

Abstract
Ligand conformational entropy plays an important role in carbohydrate recognition events. Glycans are characterized by intrinsic flexibility around the glycosidic linkages, thus in most cases, loss of conformational entropy of the sugar upon complex formation strongly affects the entropy of the binding process. By employing a multidisciplinary approach combining structural, conformational, binding energy, and kinetic information, we investigated the role of conformational entropy in the recognition of the histo blood‐group antigens A and B by human galectin‐3, a lectin of biomedical interest. We show that these rigid natural antigens are pre‐organized ligands for hGal‐3, and that restriction of the conformational flexibility by the branched fucose (Fuc) residue modulates the thermodynamics and kinetics of the binding process. These results highlight the importance of glycan flexibility and provide inspiration for the design of high‐affinity ligands as antagonists for lectins.
Keywords: blood-group antigen, conformational entropy, glycans, lectins, molecular recognition
Molecular recognition of glycans by lectins is essential biological processes with major implications for health and disease.1 From the chemical perspective, these interactions are generally weak, and except for isolated cases,2 they are characterized by a thermodynamic profile in which the favorable binding enthalpy compensates for an unfavorable entropic contribution. Indeed, despite the exo‐anomeric effect,3 which strongly influences the conformational preferences around the glycosidic linkages, the chemical nature of these glycosidic linkages endows glycans with high flexibility. The binding event usually takes place with a loss of conformational entropy, especially for the ligand,4 and as a consequence, large entropic penalties are generally observed.5, 6 Attempts to manipulate this thermodynamic balance have focused on the design of pre‐organized ligands. However, chemically induced modifications have rather unpredictable thermodynamic consequences and in fact, most of these efforts have failed7 by following the paradigm of the enthalpy/entropy compensation mechanism.8
Herein, we report the study of the molecular recognition of the A and B blood‐group tetrasaccharide antigens (A‐ and B‐BGA) by the carbohydrate recognition domain of human galectin‐3 (hGal‐3 CRD). This lectin has been related to several diseases, and it is actually a target for the development of high‐affinity ligands as antagonists for the treatment of certain cancers and fibrotic diseases.9 A‐ and B‐BGAs have been reported to be Gal‐3 binders,10 and to bind with increased affinity compared to lactose.11 However, the basis for this stronger interaction has not been clarified. In this work, we investigated this binding interaction by using a combination of NMR, ITC experiments, and MD simulations and the results demonstrate that sugar flexibility acts as a modulator of the kinetics and thermodynamics of the binding event. Specifically, the fucose (Fuc) moiety does not directly interact with the lectin, but preorganizes the ligand in the bound conformation. Interestingly, the presence of the Fuc moiety has been previously shown to restrict the conformational mobility of branched oligosaccharides, including BGA and Lex antigens.12 The preorganization of the ligand in the bound geometry eliminates the conformational selection event for complex formation, lowers the energy barrier for the association, increases the on‐rate and impacts the binding entropy. As a consequence, the fucosylated tetrasaccharides bind more strongly than their trisaccharide parent analogues, thus making BGA/hGal‐3 an ideal system to unmask the commonly hidden contribution of ligand conformational entropy in binding events.
The structural details of the interaction between N‐acetyl lactosamine (LacNAc, 1; Scheme 1) and LacNAc‐derived glycomimetics with hGal‐3 are well documented.13 The β‐Gal residue provides the key hydrogen‐bonding and π‐stacking interactions, while the other moieties provide additional stabilization to the complex. The type II A‐ and B‐BGAs (4 and 5) are LacNAc derivatives with Galα1‐3 and Fucα1‐2 terminal glycosylations. Thus, the binding of trisaccharides 2 and 3 compared to that of tetrasaccharides 4 and 5 was analyzed to dissect the effect of the different structural elements.
Scheme 1.
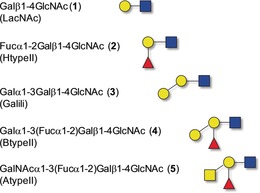
Glycan structures used in this study. Yellow circle: galactose (Gal), blue square: glucosamine (GlcNAc), red triangle: fucose (Fuc), yellow square: galactosamine (GalNAc).
1H‐STD‐NMR experiments14 showed strong saturation‐transfer difference (STD) intensities for H4, H5, and H6 of the β‐Gal residue for the four ligands (2–5), as expected. For 3–5, STDs were also observed for H1 and H2 of αGal or αGalNAc residues. On the contrary, no STD was observed for the protons of the Fuc moiety in ligands 2, 4, and 5 (Figure 1 and Figures S1–S4).
Figure 1.
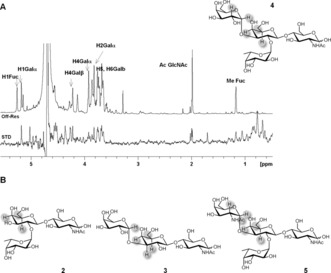
A) 1H‐STD‐NMR spectra and epitope map for the interaction of 4 with hGal‐3. B) STD epitope mapping of sugars 2, 3, and 5.
These data indicate that the four ligands share a common binding mode that is fully consistent with that described for LacNAc, where the βGal residue is the key binding element.13a The αGal/αGalNAc residues in 3, 4, and 5 provide additional contacts with the lectin. By contrast, the Fuc is fully exposed to the solvent, without establishing any direct contact with the lectin.
Additional structural information was obtained through the chemical‐shift perturbations induced by ligands 1–5 in the 1H‐15N heteronuclear single quantum coherence (HSQC) spectra of 15N‐hGal‐3. Interestingly, the addition of 10 equivalents of the Fuc‐containing 2 to hGal‐3 produced basically the same backbone chemical‐shift perturbations as LacNAc (Figure 2), involving residues in the His158‐Arg186 region. Thus, the presence of the α1‐2 Fuc in 2 does not affect any additional lectin site. In contrast, for 3–5, which contain terminal Galα and GalNAcα, additional cross‐peaks were perturbed, corresponding to amino acids located in β‐strands S3 and S4 (Figure 2 and Figures S7–S9). Furthermore, the addition of sub‐stoichiometric amounts of 3–5 to hGal‐3 evidenced two sets of 1H‐15N signals for specific amino acids of the lectin, corresponding to its free and bound forms (Figure S10). The rate of the chemical exchange between the free and bound forms of hGal‐3 is now slow in the NMR chemical‐shift timescale. This behavior differs from the titrations with ligands 1 and 2, for which progressive cross‐peak signal displacement indicated a binding event in the fast‐exchange regime.15
Figure 2.
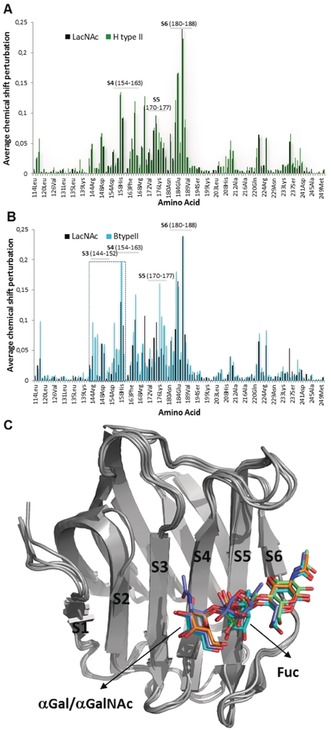
A, B) Chemical‐shift perturbations for the backbone amides of hGal‐3 upon addition of 2 (A, green) and 4 (B, blue) compared to LacNAc (black). For 3 and 5, see the Supporting Information. C) Molecular models for the complexes of hGal‐3 with 2 (green), 3 (orange), 4 (blue), and 5 (purple).
Thus, both STD and HSQC experiments agree on showing that all ligands (2–5) share the same binding mode as LacNAc. The additional αGal/αGalNAc units are located at the so‐called subsite B,16 participating in direct contacts with residues in the β‐strand S3, while the Fuc moiety in 2, 4, and 5 is exposed to the solvent. Molecular dynamics (MD) simulations for each complex built according to the NMR data were stable along the 100 ns run (Figure 2 C and the Supporting Information).
The thermodynamic parameters for the binding of 1–5 to hGal‐3 were obtained by isothermal titration calorimetry (ITC; Table 1, Figure 3). The dissociation constants (K D) for lactose and LacNAc were similar to those reported,17 with the typical negative enthalpy counteracted by a positive entropy.18 The addition of a terminal αGal residue in compound 3 increased the affinity, essentially due to the enthalpic contribution, which is in agreement with the proposed 3D model that showed additional contacts for this residue. The introduction of the α1‐2 linked Fuc moiety in 2, 4, and 5 also improved the binding affinity, as previously reported.10 However, in this case, the enthalpy was lower in comparison with the non‐fucosylated counterparts, and the improved affinity actually arises from the entropic term, with the unfavorable entropy contribution being significantly smaller. The drop in the TΔS term is especially remarkable for tetrasaccharides 4 and 5. These results contrast with previously reported thermodynamic studies, which proposed the existence of enhanced enthalpy contributions to the binding.11
Table 1.
K D and thermodynamic parameters of the interaction of hGal‐3 with 1–5, as determined by ITC.
| Glycan |
K
D
[μm] |
ΔG
[kcal mol−1] |
ΔH
[kcal mol−1] |
−TΔS
[kcal mol−1] |
|---|---|---|---|---|
| Lactose | 112.0 | −5.44 | −9.85 | 4.40 |
| LacNAc (1) | 53.2 | −5.87 | −10.57 | 4.69 |
| H type II (2) | 37.2 | −6.10 | −9.41 | 3.31 |
| Galili (3) | 18.6 | −6.51 | −11.74 | 5.23 |
| B type II (4) | 4.4 | −7.38 | −8.94 | 1.56 |
| A type II (5) | 6.2 | −7.17 | −7.85 | 0.68 |
Figure 3.
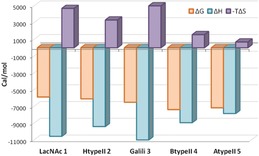
Thermodynamic profiles for the interaction of 1–5 with hGal‐3, as measured by ITC experiments.
Finally, the binding kinetics for the interaction of glycans 3–5 with hGal‐3 were characterized by taking advantage of the slow‐exchange regime in the NMR chemical‐shift timescale, which allowed the observation of separate free and bound cross‐peaks for some protons of the ligands. This is clear in the trROESY spectrum, where the chemical‐exchange cross‐peaks display opposite signs to those arising from trNOE (Figure 4 A). Indeed, for 3–5, the signals of H4, H5, and H6 of the central βGal residue are shifted dramatically up‐field in the bound form. This effect perfectly agrees with the proposed binding mode, in which the central βGal sits on top of the electron‐rich indole of W181, establishing key CH–π stacking interactions (Figure 4 B).19 These chemical exchange cross‐peaks were employed to estimate the off‐rate constants k off (see the Supporting Information) through exchange spectroscopy (EXSY) experiments.20 The association rate constants k on were deduced by using the K D determined by ITC (Table 2). Remarkably, the binding of 3–5 displayed different kinetic parameters (see the Supporting Information). Glycans 4 and 5 show similar off‐rates to 3, but bind with a faster k on, thus reducing the K D.
Figure 4.

A) 2D‐NMR trROESY spectrum of 1:10 hGal‐3/4. B) MD‐derived structure of the hGal‐3/B‐BGA (4) complex.
Table 2.
Dissociation constants and exchange rates of binding to hGal‐3.
| Compound | K D [μm] [a] | k off [s−1][b] | k on [m −1 s−1][c] |
|---|---|---|---|
| Galili (3) | 18.6 | 0.32 | 17 204 |
| B type II (4) | 4.4 | 0.44 | 10 0000 |
| A type II (5) | 6.2 | 0.43 | 69 354 |
[a] Determined by ITC. [b] Determined from EXSY. [c] Calculated from K D and k off (K D=k off/k on).
Hence, in comparison with 3, the presence of the Fuc unit in 4 and 5 strongly reduces the kinetic barrier for ligand association. This is in fact the origin of the higher binding affinity, which is, in turn, directly related to the reduced binding entropy.
It is well known that the presence of α1‐2‐Fuc reduces the internal motion in ABO blood‐group oligosaccharides.12, 21 Our NMR and MD results (see the Supporting Information for details) agree with the existence of a single conformational family for 2, 4 and 5. The situation is rather different for the non‐fucosylated trisaccharide 3, for which MD simulations predict that the Galα1‐3Galβ linkage is rather flexible, with continuous transitions along the entire trajectory between the syn‐Φ/syn(−)‐Ψ and syn‐Φ/syn(+)‐Ψ geometries (Figure 5 B). In contrast, detailed conformational analysis for the bound state showed single narrow minima for all glycosidic linkages. Thus, according to our results, trisaccharide 3 would acquire conformational stiffness upon complexation, which does not occur for the already preorganized fucosylated ligands, which display the same conformation in the free and bound states. In this way, the conformational restriction imposed by the Fuc in the branched sugars provides the proper presentation of the ligand epitope, thereby increasing the probability of successful collisions when they encounter the protein, and thus increasing the on‐rates. The energy barrier of the association event is then reduced. Likewise, the minimal rigidification taking place upon binding explains the minimal entropy penalty observed for the fucosylated tetrasaccharides.
Figure 5.
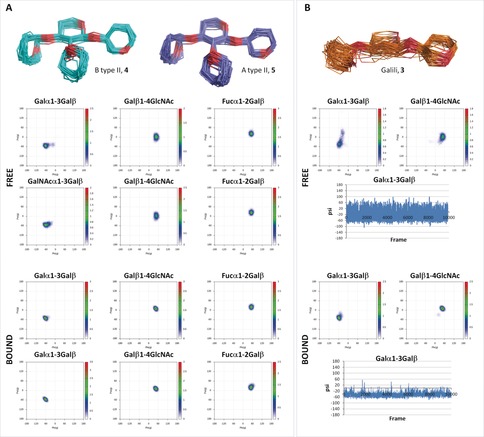
MD‐based conformational analysis of 3–5 in the free and bound states.
Therefore, this study highlights the key contribution of ligand flexibility to both equilibrium thermodynamics and binding kinetics. This contribution is probably always present, but its clear participation is difficult to address. In this particular case, the chemical nature of the partners (the architecture of the binding site for hGal‐3 and the rigidity of the histo blood antigens) has allowed us to bring to light an effect that might be usually hidden.
In conclusion, conformationally restricted fucosylated A‐ and B‐BGAs show faster association rates and less unfavorable binding entrop than non‐fucosylated analogues. As a key factor, the Fuc residue does not directly interact with the protein, but provides the required preorganization of the ligand to improve the binding affinity. These features provide inspiration for the chemical design of high affinity ligands as antagonists for hGal‐3 or for other lectins or receptors of biomedical relevance.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Avadhesha Surolia is a Science and Engineering Research Board (SERB—India) Distinguished Fellow. We thank Agencia Estatal de Investigacion and ISCIII of Spain and the European Research Council for financial support. The plasmid for hGal‐3 was kindly provided by Dr. Filipa Marcelo (Universidade Nova Lisboa). We thank Dr. Sonia Huecas (CIB‐CSIC) for discussions on ITC and Prof. H.‐J. Gabius for discussions in early stages of this research.
A. Gimeno, S. Delgado, P. Valverde, S. Bertuzzi, M. A. Berbís, J. Echavarren, A. Lacetera, S. Martín-Santamaría, A. Surolia, F. J. Cañada, J. Jiménez-Barbero, A. Ardá, Angew. Chem. Int. Ed. 2019, 58, 7268.
Contributor Information
Prof. Dr. Jesus Jiménez‐Barbero, Email: jjbarbero@cicbiogune.es.
Dr. Ana Ardá, Email: aarda@cicbiogune.es.
References
- 1. Cummings R. D., Schnaar R. L., Esko J. D., Drickamer K., Taylor M. E., Essentials of Glycobiology, 3rd ed. (Eds.: A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar, P. H. Seeberger), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 2015. –2017. [PubMed] [Google Scholar]
- 2.
- 2a. Sager C. P., Fiege B., Zihlmann P., Vannam R., Rabbani S., Jakob R. P., Preston R. C., Zalewski A., Maier T., Peczuh M. W., Ernst B., Chem. Sci. 2018, 9, 646–654; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Binder F. P. C., Lemme K., Preston R. C., Ernst B., Angew. Chem. Int. Ed. 2012, 51, 7327–7331; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7440–7444, and references therein; [Google Scholar]
- 2c. Topin J., Lelimousin M., Arnaud J., Audfray A., Pérez S., Varrot A., Imberty A., ACS Chem. Biol. 2016, 11, 2011–2020. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Tvaroska I., Bleha T., Adv. Carbohydr. Chem. Biochem. 1989, 47, 45–123; [Google Scholar]
- 3b. Lemieux R. U., Morgan A. R., Can. J. Chem. 1965, 43, 2205–2213. [Google Scholar]
- 4. Verteramo M. L., Stenström O., Ignjatović M. M., Caldararu O., Olsson M. A., Manzoni F., Leffler H., Oksanen E., Logan D. T., Nilsson U. J., Ryde U., Akke M., J. Am. Chem. Soc. 2019, 141, 2012–2026. [DOI] [PubMed] [Google Scholar]
- 5. Diehl C., Engström O., Delaine T., Håkansson M., Genheden S., Modig K., Leffler H., Ryde U., Nilsson U. J., Akke M., J. Am. Chem. Soc. 2010, 132, 14577–14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carver J. P., Pure Appl. Chem. 1993, 65, 763–770. [Google Scholar]
- 7. Navarre N., Amiot N., Van Oijen A., Imberty A., Poveda A., Jiménez-Barbero J., Cooper A., Nutley M. A., Boons G.-J., Chem. Eur. J. 1999, 5, 2281–2294. [Google Scholar]
- 8. Fox J. M., Zhao M., Fink M. J., Kang K., Whitesides G. M., Annu. Rev. Biophys. 2018, 47, 223–250. [DOI] [PubMed] [Google Scholar]
- 9. Girard A., Magnani J. L., Trends Glycosci. Glycotechnol. 2018, 30, SE211–SE220. [Google Scholar]
- 10. Stowell S. R., Arthur C. M., Mehta P., Slanina K. A., Blixt O., Leffler H., Smith D. F., Cummings R. D., J. Biol. Chem. 2008, 283, 10109–10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bachhawat-Sikdera K., Thomasa C. J., Surolia A., FEBS Lett. 2001, 500, 75–79. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Imberty A., Pérez S., Chem. Rev. 2000, 100, 4567–4588; [DOI] [PubMed] [Google Scholar]
- 12b. Zierke M., Smieško M., Rabbani S., Aeschbacher T., Cutting B., Allain F. H.-T., Schubert M., Ernst B., J. Am. Chem. Soc. 2013, 135, 13464–13472; [DOI] [PubMed] [Google Scholar]
- 12c. Battistel M. D., Azurmendi H. F., Frank M., Freedberg D. I., J. Am. Chem. Soc. 2015, 137, 13444–13447; [DOI] [PubMed] [Google Scholar]
- 12d. Aeschbacher T., Zierke M., Smiesko M., Collot M., Mallet J.-M., Ernst B., Allain F. H.-T., Schubert M., Chem. Eur. J. 2017, 23, 11598–11610. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Sörme P., Arnoux P., Kahl-Knutsson B., Leffler H., Rini J. M., Nilsson U. J., J. Am. Chem. Soc. 2005, 127, 1737–1743; [DOI] [PubMed] [Google Scholar]
- 13b. Hsieh T.-J., Lin H.-Y., Tu Z., Huang B.-S., Wu S.-C., Lin C.-H., Plos One 2015, 10, e0125946; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Atmanene C., Ronin C., Teletchea S., Gautier F. M., Djedaini-Pilard F., Ciesielski F., Vivat V., Grandjean C., Biochem. Biophys. Res. Commun. 2017, 489, 281–286; [DOI] [PubMed] [Google Scholar]
- 13d. Seetharaman J., Kanigsberg A., Slaaby R., Leffler H., Barondes S. H., Rini J. M., J. Biol. Chem. 1998, 273, 13047–13052. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Mayer M., Meyer B., J. Am. Chem. Soc. 2001, 123, 6108–6117; [DOI] [PubMed] [Google Scholar]
- 14b. Marchetti R., Perez S., Arda A., Imberty A., Jimenez-Barbero J., Silipo A., Molinaro A., ChemistryOpen 2016, 5, 274–296; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Blaum B. S., Neu U., Peters T., Stehle T., Acta Crystallogr. Sect. F 2018, 74, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Williamson M. P., Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [DOI] [PubMed] [Google Scholar]
- 16. Leffler H., Carlsson S., Hedlund M., Qian Y., Poirier F., Glycoconjugate J. 2002, 19, 433–440. [DOI] [PubMed] [Google Scholar]
- 17. Cumpstey I., Salomonsson E., Sundin A., Leffler H., Nilsson U., ChemBioChem 2007, 8, 1389–1398. [DOI] [PubMed] [Google Scholar]
- 18.The difference in binding affinity between lactose and LacNac was due mainly to the enthalpic term. This is in agreement with the structural differences found in the X-Ray crystallographic structures of the complexes hGal-3/lactose and hGal-3/lacNAc described in Ref. [13d].
- 19. Asensio J. L., Ardá A., Cañada F. J., Jiménez-Barbero J., Acc. Chem. Res. 2013, 46, 946–954. [DOI] [PubMed] [Google Scholar]
- 20. Latham M. P., Zimmermann G. R., Pardi A., J. Am. Chem. Soc. 2009, 131, 5052–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Lemieux R. U., Bock K., Delbaere L. T. J., Koto S., Rao V. S. R., Can. J. Chem. 1980, 58, 631–653; [Google Scholar]
- 21b. Rao B. N. N., Dua V. K., Bush C. A., Biopolymers 1985, 24, 2207–2229; [DOI] [PubMed] [Google Scholar]
- 21c. Bush C. A., Yan Z.-Y., Rao B. N. N., J. Am. Chem. Soc. 1986, 108, 6168–6173; [Google Scholar]
- 21d. Yan L.-Y., Bush C. A., Biopolymers 1990, 29, 799–811; [DOI] [PubMed] [Google Scholar]
- 21e. Cagas P., Bush C. A., Biopolymers 1992, 32, 277–292; [DOI] [PubMed] [Google Scholar]
- 21f. Azurmendi H. F., Bush C. A., Carbohydr. Res. 2002, 337, 905–915. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary