

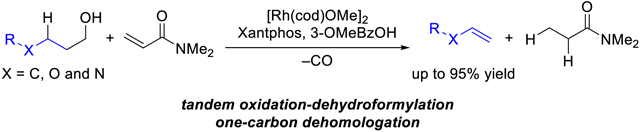
Abstract
We report a Rh-catalyst for accessing olefins from primary alcohols by a C–C bond cleavage that results in dehomologation. This functional group interconversion proceeds by an oxidation-dehydroformylation enabled by N,N-dimethylacrylamide as a sacrificial acceptor of hydrogen gas. Alcohols with diverse functionality and structure undergo oxidative dehydroxymethylation to access the corresponding olefins. Our catalyst protocol enables a two-step semisynthesis of (+)-yohimbenone and dehomologation of feedstock olefins.
Graphical Abstract
Enzymes perform one-carbon dehomologations of alcohols via the intermediacy of an aldehyde. For example, DNA demethylases oxidize alcohols to aldehyde intermediates that are decarbonylated to generate alkanes and arenes.1 Lanosterol demethylase performs a tandem oxidation and dehydroformylation to generate alkenes (Figure 1a). In contrast, while dehomologation of alcohols to generate alkanes has been achieved with various homogeneous catalysts,2 initial efforts to convert alcohols into olefins used heterogeneous catalysis and resulted in side reactions, including dehydration, olefin isomerization, and cracking, due to high reaction temperatures (>380 °C).3c Inventing ways to access olefins remains a primary focus due to their versatility as building blocks for materials and medicines.4 To achieve a mild, selective, and more general alcohol to alkene transformation, we thus focused on developing a bioinspired cascade.
Figure 1.

Inspiration for proposed alcohol oxidative dehydroxymethylation.
Our laboratory reported a dehomologation that transforms aldehydes into olefins via transfer of the formyl group and hydride onto a strained olefin acceptor, such as norbornadiene.5a Morandi coined such processes shuttle catalysis.6 Nozaki and Sorensen reported complementary dehydroformylations, through Ir-catalysis or photocatalysis, respectively (Figure 1b).5b, 3d In Sorensen’s study, he illustrated one oxidative dehydroxymethylation of a neopentylic alcohol, although a mixture of products was observed.3d Given precedence for both transfer hydrogenation7,8 and transfer dehydroformylation,5a we focused on the use of tandem Rh-catalysis to achieve a more general alcohol dehomologation to alkenes (Figure 1c).9
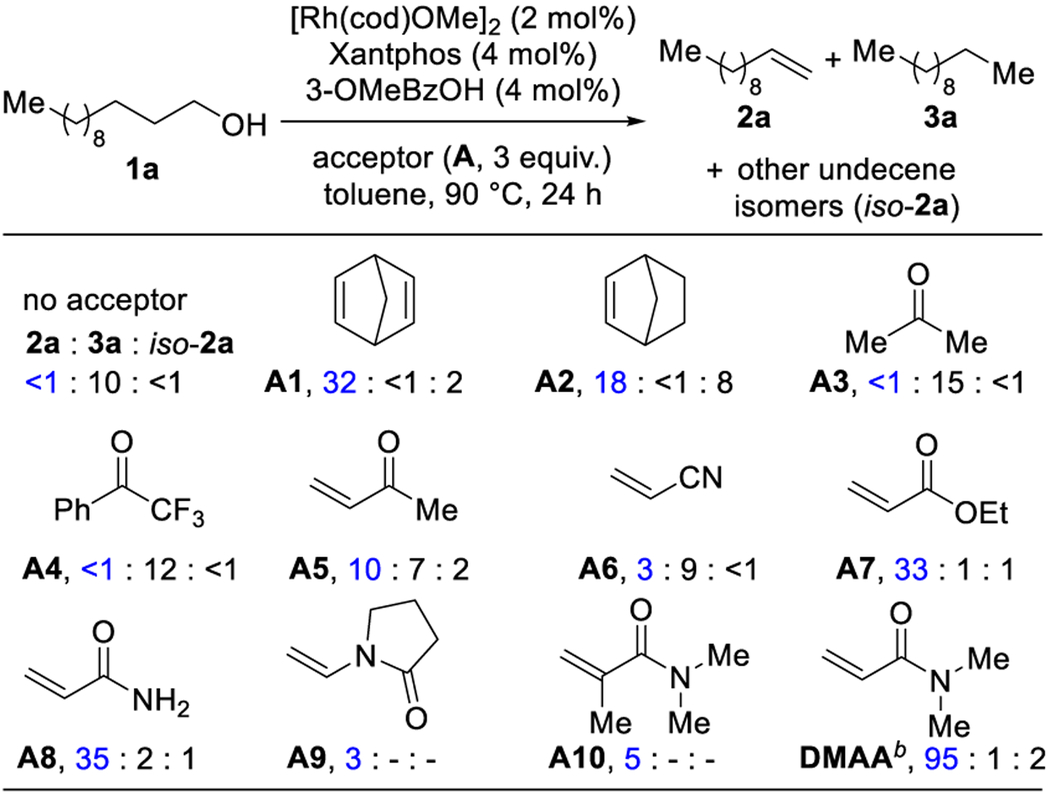
We set out to identify one catalyst capable of both transfer hydrogenation and transfer hydroformylation.10 Using 1-dodecanol 1a as a model substrate, we began our studies with a catalyst known to activate aldehyde C–H bonds ([Rh(cod)OMe]2, 3-OMeBzOH, and Xantphos, Table 1).5a Upon successful oxidation of alcohol 1a, we imagined the resulting aldehyde could undergo dehydroformylation to the alkene 2a or decarbonylation to the alkane 3a. From an initial survey, we discovered that selectivity for alkene vs alkane was influenced by the acceptor. In the absence of an acceptor, we observed undecane 3a as the only product (10% yield). In stark contrast, by using strained olefin acceptors A1 and A2, we observed 1-undecene (2a, 32% and 18% respectively), along with undecene isomers (iso-2a, 16:1 and 2.3:1, 2a:iso-2a). Using ketones as acceptors (A3–4) resulted in decarbonylation to undecane 3a. While using electron-deficient olefin acceptors, such as enone A5 or acrylonitrile A6, a mixture of 1-undecene 2a and undecane 3a was observed (1.4:1 and 1:3, 2a:3a). Using unsaturated ester or amide acceptors provided a major breakthrough in selectivity for the desired alkene 2a.
Table 1.
Effect of Acceptor on Selectivity for Oxidative Dehydroxymethylationa
|
Conditions: 1a (0.2 mmol), [Rh(cod)OMe]2 (2 mol%), 3-OMeBzOH (4 mol%), Xantphos (4 mol%) and acceptor (3 equiv.) in toluene (0.4 mL), 90 °C, 24 h. Yields were determined by GC using durene as an internal standard. b92% yield of CO by GC-TCD.
Unsaturated ester and amide acceptors (A7–A8) enabled selective formation of 1-undecene (2a, 33–35%, >20–17.5:1, 2a:3a). Use of N,N-dimethylacrylamide (DMAA) as an acceptor gave 1-undecene 2a in 95% yield and >20:1 selectivity.11 We reason DMAA affords improved reactivity because it can bind more effectively to the Rh-catalyst in comparison to other Michael acceptors (A5–A8, A10). We found that the byproduct was N,N-dimethylpropionamide, which arises from the hydrogenation of DMAA. The use of N-vinylpyrrolidone (A9) or the α-methyl substituted acrylamide A10 resulted in diminished reactivity (3–5%). Previously, we found that both CO and H2 were transferred to our strained olefin acceptor, norbornadiene A1.5a In contrast, we do not observe transfer hydroformylation, yet catalyst turnover still proceeds in the presence of CO generation, as quantified by GC-thermal conductivity detection (see SI).12
With this catalyst-acceptor combination, we performed the dehomologation of primary alcohols (Table 2a). Allylbenzene 2b was obtained (93% yield) from 4-phenyl-1-butanol, without isomerization to a conjugated olefin. 3-Phenyl-1-propanol and derivatives with electron-donating and electron-withdrawing groups gave styrenes (2c–e) in 85–93% yields. Heterocyclic alcohols, such as those with pyridine and indole, were tolerated (2f, 85%; 2g, 77%). A primary diol gave diene 2h in 88% yield, in the presence of double the amount of DMAA (6 equivalents). A β,β-disubstituted alcohol transformed to internal olefin 2i in 91% yield. Alcohols bearing alkenes and tertiary alcohols underwent dehomologation (2j, 82%; 2k, 87%). Next, we explored 1,3- or 1,4-diols and 2-, 3- or 4-amino derived alcohols (2l–s). Allylic ether 2l and amine 2o were obtained in 81% and 92% yields respectively, without allylic C–O or C–N bond cleavage or debenzylation. Enol and enamine derivatives (2m, 2n, 2p–s) can be accessed (75–83% yields). Enamine formation occurred preferentially over allyl amine formation to afford 2q (80% yield). We obtained tri-substituted enamide 2r in 75% yield from alcohol 1r. With most alcohols, excellent chemoselectivities (>20:1) were observed. In contrast, use of 3-phthalimido-1-propanol gave a 4:1 mixture of oxidation-dehydroformylation (2s) and oxidation-decarbonylation (3s). When cis- or trans-1t was used, β-hydride elimination occurred preferentially at the less substituted position to give 2t1. In addition, we found that allylic alcohols (4a-c) underwent oxidative dehydroxymethylation (75–95% yields), with only 1.5 equivalents of DMAA needed (Table 2b).13
Table 2.
Oxidative Dehydroxymethylation of Alcoholsa

|
Conditions: 1 (0.2 mmol), [Rh(cod)OMe]2 (2 mol%), 3-OMeBzOH (4 mol%), Xantphos (4 mol%) and DMAA (3 equiv.) in toluene (0.4 mL), 90 °C, 24 h. Isolated yields. bGC yields using durene as an internal standard. cDMAA (6 equiv.) used.
Next, we explored applications (Scheme 1). By combining hydroboration-oxidation with oxidative dehydroxymethylation, a one-carbon dehomologation of 1-dodecene 6 was achieved on gram scale to give 1-undecene 2a (82% yield, Scheme 1a). This two-step process provides valuable odd-numbered carbon olefins from readily available deven-numbered carbon olefins.14c A two-carbon dehomologation of olefins can be achieved by combining olefin dihydroxylation and oxidative dehydroxymethylation. For example, we found that 1-dodecene 6 could be transformed to 1-decene 2v (Scheme 1a).15 The transformation occurs efficiently with molecules that are more structurally complex (Scheme 1b). Benzyl protected deoxycholic acid derivative 8a gave olefin 9a (81% yield), with no debenzylation. We probed chemoselectivity by using triol 8b, with alcohols bearing different steric bulk. We observed oxidation-dehydroformylation of the primary alcohol and selective oxidation of the less hindered secondary alcohol to afford 9b (66% yield). Diol 8c underwent oxidative dehydroxymethylation and secondary alcohol oxidation to access (+)-yohimbenone 9c. Based on this result, we improved our previous synthesis of (+)-yohimbenone 9c by shortening the sequence to two steps.5a, 16
Scheme 1.

Synthetic Applications
While further studies are warranted, on the basis of literature reports2,5,6,11,17 and our own observations, we propose the following pathway (Scheme 3). Exchange between the benzoate counterion in Rh-complex A and an alcohol affords B. Intermediate B undergoes β-hydride elimination to give Rh-hydride C. Coordination of DMAA to C generates intermediate D. Hydrometallation of DMAA followed by protodemetalation provides the aldehyde and regenerates complex A. Oxidative addition into the aldehyde C–H bond by A generates acyl-Rh-hydride F. Reductive elimination of 3-methoxybenzoic acid generates acyl-Rh G. CO deinsertion to H, followed by β-hydride elimination, yields Rh-hydrido-carbonyl I. Olefin exchange with DMAA generates Rh-hydride J. Hydrometallation of DMAA gives complex K, and CO is extruded to make L. Finally, protodemetalation regenerates complex A.
Scheme 3.

Proposed Mechanism
To support the proposed mechanism, control experiments and deuterium-labeling experiments were carried out. Under standard conditions, neopentylic alcohol 1w oxidizes to aldehyde 10 in 90% yield (Scheme 4a). Incorporation of a quaternary carbon alpha to the carbonyl suppressed dehydroformylation. These results support the intermediacy of an aldehyde in the catalytic cycle. Of note, aldehyde 11 undergoes dehydroformylation under standard conditions (Scheme 4b), showing similar reactivity to our previous report,5a but with a more economical acceptor (i.e., norbornadiene vs DMAA). Replacing the benzoate counterion with chloride suppressed both oxidation and dehydroformylation (Scheme 4a and b). In the absence of DMAA, dehydrogenation of alcohol 1w was not observed (Scheme 4a). In contrast, decarbonylation of aldehyde 11 gave ethyl benzene 3c (78% yield, 5.5:1 3c:2c, Scheme 4b). These observations highlight the importance of both the benzoate counterion and DMAA. In support of the protonation of intermediate E (Scheme 3), we observed deuterium incorporation at the β-position of DMAA when using deuterated isopropanol D-12 (Scheme 4c). Hydrogen-deuterium exchange is possible during dehydroformylation via the benzoate counterion acting as a proton shuttle (Scheme 4c and d).5a
Scheme 4.
Probing the Mechanism.

a[Rh(cod)Cl]2 used instead of [Rh(cod)OMe]2 and 3-OMeBzOH.bYield of 2c, >20:1 2c:3c. cYield of 2c, 78% yield 3c
Using competition experiments, we studied the chemoselectivity of this cascade (Scheme 2, see SI for details). Aldehydes undergo dehydroformylation in preference to primary alcohols undergoing oxidative dehydroxymethylation, with 60:1 selectivity. Primary alcohols oxidize faster than secondary and benzylic alcohols faster than aliphatic. These observations support that alcohol oxidation is the turnover limiting cycle in this novel cascade.
Scheme 2.

Competition Experiments
Established strategies for constructing olefins, including the Wittig olefination,18 the Heck reaction,19 and olefin metathesis,20 generate carbon-carbon bonds. In contrast, our strategy contributes to emerging routes to olefins that involve C–C bond cleavage.21 These methods represent examples of a one-carbon dehomologation of carbon frameworks and thus hold promise for various applications, including the conversion of biomass into feedstocks.22 Moreover, such transformations increase retrosynthetic flexibility by allowing the interconversion of two common functional groups.14
ACKNOWLEDGMENT
Funding was provided by the National Science Foundation (CHE-1465263), the National Institutes of Health (GM105938), UC Irvine, and Chevron Phillips. F.A.C. is grateful for an NSF Graduate Research Fellowship. We thank Daniel Ess from Brigham Young University for discussion.
Footnotes
The authors declare no competing financial interests.
Supporting Information
Detailed experimental procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Ladwein KI; Jung M Angew. Chem., Int. Ed 2011, 50, 12143. [DOI] [PubMed] [Google Scholar]
- (2).For alcohol oxidation and decarbonylation, see:; (a) Ipatieff VN; Czajkowski GJ; Pines HJ Am. Chem. Soc 1951, 73, 4098. [Google Scholar]; (b) Ishige M; Sakai K; Kawai M; Hata K Bull. Chem. Soc. Jpn 1970, 43, 2186. [Google Scholar]; (c) Obora Y; Anno Y; Okamoto R; Matsu-ura T; Ishii Y Angew. Chem., Int. Ed 2011, 50, 8618. [DOI] [PubMed] [Google Scholar]; (d) Ho H-A; Manna K; Sadow AD Angew. Chem., Int. Ed 2012, 51, 8607. [DOI] [PubMed] [Google Scholar]; (e) Olsen EPK; Madsen R Chem. Eur. J 2012, 18, 16023. [DOI] [PubMed] [Google Scholar]; (f) Modak A; Naveen T; Maiti D Chem. Commun 2013, 49, 252. [DOI] [PubMed] [Google Scholar]
- (3).(a) Walling C; Humphreys RW R. J. Org. Chem 1981, 46, 1260. [Google Scholar]; (b) Kim C; Matsui Y; Orchin MJ Organometal. Chem 1985, 279, 159. [Google Scholar]; (c) Lietti L; Tronconi E; Forzatti PJ Mol. Catal. 1988, 44, 201. [Google Scholar]; (d) Abrams DJ; West JG; Sorensen EJ Chem. Sci 2017, 8, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For recent olefin constructions, see:; (a) Ludwig JR; Zimmerman PM; Gianino JB; Schindler CS Nature 2016, 533, 374. [DOI] [PubMed] [Google Scholar]; (b) Nguyen TT; Koh MJ; Shen X; Romiti F; Schrock RR; Hoveyda AH Science 2016, 352, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Koh MJ; Nguyen TT; Lam JK; Torker S; Hyvl J; Schrock RR; Hoveyda AH Nature 2017, 542, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Edwards JT; Merchant RR; McClymont KS; Knouse KW; Qin T; Malins LR; Vokits B; Shaw SA; Bao D-H; Wei F-L; Zhou T; Eastgate MD; Baran PS Nature 2017, 545, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lei C; Yip YJ; Zhou JS J. Am. Chem. Soc 2017, 139, 6086. [DOI] [PubMed] [Google Scholar]
- (5).(a) Murphy SK; Park J-W; Cruz FA; Dong VM Science, 2015, 347, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]; For other examples of aldehyde dehydroformylation, see:; (b) Kusumoto S; Tatsuki T; Nozaki K Angew. Chem., Int. Ed 2015, 54, 8458. [DOI] [PubMed] [Google Scholar]; (c) Hattori T; Takakura R; Ichikawa T; Sawama Y; Monguchi Y; Sajiki HJ Org. Chem 2016, 81, 2737. [DOI] [PubMed] [Google Scholar]
- (6).For reviews and selected examples of shuttle catalysis, see:; (a) Bhawal BN; Morandi B ACS Catal 2016, 6, 7528. [Google Scholar]; (b) Bhawal BN; Morandi Chem. Eur. J 2017, 23, 12004. [DOI] [PubMed] [Google Scholar]; (c) Fang X; Yu P Morandi B Science 2016, 351, 832. [DOI] [PubMed] [Google Scholar]; (d) Fang X; Cacherat B; Morandi B Nat. Chem 2017, 9, 1105. [DOI] [PubMed] [Google Scholar]; (e) Yu P; Morandi B Angew. Chem. Int. Ed 2017, 56, 15693. [DOI] [PubMed] [Google Scholar]
- (7).Wang D; Astruc D Chem. Rev 2015, 115, 6621. [DOI] [PubMed] [Google Scholar]
- (8).Using alcohols as surrogates for aldehydes has emerged. For a review, see:; (a) Kim SW; Zhang W; Krische MJ Acc. Chem. Res 2017, 50, 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]; For select examples, please see:; (b) Lebel H; Paquet VJ Am. Chem. Soc 2004, 126, 11152. [DOI] [PubMed] [Google Scholar]; (c) Xie X; Stahl SS J. Am. Chem. Soc 2015, 137, 3767. [DOI] [PubMed] [Google Scholar]; (d) Zultanski SL; Zhao J; Stahl SS J. Am. Chem. Soc 2016, 138, 6416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liang T; Woo SK; Krische MJ Angew. Chem. Int. Ed 2016, 55, 9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For a definition of tandem catalysis, see:; Fogg DE; dos Santos EN.; Coord. Chem. Rev 2004, 248, 2365. [Google Scholar]
- (10).The reverse process is possible. For selected examples of olefin tandem hydroformylation-hydrogenation, see:; (a) Takahashi K; Yamashita M; Ichihara T; Nakano K; Nozaki K Angew. Chem. Int. Ed 2010, 49, 4488. [DOI] [PubMed] [Google Scholar]; (b) Boogaerts IIF; White DFS; Cole-Hamilton DJ Chem. Commun 2010, 46, 2194. [DOI] [PubMed] [Google Scholar]; (c) Fuchs D Rousseau G; Diab L; Gellrich U; Breit B Angew. Chem. Int. Ed 2012, 51, 2178. [DOI] [PubMed] [Google Scholar]; (d) Diebolt O; Müller C; Vogt D Catal. Sic. Technol 2012, 2, 773. [Google Scholar]; (e) Takahashi K; Yamashita M; Nozaki KJ Am. Chem. Soc 2012, 134, 18746. [DOI] [PubMed] [Google Scholar]
- (11).We observe transfer hydroformylation and hydrogenation of A2 and A3. We observe only transfer hydrogenation of the other acceptors in Table 1. For DMAA as a transfer hydrogenation acceptor, see:; Mai VH; Nikonov GI Organometallics 2016, 35, 943. [Google Scholar]
- (12).For an example of Rh-catalyzed decarbonylation in refluxing acetone (~60 °C), see:; Bergens SH; Fairlie DP; Bosnich B Organometallics 1990, 9, 566. [Google Scholar]
- (13).For decarbonylation of allylic alcohols to give R–H, see:; Emery A; Oehlschlager AC; Unrau AM Tetrahedron Lett 1970, 50, 4401. [Google Scholar]
- (14).Breakthroughs in olefin synthesis by dehomologation have been achieved from carboxylic acid derivatives. Carboxylic acids:; (a) Gooβen LJ; Rodríguez N Chem. Commun 2004, 40, 724. [DOI] [PubMed] [Google Scholar]; (b) Liu Y; Kim KE; Herbert MB; Fedorov A; Grubbs RH; Stoltz BM Adv. Synth. Catal 2014, 356, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Y; Virgil SC; Grubbs RH; Stoltz BM Angew. Chem., Int. Ed 2015, 54, 11800. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) John A; Hillmyer MA; Tolman WB Organometallics 2017, 36, 506. Esters: [Google Scholar]; (e) Minami I; Yuhara M; Shimizu I; Tsuji JJ Chem. Soc., Chem. Commun. 1986, 118. [Google Scholar]; (f) John A; Hogan LT; Hillmyer MA; Tolman WB Chem. Commun 2015, 51, 2731 Amides: [DOI] [PubMed] [Google Scholar]; (g) Hu J; Wang M; Pu X; Shi Z Nat. Commun. 2017, 8, 14993 Acyl halides: [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Tsuji J; Ohno KJ Am. Chem. Soc 1968, 90, 94. [Google Scholar]; (i) Ohno K; Tsuji JJ Am. Chem. Soc 1968, 90, 99 Acyl cyanides: [Google Scholar]; (j) Murahashi S-I; Naota T; Nakajima NJ Org. Chem 1986, 51, 898 Thioester: [Google Scholar]; (k) Goto T; Onaka M; Mukaiyama T Chem. Lett 1980, 709. [Google Scholar]
- (15).With D-mannitol, we do not observe any dehomolgation or CO gas extrusion.
- (16).Stearman C; Wilson M; Padwa AJ Org. Chem 2009, 74, 3491. [DOI] [PubMed] [Google Scholar]
- (17).For a computational study on Rh-catalyzed dehydroformylation, see:; Luo X; Bai R; Liu S; Shan C; Chen C; Lan Y J. Org. Chem 2016, 81, 2320. [DOI] [PubMed] [Google Scholar]
- (18).For recent reviews, see:; (a) Gu Y; Tian S-K Top. Curr. Chem 2012, 327, 197. [DOI] [PubMed] [Google Scholar]; (b) Lao Z; Toy PH Beilstein J. Org. Chem 2016, 12, 2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).For recent reviews, see:; (a) Bras JL; Muzart J Chem. Rev 2011, 111, 1170. [DOI] [PubMed] [Google Scholar]; (b) Wang S-S; Yang G-Y Catal. Sci. Technol 2016, 6, 2862. [Google Scholar]
- (20).For recent reviews, see:; (a) Hoveyda AH; Zhugralin AR Nature 2007, 450, 243. [DOI] [PubMed] [Google Scholar]; (b) Fürstner, A. Science 2013, 341, 1229713. [DOI] [PubMed] [Google Scholar]
- (21).For recent reviews on C–C bond breaking, see:; (a) C–C Bond Activation. In Top. Curr. Chem; Dong G, Eds.; Springer: New York, 2014. [Google Scholar]; (b) Souilllart L; Cramer N Chem. Rev 2015, 115, 9410. [DOI] [PubMed] [Google Scholar]
- (22).Huber GW; Iborra S; Corma A Chem. Rev 2006, 106, 4044. [DOI] [PubMed] [Google Scholar]