

Abstract
No curative treatment options are available for advanced hepatocellular carcinoma (HCC). Anti‐PD1 antibody therapy can induce tumor regression in 20% of advanced HCC patients, demonstrating that co‐inhibitory immune checkpoint blockade has therapeutic potential for this type of cancer. However, whether agonistic targeting of co‐stimulatory receptors might be able to stimulate anti‐tumor immunity in HCC is as yet unknown. We investigated whether agonistic targeting of the co‐stimulatory receptor GITR could reinvigorate ex vivo functional responses of tumor‐infiltrating lymphocytes (TIL) freshly isolated from resected tumors of HCC patients. In addition, we compared GITR expression between TIL and paired samples of leukocytes isolated from blood and tumor‐free liver tissues, and studied the effects of combined GITR and PD1 targeting on ex vivo TIL responses. In all three tissue compartments, CD4+FoxP3+ regulatory T cells (Treg) showed higher GITR−expression than effector T‐cell subsets. The highest expression of GITR was found on CD4+FoxP3hiCD45RA− activated Treg in tumors. Recombinant GITR‐ligand as well as a humanized agonistic anti‐GITR antibody enhanced ex vivo proliferative responses of CD4+ and CD8+ TIL to tumor antigens presented by mRNA‐transfected autologous B‐cell blasts, and also reinforced proliferation, IFN‐γ secretion and granzyme B production in stimulations of TIL with CD3/CD28 antibodies. Combining GITR ligation with anti‐PD1 antibody nivolumab further enhanced tumor antigen‐specific responses of TIL in some, but not all, HCC patients, compared to either single treatment. In conclusion, agonistic targeting of GITR can enhance functionality of HCC TIL, and may therefore be a promising strategy for single or combinatorial immunotherapy in HCC.
Keywords: GITR, CD357, TNFRSF18, Treg, HCC, cancer immunotherapy, PD1
Short abstract
What's new?
Therapeutic antibodies that block interaction of the T cell co‐inhibitory receptor PD1 can unleash pre‐existing anti‐cancer T cell responses in hepatocellular carcinoma (HCC). However, whether agonistic targeting of co‐stimulatory receptors could stimulate anti‐tumor immunity remains unknown. This study is the first to show that agonistic targeting of the co‐stimulatory receptor GITR can reinforce the functionality of tumor‐infiltrating T cells isolated from human tumors. Combined targeting of PD1 and GITR exerts additive stimulatory effects on ex vivo functionality of tumor‐infiltrating T cells from HCC patients. Targeting of GITR thus emerges as a promising strategy for single or combinatorial immunotherapy in HCC.
Abbreviations
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- GITRL
GITR ligand
- GPC3
glypican 3
- HCC
hepatocellular carcinoma
- MAGEC2
MAGE family member C2
- PBMC
peripheral blood mononuclear cells
- PD1
programmed cell death 1
- Treg
regulatory T cells
- Th
T helper cells
- GITR
TNF receptor superfamily member 18
- TFL
tumor‐free liver
- TIL
tumor‐infiltrating lymphocytes
Introduction
Liver cancer is the second most common cause of cancer‐related mortality worldwide, with approximately 750,000 deaths per year. The most common primary liver cancer is hepatocellular carcinoma (HCC), an aggressive malignancy derived from hepatocytes.1, 2 Surgical resection and liver transplantation are curative therapies for patients with early stage disease. However, about 50% of HCC patients present with advanced disease at diagnosis and can only be offered systemic therapies which provide limited survival advantage.3, 4 Therefore, novel therapies for HCC are urgently needed.
Immune checkpoint antibodies are a new class of cancer immune therapeutics. T cells are activated upon antigen recognition via their T‐cell receptor and engagement of their co‐stimulatory immune checkpoint receptors with corresponding ligands on other cells, while they are suppressed upon interaction of their co‐inhibitory immune checkpoint receptors with their ligands. Therapeutic antibodies that block interaction of the co‐inhibitory receptor PD1 with its ligands can unleash pre‐existing anti‐cancer T‐cell responses within tumors, and have resulted in recent breakthroughs in clinical treatment of several types of advanced cancer.5, 6, 7, 8, 9, 10, 11, 12, 13 In HCC, a recent trial showed significant tumor load reduction (objective response) in response to anti‐PD1 antibody (nivolumab) therapy in about 20% of advanced HCC patients, and disease control with stable disease for ≥6 months in another 17% of patients.14 Nevertheless, more than 50% of advanced HCC patients did not respond to nivolumab. Therefore, more effective immunotherapies and optimal patient selection are still required for HCC.
Besides blockade of co‐inhibitory receptors, agonistic targeting of co‐stimulatory receptors has the potential to boost intra‐tumoral T‐cell immunity to combat cancer growth and evoke cancer regression. Importantly, in addition to activating intra‐tumoral T‐cell responses, targeting co‐stimulatory receptors can stimulate systemic anti‐tumor immunity which may protect against tumor recurrence.15 Currently, antibodies targeting different co‐stimulatory receptors are being evaluated in clinical trials for several types of solid cancer.16, 17
One of the co‐stimulatory receptors under active clinical investigation in solid malignancies is CD357, TNF receptor superfamily member 18 (TNFRSF18), also known as glucocorticoid‐induced TNFR‐related protein (GITR). We have previously revealed that tumor‐infiltrating T cells in HCC are functionally compromised. This is due to co‐inhibitory interactions,18, 19 and to high numbers of conventional CD4+FoxP3+CD25+ regulatory T cells (Treg)19 and type 1 regulatory T cells20 present in liver tumors, which inhibit functions of effector T cells. We also demonstrated that agonistic targeting of GITR can alleviate the suppressive capacity of liver tumor‐derived CD4+CD25+ Treg.21 However, the expression of GITR on cytotoxic CD8+ tumor‐infiltrating lymphocytes (TIL) and CD4+FoxP3− T helper cells (Th) in HCC patients is unknown. In addition, whether agonistic targeting of GITR can boost anti‐tumor responses of HCC patient‐derived TIL has not been studied, nor is it known whether combining GITR ligation with PD1 blockade may have synergistic effects on HCC patient‐derived TIL proliferation and activation.
Therefore, in a proof‐of‐concept preclinical study, we investigated whether agonistic targeting of the co‐stimulatory receptor GITR could reinvigorate ex vivo functional responses of TIL from HCC patients. In addition, we compared GITR expression between TIL and paired samples of leukocytes isolated from peripheral blood and tumor‐free liver (TFL) tissues, and studied the effects of combined GITR and PD1 targeting on ex vivo TIL responses.
Patients and Methods
Patients
Thirty‐five patients who were eligible for surgical resection of HCC and two HCC patients who were eligible for liver transplantation were enrolled in the study from January 2014 to December 2017. Paired fresh tissue samples from tumors and surrounding (minimum 1 cm distance from the tumor) tumor‐free liver tissues were obtained, and tumor‐infiltrating leukocytes and intra‐hepatic leukocytes were isolated, respectively. From part of the patients peripheral blood was collected on the day of resection. None of the patients received chemotherapy or immunosuppressive treatment within three months prior to the surgery. The clinical characteristics of the patients are summarized in Table 1. Eighty percent of the patients included in the study are from Caucasian origin. The study was approved by the local ethics committee, and all the patients signed the informed consent before tissue and blood donation.
Table 1.
Patient characteristics
| HCC (n = 37) | |
|---|---|
| Sex (male/female) | 25 / 12 |
| Age (years)1 | 61.2 ± 3.2 |
| Race (Caucasian/Asian/African) | 32 / 2 / 3 |
| Liver fibrosis (metavir score) F0 / F2 / F3 / F4‐cirrhosis | 17 / 5 / 3 / 12 |
| AFP (μg/L): <20 /20–400/ >400 | 26 / 3 / 8 |
| Stage of disease (TNM) | St I n = 16, St II n = 16, St IIIa n = 3, St IVa n = 1, unknown 1 |
Abbreviations: AFP, alpha fetoprotein; TNM, tumor‐node‐metastasis.
Etiology of liver disease: no known liver disease (n = 14), HBV/HCV (n = 4/5), alcohol‐related liver disease (n = 4), HBV + alcohol (n = 1), non‐alcoholic steatohepatitis (NASH)/non‐alcoholic fatty liver disease (NAFLD) (n = 2/3), HBV + NASH (n = 1), Abernathy (n = 1), hemochromatosis (n = 2).
Mean ± SEM.
Cell preparation
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll density gradient centrifugation. Single cell suspensions from tumors and TFL were obtained by tissue digestion. Briefly, fresh tissues were cut into small pieces and digested with 0.125 mg/ml collagenase IV (Sigma‐Aldrich, St. Louis, MO) and 0.2 mg/ml of DNAse I (Roche, Indianapolis, IN) in Hanks' Balanced Salt solution with Ca2+ and Mg2+ (Sigma, Zwijndrecht, The Netherlands) for 30–60 min under continuous stirring at 37°C. Cell suspensions were filtered through 100 μm pore cell strainers (BD Biosciences, Erembodegem, Belgium) and mononuclear leukocytes were obtained by Ficoll density gradient centrifugation. Viability was determined by trypan blue exclusion.
Flow cytometric analysis
Fresh PBMC and mononuclear leukocytes isolated from tumors and TFL were analyzed for expression of surface and intracellular markers using specific antibodies (Supporting Information Table 1). Dead cells were excluded using a LIVE/DEAD fixable dead cell stain kit with aqua fluorescent reactive dye (Invitrogen, Paisley, UK) or Live/dead stain eFluor506 (eBioscience, Vienna, Austria). Cell surface staining with fluorochrome‐conjugated antibodies was performed in the dark at 4°C for 30 min, after which cells were fixed and permeabilized using the FoxP3 staining buffer set (eBioscience) and stained for intracellular antigens. Cells were measured by a FACSCanto II or a FACSAria SORP II flow cytometer (BD Biosciences, San Diego, USA) and analyzed using FlowJo software version X.0.7 (TreeStar Inc.). Appropriate isotype control antibodies were used for gating purposes.
Ex vivo polyclonal T‐cell activation assay
All TIL cultures were performed in RPMI 1640 (Lonza, Breda, The Netherlands) supplemented with 10% human AB serum (Sigma), 2 mM L‐glutamine (Invitrogen), 50 mM Hepes Buffer (Lonza), 1% penicillin–streptomycin (Life Technologies), 5 mM Sodium Pyruvate (Gibco) and 1% minimum essential medium nonessential amino acids (MEM NEAA) (complete medium) at 37°C. TIL were cultured in 96‐well round‐bottom culture plates (106 cells/mL) and typically stimulated with anti‐human CD3/CD28 Dynabeads (Gibco‐Life Technologies AS, Norway) at a cell: bead ratio of 10:1. This ratio provides suboptimal T‐cell stimulation.18 To test the effects of GITR stimulation, 1 μg/mL azide‐free and low endotoxin soluble GITR ligand (GITRL, RnD systems) crosslinked with 2.5 μg/mL anti‐HA antibody (RnD systems), or 10 μg/mL humanized IgG1 agonistic antibody against GITR (Pfizer Inc.), or the corresponding isotype control antibody (anti‐HA hIgG1 clone 4FNL; Pfizer Inc.) were added. After 4–5 days, culture supernatant was collected and production of IFN‐γ and granzyme B was quantified by LegendPlex (BioLegend, San Diego, USA). Cells were harvested and stained with anti‐CD8, anti‐CD4, and anti‐CD3 antibodies (surface staining) and anti‐Ki‐67 antibody (nuclear staining). Dead cells were excluded using the LIVE/DEAD fixable dead cell stain kit with aqua fluorescent reactive dye, and T‐cell proliferation was determined based on Ki‐67 positivity by flow cytometry analysis.
Ex vivo mRNA‐encoded full length tumor antigen‐specific T‐cell stimulation assay
Expansion of patient B cells from PBMC by stimulation with trimeric CD40 ligand and IL‐4, in vitro generation of eGFP, glypican 3 (GPC3) and MAGEC2 mRNA, and mRNA electroporation of B‐cell blasts were performed as previously described.18
Tumor‐infiltrating leukocytes were labeled with 0.1 μM carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen), after which 105 TIL in 100 μl complete medium were transferred to each well of 96‐well round‐bottom culture plates. GPC3 mRNA‐, MAGEC2 mRNA‐ or (as a negative control) eGFP mRNA‐transfected autologous CD40‐activated B cells (B cell blasts) in 100 μl of the same medium were added at a TIL:B‐cell ratio of 1:1. TIL were co‐cultured with B‐cell blasts in the presence or absence of 1 μg/mL GITRL crosslinked with 2.5 μg/mL anti‐HA antibody, or 10 μg/mL humanized agonistic antibody against GITR (Pfizer Inc.), or 10 μg/mL nivolumab (BMS, obtained from Erasmus MC hospital pharmacy), or corresponding isotype control antibodies (hIgG1 and anti‐BHV hIgG4 clone 26H6 (Pfizer Inc.), respectively). After 6 days, supernatants were stored for later cytokine analysis, while cells were harvested, and stained with CD3, CD4, and CD8 antibodies. Dead cells were excluded using the LIVE/DEAD fixable dead cell stain kit with aqua fluorescent reactive dye, and T‐cell proliferation was determined based on CFSE dilution by flow cytometry analysis.
Statistical analysis
All data set distributions were analyzed for normality by the Shapiro–Wilk normality test. Differences between paired groups of data were analyzed according to their distribution by either paired t test or Wilcoxon matched pairs test. The statistical analysis was performed using GraphPad Prism Software (version 5.0). p Values less than 0.05 were considered statistically significant in Figures 3 (except 3C), 4, 5, 7 and Supporting Information Figure 2 (* = p < 0.05; ** = p < 0.01; *** = p < 0.001). Bonferroni correction was used to correct for multiple hypotheses testing, so p values less than 0.025 (0.05 divided by 2) were considered statistically significant in Figures 1, 2, 3 c and Supporting Information Figure 1 (* = p < 0.025; ** = p < 0.01; *** = p < 0.001).
Figure 3.
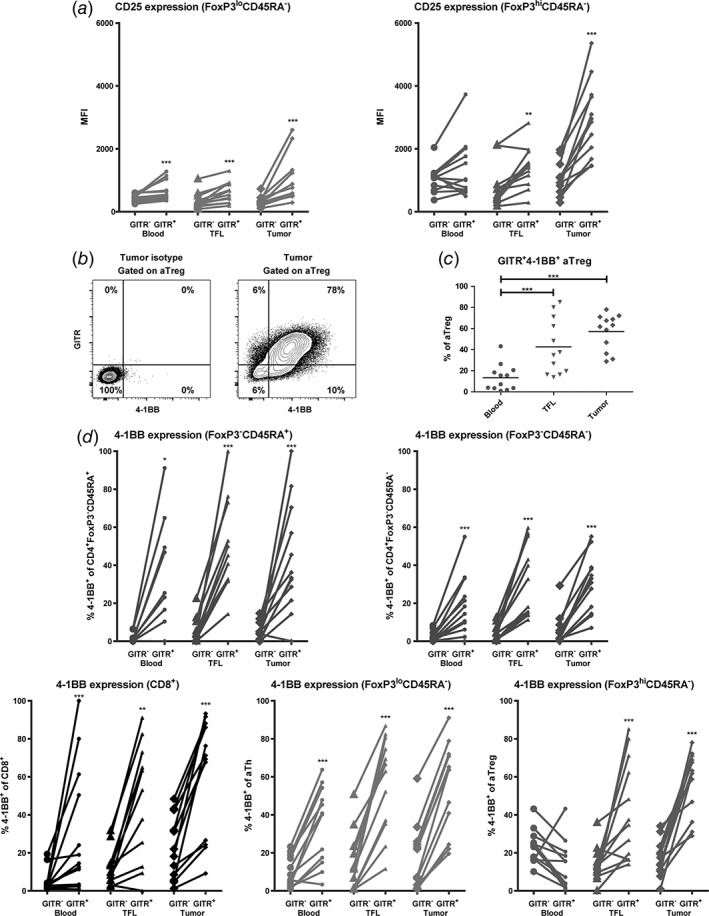
GITR expression on activated T cell subsets is accompanied by increased CD25 and 4‐1BB expression. (a) GITR+ activated T helper (CD4+FoxP3loCD45RA−) and activated regulatory T cell (CD4+FoxP3hiCD45RA−) subsets express higher levels of CD25 than their GITR− counterparts. (b) Representative example of flow cytometric analysis, showing co‐expression of GITR and 4‐1BB on intra‐tumoral activated Treg (aTreg) (fraction II). (c) Percentages of GITR+4‐1BB+ activated Treg in blood, TFL, and tumor. Dots represent individual patients and lines present means. (d) 4‐1BB+ frequencies within GITR− versus GITR+ cells of CD8+ T cells and four different CD4+ T cell subsets in blood, TFL, and tumor. N = 12 patients. * = p < 0.05; ** = p < 0.01; *** = p < 0.001.
Figure 1.
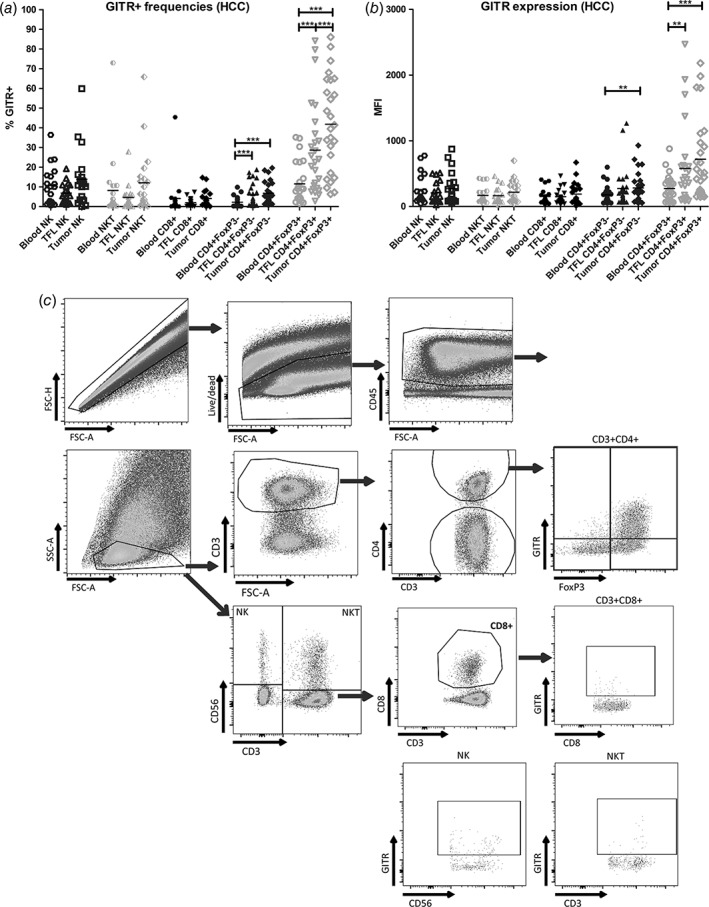
CD4+FoxP3+ TIL from HCC patients show highest GITR expression. (a) GITR+ cell frequencies within NK cells, NKT cells, CD8+, CD4+FoxP3− and CD4+FoxP3+ T cell subsets in mononuclear leukocytes isolated from blood, tumor‐free liver (TFL), and tumor tissues from HCC patients. (b) As in (a), showing median fluorescence intensities (MFI) of GITR expression on the indicated immune cell subsets. Dots represent individual patients and lines present means. N = 19–26 patients. ** = p < 0.01; *** = p < 0.001. (c) Representative FACS dot plots of one patient display the gating strategies.
Figure 2.
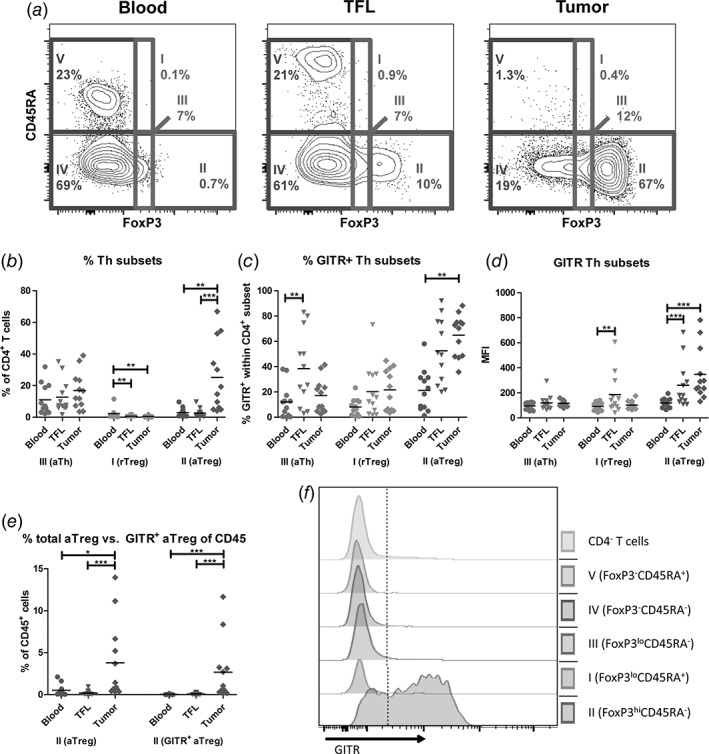
Activated Treg are highly enriched in HCC tumors and express the highest levels of GITR. (a) Representative example of flow cytometric analysis of CD4+ T cell subsets by FoxP3 and CD45RA, identifying five fractions: FoxP3loCD45RA+ resting Treg (fraction I), FoxP3hiCD45RA− activated Treg (fraction II), FoxP3loCD45RA− activated Th (fraction III), FoxP3−CD45RA− memory Th (fraction IV), and FoxP3−CD45RA+ naïve Th (fraction V). (b) Percentages of fractions I‐III within CD4+ T cells. (c) GITR+ frequencies within fractions I‐III. (d) Median fluorescence intensities (MFI) of GITR expression on cells within fractions I‐III. (e) Percentages of total activated Treg (left) and GITR+ activated Treg (right) within CD45+ mononuclear leukocytes. Dots represent individual patients and lines present means. N = 12 patients. * = p < 0.025; ** = p < 0.01; *** = p < 0.001. (f) Representative FACS histograms of one patient display the expression of GITR in CD4+ T cell subsets.
Results
CD4+FoxP3+ TIL show high GITR expression
First, we determined frequencies of CD3−CD56+ NK cells, CD3+CD56+ NKT cells, CD8+ T cells, total CD4+ T cells, CD4+FoxP3− T cells, and CD4+FoxP3+ T cells within the CD45+ cells isolated from blood, TFL tissues, and tumors of HCC patients. While tumors contained lower frequencies of NK cells and NKT cells, they contained more T cells within CD45+ cells than TFL (Supporting Information Fig. 1A). The distribution of T‐cell subsets further showed that CD4+FoxP3+ T cells accumulated within tumors but CD4+FoxP3− T helper cells not, while proportions of CD8+ T cells in tumors were reduced compared to TFL (Supporting Information Fig. 1B). We then analyzed GITR expression within these immune cell subsets. The proportions of GITR‐expressing NK cells (on average 12% in the tumor), NKT cells (on the average 12% in the tumor), and CD8+ T cells (on average 5% in the tumor) did not differ among tumor, TFL and blood (Fig. 1a‐c). In contrast, GITR expression was slightly increased on CD4+FoxP3− T cells in tumor and TFL as compared to blood, with on average 7% of CD4+FoxP3− cells expressing GITR (Fig. 1a‐c). The highest expression of GITR was observed on CD4+FoxP3+ TIL (Fig. 1b), with an average of 45% of cells expressing GITR, which was significantly higher than CD4+FoxP3+GITR+ T‐cell proportions in blood and TFL (Fig. 1a). No significant differences in GITR expression on immune cell subsets in tumors were observed between patients with ‘no underlying liver disease’, HBV or HCV viral hepatitis, or other diseases i.e. alcohol‐related, NASH, NAFLD etc., or between patients with no liver fibrosis and patients with liver fibrosis (metavir score F2‐F4) (data not shown).
CD4+FoxP3hiCD45RA− activated Treg are highly abundant in the tumor and display highest GITR expression
Because functional Treg subsets can be defined by CD45RA expression,22 we included CD45RA to further define FoxP3‐expressing CD4+ T‐cell populations. The combination of FoxP3 and CD45RA leads to the identification of five CD4+ T‐cell subsets: FoxP3loCD45RA+ resting Treg (fraction I), FoxP3hiCD45RA− activated Treg (fraction II), FoxP3loCD45RA− activated Th (fraction III), FoxP3−CD45RA− memory Th (fraction IV), and FoxP3−CD45RA+ naïve Th (fraction V; Fig. 2a). We evaluated the CD4+FoxP3+ subset distributions in blood, TFL, and tumor. There was no significant difference in the frequencies of activated Th (fraction III) within CD4+ T cells among the three compartments (Fig. 2b). Resting Treg (fraction I) were present at low frequencies in blood but rare in liver tissues, whereas activated Treg (fraction II) were about nine‐fold more abundant in tumors than in TFL or blood (Fig. 2b).
We then set out to analyze GITR expression on FoxP3‐expressing CD4+ T‐cell subsets (fraction I, II, and III). All fractions expressed GITR, but the highest GITR expression was found on intra‐tumoral activated Treg (fraction II), with on average nearly 70% of cells expressing GITR (Fig. 2c, d, and f). Because of the increased proportion of activated Treg in tumors (Fig. 2b) and the high GITR expression on these cells (Fig. 2c), we observed an approximate 30‐fold increase of GITR+ activated Treg within CD45+ immune cells in tumors over TFL and an approximate 50‐fold increase of GITR+ activated Treg within CD45+ immune cells in tumors over blood (Fig. 2e).
GITR is expressed on activated T‐cell subsets
Subsequently, we aimed to identify whether GITR expression coincided with expression of the activation markers CD25 and 4‐1BB (also known as CD137, an accepted marker for detection of antigen‐specific T‐cell responses).23, 24 We found that GITR+ activated Th and activated Treg in liver tissues expressed significantly higher levels of CD25 than their GITR− counterparts (Fig. 3a). Similarly, GITR+CD4+FoxP3− Th cells expressed higher levels of CD25 than GITR−CD4+FoxP3− Th cells (data not shown).
We further found that on average 60% of activated Treg in tumors co‐expressed GITR and 4‐1BB. Proportions of activated Treg that co‐expressed GITR and 4‐1BB were significantly higher in tumors than in blood and showed a higher trend in tumors than in TFL (p = 0.04) (Fig. 3b‐c), which was also observed for CD8+ T cells, CD4+FoxP3− T‐cell subsets, and activated Th (data not shown). GITR‐expressing CD8+ T cells, CD4+FoxP3−CD45RA+ naïve Th, CD4+FoxP3−CD45RA− memory Th, and activated Th cells in liver tissues and blood showed a more activated phenotype than their GITR− counterparts, whereas GITR+ activated Treg in liver tissues, but not in blood, showed a more activated phenotype, as marked by increased 4‐1BB expression (Fig. 3d), which is in line with enhanced CD25 expression on GITR+ T cells.
GITR ligation enhances CD4+ and CD8+ TIL proliferation
We previously showed that engagement of GITR by GITRL reduced the suppression that HCC tumor‐derived Treg exert on blood‐derived CD4+CD25− T cells in ex vivo cultures.19, 21 We now evaluated whether GITR ligation can enhance effector responses of HCC‐derived CD4+ and CD8+ TIL. Because monoclonal antibodies are more amenable to therapeutic development than recombinant proteins, we also assessed the effect of a humanized agonistic antibody against GITR (human IgG1, Pfizer Inc.). To test the effect of GITR ligation, we used two T‐cell stimulation assays.
First, we analyzed TIL proliferation and cytokine secretion after 4‐ to 5‐day culture with anti‐CD3/anti‐CD28 beads in the absence or presence of GITRL or anti‐GITR agonistic antibody. GITRL robustly enhanced CD8+ T‐cell proliferation and granzyme B production and slightly enhanced CD4+ T‐cell proliferation, whereas anti‐GITR antibody significantly enhanced CD8+ T‐cell proliferation, granzyme B and IFN‐γ production (Fig. 4). No enhanced proliferation or cytokine secretion was observed in the presence of the corresponding hIgG1 isotype control (Fig. 4).
Figure 4.
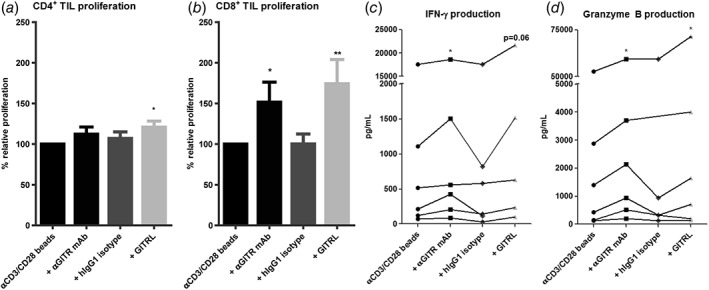
GITR ligation enhances ex vivo proliferation and effector molecule production of CD4+ and CD8+ TIL. Effects of soluble GITRL (1 μg/mL crosslinked with anti‐HA antibody) or 10 μg/mL humanized agonistic anti‐GITR antibody on proliferation of (a) CD4+ TIL, and (b) CD8+ TIL upon 4 to 5 days’ culture of tumor‐derived mononuclear cells with a suboptimal amount of anti‐CD3/CD28 beads. Proliferation was measured by determination of percentages of CD4+ or CD8+ T cells expressing Ki‐67 at the end of the culture. Baseline proliferation (= % of Ki‐67+ T cells in the absence of GITRL or antibodies) was normalized to 100% for each tested patient. Bars show mean percentages of Ki‐67+ CD4+ or CD8+ T cells in cultures relative to baseline proliferation in cultures derived from n = 9 patients with SEM. An irrelevant human IgG1 antibody served as an isotype‐matched control antibody for the anti‐GITR antibody. (c) Production of IFN‐γ and d) granzyme B in culture supernatant was quantified. Data of patients with detectable amounts of IFN‐γ or granzyme B are shown, each line represents one patient. * = p < 0.05; ** = p < 0.01.
In order to evaluate tumor antigen‐specific responses, we expanded autologous B‐cell blasts from patient PBMC by a 2‐ to 3‐week culture in the presence of trimeric CD40 ligand and IL‐4. These B‐cell blasts were then transfected with mRNA encoding the tumor antigens GPC3 or MAGEC2, or eGFP as a negative control, and co‐cultured with CFSE‐labeled TIL for 6 days, as previously described.18 Proliferation of CD4+ TIL and CD8+ TIL in response to GPC3 and/or MAGEC2 was significantly enhanced after engagement of GITR by GITRL or anti‐GITR antibody (Fig. 5a‐c), in contrast to the corresponding hIgG1 isotype control.
Figure 5.
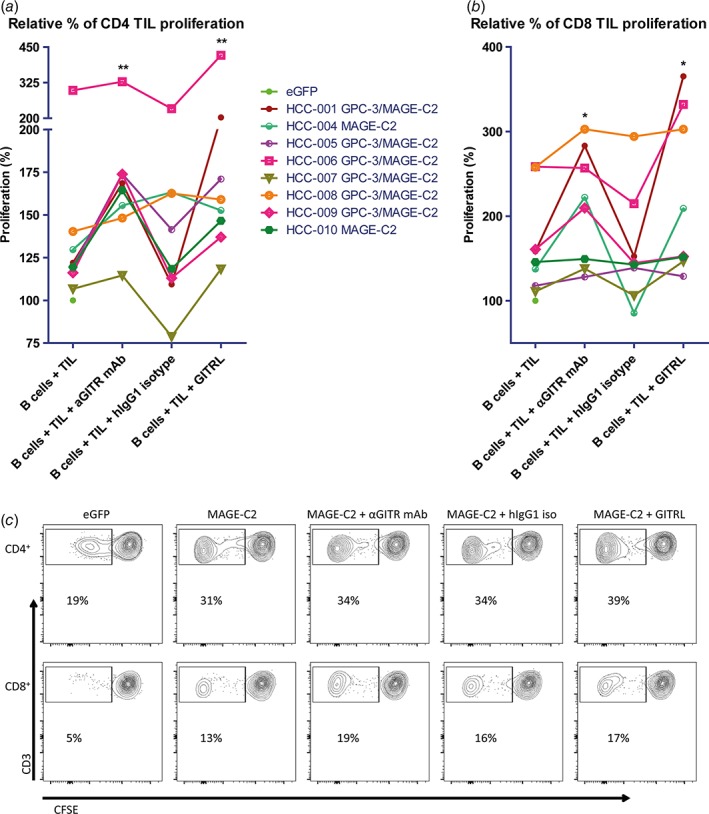
GITR ligation increases ex vivo proliferation of CD4+ and CD8+ TIL in response to tumor antigens presented by mRNA‐transfected autologous B cells. Effects of soluble GITRL (1 μg/mL crosslinked with anti‐HA antibody) or 10 μg/mL humanized agonistic anti‐GITR antibody on proliferation of (a) CD4+ TIL, and (b) CD8+ TIL upon 6 days’ culture of CFSE‐labeled tumor‐derived mononuclear cells with B cell blasts electroporated with mRNA encoding tumor antigens GPC3 or MAGEC2 (or eGFP as a negative control). Proliferation was measured by determination of percentages of CFSElow CD4+ or CD8+ T cells at the end of the culture. Baseline proliferation (= % of CFSElow T cells in the presence of eGFP‐electroporated B cells) was normalized to 100% for each tested patient, and is indicated by a closed green circle. The data depicted as ‘B cells + TIL’ demonstrate that TIL from all patients showed enhanced responses to MAGEC2 as compared to eGFP, while TIL of 6 patients also responded to GPC3. For those patients whose TIL responded to both tumor antigens, the average response to GPC3‐ and MAGEC2‐electroporated B cells was depicted. Each line represents one patient. An irrelevant human IgG1 antibody served as an isotype‐matched control antibody for the anti‐GITR antibody. Data show responses in TIL cultures of n = 8 patients. * = p < 0.05; ** = p < 0.01 compared to baseline (B cells + TIL). (c) Representative FACS contour plots of one patient display the proliferation of CD4+ and CD8+ TIL in response to autologous eGFP or MAGEC2‐electroporated B cells.
PD1 blockade in combination with GITR ligation further enhances tumor antigen‐specific responses of TIL from some patients
Because PD1 blockade with the humanized anti‐PD1 antagonistic antibody nivolumab resulted in tumor regression (objective response) in about 20% of patients with advanced HCC,14 we assessed the co‐expression of GITR and PD1 on T‐cell subsets and the effects of combined targeting of GITR and PD1. Within all T‐cell subsets the majority of GITR+ cells in the tumor, TFL and blood co‐expressed PD‐1. However, PD‐1 was expressed on larger proportions of CD8+ T cells, FoxP3−CD45RA+ naïve Th, FoxP3−CD45RA− memory Th, activated Th than GITR, and hence only part of PD1+ cells in these subsets co‐expressed GITR. In contrast, the majority of PD1+ activated Treg in TFL and tumor and of PD1+ resting Treg in TFL co‐expressed GITR (Fig. 6a–b).
Figure 6.
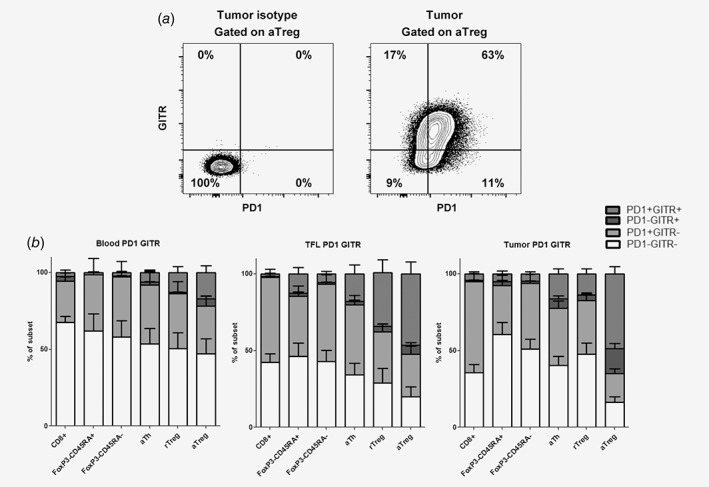
Co‐expression of GITR and PD1. (a) Representative example of flow cytometric analysis of GITR and PD1 expression on tumor‐derived activated Treg. (b) Mean frequencies of GITR−PD1−, GITR−PD1+, GITR+PD1− and GITR+PD1+ within CD8+ T cells, FoxP3−CD45RA+ Th, FoxP3−CD45RA− Th, activated Th, resting Treg and activated Treg in blood, TFL, and tumor with SEM. N = 12 patients.
Addition of nivolumab significantly enhanced ex vivo proliferative responses of CD4+ TIL and almost significantly enhanced CD8+ TIL proliferation to tumor antigens (Fig. 7a‐c). Although the combination of nivolumab with GITRL did not significantly enhance proliferation of tumor antigen‐specific CD4+ or CD8+ T cells compared to single nivolumab or GITRL treatment (Supporting Information Fig. 2), individual patients’ TIL responses (HCC‐001, HCC‐004, HCC‐005, HCC‐006) benefited from the combination of nivolumab and GITRL (Fig. 7a‐c). In addition, we observed that the average proliferative response to the combination of nivolumab and GITRL was 19–20% higher in CD4+ T cells and 27–34% higher in CD8+ T cells, compared to nivolumab alone or GITRL alone, respectively (Supporting Information Fig. 2).
Figure 7.
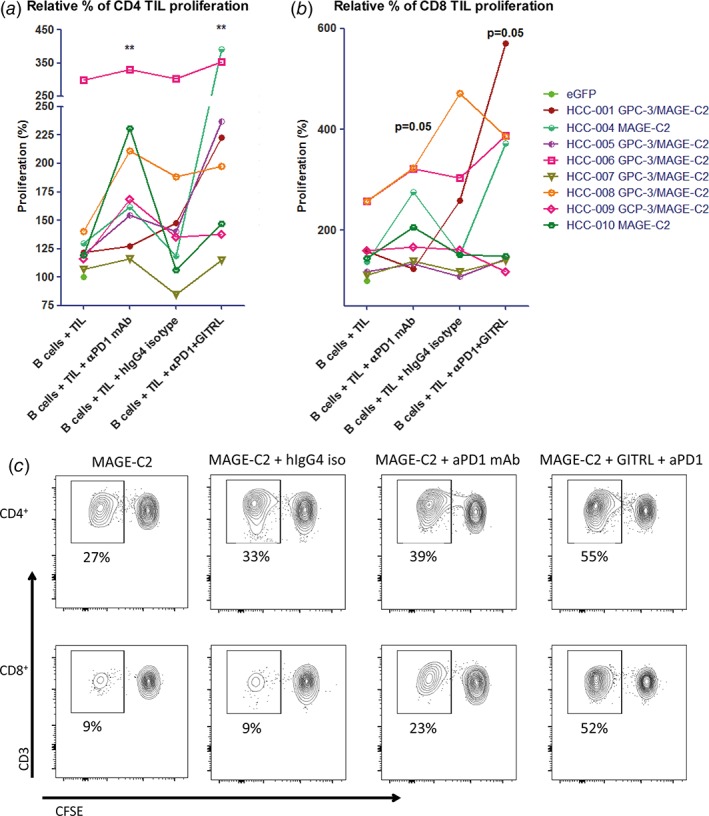
Combined targeting of GITR and PD1 increases ex vivo proliferation of CD4+ and CD8+ TIL in response to tumor antigens presented by mRNA‐transfected autologous B cells in some patients. Effects of 10 μg/mL antagonistic anti‐PD1 antibody (nivolumab) or anti‐PD1 antibody combined with soluble GITRL (1 μg/mL crosslinked with anti‐HA antibody) on proliferation of (a) CD4+ TIL, and (b) CD8+ TIL upon 6 days’ culture of CFSE‐labeled tumor‐derived mononuclear cells with B cell blasts electroporated with mRNA encoding tumor antigens GPC3 or MAGEC2 (or eGFP as a negative control). Proliferation was measured by determination of percentages of CFSElow CD4+ or CD8+ T cells at the end of the culture. Baseline proliferation (= % of CFSElow T cells in the presence of eGFP‐electroporated B cells) was normalized to 100% for each tested patient, and is indicated by a closed green circle. For those patients whose TIL responded to both tumor antigens, the average response to GPC3‐ and MAGEC2‐electroporated B cells was depicted. Each line represents one patient. An irrelevant human IgG4 antibody served as an isotype‐matched control antibody for the anti‐PD1 antibody. Data show responses in cultures of n = 8 patients. * = p < 0.05; ** = p < 0.01 compared to baseline (B cells + TIL). (c) Representative FACS contour plots of one patient display the proliferation of CD4+ and CD8+ TIL in response to autologous MAGEC2‐electroporated B cells.
Discussion
Cancer immunotherapy by targeting GITR has shown anti‐tumor immune responses and tumor regression in a variety of cancer mouse models, including bladder cancer, colon cancer, and melanoma.16 Several clinical trials to target GITR by GITRL or anti‐GITR‐antibodies in solid tumors are starting, ongoing, or just completed (a.o. NCT02697591, NCT01239134, NCT03295942). However, whether GITR targeting can enhance responsiveness of TIL isolated from human tumors is as yet unknown. We have previously shown that GITR ligation decreases the suppressive capacity of conventional Treg present in HCC tumors.19, 21 Based on this observation, we hypothesized that GITR ligation may be able to enhance the responsiveness of TIL from HCC patients. In this study, we tested this hypothesis. We show that GITR ligation indeed enhances ex vivo TIL proliferation in polyclonal and tumor antigen‐specific stimulations of TIL isolated from HCC tumors. Moreover, GITRL and anti‐GITR antibody enhance effector cytokine and granzyme B production in TIL cultures (Figs. 4 and 5).
Extending our earlier studies,19, 21 in which we showed enhanced GITR expression on tumor‐infiltrating Treg compared to their counterparts in TFL and blood in HCC patients, we now extensively evaluated GITR expression on different lymphocyte subsets (Figs. 1 and 2). Our data show that intra‐tumoral CD4+FoxP3+ T cells, particularly CD4+FoxP3hiCD45RA− activated Treg, express the highest levels of GITR, while CD4+Foxp3− Th cells, CD8+ T cells, NK cells and NKT cells express lower levels. CD4+FoxP3loCD45RA− T cells (fraction III), an activated Th subset that is nonsuppressive, produces pro‐inflammatory cytokines, and can contribute to anti‐tumor immunity,22, 25 also expressed lower levels of GITR compared to activated Treg. GITR+ Treg showed an activated phenotype, as evidenced by elevated CD25 and 4‐1BB expression (Fig. 3). Together with the high proportions of CD4+FoxP3hiCD45RA− activated Treg in TIL (Fig. 2b, e), these data suggest that the preferential target of GITR ligand and anti‐GITR antibody in our TIL cultures were probably highly activated Treg. However, we do not exclude that targeting of activated effector T cells which expressed GITR (Fig. 3) contributed to the functional effects of GITRL and anti‐GITR antibody in our TIL cultures. Indeed, a recent study using an experimental mouse tumor model showed that anti‐GITR antibody therapy targets both activated highly suppressive Treg as well as effector T cells.26
GITRL and anti‐GITR antibody resulted in comparable enhancements of ex vivo TIL proliferation, effector cytokine and granzyme B production, showing that in our ex vivo culture conditions a fully humanized anti‐GITR antibody is as effective as the natural trimeric ligand. In vivo targeting of GITR can boost intra‐tumoral T‐cell immunity via several mechanisms: 1) by selective depletion of Treg via co‐engagement of activating Fcγ‐receptors on innate immune cells;26, 27, 28 2) by GITR‐signaling into Treg, leading to lineage destabilization and reduction of their suppressive capacity;29 3) by GITR‐signaling into effector T cells, thereby increasing their resistance to Treg suppression.26, 30 Activation of human GITR‐signaling requires trimerization of GITR.16 We used a trimeric GITR‐ligand that we crosslinked using an anti‐HA tag antibody, thereby further increasing its ability to multimerize GITR and activate GITR‐signaling. Induction of signaling of TNF superfamiliy receptors such as GITR by agonistic antibodies is thought to require co‐engagement of inhibitory Fcγ‐receptors.31, 32 Since the anti‐GITR antibody that we used has a human IgG1 Fc part which has a high binding affinity for all human Fcγ‐receptors, including the inhibitory Fcγ‐receptor IIb,33 and we deliberately used unfractionated TIL which we previously showed to contain innate immune cells18 that express both activating and inhibitory Fcγ‐receptors (van Beek, unpublished results) to study the effects of the anti‐GITR antibody on TIL responses, we suppose that its effect on TIL responses is also mediated by induction of GITR‐signaling in Treg and effector T cells. The robust T cell stimulatory activity of the anti‐GITR antibody may be co‐determined by its ligand blocking nature, which prevents reverse signaling of GITR through GITRL to induce indoleamine 2,3‐dioxygenase expression in antigen‐presenting cells and lead to T‐cell suppression.34 Whether this anti‐GITR antibody can mediate depletion of Treg in our TIL cultures remains to be elucidated.
In addition, we evaluated the combination of GITR ligation and PD1 blockade (Fig. 7 and Supporting Information Figure 2) because single PD1 blockade with nivolumab shows clinical efficacy in only a minority of HCC patients.14 In a mouse ovarian tumor model, the combination of GITR ligation and PD1 blockade induced potent antitumor immunity and tumor regression, while anti‐GITR or anti‐PD1 antibodies alone did not have any therapeutic effect.35 Several clinical studies in cancer patients are currently studying combinatorial targeting of GITR and the PD1/PDL1 pathway.17, 36 Addition of nivolumab alone enhanced ex vivo TIL responses to a similar extent as GITRL (Supporting Information Fig. 2), while the combination of GITRL and nivolumab further enhanced tumor antigen‐specific proliferative responses of TIL from some, but not all patients (Fig. 7). One reason for the limited additive effect of combined PD‐1 and GITR‐targeting on TIL responses may be related to the extensive co‐expression of both target molecules on Treg. About 50% of intra‐tumoral Treg co‐expressed GITR and PD‐1 (Fig. 6). Importantly, blockade of PD‐L1/PD‐1 interaction has been shown to enhance the suppressive capacity of human Treg,37, 38 and might therefore counteract the reduction of Treg suppressive function by GITR ligation.
Our data show that about 10% of NK‐cells in tumors, TFL and blood express GITR, although at a lower expression level than Treg. Accumulating evidence indicates that agonistic GITR‐targeting can suppress human NK cell functions. In vitro engagement of GITR on human NK cells by GITR‐ligand expressing tumor cells, GITR‐ligand‐Ig fusion protein, or soluble GITR‐ligand produced by tumor cells or present in human sera, inhibits their cytotoxic functions and IFN‐γ production.39, 40, 41 In addition, targeting of purified human NK cells with an agonistic anti‐GITR antibody inhibits human NK‐cell proliferation and pro‐inflammatory cytokine production.42 Therefore, whereas our data show that agonistic targeting of GITR enhances ex vivo reactivity of tumor‐infiltrating T cells of HCC patients, clinical effects of agonistic GITR targeting in cancer patients may be counteracted by suppression of anti‐cancer responses of tumor‐infiltrating NK cells. The net clinical effect of GITR‐targeting in cancer patients can only be studied in clinical trials. Whether IFN‐γ and granzyme B released into the supernatants of our ex vivo cultures of human TIL (Fig. 4c‐d) were exclusively derived from T cells or also from NK cells, is uncertain. Therefore, we cannot exclude that inhibitory effects of GITR‐ligation on cytokine and granzyme B production by NK cells that were present in the TIL cultures have partially obscured stimulatory effects on their production by T cells.
Our study has some limitations: 1) since we could often isolate limited numbers of TIL from patient tumor samples and the required numbers of TIL in the assays are large, we could include only a fraction of patients for functional assays; 2) for the same reason, we were not able to investigate the mechanism of action by which GITRL and anti‐GITR antibody enhanced ex vivo TIL responses, neither the involvement of Fcγ‐receptor‐expressing innate immune cell subsets in the effects of the anti‐GITR antibody; 3) because most of our patients underwent resection, and a few underwent liver transplantation, they only represent (very) early stages of HCC patient population; 4) most patients in our cohort have no preexisting severe liver fibrosis, in contrast to HCC patients in many other studies in e.g. Asia.
On the other hand, our study has several strengths: 1) we are the first to show that GITR ligation can enhance responsiveness of TIL isolated from human tumors; 2) we used total tumor‐infiltrating leukocytes to mimic the tumor immune environment as close as possible; 3) we used an antigen‐specific assay that included two tumor‐associated antigens (GPC3 and MAGEC2) that are selectively and prevalently expressed in HCC tumors;43 4) we directly compared GITRL with an agonistic anti‐GITR antibody. Since GITR targeting as single treatment or in combination with targeting of another immune checkpoint was effective in a variety of cancer models in mice, our data showing enhanced responses of human HCC TIL upon GITR ligation may also have relevance for other human cancers.
In sum, our study demonstrates that GITR ligation can enhance the functionality of tumor‐infiltrating T cells in HCC, and therefore may be a promising immunotherapeutic target for patients with HCC. In addition, our results suggest that combined targeting of GITR and PD1 may improve anti‐tumor T‐cell responses in some, but not all, HCC patients.
Authors’ Contributions
A.v.B. contributed to study concept and design, acquisition, analysis and interpretation of data, statistical analysis, drafting of the manuscript, critical revision of the manuscript for important intellectual content.
G.Z. contributed to study concept and design, acquisition, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content and obtained funding.
M.D. contributed to material support and critical revision of the manuscript for important intellectual content.
P.B., S.M. and M.v.d.H‐M. contributed to acquisition of data.
L.N. and L.C.C. contributed to acquisition of data and critical revision of the manuscript for important intellectual content.
W.P., J.I. and C.H. contributed to material support.
Q.P. contributed to critical revision of the manuscript for important intellectual content.
A.M. and S.B. contributed to study concept and design, study supervision and obtained funding.
M.B. contributed to critical revision of the manuscript for important intellectual content and obtained funding.
D.S. contributed to study concept and design, critical revision of the manuscript for important intellectual content, material support, study supervision and obtained funding.
J.K. contributed to study concept and design, writing of the manuscript, study supervision and obtained funding.
Supporting information
Supplementary Figure 1: Distribution of NK, NKT, and T cell subsets in blood, tumor‐free liver, and HCC tumor tissues. A) Frequencies of CD3−CD56+ NK cells, CD3+CD56+ NKT cells, and CD3+CD56− T cells within CD45+ mononuclear leukocytes. N = 11–12 patients. B) Frequencies of CD8+ T cells, CD4+FoxP3− T cells, and CD4+FoxP3+ T cells within CD3+ cells. N = 16–20 patients. Dots represent individual patients and lines present means. * = p < 0.025; ** = p < 0.01; *** = p < 0.001.
Supplementary Figure 2: Effects of single targeting of PD1 or GITRL and combined targeting of PD1 and GITR on TIL proliferation in response to tumor antigens presented by mRNA‐transfected autologous B cells. Effects of 10 μg/mL antagonistic anti‐PD1 antibody (nivolumab), soluble GITRL (1 μg/mL crosslinked with anti‐HA antibody), or anti‐PD1 antibody combined with soluble GITRL on proliferation of A) CD4+ TIL, and B) CD8+ TIL upon 6 days’ culture of CFSE‐labeled tumor‐derived mononuclear cells with B cell blasts electroporated with mRNA encoding tumor antigens GPC3 or MAGEC2 (or eGFP as a control). Proliferation was measured by determination of percentages of CFSElow CD4+ or CD8+ T cells at the end of the culture. Baseline proliferation (= % of CFSElow T cells in the presence of eGFP‐electroporated B cells) was normalized to 100% for each tested patient. Proliferation in response to tumor antigen is shown as percentage of CFSElow cells compared to baseline proliferation in response to eGFP. For those patients whose TIL responded to both tumor antigens, the average response to GPC3‐ and MAGEC2‐electroporated B cells was depicted. Bars show mean percentages in cultures derived from n = 8 patients with SEM.
Supplementary Table 1: Anti‐human antibodies used for flow cytometry.
Acknowledgement
This study was supported by a research grant of Pfizer Inc. to Dave Sprengers and Jaap Kwekkeboom, and by a PhD‐fellowship of the China Scholarship Council to Guoying Zhou (number 201306270017).
A.A.v.B and G.Z. shared first authorship.
D.S. and J.K. shared last authorship.
Conflict of interest: None of the authors has any conflict of interest.
References
- 1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 2. Mazzanti R, Gramantieri L, Bolondi L. Hepatocellular carcinoma: epidemiology and clinical aspects. Mol Aspects Med 2008;29:130–43. [DOI] [PubMed] [Google Scholar]
- 3. El‐Serag HB, Marrero JA, Rudolph L, et al. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology 2008;134:1752–63. [DOI] [PubMed] [Google Scholar]
- 4. European Association for the Study of the Liver . EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2018;69:182–236. [DOI] [PubMed] [Google Scholar]
- 5. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open‐label phase 3 study (KEYNOTE‐006). Lancet 2017;390:1853–62. [DOI] [PubMed] [Google Scholar]
- 6. Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro‐oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO‐4538‐12, ATTRACTION‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2017;390:2461–71. [DOI] [PubMed] [Google Scholar]
- 7. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet 2017;389:255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second‐line therapy for advanced Urothelial carcinoma. N Engl J Med 2017;376:1015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Larkin J, Chiarion‐Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or Monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McDermott DF, Drake CG, Sznol M, et al. Survival, durable response, and Long‐term safety in patients with previously treated advanced renal cell carcinoma receiving Nivolumab. J Clin Oncol 2015;33:2013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long‐term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus Docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med 2015;373:1627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti‐PD‐1) in melanoma. N Engl J Med 2013;369:134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. El‐Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open‐label, non‐comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sagiv‐Barfi I, Czerwinski DK, Levy S, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 2018;10:eaan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Knee DA, Hewes B, Brogdon JL. Rationale for anti‐GITR cancer immunotherapy. Eur J Cancer 2016;67:1–10. [DOI] [PubMed] [Google Scholar]
- 17. Waight JD, Gombos RB, Wilson NS. Harnessing co‐stimulatory TNF receptors for cancer immunotherapy: current approaches and future opportunities. Hum Antibodies 2017;25:87–109. [DOI] [PubMed] [Google Scholar]
- 18. Zhou G, Sprengers D, Boor PPC, et al. Antibodies against immune checkpoint molecules restore functions of tumor‐infiltrating T cells in hepatocellular carcinomas. Gastroenterology 2017;153:1107–19. e10. [DOI] [PubMed] [Google Scholar]
- 19. Pedroza‐Gonzalez A, Verhoef C, Ijzermans JN, et al. Activated tumor‐infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology 2013;57:183–94. [DOI] [PubMed] [Google Scholar]
- 20. Pedroza‐Gonzalez A, Zhou G, Vargas‐Mendez E, et al. Tumor‐infiltrating plasmacytoid dendritic cells promote immunosuppression by Tr1 cells in human liver tumors. Oncoimmunology 2015;4:e1008355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pedroza‐Gonzalez A, Zhou G, Singh SP, et al. GITR engagement in combination with CTLA‐4 blockade completely abrogates immunosuppression mediated by human liver tumor‐derived regulatory T cells ex vivo. Oncoimmunology 2015;4:e1051297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009;30:899–911. [DOI] [PubMed] [Google Scholar]
- 23. Wolfl M, Kuball J, Ho WY, et al. Activation‐induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007;110:201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shi XL, de Mare‐Bredemeijer EL, Tapirdamaz O, et al. CMV primary infection is associated with donor‐specific T cell Hyporesponsiveness and fewer late acute rejections after liver transplantation. Am J Transplant 2015;15:2431–42. [DOI] [PubMed] [Google Scholar]
- 25. Saito T, Nishikawa H, Wada H, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 2016;22:679–84. [DOI] [PubMed] [Google Scholar]
- 26. Mahne AE, Mauze S, Joyce‐Shaikh B, et al. Dual roles for regulatory T‐cell depletion and Costimulatory signaling in agonistic GITR targeting for tumor immunotherapy. Cancer Res 2017;77:1108–18. [DOI] [PubMed] [Google Scholar]
- 27. Coe D, Begom S, Addey C, et al. Depletion of regulatory T cells by anti‐GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol Immunother 2010;59:1367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bulliard Y, Jolicoeur R, Windman M, et al. Activating fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor‐targeting antibodies. J Exp Med 2013;210:1685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schaer DA, Budhu S, Liu C, et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res 2013;1:320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nishikawa H, Kato T, Hirayama M, et al. Regulatory T cell‐resistant CD8+ T cells induced by glucocorticoid‐induced tumor necrosis factor receptor signaling. Cancer Res 2008;68:5948–54. [DOI] [PubMed] [Google Scholar]
- 31. Li F, Ravetch JV. Antitumor activities of agonistic anti‐TNFR antibodies require differential FcgammaRIIB coengagement in vivo. Proc Natl Acad Sci U S A 2013;110:19501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dahan R, Ravetch JV. Co‐targeting of adenosine signaling pathways for immunotherapy: potentiation by fc receptor engagement. Cancer Cell 2016;30:369–71. [DOI] [PubMed] [Google Scholar]
- 33. Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol 2010;10:328–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grohmann U, Volpi C, Fallarino F, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med 2007;13:579–86. [DOI] [PubMed] [Google Scholar]
- 35. Lu L, Xu X, Zhang B, et al. Combined PD‐1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J Transl Med 2014;12:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cabo M, Offringa R, Zitvogel L, et al. Trial watch: Immunostimulatory monoclonal antibodies for oncological indications. Oncoimmunology 2017;6:e1371896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franceschini D, Paroli M, Francavilla V, et al. PD‐L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT‐5 phosphorylation in patients chronically infected with HCV. J Clin Invest 2009;119:551–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peligero C, Argilaguet J, Guerri‐Fernandez R, et al. PD‐L1 blockade differentially impacts regulatory T cells from HIV‐infected individuals depending on plasma Viremia. PLoS Pathog 2015;11:e1005270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baltz KM, Krusch M, Bringmann A, et al. Cancer immunoediting by GITR (glucocorticoid‐induced TNF‐related protein) ligand in humans: NK cell/tumor cell interactions. FASEB J 2007;21:2442–54. [DOI] [PubMed] [Google Scholar]
- 40. Baltz KM, Krusch M, Baessler T, et al. Neutralization of tumor‐derived soluble glucocorticoid‐induced TNFR‐related protein ligand increases NK cell anti‐tumor reactivity. Blood 2008;112:3735–43. [DOI] [PubMed] [Google Scholar]
- 41. Baessler T, Krusch M, Schmiedel BJ, et al. Glucocorticoid‐induced tumor necrosis factor receptor‐related protein ligand subverts immunosurveillance of acute myeloid leukemia in humans. Cancer Res 2009;69:1037–45. [DOI] [PubMed] [Google Scholar]
- 42. Liu B, Li Z, Mahesh SP, et al. Glucocorticoid‐induced tumor necrosis factor receptor negatively regulates activation of human primary natural killer (NK) cells by blocking proliferative signals and increasing NK cell apoptosis. J Biol Chem 2008;283:8202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sideras K, Bots SJ, Biermann K, et al. Tumour antigen expression in hepatocellular carcinoma in a low‐endemic western area. Br J Cancer 2015;112:1911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Distribution of NK, NKT, and T cell subsets in blood, tumor‐free liver, and HCC tumor tissues. A) Frequencies of CD3−CD56+ NK cells, CD3+CD56+ NKT cells, and CD3+CD56− T cells within CD45+ mononuclear leukocytes. N = 11–12 patients. B) Frequencies of CD8+ T cells, CD4+FoxP3− T cells, and CD4+FoxP3+ T cells within CD3+ cells. N = 16–20 patients. Dots represent individual patients and lines present means. * = p < 0.025; ** = p < 0.01; *** = p < 0.001.
Supplementary Figure 2: Effects of single targeting of PD1 or GITRL and combined targeting of PD1 and GITR on TIL proliferation in response to tumor antigens presented by mRNA‐transfected autologous B cells. Effects of 10 μg/mL antagonistic anti‐PD1 antibody (nivolumab), soluble GITRL (1 μg/mL crosslinked with anti‐HA antibody), or anti‐PD1 antibody combined with soluble GITRL on proliferation of A) CD4+ TIL, and B) CD8+ TIL upon 6 days’ culture of CFSE‐labeled tumor‐derived mononuclear cells with B cell blasts electroporated with mRNA encoding tumor antigens GPC3 or MAGEC2 (or eGFP as a control). Proliferation was measured by determination of percentages of CFSElow CD4+ or CD8+ T cells at the end of the culture. Baseline proliferation (= % of CFSElow T cells in the presence of eGFP‐electroporated B cells) was normalized to 100% for each tested patient. Proliferation in response to tumor antigen is shown as percentage of CFSElow cells compared to baseline proliferation in response to eGFP. For those patients whose TIL responded to both tumor antigens, the average response to GPC3‐ and MAGEC2‐electroporated B cells was depicted. Bars show mean percentages in cultures derived from n = 8 patients with SEM.
Supplementary Table 1: Anti‐human antibodies used for flow cytometry.