritis (SpA) is a chronic inflammatory disease predominantly affecting the sacroiliac joints and spine. The disease comprises 2 subpopulations: those with radiographic axial SpA (also known as ankylosing spondylitis) and those with nonradiographic axial SpA, who have been reported to have a similar disease burden 1, 2, 3, 4, 5, 6. Conventional therapy for axial SpA comprises nonpharmacologic management, such as physical therapy, and nonsteroidal antiinflammatory drugs (NSAIDs) as first‐line treatment. In routine clinical practice, patients are also treated with conventional disease‐modifying antirheumatic drugs (DMARDs), corticosteroids, and analgesics. However, for patients with active disease and objective signs of inflammation (sacroiliac joint inflammation on magnetic resonance imaging [MRI] or elevated C‐reactive protein [CRP] level) despite treatment with NSAIDs, treatment with anti–tumor necrosis factor (anti‐TNF) agents has been recommended 7, 8, 9, 10.
There is a lack of understanding of disease presentation, progression, and prognosis of nonradiographic axial SpA 11, 12, 13, 14. It has been suggested that nonradiographic axial SpA could be a self‐limiting disease, with the potential for spontaneous remission 15, 16, 17, 18. This, in part, has resulted in the lack of access to anti‐TNF agents for many patients with nonradiographic axial SpA in several countries.
The C‐axSpAnd Study is the first to incorporate a 52‐week placebo‐controlled time period to investigate the efficacy of an anti‐TNF agent, certolizumab pegol (CZP), in a population of nonradiographic axial SpA patients. This study employed a unique design that incorporated a long placebo‐controlled phase, which permitted optimization of nonbiologic treatment over a sufficiently long time frame to assess disease remission.
Patients and methods
Study design
The C‐axSpAnd Study (ClinicalTrials.gov identifier: NCT02552212) is a 3‐year, phase III multicenter study investigating the efficacy and safety of CZP in patients with nonradiographic axial SpA. Patients were randomized to receive either CZP or placebo in addition to their current nonbiologic background medication (NBBM) at 80 sites located in Australia, Europe, North America, and Taiwan, in a 52‐week, parallel‐group, double‐blind, placebo‐controlled study. The primary objective of the study was to assess the efficacy of CZP 200 mg every 2 weeks on signs and symptoms in patients with active nonradiographic axial SpA who had previously been treated unsuccessfully with ≥2 NSAIDs.
This study was approved by the national, regional, or independent ethics committees or institutional review boards at participating sites and was conducted in accordance with applicable regulatory and International Conference on Harmonization Good Clinical Practice requirements, based on the Declaration of Helsinki and local laws. Protocol amendments after the commencement of the trial are provided in Supplementary Appendix A, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract.
Patients
Eligible patients were required to 1) be age ≥18 years, 2) have a documented diagnosis of adult‐onset axial SpA, meeting the Assessment of SpondyloArthritis international Society (ASAS) classification criteria 19, 3) have ≥12 months of symptom duration, and 4) have active disease at screening and baseline (defined as a Bath Ankylosing Spondylitis Disease Activity Index [BASDAI] 20 score of ≥4 and spinal pain score of ≥4 on a 0–10 scale) despite treatment with NBBMs, including ≥2 NSAIDs. Patients with radiographic sacroiliitis meeting the modified New York classification criteria 21 were excluded from the trial. Patients were also required to have objective signs of inflammation, i.e., either active sacroiliitis as evidenced by MRI (based on the ASAS definition of a positive MRI 22) at screening or a baseline CRP level above the upper limit of normal (defined as 10.0 mg/liter). Pelvic radiographs and MRI scans were read centrally by 2 readers (and by an adjudicator, if necessary). For full patient selection criteria, see Supplementary Appendix B, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract. All patients provided written informed consent to participate in the study.
Randomization and masking
Patients were randomized 1:1 using an interactive response system that allocated and maintained all treatment details. Due to the differences in presentation and viscosity between CZP and placebo, special precautions were taken to maintain the blinding, including provision of the study treatment in a sealed box with a prefilled syringe containing either CZP 200 mg or placebo, and there was no information about the study treatment on packaging or labeling. Members of the study team were blinded with regard to the randomization schedule until after database lock and unblinding. For patients who switched to open‐label treatment with CZP or any other treatment, efforts were made to maintain blinding with regard to their prior double‐blind study treatment. Randomization was stratified based on MRI and CRP status (MRI+/CRP+, MRI+/CRP−, and MRI−/CRP+), as well as by geographic region (Asia/Australia, Europe, and North America).
Procedures
Study treatment (CZP or placebo) was administered via a prefilled syringe. CZP was given at a dose of 400 mg at weeks 0, 2, and 4 (loading dose) followed by 200 mg every 2 weeks (Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract). At the discretion of the treating rheumatologist, NBBM for this disease could be adjusted at any time during the trial; however, changes before week 12, or within 4 weeks prior to weeks 24 and 52, were discouraged. NBBMs allowed in this study included NSAIDs, conventional DMARDs such as sulfasalazine, methotrexate, hydrochloroquine, oral and intravenous corticosteroids, and opioid and nonopioid analgesics (Table 1 and Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract). To reflect the provision of NBBM, the treatment groups are hereafter referred to as placebo plus NBBM and CZP 200 mg with NBBM (CZP plus NBBM).
Table 1.
Patient demographic and clinical characteristics at baseline (n = 317)a
| Placebo plus NBBM group (n = 158) | CZP plus NBBM, 200 mg every 2 weeks (n = 159) | |
|---|---|---|
| Demographic characteristic | ||
| Age, mean ± SD years | 37.4 ± 10.8 | 37.3 ± 10.5 |
| Female | 82 (51.9) | 81 (50.9) |
| HLA–B27 positive | 132 (83.5) | 128 (80.5) |
| White race | 148 (93.7) | 152 (95.6) |
| Geographic region | ||
| North America | 13 (8.2) | 15 (9.4) |
| Europe | 130 (82.3) | 130 (81.8) |
| Asia/Australia | 15 (9.5) | 14 (8.8) |
| Disease characteristic | ||
| Symptom duration, mean ± SD years | 8.0 ± 7.5 | 7.8 ± 7.7 |
| Time since first diagnosis, years | ||
| Mean ± SD | 4.0 ± 5.4 | 3.6 ± 4.8 |
| Median (range) | 2.1 (0.0–38.2) | 1.7 (0.1–29.2) |
| CRP, mean ± SD mg/liter | 15.8 ± 17.7 | 15.8 ± 17.8 |
| Elevated CRP at baseline (CRP > ULN) | 83 (52.5) | 89 (56.0) |
| ASDAS, mean ± SD | 3.8 ± 0.9 | 3.8 ± 0.8 |
| BASDAI score, mean ± SD | 6.8 ± 1.3 | 6.9 ± 1.4 |
| BASFI score, mean ± SD | 5.4 ± 2.2 | 5.4 ± 2.1 |
| BASMI score, mean ± SD | 2.8 ± 1.4 | 3.0 ± 1.3 |
| Sacroiliac joint SPARCC score, mean ± SD | 8.5 ± 12.3 | 7.8 ± 10.8 |
| Nocturnal spinal pain score, mean ± SD (scale 0–10) | 6.6 ± 2.1 | 6.6 ± 2.3 |
| ASQoL score, mean ± SD | 12.1 ± 4.3 | 11.7 ± 4.3 |
| Uveitis | ||
| History | 25 (15.8) | 22 (13.8) |
| Current | 11 (7.0) | 6 (3.8) |
| Enthesitis | 122 (77.2) | 125 (78.6) |
| MASES, mean ± SD | 4.8 ± 3.5 | 4.8 ± 3.2 |
| ASAS‐NSAID score, mean ± SD | 66.3 ± 48.7 | 69.6 ± 48.0 |
| MRI/CRP stratificationb | ||
| MRI+/CRP+ | 42 (26.6) | 45 (28.3) |
| MRI+/CRP− | 76 (48.1) | 74 (46.5) |
| MRI−/CRP+ | 39 (24.7) | 38 (23.9) |
| Prior and concomitant medications | ||
| NSAIDs | ||
| Prior | 154 (97.5) | 157 (98.7) |
| Concomitant | 138 (87.3) | 138 (86.8) |
| DMARDs | ||
| Prior | 73 (46.2) | 77 (48.4) |
| Concomitant | 48 (30.4) | 55 (34.6) |
| Corticosteroids | ||
| Prior | 36 (22.8) | 31 (19.5) |
| Concomitant | 16 (10.1) | 16 (10.1) |
| Anti‐TNF medication, any prior usage | 11 (7.0) | 7 (4.4) |
Except where indicated otherwise, values are the number (%) of patients in the analysis population. NBBM = nonbiologic background medication; CZP = certolizumab pegol; ULN = upper limit of normal; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index (0–10 scale; higher scores indicate higher disease activity); BASFI = Bath Ankylosing Spondylitis Functional Index (0–10 scale; higher scores indicate worse function); BASMI = Bath Ankylosing Spondylitis Metrology Index (0–10 scale; higher scores indicate more severe spinal mobility impairment); SPARCC = Spondyloarthritis Research Consortium of Canada; ASQoL = ankylosing spondylitis quality of life (0–18 scale; higher scores indicate worse quality of life); MASES = Maastricht Ankylosing Spondylitis Enthesitis Score (0–13 scale; higher scores indicate more severe enthesitis); ASAS‐NSAID score = Assessment of SpondyloArthritis international Society–nonsteroidal antiinflammatory drug score (0–100 scale; higher scores indicate greater NSAID intake); DMARDs = disease‐modifying antirheumatic drugs; anti‐TNF = anti–tumor necrosis factor.
Three patients were classified as magnetic resonance imaging negative (MRI−)/C‐reactive protein negative (CRP−) and were determined to be protocol deviations.
In addition, patients were able to switch to open‐label CZP treatment or any other biologic at any point during the trial if their disease activity required escalation of treatment. Patients who had transitioned to open‐label treatment continued to be followed up during the study. At the completion of the week‐52 visit, eligible patients could receive CZP treatment for an additional 2 years in an open‐label follow‐up extension period. At the time of publication, patients in this study are receiving treatment as part of the open‐label follow‐up period. This study describes the results of the 52‐week placebo‐controlled period.
Outcome measures
The primary efficacy end point was defined as a composite outcome measure that was achieved if all of the following 3 criteria were fulfilled: 1) the patient remained in the study until week 52, 2) the patient continued taking double‐blind study treatment throughout, and 3) the patient achieved major improvement (MI) in the Ankylosing Spondylitis Disease Activity Score (ASDAS) 23 at week 52. ASDAS‐MI is defined as a ≥2.0‐point decrease from the baseline score in the ASDAS or achievement of the lowest possible ASDAS value (0.6) 24. The ASDAS comprises the following 5 elements, which are algorithmically combined into a continuous measure of disease activity: patient's self‐reported back pain, peripheral pain/swelling in joints and duration of morning stiffness (all from the BASDAI questionnaire), Patient's Global Assessment of Disease Activity (PGADA), and CRP level 25. All patient‐reported outcome assessments were graded on a numerical rating scale (0–10). The measured CRP level (mg/liter; measured at a central laboratory) was used for ASDAS calculations, with a minimum value of 2 mg/liter as validated previously 24. Details regarding scoring and time points for each efficacy end point are provided in Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract).
Key secondary efficacy end points within the statistical testing hierarchy included the following: achievement of 40% improvement according to the ASAS (ASAS40) at weeks 12 and 52, change from the baseline BASDAI score at weeks 12 and 52, change from the baseline Bath Ankylosing Spondylitis Functional Index (BASFI) score 26 at weeks 12 and 52, the Spondyloarthritis Research Consortium of Canada (SPARCC) score for sacroiliac joints 27 at week 12, the Ankylosing Spondylitis Quality of Life (ASQoL) score 28 at week 52, nocturnal back pain at week 52, and the number of patients who had new or recurrent anterior uveitis flares at week 52 (Supplementary Table 2, at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract). Exposure‐adjusted incidence rates for postbaseline uveitis flares at week 52 were calculated post hoc.
Additional outcomes not included in the hierarchical testing procedure were also evaluated. These included the EuroQoL 5‐domain Health State Profile 29, physical and mental component scores of the Short Form 36 health survey (SF‐36) 30, the ASAS‐NSAID score (a quantification of cumulative NSAID intake on a scale of 0–100, with 0 representing no intake and 100 representing maximum intake), the Bath Ankylosing Spondylitis Metrology Index (BASMI) linear score 31, the week‐52 SPARCC score, and enthesitis as measured by the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) 32.
In addition, post hoc analyses of patients who continued taking placebo up to week 52 were conducted, including assessment of ASDAS‐MI, ASAS40, and BASDAI. Finally, post hoc analyses were conducted to investigate the disease activity of patients at the point of switching from placebo to open‐label CZP treatment.
Statistical analysis
The sample size was calculated using a chi‐square test of equal proportions with continuity correction. A total sample size of 300 subjects (150 subjects per treatment group) was planned to provide 95% power to detect a significant difference in the ASDAS‐MI response rate at week 52 between CZP and placebo groups, based on a 2‐sided significance level of 0.05. This was based on the assumption of expected response rates for ASDAS‐MI at week 52 of 40% and 20% for CZP and placebo groups, respectively.
The population studied in all efficacy analyses included all patients who underwent randomization and received ≥1 dose of study medication (full analysis set), analyzed based on the intent‐to‐treat principle. Safety outcomes are reported for the safety set, which consisted of all patients treated with ≥1 dose of study medication.
The primary efficacy end point and the 10 key secondary efficacy end points were tested for statistical significance according to a hierarchical testing procedure to maintain a familywise error rate of 5% (the full hierarchy is included in Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract). Testing was conducted at a significance level of 0.05 (2‐sided), starting with the primary efficacy end point and, if the null hypothesis was rejected, proceeding with the secondary efficacy end points in their prespecified order.
The primary efficacy end point was analyzed using a logistic regression model with treatment group, region, and MRI/CRP stratification as fixed effects. The odds ratio (OR) of the ASDAS‐MI at week 52 was estimated and tested between treatment groups. Patients completing week 52 taking double‐blind treatment but with no available ASDAS score at this time point were classified as nonresponders.
Sensitivity analyses of the primary efficacy end point used alternate approaches for handling of missing data with various assumptions for missingness mechanisms, including re‐analysis after multiple imputation, re‐analysis with all available data regardless of whether a subject continued to receive the randomized treatment (i.e., including post‐escape data), tipping point analysis (i.e., various delta adjustments for the missing data were conducted to identify assumptions about the missing data, under which there is no longer evidence of a treatment effect), and observed case analysis (in which only observed data from patients who continued the original double‐blind study treatment were included).
Continuous efficacy end points were analyzed using an analysis of covariance model with treatment group, region, and MRI/CRP classification as fixed effects and the baseline score as a covariate. The primary analysis used a reference‐based multiple imputation procedure (in which missing values for both treatment groups were imputed using an imputation model developed using data from the placebo group only). Sensitivity analyses were conducted using multiple imputation, last observation carried forward, and observed case analysis.
Dichotomous secondary end points were analyzed via logistic regression based on a model similar to that used for the primary efficacy end point analysis. Safety outcomes were summarized using descriptive statistics. All statistical analyses were performed using SAS, version 9.4.
Results
From September 2015, 941 patients with nonradiographic axial SpA were screened at 104 sites, and 624 of these patients were excluded (Figure 1). A total of 317 patients were randomized to receive treatment as follows: 158 patients to the placebo plus NBBM group and 159 to the CZP plus NBBM group. In total, 260 patients from Europe, 29 patients from Taiwan/Australia, and 28 patients from North America were included. Baseline characteristics of the patients were well‐balanced between treatment groups (Table 1). Full details of NBBM at baseline in the 2 treatment groups are provided in Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract.
Figure 1.
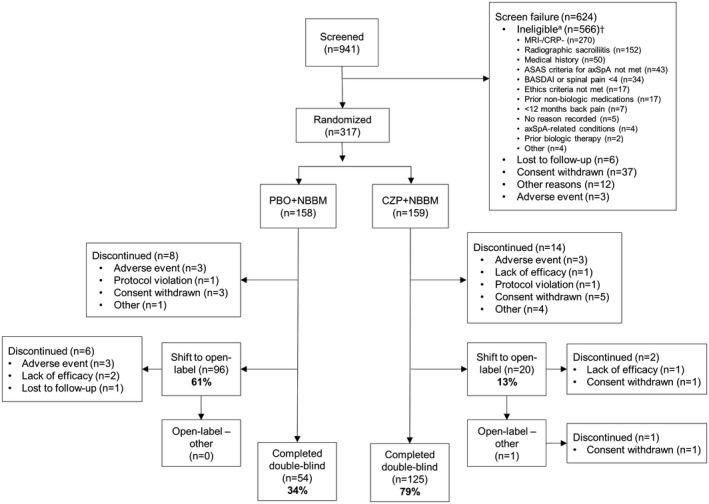
Disposition of the patients. † = some patients met/did not meet multiple eligibility criteria; therefore, patient numbers in the disposition of patients do not add up to the total number of ineligible patients. MRI = magnetic resonance imaging; CRP = C‐reactive protein; ASAS = Assessment of SpondyloArthritis international Society; axSpA = axial spondyloarthritis; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; PBO = placebo; NBBM = nonbiologic background medication; CZP = certolizumab pegol 200 mg.
The primary end point, ASDAS‐MI at week 52, was reached by 47.2% (75 of 159) of CZP plus NBBM patients and 7.0% (11 of 158) of placebo plus NBBM patients (P < 0.0001) (OR for CZP plus NBBM versus placebo plus NBBM 15.2 [95% confidence interval 7.3–31.6]) (Figure 2A). All sensitivity analyses of the primary end point, performed in order to assess the impact of various missing data assumptions, supported the results of the primary analysis, with ORs ranging from 2.2 to 16.1 (Figure 3).
Figure 2.
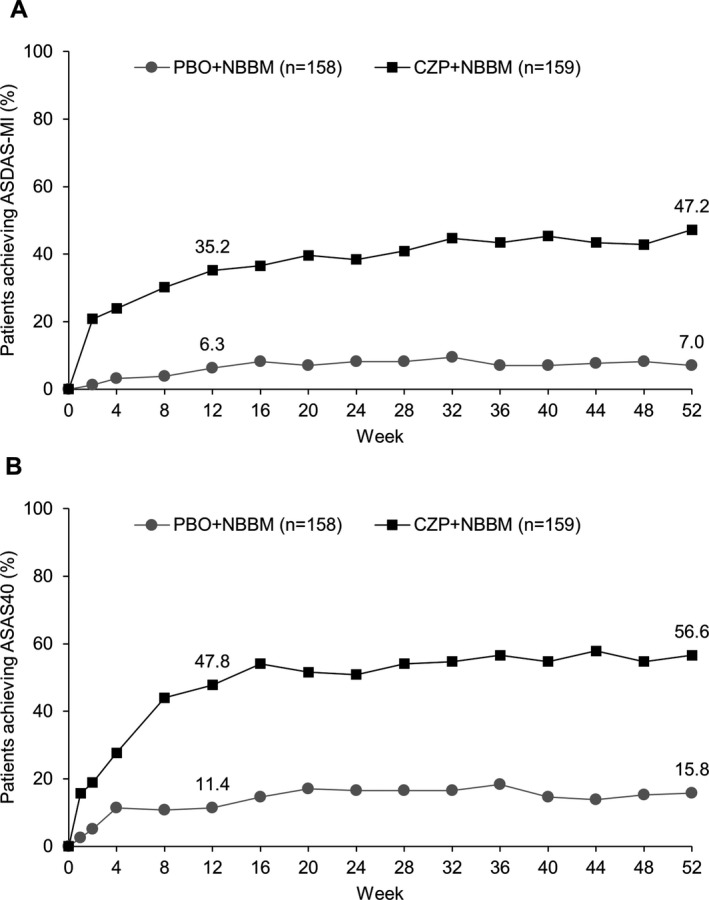
Proportion of patients achieving A, major improvement in the Ankylosing Spondylitis Disease Activity Score (ASDAS‐MI) and B, 40% improvement in disease activity according to the Assessment of SpondyloArthritis international Society criteria (ASAS40) by week 52. P < 0.0001 for certolizumab pegol (CZP) versus placebo (PBO) at week 12 and week 52 for both ASDAS‐MI and ASAS40. NBBM = nonbiologic background medication.
Figure 3.
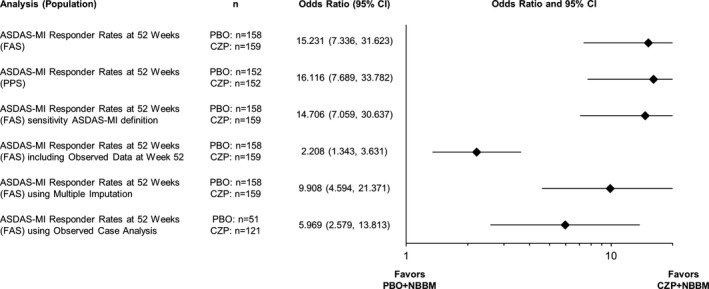
Forest plot of major improvement in the Ankylosing Spondylitis Disease Activity Score (ASDAS‐MI) sensitivity analyses at week 52. 95% CI = 95% confidence interval; FAS = full analysis set; PBO = placebo; CZP = certolizumab pegol 200 mg; PPS = per protocol set; NBBM = nonbiologic background medication.
By week 52, 60.8% (96 of 158) of placebo plus NBBM patients compared to 12.6% (20 of 159) of CZP plus NBBM patients had switched to open‐label treatment. No switches occurred between baseline and week 12; the majority of switches occurred between weeks 12 and 24. Of the 116 patients who switched to open‐label CZP treatment prior to week 52, 1 patient subsequently switched to an alternative open‐label anti‐TNF treatment and then later discontinued the study prior to week 52. In total, 69.6% (110 of 158) of placebo plus NBBM patients had changes to their background medication during the study, compared to 27.7% (44 of 159) of CZP plus NBBM patients. During the 52‐week placebo‐controlled period, 6.9% (22 of 317) of patients withdrew from the study for reasons outlined in Figure 1: 5.1% (8 of 158) withdrew from the placebo plus NBBM treatment group and 8.8% (14 of 159) from the CZP plus NBBM group. At week 52, 78.6% (125 of 159) of CZP plus NBBM patients had completed the 52‐week placebo‐controlled period taking double‐blind study medication, compared to 34.2% (54 of 158) of placebo plus NBBM patients.
All secondary end points within the prespecified hierarchical testing procedure could be tested (Table 2 and Supplementary Table 4, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract). At week 12, 47.8% (76 of 159) of CZP plus NBBM patients had achieved an ASAS40 response, compared to 11.4% (18 of 158) of placebo plus NBBM patients (P < 0.0001) (Figure 2B). By week 52, 56.6% (90 of 159) of CZP plus NBBM patients and 15.8% (25 of 158) of placebo plus NBBM patients had achieved an ASAS40 response (P < 0.0001).
Table 2.
Study outcomesa
| Placebo plus NBBM group (n = 158) | CZP plus NBBM group (n = 159)b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Week 12, observed case analysis | Week 12, imputed | Week 52, observed case analysis | Week 52, imputed | Baseline | Week 12, observed case analysis | Week 12, imputed | Week 52, observed case analysis | Week 52, imputed | |
| Hierarchy outcome measure | ||||||||||
| ASDAS‐MI, no./total no. (%)c | 0.0 | 10/155 (6.5) | 10/158 (6.3) | 11/51 (21.6) | 11/158 (7.0) | 0.0 | 56/155 (36.1) | 56/159 (35.2) | 75/121 (62.0) | 75/159 (47.2) |
| ASAS40, no./total no. (%)c | 0.0 | 18/155 (11.6) | 18/158 (11.4) | 25/51 (49.0) | 25/158 (15.8) | 0.0 | 76/155 (49.0) | 76/159 (47.8) | 90/121 (74.4) | 90/159 (56.6) |
| BASDAI score, mean ± SD (total no.)d | 6.79 ± 1.28 (158) | 5.68 ± 2.08 (155) | 5.71 ± 2.09 (158) | 3.47 ± 1.73 (51) | 5.47 ± 2.30 (158) | 6.88 ± 1.40 (159) | 3.89 ± 2.19 (155) | 3.93 ± 2.21 (157) | 2.64 ± 2.08 (121) | 3.26 ± 2.47 (157) |
| BASFI score, mean ± SD (total no.)d | 5.44 ± 2.18 (158) | 4.89 ± 2.44 (155) | 4.95 ± 2.46 (158) | 2.88 ± 1.94 (51) | 4.71 ± 2.60 (158) | 5.41 ± 2.12 (158) | 3.19 ± 2.34 (155) | 3.20 ± 2.33 (157) | 2.13 ± 2.09 (121) | 2.68 ± 2.42 (157) |
| SPARCC SI joint score, mean ± SD (total no.) | 8.46 ± 12.31 (153) | 8.43 ± 12.39 (139) | – | 5.84 ± 10.99 (46) | – | 7.79 ± 10.82 (154) | 2.81 ± 5.44 (138) | – | 1.92 ± 3.96 (110) | – |
| Nocturnal spinal pain (NRS), mean ± SD (total no.)d | 6.6 ± 2.1 (158) | 5.5 ± 2.6 (155) | 5.6 ± 2.6 (158) | 3.5 ± 2.4 (51) | 5.4 ± 2.8 (51) | 6.6 ± 2.3 (158) | 3.3 ± 2.6 (155) | 3.4 ± 2.7 (157) | 2.0 ± 2.1 (121) | 2.7 ± 2.7 (157) |
| ASQoL score, mean ± SD (total no.)d | 12.11 ± 4.25 (158) | 10.52 ± 5.18 (155) | 10.61 ± 5.17 (158) | 6.76 ± 5.14 (51) | 10.34 ± 5.47 (158) | 11.70 ± 4.34 (158) | 6.70 ± 5.47 (155) | 6.67 ± 5.45 (157) | 4.14 ± 4.67 (121) | 5.38 ± 5.42 (157) |
| Uveitis | ||||||||||
| Patients with flares, no./total no. (%)c | 11/158 (7.0) | – | NA | 8/158 (5.1) | NA | 6/159 (3.8) | – | NA | 4/159 (2.5) | NA |
| EAIR, 95% CIe | – | – | NA | 7.21 (3.11–14.20) | NA | – | – | NA | 2.50 (0.68–6.39) | NA |
| Additional outcome measures | ||||||||||
| SF‐36 PCS, mean ± SD (total no.)d | 33.7 ± 7.0 (157) | 36.1 ± 7.8 (155) | 35.9 ± 7.8 (157) | 42.2 ± 6.7 (51) | 37.0 ± 8.2 (157) | 34.6 ± 7.1 (157) | 42.6 ± 8.5 (155) | 42.6 ± 8.5 (157) | 47.4 ± 7.8 (121) | 44.9 ± 9.5 (157) |
| SF‐36 MCS, mean ± SD (total no.)d | 41.2 ± 10.1 (157) | 43.3 ± 10.8 (155) | 43.2 ± 10.8 (157) | 47.8 ± 11.2 (51) | 42.7 ± 11.4 (157) | 42.0 ± 11.0 (157) | 46.3 ± 10.5 (155) | 46.4 ± 10.5 (157) | 48.2 ± 10.4 (121) | 47.2 ± 10.8 (157) |
| EQ‐5D, mean ± SD (total no.)d | 44.6 ± 19.9 (157) | 53.5 ± 21.7 (155) | 53.3 ± 21.7 (157) | 65.5 ± 18.0 (51) | 53.4 ± 22.4 (157) | 47.7 ± 20.1 (157) | 65.1 ± 18.5 (155) | 65.0 ± 18.5 (157) | 74.2 ± 18.3 (121) | 68.4 ± 22.0 (157) |
| ASAS‐NSAID score, mean ± SD (total no.)d | 66.3 ± 48.7 (158) | 65.8 ± 49.3 (147) | 65.1 ± 49.0 (158) | 56.5 ± 59.0 (48) | 63.4 ± 50.3 (158) | 69.6 ± 48.0 (159) | 63.7 ± 48.2 (153) | 62.8 ± 48.4 (158) | 52.7 ± 50.7 (109) | 52.3 ± 50.4 (158) |
| CRP, mean ± SD (total no.)d | 15.8 ± 17.7 (158) | 13.2 ± 17.2 (155) | 14.6 ± 23.9 (158) | 8.7 ± 10.8 (51) | 12.9 ± 21.8 (158) | 15.8 ± 17.8 (159) | 6.7 ± 15.1 (154) | 6.6 ± 15.0 (157) | 6.2 ± 15.6 (122) | 6.1 ± 14.0 (157) |
| BASMI, mean ± SD (total no.)d | 2.80 ± 1.40 (156) | 2.73 ± 1.36 (151) | 2.75 ± 1.41 (156) | 2.47 ± 1.16 (47) | 2.80 ± 1.39 (158) | 2.96 ± 1.29 (157) | 2.58 ± 1.37 (148) | 2.55 ± 1.35 (154) | 2.12 ± 1.25 (116) | 2.37 ± 1.36 (156) |
| MASES, mean ± SD (total no.)d | 4.8 ± 3.5 (122) | 4.4 ± 3.7 (119) | 4.4 ± 3.7 (122) | 2.2 ± 3.1 (36) | 4.3 ± 3.8 (122) | 4.8 ± 3.2 (125) | 2.7 ± 3.4 (122) | 2.7 ± 3.4 (123) | 1.3 ± 2.2 (91) | 2.1 ± 3.1 (123) |
NBBM = nonbiologic background medication; ASDAS‐MI = major improvement in Ankylosing Spondylitis Disease Activity Score; ASAS40 = achievement of 40% improvement according to the Assessment of SpondyloArthritis international Society criteria; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index (0–10 scale; higher scores indicate higher disease activity); BASFI = Bath Ankylosing Spondylitis Functional Index (0–10 scale; higher scores indicate worse function); SPARCC = Spondyloarthritis Research Consortium of Canada; SI = sacroiliac; NRS = numeric rating scale; ASQoL = ankylosing spondylitis quality of life (0–18 scale; higher scores indicate worse quality of life); NA = not applicable; EAIR = exposure‐adjusted incidence rate; 95% CI = 95% confidence interval; SF‐36 = Short Form 36 health survey (0–100 scale; higher scores indicate better quality of life); PCS = physical component summary score; MCS = mental component score; EQ‐5D: EuroQol 5‐domain questionnaire (0–100 scale; higher scores indicate better health status); ASAS‐NSAID score = ASAS–nonsteroidal antiinflammatory drug score (0–100 scale; higher scores indicate greater NSAID intake); CRP = C‐reactive protein; BASMI = Bath Ankylosing Spondylitis Metrology Index (0–10 scale; higher scores indicate more severe spinal mobility impairment); MASES = Maastricht Ankylosing Spondylitis Enthesitis Score (0–13 scale; higher scores indicate more severe enthesitis).
This treatment group received certolizumab pegol (CZP) 200 mg every other week.
Imputed using nonresponder imputation.
Imputed using last observation carried forward.
Exposure per 100 subject‐years.
Clinically meaningful and statistically significant responses for the BASDAI and the BASFI were achieved in CZP plus NBBM–treated patients and are shown in Table 2. MRI inflammation in the sacroiliac joints significantly improved in CZP plus NBBM–treated patients, decreasing from a mean ± SD sacroiliac joint SPARCC score of 7.79 ± 10.82 at baseline to 1.92 ± 3.96 at week 52, compared to 8.46 ± 12.31 in the placebo‐treated population at baseline to 5.84 ± 10.99 at week 52 (P < 0.0001) (Table 2). There were corresponding improvements in nocturnal spinal pain in CZP plus NBBM–treated patients that were maintained to week 52 (Table 2). Similar improvements and sustained responses were observed in health‐related quality of life outcomes such as the ASQoL and SF‐36 (Table 2). Postbaseline uveitis was observed in 2.5% (4 of 159) of CZP plus NBBM patients compared to 5.1% (8 of 158) of placebo plus NBBM patients, but this difference did not achieve statistical significance. Improvements were also observed in CZP plus NBBM–treated patients for additional end points tested (Table 2).
Treatment‐emergent adverse events during the placebo‐controlled period of the study are shown in Table 3. One malignancy was reported in the placebo plus NBBM group, and 2 malignancies were reported in the CZP plus NBBM group. There were no deaths, serious cardiovascular events, or opportunistic infections (including tuberculosis) during the trial. The adverse events reported were consistent with those already identified for CZP and other anti‐TNF agents 33, 34.
Table 3.
Safety outcomes for all patients (n = 317)a
| Placebo plus NBBM group (n = 158) | CZP plus NBBM group (n = 159)b | |
|---|---|---|
| Any TEAE | 101 (63.9) | 120 (75.5) |
| Serious TEAEs | 3 (1.9) | 8 (5.0) |
| Drug‐related TEAEs | 23 (14.6) | 48 (30.2) |
| Deaths | 0 | 0 |
| TEAEs of interest | ||
| Opportunistic infections | 0 | 0 |
| Tuberculosis | 0 | 0 |
| Malignant or unspecified tumors | 1 (0.6) | 2 (1.3) |
| Serious cardiovascular events | 0 | 0 |
| Hematopoietic cytopenia | 0 | 0 |
| Serious bleeding events | 0 | 0 |
| Hepatic events | 4 (2.5) | 9 (5.7) |
| Hypersensitivity/anaphylactic reactions | 0 | 0 |
| Demyelinating disorders | 0 | 0 |
The safety set consisted of all patients treated with ≥1 dose of study medication. Values are the number (%). NBBM = nonbiologic background medication; TEAE = treatment‐emergent adverse event.
Certolizumab pegol (CZP) 200 mg every 2 weeks.
Fifty‐four patients continued taking placebo plus NBBM throughout the 52‐week period. Of these patients, 51 had assessments at week 52 for ASDAS‐MI and ASAS40. Post hoc analysis revealed that 21.6% (11 of 51) reached ASDAS‐MI, while 49.0% (25 of 51) achieved an ASAS40 response. Demographic details and NBBM use in the 11 placebo plus NBBM patients who reached ASDAS‐MI are provided in Supplementary Table 5, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40866/abstract. The mean change from baseline in the BASDAI score in the 54 patients who continued taking placebo throughout the 52‐week period was −2.90 ± 2.19, compared to −4.28 ± 2.13 in the CZP‐treated patients.
In post hoc analyses investigating the disease activity of patients switching from placebo to open‐label CZP treatment, 96.9% (93 of 96) of patients did not demonstrate an ASDAS‐MI response at the time of switching. Additionally, 93.8% (90 of 96) and 87.5% (84 of 96) of patients were classified as ASAS40 and ASAS20 nonresponders, respectively. At the time of switching, the mean ± SD ASDAS and BASDAI values for these patients were 3.60 ± 0.93 and 6.56 ± 1.69, respectively.
Discussion
The results from this 52‐week placebo‐controlled trial, conducted in patients with active nonradiographic axial SpA with objective signs of inflammation, highlight the efficacy of CZP in the nonradiographic axial SpA population, with clinically relevant and statistically significant differences observed in the primary end points and all but 1 of the secondary end points, which addressed different aspects of the disease (e.g., disease activity, physical function, and pain). All sensitivity analyses, which used alternate approaches to handling missing data, confirmed these results. The only exception was the number of patients with anterior uveitis flares, although this should be interpreted with caution due to the small number of affected patients in each treatment group. Relevant improvements with CZP plus NBBM treatment were also observed for additional outcomes, including objective signs of inflammation (MRI and CRP level), SPARCC at week 52, BASMI, MASES, and patient‐reported quality of life outcomes. The favorable outcomes observed in this study are in accordance with previous phase III placebo‐controlled trials that have evaluated the use of anti‐TNF agents in nonradiographic axial SpA over shorter time frames, ranging from 12 to 16 weeks 1, 35, 36, 37. In addition, safety outcomes were in accordance with those published previously 34.
The unique study design of the trial, which comprised a 1‐year placebo‐controlled period, the ability to change background medication, and the ability to switch to biologic treatment, enabled evaluation of the course of nonradiographic axial SpA and optimization of NBBMs. During the study, substantially more patients randomized to receive placebo switched to open‐label CZP treatment compared to those randomized to receive CZP, although no switches occurred before 12 weeks. The disease activity in placebo patients switching to open‐label CZP treatment remained high at the time of switching, as illustrated by the high ASDAS and BASDAI scores observed at this time point. This high level of disease activity suggests that in the majority of patients with active disease, nonradiographic axial SpA does not spontaneously remit and cannot be controlled with nonbiologic medications. This is further supported by the fact that only 11 of the 158 patients who were randomized to receive placebo could complete 52 weeks and achieve ASDAS‐MI. These findings also highlight the limitations of nonbiologic treatments in patients with active disease with objective signs of inflammation in whom treatment with at least 2 NSAIDs was unsuccessful. This is supported by the fact that 69.6% of placebo plus NBBM patients had changes in their background medication over the course of the study, compared to 27.7% of CZP plus NBBM patients. There were also greater reductions in ASAS‐NSAID scores for CZP plus NBBM patients versus placebo plus NBBM patients over the 52‐week trial period, reflecting reduced use of nonbiologic background medications such as NSAIDs.
The C‐axSpAnd trial was designed to enroll patients with nonradiographic axial SpA who would derive the greatest benefit from treatment with an anti‐TNF agent. To achieve this, patients were included in the study only if they exhibited objective signs of inflammation despite therapy with NSAIDs, and if they had a high level of clinical symptoms, including elevated BASDAI and spinal pain scores. Furthermore, screening radiographs were assessed centrally by 2 experienced readers to ensure that no patients with radiographic axial SpA (ankylosing spondylitis) were included.
Another strength of the C‐axSpAnd study design is its use of ASDAS‐MI as a primary end point. This outcome is a validated, highly discriminatory measure used to assess disease activity, incorporating both patient self‐reported and objective measures of disease activity 8. However, due to the weighting of its constituent measures, the ASDAS may be impacted by substantial changes in CRP level. To address this, improvement in signs and symptoms of disease was also measured using ASAS40, for which a similar effect was observed.
In conclusion, the results from this 52‐week placebo‐controlled trial indicate that nonradiographic axial SpA does not demonstrate spontaneous remission in the majority of patients with active disease. Since improvements in clinical efficacy and objective signs of inflammation were maintained to week 52 in CZP‐treated nonradiographic axial SpA patients, this study demonstrates the significant benefit of using CZP, an anti‐TNF agent, in addition to NBBMs in patients with nonradiographic axial SpA exhibiting objective signs of inflammation.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be submitted for publication. Dr. Deodhar had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Deodhar, Gensler, Kay, Maksymowych, Haroon, Landewé, Rudwaleit, Hall, Bauer, Hoepken, Killgallen, van der Heijde.
Acquisition of data
Deodhar, Gensler, Kay, Maksymowych, Haroon, Hall, Bauer, Hoepken, Killgallen.
Analysis and interpretation of data
Deodhar, Gensler, Kay, Maksymowych, Haroon, Landewé, Rudwaleit, Hall, Bauer, Hoepken, de Peyrecave, Killgallen, van der Heijde.
Role of the study sponsor
UCB Pharma was involved in study conduct, data analysis, data interpretation and writing of the final report. Data collection was performed by a contract research organization. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit. Owen Davies, PhD, Lucian Ionescu, MD, and Victor Sloan, MD (UCB Pharma, Belgium) contributed to study protocol development, and Raphael Teichmann, PhD (UCB Pharma, Germany) contributed to overall study conduct. Simone E. Auteri, MSc, EMS (UCB Pharma, Belgium) assisted with publication coordination, and Jessica Patel, PhD and Eleanor Thurtle, MChem (Costello Medical, UK) provided writing and editorial assistance. Publication of this article was contingent upon approval by UCB Pharma.
Supporting information
Supplementary Appendix A
Supplementary Appendix B
ClinicalTrials.gov identifier: NCT02552212.
Supported by UCB Pharma.
Dr. Deodhar has received consulting fees from AbbVie, Janssen, Pfizer, and UCB Pharma (less than $10,000 each), and from Eli Lilly and Novartis (more than $10,000 each) and research support from those companies. Dr. Gensler has received consulting fees from Galapagos, Eli Lilly, Janssen, and Pfizer (less than $10,000 each) and research support from UCB Pharma, AbbVie, Amgen, and Novartis. Dr. Kay has received consulting fees from AbbVie, Amgen, Boehringer Ingelheim, Celltrion Healthcare, Janssen, Merck, Novartis, Pfizer, Roche, Samsung Bioepis, Sandoz, and UCB Pharma (less than $10,000 each) and research support from AbbVie, Gilead Sciences, Pfizer, Roche, and UCB Pharma. Dr. Maksymowych has received consulting fees and/or honoraria from AbbVie, Eli Lilly, Boehringer, Galapagos, Janssen, Novartis, Pfizer, and UCB Pharma (less than $10,000 each), research support from AbbVie, Pfizer, Janssen, and Novartis, and is Chief Medical Officer of Canadian Research and Education Arthritis. Dr. Haroon has received consulting fees from AbbVie, Amgen, Janssen, Merck, Novartis, and UCB Pharma (less than $10,000 each). Dr. Landewé has received consulting fees from AbbVie, Ablynx, Amgen, AstraZeneca, Bristol‐Myers Squibb, Centocor, Galapagos, GlaxoSmithKline, Janssen, Eli Lilly, Merck, Novartis, Pfizer, Roche, Schering, and UCB Pharma (less than $10,000 each) and research support from those companies. Dr. Rudwaleit has received consulting fees, speaking fees, and/or honoraria from AbbVie, Bristol‐Myers Squibb, Celgene, Janssen, Eli Lilly, MSD, Novartis, Pfizer, Roche, and UCB Pharma (less than $10,000 each). Dr. Hall has received consulting fees from AbbVie, Eli Lilly, Novartis, and UCB Pharma (less than $10,000 each) and research support from those companies. Dr. van der Heijde has received consulting fees from AbbVie, Amgen, Astellas, AstraZeneca, Bristol‐Myers Squibb, Boehringer Ingelheim, Celgene, Daiichi, Eli Lilly, Galapagos, Gilead, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, and UCB Pharma (less than $10,000 each) and is director of Imaging Rheumatology BV. No other disclosures relevant to this article were reported.
UCB Pharma will share anonymized patient‐level data and additional information including clinical study report, study protocols, and statistical analysis plan.
References
- 1. Landewé R, Braun J, Deodhar A, Dougados M, Maksymowych WP, Mease PJ, et al. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24‐week results of a double‐blind randomised placebo‐controlled phase 3 study. Ann Rheum Dis 2014;73:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sieper J, Kivitz AJ, van Tubergen AM, Deodhar AA, Coteur G, Woltering F, et al. Rapid improvements in patient‐reported outcomes with certolizumab pegol in patients with axial spondyloarthritis, including ankylosing spondylitis and non‐radiographic axial spondyloarthritis: 24‐week results of a phase 3 double blind randomized placebo‐controlled study [abstract]. Value Health 2013;16 Suppl:A227. [Google Scholar]
- 3. Baraliakos X, Braun J. Non‐radiographic axial spondyloarthritis and ankylosing spondylitis: what are the similarities and differences? RMD Open 2015;Suppl 1:e000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taurog JD, Chhabra A, Colbert RA. Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med 2016;374:2563–74. [DOI] [PubMed] [Google Scholar]
- 5. Keat A, Bennett AN, Gaffney K, Marzo‐Ortega H, Sengupta R, Everiss T. Should axial spondyloarthritis without radiographic changes be treated with anti‐TNF agents? Rheumatol Int 2017;37:327–36. [DOI] [PubMed] [Google Scholar]
- 6. Wallman JK, Kapetanovic MC, Petersson IF, Geborek P, Kristensen LE. Comparison of non‐radiographic axial spondyloarthritis and ankylosing spondylitis patients: baseline characteristics, treatment adherence, and development of clinical variables during three years of anti‐TNF therapy in clinical practice. Arthritis Res Ther 2015;17:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ward MM, Deodhar A, Akl EA, Lui A, Ermann J, Gensler L, et al. American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network 2015 recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol 2016;68:282–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van der Heijde D, Ramiro S, Landewé R, Baraliakos X, van den Bosch F, Sepriano A, et al. 2016 update of the ASAS‐EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis 2017;76:978–91. [DOI] [PubMed] [Google Scholar]
- 9. Sepriano A, Regel A, van der Heijde D, Braun J, Baraliakos X, Landewé R, et al. Efficacy and safety of biological and targeted‐synthetic DMARDs: a systematic literature review informing the 2016 update of the ASAS/EULAR recommendations for the management of axial spondyloarthritis. RMD Open 2017;3:e000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sieper J, Braun J, Dougados M, Baeten D. Axial spondyloarthritis. Nat Rev Dis Primers 2015;1:15013. [DOI] [PubMed] [Google Scholar]
- 11. Rudwaleit M, Sieper J. Referral strategies for early diagnosis of axial spondyloarthritis. Nat Rev Rheumatol 2012;8:262–8. [DOI] [PubMed] [Google Scholar]
- 12. US Food and Drug Administration . FDA briefing document: Assessment of SpondyloArthritis international Society (ASAS) classification criteria for axial spondyloarthritis and the implications of using these criteria for drug approval. 2013. URL: https://www.pharmamedtechbi.com/~/media/Supporting%20Documents/The%20Pink%20Sheet%20DAILY/2013/July/Spondyloarthritis_AC_FDA_brfg.pdf.
- 13. Kiltz U, Baraliakos X, Karakostas P, Igelmann M, Kalthoff L, Klink C, et al. Do patients with nonradiographic axial spondylarthritis differ from patients with ankylosing spondylitis? Arthritis Care Res (Hoboken) 2012;64:1415–22. [DOI] [PubMed] [Google Scholar]
- 14. Robinson PC, Bird P, Lim I, Saad N, Schachna L, Taylor AL, et al. Consensus statement on the investigation and management of non‐radiographic axial spondyloarthritis (nr‐axSpA). Int J Rheum Dis 2014;17:548–56. [DOI] [PubMed] [Google Scholar]
- 15. Sampaio‐Barros PD, Bortoluzzo AB, Conde RA, Costallat LT, Samara AM, Bertolo MB. Undifferentiated spondyloarthritis: a longterm followup. J Rheumatol 2010;37:1195–9. [DOI] [PubMed] [Google Scholar]
- 16. US Food and Drug Administration . Summary minutes of the Arthritis Advisory Committee meeting. 2013. URL: https://wayback.archive-it.org/7993/20170404145509/https://www.fda.gov/AdvisoryCommittees/Calendar/ucm355358.htm.
- 17. Deodhar A, Reveille JD, van den Bosch F, Braun J, Burgos‐Vargas R, Caplan L, et al. The concept of axial spondyloarthritis: joint statement of the Spondyloarthritis Research and Treatment Network and the Assessment of SpondyloArthritis international Society in response to the US Food and Drug Administration's comments and concerns. Arthritis Rheumatol 2014;66:2649–56. [DOI] [PubMed] [Google Scholar]
- 18. Poddubnyy D, Gensler LS. Spontaneous, drug‐induced, and drug‐free remission in peripheral and axial spondyloarthritis. Best Pract Res Clin Rheumatol 2014;28:807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rudwaleit M, van der Heijde D, Landewé R, Akkoc N, Brandt J, Chou CT, et al. The Assessment of SpondyloArthritis international Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann Rheum Dis 2011;70:25–31. [DOI] [PubMed] [Google Scholar]
- 20. Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol 1994;21:2286–91. [PubMed] [Google Scholar]
- 21. Van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis: a proposal for modification of the New York criteria. Arthritis Rheum 1984;27:361–8. [DOI] [PubMed] [Google Scholar]
- 22. Lambert RG, Bakker PA, van der Heijde D, Weber U, Rudwaleit M, Hermann KG, et al. Defining active sacroiliitis on MRI for classification of axial spondyloarthritis: update by the ASAS MRI working group. Ann Rheum Dis 2016;75:1958–63. [DOI] [PubMed] [Google Scholar]
- 23. Lukas C, Landewé R, Sieper J, Dougados M, Davis J, Braun J, et al, for the Assessment of SpondyloArthritis international Society . Development of an ASAS‐endorsed disease activity score (ASDAS) in patients with ankylosing spondylitis. Ann Rheum Dis 2009;68:18–24. [DOI] [PubMed] [Google Scholar]
- 24. Machado P, Navarro‐Compán V, Landewé R, van Gaalen FA, Roux C, van der Heijde D. Calculating the Ankylosing Spondylitis Disease Activity Score if the conventional C‐reactive protein level is below the limit of detection or if high‐sensitivity C‐reactive protein is used: an analysis in the DESIR cohort. Arthritis Rheumatol 2015;67:408–13. [DOI] [PubMed] [Google Scholar]
- 25. Van der Heijde D, Lie E, Kvien TK, Sieper J, van den Bosch F, Listing J, et al. ASDAS, a highly discriminatory ASAS‐endorsed disease activity score in patients with ankylosing spondylitis. Ann Rheum Dis 2009;68:1811–8. [DOI] [PubMed] [Google Scholar]
- 26. Calin A, Garrett S, Whitelock H, Kennedy LG, O'Hea J, Mallorie P, et al. A new approach to defining functional ability in ankylosing spondylitis: the development of the Bath Ankylosing Spondylitis Functional Index. J Rheumatol 1994;21:2281–5. [PubMed] [Google Scholar]
- 27. Maksymowych WP, Inman RD, Salonen D, Dhillon S, Williams M, Stone M, et al. Spondyloarthritis Research Consortium of Canada magnetic resonance imaging index for assessment of sacroiliac joint inflammation in ankylosing spondylitis. Arthritis Rheum 2005;53:703–9. [DOI] [PubMed] [Google Scholar]
- 28. Doward LC, Spoorenberg A, Cook SA, Whalley D, Helliwell PS, Kay LJ, et al. Development of the ASQoL: a quality of life instrument specific to ankylosing spondylitis. Ann Rheum Dis 2003;62:20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hurst NP, Kind P, Ruta D, Hunter M, Stubbings A. Measuring health‐related quality of life in rheumatoid arthritis: validity, responsiveness and reliability of EuroQol (EQ‐5D). Br J Rheumatol 1997;36:551–9. [DOI] [PubMed] [Google Scholar]
- 30. Ware JE Jr, Snow KK, Kosinski M, Gandek B. SF‐36 health survey: manual and interpretation guide. Boston: The Health Institute, New England Medical Center; 1993. [Google Scholar]
- 31. Jenkinson TR, Mallorie PA, Whitelock HC, Kennedy LG, Garrett SL, Calin A. Defining spinal mobility in ankylosing spondylitis (AS): the Bath AS Metrology Index. J Rheumatol 1994;21:1694–8. [PubMed] [Google Scholar]
- 32. Heuft‐Dorenbosch L, Spoorenberg A, van Tubergen A, Landewe R, van ver Tempel H, Mielants H, et al. Assessment of enthesitis in ankylosing spondylitis. Ann Rheum Dis 2003;62:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maxwell LJ, Zochling J, Boonen A, Singh JA, Veras MM, Tanjong Ghogomu E, et al. TNF‐α inhibitors for ankylosing spondylitis [review]. Cochrane Database Syst Rev 2015;CD005468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van der Heijde D, Dougados M, Landewe R, Sieper J, Maksymowych WP, Rudwaleit M, et al. Sustained efficacy, safety and patient‐reported outcomes of certolizumab pegol in axial spondyloarthritis: 4‐year outcomes from RAPID‐axSpA. Rheumatology (Oxford) 2017;56:1498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sieper J, van der Heijde D, Dougados M, Mease PJ, Maksymowych WP, Brown MA, et al. Efficacy and safety of adalimumab in patients with non‐radiographic axial spondyloarthritis: results of a randomised placebo‐controlled trial (ABILITY‐1). Ann Rheum Dis 2013;72:815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sieper J, van der Heijde D, Dougados M, Maksymowych WP, Scott BB, Boice JA, et al. A randomized, double‐blind, placebo‐controlled, sixteen‐week study of subcutaneous golimumab in patients with active nonradiographic axial spondyloarthritis. Arthritis Rheumatol 2015;67:2702–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dougados M, van der Heijde D, Sieper J, Braun J, Maksymowych WP, Citera G, et al. Symptomatic efficacy of etanercept and its effects on objective signs of inflammation in early nonradiographic axial spondyloarthritis: a multicenter, randomized, double‐blind, placebo‐controlled trial. Arthritis Rheumatol 2014;66:2091–102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Appendix A
Supplementary Appendix B