

Abstract
To understand the evolution and molecular characteristics of Jiangxi H9N2 viruses, we isolated 17 viruses in 2011 and analyzed their characteristics. Phylogenetic analyses revealed that their hemagglutinin genes originate from JS/1/00‐like sublineage, neuraminidase genes originate from BJ/94‐like sublineage, PB1, PA, NP, and NS genes all come from SH/F/98‐like sublineage, PB2 genes originate from ST/163/04‐like sublineage, while M genes come from G1‐like sublineage. Genotype analysis showed that our isolates were classified as genotype 57. Molecular analyses indicated that our strains contained specific sites characteristic of low‐pathogenic viruses. The current study once again highlights the necessity for continued surveillance of novel H9N2 viruses.
Keywords: genotype analysis, H9N2 influenza virus, molecular analyses, phylogenetic analyses
1. INTRODUCTION
Influenza A virus (AIV) is a member of the orthomyxoviridae, and it consists of eight gene segments encoding 16 proteins.1 AIVs are classified into 18 hemagglutinin (HA) and 11 neuraminidase (NA) subtypes according to the antigenicity of surface glycoproteins.2 The H9N2 subtype virus is a prominent member of the influenza A family and has been found in chicken, duck, and other poultry species.3 Epidemiological research demonstrated that the first isolation of H9N2 influenza viruses from turkeys occurred in North America in 1966.4 In China, the H9N2 virus was firstly isolated from chickens in 1994 in Guangdong and then became the most prevalent subtype of influenza viruses.5 Notably, H9N2 subtype viruses could infect many poultry species in markets and caused severe economic losses.6 What's more, infections of H9N2 viruses in humans also caused great attention by its ability to transmit from avian species to mammals.7 So H9N2 influenza viruses have caused a great threat to public health.
Genetic studies proved that HA and NA genes of H9N2 influenza viruses were divided into North America and Eurasian lineages. The North America lineage was represented by A/Turkey/Wisconsin/1966 (WI/66‐like), while the Eurasian lineage included several distinct sublineages: A/chicken/Beijing/1/1994‐like (BJ/94‐like), A/quail/Hong Kong/G1/1997‐like (G1‐like), A/Chicken/Korea/38349‐p96323/1996‐like (Korea‐like), or A/Duck/Hong Kong/Y439/1997‐like (Y439‐like). Internal genes from H9N2 viruses displayed a greater diversity, including G1‐like, A/chicken/Shanghai/F/1998‐like (F/98‐like), and H5N1‐like sublineages.8 In China, BJ/94‐like and G1‐like viruses have been predominant sublineages since the mid‐1990s. And A/duck/Shantou/163/2004‐like (ST/163/04‐like) viruses had Korea‐like HA genes.3 Above all, H9N2 viruses are still continuously evolving and they deserve our close attention.
To date, at least 98 genotypes have been identified.9 Moreover, since 2007 genotype 57 (G57) has been prevalent and became the dominant lineage in eastern China, which bear the backbone of F/98‐like viruses by integrating to PB2 and M genes of G1‐like viruses.10 Recent research demonstrated that G57 had the ability to donate gene segments to other highly pathogenic AIVs like H5N2, the 2013 H7N9, and H10N8, which has caused human infections and deaths.11
Despite the genesis of H9N2 viruses in China being well analyzed,3, 9, 10 genetic characterization of H9N2 in Jiangxi province has not been reported in recent years. In 2011, we isolated 17 H9N2 viruses from poultry. Eight gene segments of 17 viruses were sequenced and characterized comparing with other available sequences in the NCBI database.
2. MATERIALS AND METHODS
2.1. Virus isolation and identification
Cloacal and tracheal swabs were collected in Jiangxi of China from an epidemiological survey of 12 live‐poultry markets throughout the whole year in 2011. According to the standard of International Enzootic Office, virus isolation was performed in 10‐day‐old specific‐pathogen‐free (SPF) embryonated chicken eggs via allantoic sac route and was incubated for 72 hours at 37°C. The allantoic fluids of the live embryos were collected and subjected to hemagglutination (HA) and hemagglutination inhibition (HI) tests to identify HA subtypes. NA subtypes were identified by nucleotide sequencing and National Center for Biotechnology Information (NCBI) BLAST.
2.2. Sequencing
Briefly, virus RNA extraction from infective allantoic fluid was performed using a TIANamp Virus RNA Kit (Tiangen Biotech, Beijing, China). After reverse transcription, polymerase chain reaction (PCR) was performed using specific primers to amplify complementary DNA, as described.5 PCR products were purified with an AxyPrep DNA Gel Extraction Kit (Axygen Inc., Hangzhou, China) and were sequenced by using the BigDye Terminator v3.1 Cycle sequencing Kit on an ABI Genetic analyzer 3500xL (Applied Biosystems, Foster City, CA). The nucleotide sequences for influenza virus genes obtained from this study have been deposited to GenBank under accession numbers MF085026‐MF085033, MF113058‐MF113161, and MF372884‐MF372907.
2.3. Phylogenetic analysis
The sequences selected from NCBI include all the Jiangxi H9N2 isolates, representative strains reported from the previous study, and highly pathogen avian influenza viruses (HPAIVs) sharing high homology with internal genes of strains in this study. By using the Lasergene sequence analysis software package (DNAStar, Inc, Madison, WI), all sequence data were compiled and edited. Pairwise sequence alignments were also performed by using Clustal W. To determine the robustness and evaluate the consistency of the tree topologies, 1000 bootstrap replicates and the neighbor‐joining trees were constructed using MEGA7.1.
2.4. Genotype definition
Genotypes were defined by gene phylogeny.12 And the genotype of H9N2 influenza viruses used in this study was determined as previously described.11
2.5. Molecular analysis
The deduced amino acid sequences were also aligned and analyzed with Megalign module of the Lasergene sequence and MEGA version 7.1. Potential glycosylation sites of HA and NA were analyzed by an online software called NetNglyc 1.0 Server.
3. RESULTS AND DISCUSSION
3.1. Virus isolation and identification
The collection of tracheal and cloacal swabs in Jiangxi live‐poultry markets was conducted in 2011. After propagation in SPF embryonated chicken eggs and HA tests, 17 virus isolates were obtained, of which 15 isolates were from chickens and 2 isolates from ducks. On the basis of HI tests, 17 H9 subtype influenza isolates were identified. To identify the NA subtype of these strains, we sequenced their NA genes and performed the NCBI BLAST, discovering that all isolates belonged to the N2 subtype. Results of virus isolation and identification are shown in Supporting Information Table S1.
3.2. Phylogenetic analysis
The trees of HA genes showed that Eurasian lineage consisted of three sublineages including BJ/94‐like, G1‐like, and JS/1/00‐like. All of the tested H9N2 isolates clustered into the JS/1/00‐like sublineage (Figure 1A). Phylogenetic analysis of NA genes showed that there were two distinct lineages: North American and Eurasian lineages (Figure 1B). Eurasian lineage included BJ/94‐like, G1‐like, and G9‐like sublineages. The NA genes analyzed in this study showed high gene homology (92.9%‐93.6%) and all belonged to BJ/94‐like sublineage (Figure 1).
Figure 1.
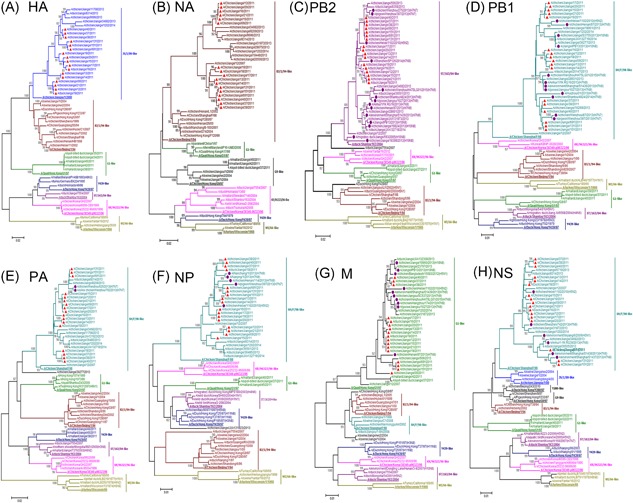
Phylogenetic trees for the HA (A) and NA (B) PB2 (C), PB1 (D), PA (E), NP (F), M (G), and NS (H) genes of all the representative influenza A viruses of the H9N2 subtype. Trees were generated by the neighbor‐joining method in the MEGA 7.1 program. Numbers above or below branches demonstrate neighbor‐joining bootstrap values. Analysis was based on nucleotides 1 to 1683 of the HA gene, 1 to 1410 of the NA gene, 1 to 2280 of the PB2 gene, 1 to 2274 of the PB1 gene, 1 to 2151 of the PA gene, 1 to 1497 of the NP gene, 1 to 982 to the M gene, and 1 to 838 of the NS gene. The sequences labeled with purple dots are HPAIVs sharing high homology with the H9N2 isolates in this study. The lengths of the horizontal lines are relative to the minimum number of nucleotide differences required to join nodes. Vertical lines are for spacing and labeling. The viruses isolated in Jiangxi were highlighted in red trigon. The names of the viruses could be found in Supporting Information Table S1. HA,hemagglutinin; HPAIV, highly pathogen avian influenza viruses; NA, neuraminidase
Phylogenetic analyses of six internal genes showed that they had more diverse origins than surface genes (Figure 1). Analysis of the PB2 genes showed two evolutionary lineages: North American and Eurasian lineages. Four sublineages including BJ/94‐like, G1‐like, ST/163/04‐like, and Korea‐like fell into Eurasian lineage (Figure 1C). PB2 genes from 17 H9N2 isolates clustered into the ST/163/04‐like sublineage. Interestingly, seven virus isolates in this study showed high gene homologies (>99%) with H7N7‐like (A/chicken/Wenzhou/87/2013 and CK/WZ/H7N7) influenza viruses in Wenzhou. The previous study revealed that the generation of novel CK/WZ/H7N7 viruses was by reassortment of prevailing H9N2 viruses with H7N7 viruses.13 Moreover, the PB2 phylogenetic tree showed that all 17 H9N2 viruses had a close relationship to the subtype H7N9 and H5N2 isolating from Hebei and southern China (Figure 1C). In the phylogenetic tree of PB1 genes, H9N2 viruses formed seven distinct sublineages, including SH/F/98‐like, G1‐like, Korea‐like, BJ/94‐like, WI/66‐like, ST/163‐like, and Y439‐like (Figure 1D). Our analyzed H9N2 virus isolates fell into SH/F/98‐like sublineage, while most other H9N2 viruses in Jiangxi isolated from wild birds clustered into G1‐like sublineage. As with PB2 genes, PB1 genes of all 17 H9N2 isolates exhibited high gene homologies (>97%) with those of CK/WZ/H7N7 viruses. Notably, they were also closely connected with those of H7N9 viruses isolated from a human in Anhui (Figure 1D). In PA phylogenetic tree (Figure 1E), seven different sublineages of H9N2 influenza viruses were recognized, including SH/F/98‐like, BJ/94‐like, Y439‐like, ST/163/04‐like, Korea‐like, WI/66‐like, and G1‐like. Our H9N2 isolates belonged to SH/F/98‐like sublineage. It was interesting that three isolates also showed high gene homologies (>99%) with CK/WZ/H7N7 viruses. NP genes of H9N2 viruses formed seven sublineages: BJ/94‐like, G1‐like, SH/F/98‐like, Korea‐like, WI/66‐like, ST/163‐like, and Y439‐like. Our H9N2 virus isolates clustered into SH/F/98‐like sublineage (Figure 1F). As with polymerase genes, NP genes from 17 H9N2 isolates also showed high gene homologies with those of CK/WZ/H7N7 viruses. In addition, 14 viruses had a close relationship with H7N9 viruses labeled with purple dots that have resulted in human fatalities.14 M genes of 17 Jiangxi viruses obtained in this study clustered into G1‐like sublineage (Figure 1G). And results from genome comparison displayed that the similarity in M genes was found between H9N2 Jiangxi isolates and HPAI viruses including H7N9, H10N8, H5N1, and H1N2 subtypes. As shown in NS phylogenetic tree (Figure 1H), all Jiangxi virus isolates shaped a well‐supported cluster in the SH/F/98‐like sublineage and displayed a nucleotide homology of 94.8% to 97.1% with SH/F/98‐like sublineage. Notably, NS genes of HPAI subtype H5N1, H5N2, and H7N9 viruses also clustered into the SH/F/98‐like sublineage. The fact that the internal genes of these H9N2 viruses have similarities to those of H5N1, H7N9, and H10N8 viruses deserve our attention.
3.3. Genotyping
Moreover, the results of phylogenetic trees showed that our isolates were from a recently discovered genotype classified as G57. Some reports revealed that G57 had replaced multiple genotypes and became the predominant genotype in 2010‐2013.11 It was reasonable for us to posit that G57 had been prevalent in Jiangxi till 2011 since it appeared in 2007. Furthermore, promptly effective measures should be taken to control further spread of the G57 influenza viruses among poultry in China, and a continuous extensive virus surveillance was also needed.
3.4. Predicted molecular characteristics
To understand the molecular characterization of H9N2 Jiangxi virus isolates, deduced amino acid sequences of virus proteins were aligned and analyzed with those of representative H9N2 and HPAI viruses from the NCBI influenza database (Table 1, Supporting Information Tables S2 and S3). By comparison, we identified that 17 H9N2 Jiangxi influenza isolates with 561 amino acids (1683 nucleotide). As shown in Table 1, the amino acid sequences at the cleavage sites of the HA protein represented the low pathogenicity feature. Notably, at the receptor binding sites (H9 numbering), all isolated H9N2 viruses possessed leucine (L) at position 234, which was reported to contribute to the human‐type receptor binding of the H9N2 virus, and glycine (G) at position 236 also displaying human virus‐like cell tropisms.15 According to results predicted by NetNGlyc 1.0 Server, all 17 Jiangxi isolates possessed 8 N‐linked potential glycosylation sites in the HA protein at positions (29, 141, 218, 298, 305, 313, 492, and 551). Increase and lack of potential glycosylation sites were displayed in Supporting Information Table S2. This phenomenon may symbolize an adaptation of H9N2 within poultry since a change in glycosylation pattern.16 Likewise, analysis of the NA protein sequences of viruses also displayed that five Jiangxi isolates possessed seven potential glycosylation sites, while others maintained six potential glycosylation sites (Supporting Information Table S2). Moreover, amino acids in the enzyme active sites in all Jiangxi isolates were conserved. Besides, each Jiangxi isolates contained a 3‐amino acid deletion (residues 63‐65) in the NA stalk region (Table 1). It is accepted that deletion within the NA stalk region may be a significant characteristic equilibrizing the supplementary activities of the HA and NA on the adaption of the virus to poultry.17 Moreover, when deletion of NA stalk combined with substitution A316S of HA protein, HA cleavage efficiency increased.18 It is reported that when deletion of NA stalk combined with substitution A316S of HA protein, HA cleavage efficiency increased.18 And in our study, 17 viruses had a substitution at position 316 of HA protein, and we thought that this phenomenon had been stable in Jiangxi by comparison with other H9N2 isolates in Jiangxi.
Table 1.
Analysis of amino acid sequences of surface and internal proteins
| HA clevage site | HA receptor binding sites | NA stalk deletion | PB2 | PB1‐F2 | PB1 | PA | NP | M2 | NS1 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 335‐339 | 234 | 236 | 63‐65 | 627E | 66N | 368V | 436E | 136L | 16E | 42S | |
| Virus strains | RSSR↓G | L | G | ||||||||
| CK/BJ/94 | + | Q | + | − | + | + | I | + | + | G | + |
| DK/HK/Y280 | + | + | + | + | + | + | I | + | + | G | + |
| CK/SH/F/98 | + | Q | + | + | + | + | I | + | + | G | + |
| QA/HK/Y439 | ASNR↓G | Q | + | − | + | + | I | + | + | + | + |
| QA/HK/G1 | + | + | + | 38‐40 | + | S | I | + | + | + | + |
| TK/WI/66 | VSSR↓G | Q | + | − | + | + | I | + | M | G | + |
| CK/KR/96 | ASYR↓G | Q | + | − | + | + | I | + | + | + | + |
| CK/JS/1/00 | + | + | + | − | + | + | I | + | + | + | + |
| CK/JX/09 | KSSR↓G | + | + | + | + | + | + | + | + | + | + |
| CK/JX/12 | KSSR↓G | + | + | + | + | + | + | + | + | + | + |
| CK/JX/13 | + | + | + | + | + | + | I | + | + | + | + |
| CK/JX/14 | + | + | + | + | + | + | I | + | + | + | + |
| CK/JX/15 | + | + | + | + | + | + | + | + | + | + | + |
| DK/JX/16 | + | + | + | + | + | + | + | + | + | + | + |
| CK/JX/17 | + | + | + | + | + | + | + | + | + | + | + |
| DK/JX/18 | KSSR↓G | + | + | + | + | + | + | + | + | + | + |
| CK/JX/19 | + | + | + | + | + | + | + | + | + | + | + |
| CK/JX/20 | + | + | + | + | + | + | + | + | + | + | + |
| CK/JX/21 | KSSR↓G | + | + | + | + | + | + | + | + | + | + |
| CK/JX/37 | + | + | + | + | + | + | + | + | + | + | + |
| CK/JX/38 | + | + | + | + | + | + | + | + | + | + | + |
| CK/JX/40 | + | + | + | + | + | + | + | + | + | + | + |
| CK/JX/13Y | KSSR↓G | + | + | + | + | + | + | + | + | V | + |
| CK/JX/15Y | KSSR↓G | + | + | + | + | + | + | + | + | V | + |
| CK/JX/69Y | KSSR↓G | + | + | + | + | + | + | + | + | V | + |
A number of important residues in PB2, PB1, PA, and NP proteins were known as playing a vital role for the adaptation of avain influenza viruses to a new host species.14 As shown in Table 1, most Jiangxi isolates contained typical avian residues at these sites.
Some reports demonstrated that the vital residues at T37A, R95K, S224N, K242N of M1, and D21G of M2 played a crucial role in increasing virus virulence.19 In our study, those mutations occurred in all 17 Jiangxi isolates. As is reported,20 substitution of S for P at residue 42 (P42S) of NS1 could cause virus virulence to increase. All Jiangxi isolates possessed S at residue 42 of NS1.
By analyzing the evolution and molecular characteristics, the current study once again highlights the necessity for continued systematic influenza surveillance in poultry in countries in which the virus is endemic to track the emergence of novel H9N2 viruses with pandemic potential.
AUTHOR CONTRIBUTIONS
GYD, LH conceived and designed the manuscript. GYD, LH, and WJH analyzed the data and drafted the manuscript. HXY, XLL, CMW, QMS, and TZ participated in data analysis. All authors read and approved the final manuscript.
COMPLIANCE WITH ETHICS GUIDELINES
This study does not contain any studies with human or animal subjects performed by any of the authors.
Supporting information
Table S1 Results of viruses isolation and identification
Table S2. Analysis of amino acid sequences of HA and NA proteins
Table S3. Analysis of the amino acid sequences of the internal proteins of Jiangxi isolates in our study in 2011
Table S4 Accession numbers ### in NCBI
ACKNOWLEDGMENT
This study was supported by grants from the National High Technology Research and Development Program (863 Program) of China (2012AA12A407).
Han L, He W, Yan H, et al. The evolution and molecular characteristics of H9N2 avian influenza viruses in Jiangxi of China. J Med Virol. 2019;91:711‐716. 10.1002/jmv.25363
References
REFERENCES
- 1. Vasin AV, Temkina OA, Egorov VV, Klotchenko SA, Plotnikova MA, Kiselev OI. Molecular mechanisms enhancing the proteome of influenza A viruses: an overview of recently discovered proteins. Virus Res. 2014;185(7):53‐63. http://111.com [DOI] [PubMed] [Google Scholar]
- 2. Wu Y, Wu Y, Tefsen B, Shi Y, Gao GF. Bat‐derived influenza‐like viruses H17N10 and H18N11. Trends Microbiol. 2014;22(4):183‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu KM, Smith GJD, Bahl J, et al. The genesis and evolution of H9N2 influenza viruses in poultry from southern China, 2000 to 2005. J Virol. 2007;81(19):10389‐10401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Homme PJ, Easterday BC. Avian influenza virus infections. I. Characteristics of influenza A‐turkey‐Wisconsin‐1966 virus. Avian Dis. 1970;14(1):66‐74. [PubMed] [Google Scholar]
- 5. Guo YJ, Krauss S, Senne DA, et al. Characterization of the pathogenicity of members of the newly established H9N2 influenza virus lineages in Asia. Virology. 2000;267(2):279‐288. [DOI] [PubMed] [Google Scholar]
- 6. Li KS, Xu KM, Peiris JSM, et al. Characterization of H9 subtype influenza viruses from the ducks of Southern China: a candidate for the next influenza pandemic in humans? J Virol. 2003;77(12):6988‐6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Butt KM, Smith GJD, Chen H, et al. Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. J Clin Microbiol. 2005;43(11):5760‐5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cameron KR, Gregory V, Banks J, et al. H9N2 subtype influenza A viruses in poultry in pakistan are closely related to the H9N2 viruses responsible for human infection in Hong Kong. Virology. 2000;278(1):36‐41. [DOI] [PubMed] [Google Scholar]
- 9. Dong G, Xu C, Wang C, et al. Reassortant H9N2 influenza viruses containing H5N1‐Like PB1 genes isolated from black‐billed magpies in Southern China. PLoS One. 2011;6(9):e25808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gu M, Chen H, Li Q, et al. Enzootic genotype S of H9N2 avian influenza viruses donates internal genes to emerging zoonotic influenza viruses in China. Vet Microbiol. 2014;174(3‐4):309‐315. [DOI] [PubMed] [Google Scholar]
- 11. Pu J, Wang S, Yin Y, et al. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc Natl Acad Sci USA. 2015;112(2):548‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu KM, Li KS, Smith GJD, et al. Evolution and molecular epidemiology of H9N2 influenza A viruses from quail in southern China, 2000 to 2005. J Virol. 2007;81(6):2635‐2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lam TTY, Wang J, Shen Y, et al. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature. 2013;502(7470):241‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santos‐Pardo I, Martínez‐Morillo M, Villuendas R, Bayes‐Genis A. Anti‐Ro antibodies and reversible atrioventricular block. N Engl J Med. 2013;368(24):2335‐2337. [DOI] [PubMed] [Google Scholar]
- 15. Zhu Y, Yang D, Ren Q, et al. Identification and characterization of a novel antigenic epitope in the hemagglutinin of the escape mutants of H9N2 avian influenza viruses. Vet Microbiol. 2015;178(1‐2):144‐149. [DOI] [PubMed] [Google Scholar]
- 16. Baigent SJ, Mccauley JW. Influenza type A in humans, mammals and birds: determinants of virus virulence, host‐range and interspecies transmission. Bioessays News Rev Mol Cell Dev Biol. 2003;25(7):657‐671. [DOI] [PubMed] [Google Scholar]
- 17. Matrosovich M, Zhou N, Kawaoka Y, Webster R. The surface glycoproteins of H5 influenza viruses isolated from humans, chickens, and wild aquatic birds have distinguishable properties. J Virol. 1999;73(2):1146‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun Y, Tan Y, Wei K, et al. Amino Acid 316 of hemagglutinin and the neuraminidase stalk length influence virulence of H9N2 influenza virus in chickens and mice. J Virol. 2013;87(5):2963‐2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iqbal M, Yaqub T, Reddy K, Mccauley JW. Novel genotypes of H9N2 influenza A viruses isolated from poultry in Pakistan containing NS genes similar to highly pathogenic H7N3 and H5N1 viruses. PLoS One. 2009;4(6):e5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiao P, Tian G, Li Y, et al. A single‐amino‐acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J Virol. 2008;82(3):1146‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Results of viruses isolation and identification
Table S2. Analysis of amino acid sequences of HA and NA proteins
Table S3. Analysis of the amino acid sequences of the internal proteins of Jiangxi isolates in our study in 2011
Table S4 Accession numbers ### in NCBI