

SUMMARY
Insulin stimulates the conversion of phosphatidylino-sitol-4,5-bisphosphate (PI(4,5)P2) to phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3), which mediates downstream cellular responses. PI(4,5)P2 is produced by phosphatidylinositol-4-phosphate 5-kinases (PIP5Ks) and by phosphatidylinositol-5-phos-phate 4-kinases (PIP4Ks). Here, we show that the loss of PIP4Ks (PIP4K2A, PIP4K2B, and PIP4K2C) in vitro results in a paradoxical increase in PI(4,5)P2 and a concomitant increase in insulin-stimulated production of PI(3,4,5)P3. The reintroduction of either wild-type or kinase-dead mutants of the PIP4Ks restored cellular PI(4,5)P2 levels and insulin stimulation of the PI3K pathway, suggesting a catalytic-independent role of PIP4Ks in regulating PI(4,5)P2 levels. These effects are explained by an increase in PIP5K activity upon the deletion of PIP4Ks, which normally suppresses PIP5K activity through a direct binding interaction mediated by the N-terminal motif VMLϕFPDD of PIP4K. Our work uncovers an allosteric function of PIP4Ks in suppressing PIP5K-mediated PI(4,5)P2 synthesis and insulin-dependent conversion to PI(3,4,5)P3 and suggests that the pharmacological depletion of PIP4K enzymes could represent a strategy for enhancing insulin signaling.
Graphical Abstract
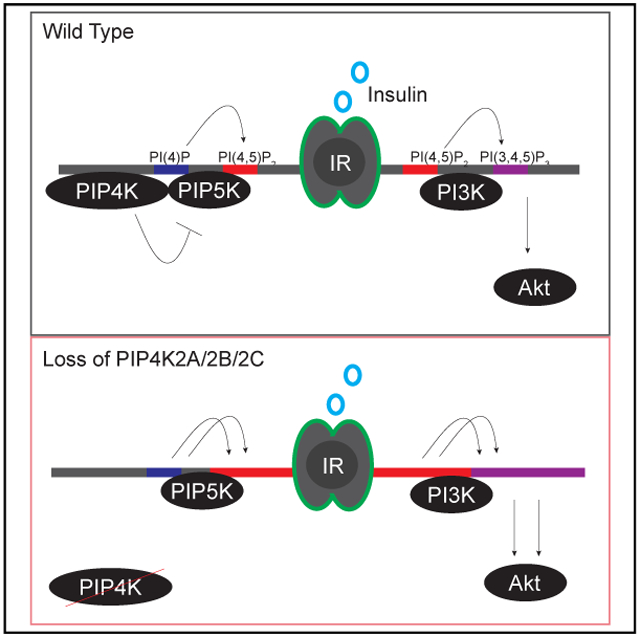
In Brief
PI(4,5)P2 is produced by both phosphatidylinositol-4-phosphate 5-kinases (PIP5Ks) and by phosphatidylinositol-5-phosphate 4-kinases (PIP4Ks). Wang et al. report an allosteric function of a conserved N-terminal motif of PIP4Ks in suppressing PIP5K-mediated PI(4,5)P2 synthesis and insulin-dependent conversion to PI(3,4,5) P3. This non-catalytic role has implications for the development of PIP4K targeted therapies.
INTRODUCTION
Phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) plays numerous roles in cellular regulation. It mediates actin remodeling at the plasma membrane, modulates vesicle trafficking, and is the substrate that hormone-stimulated phospholipases type C (PLC) use to generate the second messengers diacylglycerol and inositol-1,4,5-trisphosphate (Balla, 2013; Sun et al., 2013). PI(4,5) P2 is also the substrate that class 1 phosphoinositide 3-kinases use (Saito et al., 2003) to generate the second messenger phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3) in response to insulin and other growth factors (Fruman et al., 2017).
Yeasts have a single enzyme for generating PI(4,5)P2 encoded by MSS4 (Homma et al., 1998), whereas mammals have six genes encoding enzymes that generate PI(4,5)P2. PIP5K1A, PIP5K1B, and PIP5K1C produce PI(4,5)P2 from phosphati-dylinositol-4-phosphate (PI(4)P), while PIP4K2A, PIP4K2B, and PIP4K2C generate PI(4,5)P2 from phosphatidylinositol-5-phos-phate (PI(5)P) (Rameh et al., 1997; van den Bout and Divecha, 2009). All multicellular organisms have genes from both families. The enzymes encoded by these two families of genes have sequence and structural similarities, but the activation loop of the PIP4Ks confers strict substrate selectivity for PI(5)P over PI(4)P (Kunz et al., 2000, 2002).
In settings where phosphoinositides have been quantified, PI(4)P and PI(4,5)P2 each constitute between 30% and 50% of total cellular phosphoinositides, respectively. Whereas local levels of PI(4,5)P2 can transiently drop when cells are stimulated with growth factors or hormones that activate PLC or phosphoinositide 3-kinases (PI3Ks), the total levels of PI(4,5)P2 and PI(4)P remain remarkably constant. PI(4)P is >100-fold more abundant than PI(5)P in cells, and it is generally assumed that most PI(4,5)P2 in mammalian cells is generated from PI(4)P via PIP5Ks (Balla, 2013). In B cell activation, transient recruitment of PIP5K1A is necessary for the generation of PI(3,4,5)P3 for signal transduction (Saito et al., 2003).
Because PI(5)P constitutes ~1% of cellular phosphoinositides, it is uncertain whether this lipid contributes substantially to total cellular PI(4,5)P2, raising speculation that the function of the PIP4Ks is primarily to decrease the level of PI(5)P (Jones et al., 2006; Wilcox and Hinchliffe, 2008). Recent work from our laboratory showed that the conversion of PI(5)P to PI(4,5)P2 by PIP4K2A and PIP4K2B, likely on lysosomes, is critical to mediate the fusion between autophagosomes and lysosomes (Lundquist et al., 2018). This study argued that while the PIP4Ks generate only a small fraction of cellular PI(4,5)P2, the location of the PI(4,5)P2 generated by these enzymes plays an important role in the completion of autophagy. There is also evidence for pools of PIP4K2A/PIP4K2B/PIP4K2C in the plasma membrane, Golgi, and nucleus, so the PIP4K enzymes are likely to have many other functions beyond autophagy regulation (Bultsma et al., 2010; Jones et al., 2006; Mackey et al., 2014).
PIP4K family members may also suppress insulin-PI3K-mammalian target of rapamycin complex 1 (mTORC1) signaling in vivo, despite their enzymatic function to synthesize PI(4,5)P2. Homozygous germline deletion of Pip4k2b−/− in mice causes reduced adiposity and increased insulin sensitivity in muscle (Lamia et al., 2004), and Pip4k2c−/− mice have enhanced TORC1 signaling (Shim et al., 2016). In addition, the accumulation of PI(5)P, resulting from the loss of the highly active forms of PIP4K, increases signaling through the PI3K pathway (Carri-caburu et al., 2003; Pendaries et al., 2006). While this is one mechanism by which PIP4Ks could influence PI3K activity, additional possibilities remain to be explored. For example, it was previously shown that overexpressed PIP4K2A can interact with all three PIP5K isoforms, but the nature of this binding remains a mystery (Hinchliffe et al., 2002), Overall, it is not clear how membrane lipid changes underlie the molecular mechanism by which the PIP4Ks regulate the PI3K pathway, and it is unknown whether one PIP4K isoform can compensate for others to suppress growth factor signal transduction.
In this study, we demonstrate that PIP4Ks have distinct catalytic and non-catalytic functions in controlling cellular metabolism and that it is the loss of catalytic-independent functions of PIP4Ks that underlie enhanced insulin signaling. We report our findings in parallel with an independent study in Drosphila, placing PIP4K as an evolutionarily conserved modulator of the PI3K pathway (Sharma et al., 2019).
RESULTS
Loss of PIP4K Family Members Does Not Affect Cell Viability
To investigate the role of PIP4K enzymes in cellular signaling, we generated tools to systematically deplete individual members of the PIP4K family. We cloned a series of lentiviral-based tandem miR-E-based short hairpin RNAs (shRNAs) (Tables S1 and S2) and induced the stable knockdown of PIP4K family isoforms in HeLa cells and other human immortalized or transformed cells (Figures 1A, S1A-S1C, and S2A-S2E; Fellmann et al., 2011, 2013; Pelossof et al., 2017). shRNA-mediated silencing of one or all of the PIP4Ks did not result in gross changes in morphology or cause a major change in the growth rate of the cells examined (Figure S1D). As an orthogonal approach, we used CRISPR/Cas9 to delete PIP4K2A, PIP4K2B, and PIP4K2C in HEK293T cells (Figure 1B; Tables S1 and S2), with similar results (Ran et al., 2013).
Figure 1. Validation of Tools to Eliminate All Three PIP4K Isoforms Reveals Paradoxical Increase in PI(4,5)P2.

(A) Western blots showing the efficiency of knockdown of PIP4K isoforms in HeLa cells. Cell line notation and their descriptions are listed in Table S2.
(B) Western blots showing 293T clones with CRISPR-mediated knockout of PIP4K2A, PIP4K2B, and PIP4K2C, with cell line descriptions in Table S2. Parental cell line is in the first lane, and the CRISPR clonal cell line with intact PIP4K is designed as WT. A faint non-specific band that runs slightly faster than PIP4Ks is just visible.
(C and D) Quantification by HPLC of PI(5)P, which increases in cells with the loss of PIP4K, shown in HeLa TKD cells (C) as well as 293T TKO cells (D).
(E) Knock down of PIP4K enhances insulin signaling in HeLa cells, as shown by western blot. Quantification by Li-Cor indicated the respective bands below.
(F) Immunofluorescence detection of lysosomal marker Lamp1 showing increased lysosomal accumulation in 293T TKO versus control cells. Representative images shown.
(G and H) PI(4,5)P2 increases in cells with loss of PIP4K, shown in HeLa TKD cells (G) and 293T TKO cells (H), measured by HPLC.
(I and J) PI(4)P decreases in cells with loss of PIP4K, shown in HeLa TKD cells (I) and 293T TKO cells (J), measured by HPLC.
(K) 293T TKO cells have higher levels of PI(3,4,5)P3, as measured by HPLC.
Significance calculated using ANOVA with Holm-Sidak multiple comparisons to control cell line with intact PIP4K. **p < 0.01, ***p < 0.001, and ****p < 0.0001. Data are represented as means ± SEMs. n = 3.
Loss of PIP4Ks Results in Increased PI(5)P levels, Increased PI3K Activation, and Defects in Autophagy
PIP4K2A/B/C triple knockout 293T cells (hereafter TKO) and triple knockdown HeLa cells (hereafter TKD) recapitulated previously reported phenotypes in which PIP4K enzymes were depleted. As reported (Gupta et al., 2013), the loss of all three enzymes conferred a 2-fold increase in their substrate, PI(5)P (Figures 1C and 1D). In addition, TKD cells exhibited increased PI3K signaling upon insulin stimulation (Figure 1E), which is consistent with observations of increased insulin sensitivity in Pip4k2b−/− mice (Carricaburu et al., 2003; Lamia et al., 2004). Furthermore, 293T TKO cells accumulated LAMP1 lysosomes, which we attributed to defects in autophagosome-lysosome fusion as described in Pip4k2a−/−, Pip4k2b−/−mouse embryonic fibroblasts (MEFs) (Figure 1F; Lundquist et al., 2018).
Cellular PI(4,5)P2 Is Increased upon Depletion of PIP4K
Depletion of the three PIP4Ks in cells altered the levels of phos-phoinositides beyond PI(5)P. In both TKD and TKO cells, we observed a surprising increase in PI(4,5)P2 and concomitant decrease in PI(4)P (Figures 1G-1J and S2F-S2P). Basal levels of PI(3,4,5)P3 during growth in culture are typically below the detection limit in high-performance liquid chromatography (HPLC)-based assays. However, we were able to detect the basal elevation of PI(3,4,5)P3 in TKO cells, suggesting that the increased PI(4,5)P2 is able to be used by PI3K (Figure 1K). PI(4,5)P2 constitutes ~50% of all phosphoinositides, although the majority is bound to the cytoskeletal and scaffolding proteins required for cell structure, migration, and adhesion (Brown, 2015; Choi et al., 2015). While PI(4,5)P2 is highly abundant, local production of PI(4,5)P2 by PIP5Ks offers spatiotemporal control of signaling through the PI3K and PLC pathways (Choi et al., 2015; Saito et al., 2003; Xie et al., 2009).
Identification of a Catalytic-Independent Function of PIP4K
Members of the PIP4K family have dramatically different catalytic rates. For instance, PIP4Kα (PIP4K2A) has 1,000-fold higher activity than PIP4Kγ (PIP4K2C), which is considered near-enzy-matically dead (Clarke and Irvine, 2013). Analysis of PI(4,5)P2 levels in cells with single or double knockdown of PIP4K isoforms revealed an additive effect among all three isoforms that does not correlate with their relative catalytic activities (Figures 2A and 2B). Double knockdown of the most active isoforms, PIP4Kα/β, did not phenocopy the triple knockdown, suggesting that catalytic activity is not the most important factor in regulating PI(4,5)P2 levels. To evaluate whether the catalytic activity of PIP4K enzymes is important for PI(4,5)P2 regulation, we generated shRNA-resistant hemagglutinin (HA)-tagged kinase-active or kinase-dead PIP4K isoforms (Figures 2C and 2D).
Figure 2. Identification of a Catalytic-Independent Function of PIP4K.

(A and B) Quantification of PI(4)P and PI(4,5)P2 levels by HPLC upon knockdown of one or multiple isoforms of PIP4K in 293T cells (A) or HeLa cells (B) using tandem shRNA constructs. Hairpin constructs are indicated on the × axis, with descriptions in Table S2.
(C and D) Western blot validation of rescue cell line panel with reconstitution of kinase-active or kinase-dead PIP4K2AKD or kinase-active PIP4K2C into HeLa TKD cells (C) or 293T TKO cells (D) at near-endogenous levels. Tagged 3xHA-PIP4K proteins are larger than the endogenous protein. A faint non-specific band is visible running slightly faster than the endogenous protein.
(E–J) Quantification by HPLC of PI(4,5)P2 in HeLa TKD rescue cell lines (E) and 293T TKO rescue cell lines (F). Quantification of PI(4)P in HeLa TKD rescue cell lines (G) and 293T TKO rescue cell lines (H). Quantification of PI(5)P in HeLa TKD rescue cell lines (I) and 293T TKO rescue cell lines (J).
Significance calculated using ANOVA with Holm-Sidak multiple comparisons to TKD or TKO cell lines. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Data are represented as means ± SEMs. n = 3.
The expression of either kinase-active or kinase-dead (PIP4K2AkD) PIP4Ks rescued PI(4,5)P2 levels (Figures 2E and 2F). Moreover, PI(4)P levels also returned to baseline with the expression of either kinase-active or kinase-dead PIP4K2A in both TKD and TKO cell lines (Figures 2G and 2H). In contrast, only the active PIP4K2A restored normal PI(5)P levels, indicating that the elevation of this lipid species in knockdown cells, in contrast to PI(4,5)P2, is due to the loss of PIP4K enzymatic activity (Figures 2I and 2J). We used a 3′ internal ribosome entry site (IRES)-GFP tag to sort for cell populations with low and high expression of PIP4K2AKD populations, PIP4K2AKD_low and PIP4K2AKD_high, respectively (Figure S3A). HPLC analysis of these cell lines shows a dose-dependent decrease in PI(4,5)P2 and increase in PI(4)P correlated with the levels of PIP4K2AKD expression (Figures S3B and S3C). Thus, we conclude that PIP4Ks have a catalytic-independent role in maintaining the homeostasis of cellular PI(4)P and PI(4,5)P2 levels, distinct from their enzymatic function in converting PI(5)P to PI(4,5)P2.
PIP5K Activity Is Elevated in Cells Depleted of PIP4K
The observed increase in PI(4,5)P2 relative to PI(4)P is consistent with a robust production of PI(4,5)P2 by the PIP5Ks. We measured PIP5K activity in mechanically disrupted cells and found that TKO cells exhibit elevated PIP5K activity, which is consistent with the model that PIP5K is more active in the absence of PIP4K. The increased PIP5K activity was reversed in cells expressing either kinase-active or kinase-dead PIP4K isoforms, indicating that it is not dependent on the enzymatic activity of these proteins (Figure 3A).
Figure 3. PIP4K Inhibits PIP5K Activity.

(A) Changes in PIP5K activity in 293T cells. Cell pellets were normalized to cell number and sonicated in the presence of excess PI(4)P. Radioactive ATP32 was added for kinase reaction. Lipids were extracted and quantified by TLC, with confirmation by HPLC after deacylation.
(B–D) Quantification of in vitro kinase assay measuring conversion of PI(4)P to radiolabeled PI(4,5)P2 by PIP5K1A. PIP5K1A activity is inhibited in vitro by addition of 2- to 10-fold molar excess of PIP4K2A, PIP4K2B, or PIP4K2C (B). Once denatured, PIP4K2C no longer inhibits PIP5K1A (C). In the presence of 0.1% Triton X-100, PIP4K2A no longer inhibits PIP5K1A activity (D).
(E) Western blot validation of near-endogenous level expression of 3xHA-PIP5K1A levels in 293T control and TKO cells. Tagged 3xHA-PIP5K1A is slightly larger than the endogenous protein.
(F) Pull-down using anti-HA beads from 293T WT cells expressing empty 3xHA vector (empty) or 3xHA-PIP5K1A (PIP5K1A). Western blot of input (first two lanes) and immunoprecipitation of endogenous PIP4K2A with PIP5K1A (second two lanes).
(G and H) 293T lysates were incubated with beads pre-incubated with either GST or GST-PIP4K2C, used as bait. Pull-downs analyzed by western blot strongly to detect the enrichment of PIP5K1A and PIP5K1C(G). Analysis of flow-through reflects the extent to which lysate is depleted of PIP5K1A and PIP5K1C by bait (H).
(I) Sequence homology of N-terminal region in PIP4K. Mutated residues of PIP4K2C aa69–75 are indicated in red.
(J) 293T lysates were incubated with beads conjugated to GST-PIP4K2C or GST-PIP4K2CVD, used as bait. Bead pull-downs were analyzed by western blot for the presence of PIP5K1A, which is able to bind WT PIP4K2C but not PIP4K2CVD.
(K) Quantification of in vitro kinase assay measuring the conversion of PI(4)P to radiolabeled PI(4,5)P2 by PIP5K1A, which is inhibited by PIP4K2C but not PIP4K2CVD.
(L and M) Quantification by HPLC of PI(4)P (L) and PI(4,5)P2 (M) in HEK293T TKO cell lines rescued with PIP4K2C and PIP4K2CVD.
Significance calculated using ANOVA with Holm-Sidak multiple comparisons to control cell line. **p < 0.01, ****p < 0.0001, and n.s., not significant. Data are represented as means ± SEMs. n = 3.
All three isoforms of PIP5K are stimulated by phosphatidic acid (PA) and by association with G-proteins such as Rac (Jenkins et al., 1994; Weernink et al., 2004). However, PIP5K activation in PIP4K-depleted cells is not due to these upstream effectors, as cells with PIP4K knockdown did not exhibit increased PA levels (Figure S3D), and the knockout of Rac1 did not abrogate the ability of PIP4K to modulate levels of PI(4)P and PI(4,5)P2 (Figures S3E-S3G). Furthermore, protein levels of PIP5K1A and PIP5K1C were not consistently altered in response to knockdown or knockout of the PIP4K family (Figures S3H and S3I).
Lastly, we considered the possibility that PIP4K has a catalytic-independent role in masking PI(5)P, thereby reducing the availability of adaptor proteins that may inhibit PIP5K activity. To test whether the increased availability of PI(5)P can enhance PIP5K activity to decrease the PI(4)P to PI(4,5)P2 ratio, we transfected cells with Ipgd, a bacterial 4-phosphatase recognizing PI(4,5)P2. While transfection of Ipgd into 293T cells caused a dramatic increase in cellular PI(5)P, HPLC analysis of cellular phos-phoinositides did not suggest a concomitant increase in PIP5K activity. The increased availability of PI(5)P in cells with intact PIP4K (wild type [WT]) did not cause a decreased ratio of PI(4)P to PI(4,5)P2. In addition, the introduction of Ipgd into TKO cells caused a large increase in PI(5)P, but it did not change the measured PI(4)P or PI(4,5)P2 levels (Figures S3J-S3L). These results suggest that the availability of PI(5)P does not mediate the observed change in PIP5K activity.
PIP4K Can Inhibit PIP5K through Direct Interactions on the Surface of Negatively Charged Membranes
Members of the PIP4K and PIP5K family have been reported to physically interact (Hinchliffe et al., 2002; Huttlin et al., 2017). Such an interaction would be an opportunity for PIP4K to directly inhibit PIP5K. Notably, PIP4K family members are considerably more abundant than PIP5K family members, making the stoichiometric ratio favorable for PIP4Ks to inhibit PIP5Ks (Itzhak et al., 2016; Wiśniewski et al., 2014). To test whether the altered PIP5K activity in PIP4K-depleted cells could be a result of a disrupted physical interaction between these two kinase families, we measured in vitro kinase activity with purified proteins. We found that all three PIP4K isoforms could directly inhibit the PIP5K2A-catalyzed phosphorylation of PI(4)P into PI(4,5)P2 (Figure 3B). To eliminate the possibility that a small molecule contaminant from our PIP4K purifications could inhibit PIP5K1A, we boiled PIP4K2C and found that denatured PIP4K2C could no longer inhibit PIP5K1A (Figure 3C). To investigate whether the interactions between PIP4K and PIP5K occur at the membrane bilayer, we repeated the experiment with detergent to eliminate the formation of PI(4)P liposomes. Presenting phosphoinositides in Triton X-100 micelles enhances the activity of some PI kinases, such as the PI4Ks (Guo et al., 2003), but we found that it reduced PIP5K1A activity overall. The addition of detergent eliminated the ability of PIP4K2A to inhibit PIP5K1A (Figure 3D). These data suggest that recruitment of both proteins to the membrane surface is required to mediate the inhibitory interaction. As we performed this assay using vast molar excess of the substrate PI(4)P relative to the levels of both enzymes, we postulate that sequestration of PI(4)P by PIP4Ks is unlikely to account for this inhibition. As expected, given the membrane-dependence of PIP4K and PIP5K association, attempts to co-immunoprecipi-tate PIP4K with PIP5K in the presence of detergent were unsuccessful. However, by chemical crosslinking in intact cells, we demonstrated the interaction of 3xHA-tagged PIP5K1A expressed at near-endogenous levels with endogenous PIP4K2A (Figures 3E and 3F). Furthermore, glutathione S-transferase (GST)-PIP4K2C-conjugated glutathione beads pulled down the majority of PIP5K1A in 293T lysates, but only a tiny fraction of PIP5K1C (Figures 3G and 3H), supporting the hypothesis that PIP4Ks interact with PIP5K1A in cells to regulate PI(4,5)P2 levels.
To further define the nature of this interaction, we generated PIP4K2C variants and assessed their ability to inhibit PIP5K1A activity in vitro. Human PIP4K2A (PDB: 2YBX) and PIP4K2C (PDB: 2GK9) have been crystallized, while the only PIP5K family member that has been crystallized is from Danio rerio, and it forms a crystal structure similar to that of the PIP4K proteins (PDB: 5E3S) (Muftuoglu et al., 2016). PIP4K2C crystallizes as a tetramer via side-by-side interactions of two dimers in which all four catalytic pockets appear on the same side of the positively charged tetramer. Thus, we focused our mutagenesis efforts on PIP4K2C, beginning with a truncation mutant from the COSMIC database carrying a frameshift mutation after amino acid (aa) 132 of PIP4K2C (https://cancer.sanger.ac.uk/cosmic). This truncated mutant, which maintains the residues involved in both dimeric interactions and tetrameric interactions was as potent as full-length PIP4K2C in its ability to inhibit PIP5K1A activity in vitro (Figure S4A). Hence, we explored the first 132 residues of PIP4K2C and identified 2 regions that face the opposing dimer in the tetrameric structure: aas 69–75 (VMLLPDD) and aas 89–95 (FHRENLP) (Figures 3I and S4B). Mutating 74DD → NN or 91REN → EED of GST-PIP4K2C did not prevent this protein from inhibiting PIP5K1A (Figure S4C). However, introducing mutations into this region that are likely to impair both hydrophobic and charge interactions across the dimer-dimer interaction (VMLLPDD → EIFLPNN), hereafter called PIP4K2CVD, resulted in a loss of interaction with PIP5K1A (Figure 3J). Furthermore, PIP4K2CVD failed to inhibit PIP5K1A activity (Figure 3K) while maintaining PIP4K activity (Figures S4D and S4E), confirming that the protein is folded and functional. To confirm that this interaction between PIP4Ks and PIP5K1A is the physiologic mechanism of increased PI(4,5)P2 production observed in our CRISPR knockout cells, we reconstituted the TKO cells with PIP4K2CVD or WT PIP4K2C, as previously described (Figure S4F). Notably, the PIP4K2CVD mutant failed to reverse the decreased PI(4)P and increased PI(4,5)P2 observed in the TKO cells (Figures 3L and 3M), which is consistent with a failure to interact with or inhibit PIP5K1A, as we had observed in vitro.
These results reveal a mechanism for the regulation of PI(4,5)P2 production, whereby PIP5K1A is negatively regulated by a direct interaction with the N-terminal motif VMLΦPDD of PIP4Ks. We propose that a fraction of cellular PIP4Ks directly interact with PIP5K enzymes at membranes enriched in PI(4)P and function to attenuate the PIP5K-mediated conversion of PI(4)P to PI(4,5)P2. Although historically, attempts to directly visualize the subcellular localization of PIP4Ks have been inconsistent, recent global quantitative mapping of protein subcellular localization suggests that both PIP5Ks and PIP4Ks are largely bound to plasma membranes (Bultsma et al., 2010; Hinchliffe et al., 1999, 2002; Itoh et al., 1998; Itzhak et al., 2016; Kunz et al., 2000).
Structural Role of PIP4K in Regulating PIP5K and PI3K Pathway Is Distinct from Its Catalytic Role in Autophagy
Levels of PI(4,5)P2 are remarkably stable to perturbations of its precursor PI(4)P (Nakatsu et al., 2012), yet we find that depletion of the PIP4K family members results in nearly 2-fold changes in PI(4)P and PI(4,5)P2 (Figures 1G-1J). Insulin activation of the PI3K pathway is initiated at the plasma membrane, where conversion of PI(4,5)P2 into PI(3,4,5)P3 recruits effector proteins Akt and Pdk1 to propagate signaling cascades. We hypothesized that, in PIP4K-depleted cells, increased PI(3,4,5)P3 may be attributed to the activation of the PI3K pathway due to elevated production of its substrate, PI(4,5)P2. During acute stimulation, local production of PI(4,5)P2 by PIP5K1A sustains the activation of the PI3K pathway in B cells, keratinocytes, and breast adenocarcinoma (Choi et al., 2016; Saito et al., 2003; Xie et al., 2009). HPLC analysis of lipids showed that PIP4K knockout cells exhibited a 4-fold increase in PI(3,4,5)P3 upon acute insulin stimulation (Figure 4A). Consistent with the model that PIP4Ks suppress insulin signaling by their catalytic-independent suppression of PI(4,5)P2 synthesis by the PIP5Ks, expression of the kinase-dead PIP4K2A or the near-kinase-dead PIP4K2C in TKO cells was able to reverse the insulin-dependent increase in PI(3,4,5)P3 accumulation. Expression of the PIP4K2CVD mutant failed to rescue PI(3,4,5)P3 levels (Figure 4A), which is consistent with our model that the VMLΦPDD motif mediates negative regulation of the PI3K pathway.
Figure 4. Structural Role of PIP4K in Regulating PIP5K and the PI3K Pathway Is Distinct from Its Catalytic Role in Autophagy.

(A) Insulin stimulation of PI(3,4,5)P3 synthesis by PI3K in 293T rescue cell lines, quantified by HPLC. Cells were serum starved for 20 h, then stimulated with 50 ng/mL insulin for 5 min.
(B) Insulin stimulation of Akt activation in HeLa rescue cell line panel. Cells were serum starved for 12 h, then stimulated with 250 ng/mL insulin for 10min. Western blot band intensities were measured by Li-Cor and are indicated under the relevant band.
(C) Quantification of insulin-stimulated increase in pAkt-473in HeLa rescue cell line panel, normalized to total Akt. n = 3.
(D and E) Immunofluorescence detection of lysosomal marker Lamp1 in the 293T rescue cell line panel shows dependence on catalytic activity to rescue increased lysosomal accumulation. Representative images are shown (D), and full quantification is detailed (E).
(F) Quantification of TFEB target gene expression by qRT-PCR in 293T rescue cell line panel.
Significance calculated using ANOVA with Holm-Sidak multiple comparisons to control cell line. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and n.s., not significant.
Furthermore, HeLa TKD cells displayed enhanced PI3K pathway activation, as judged by S473 phosphorylation of AKT as well as AKT-mediated phosphorylation of PRAS40 (Manning and Toker, 2017; Figures 4B and 4C). Expression of either PIP4K2AKD or PIP4K2C reversed the enhanced insulin sensitivity, whether measured by PI(3,4,5)P3 levels or AKT activation. Our results indicate that increasing PIP5K activity by eliminating PIP4K isoforms drives the overproduction of PI(4,5)P2 in plasma membrane pools that may be used by PI3K during insulin signaling.
Finally, we asked whether the non-catalytic function of PIP4Ks was required for the previously reported role of PIP4K2A in mediating autophagy (Lundquist et al., 2018). As expected, loss of PIP4Ks resulted in the accumulation of LAMP1+ lysosomes and increased transcription factor EB (TFEB) transcriptional targets. However, unlike the changes observed in phosphoinositides, only reconstitution with kinase-active but not kinase-dead PIP4K2A showed restored regulation of autophagy (Figure 4D-4F). This work highlights the complexity and multi-functionality of PIP4K enzymes.
DISCUSSION
In this study, we show that the proteins encoded by the PIP4K2A, PIP4K2B,and PIP4K2C genes have distinct catalytic and non-catalytic functions and provide insight into the phenotypes observed in Pip4k-deficient mice. The roles of the PIP4K2A and PIP4K2B isoforms in autophagosome-lysosome fusion that were recently reported in MEFs and in vivo using mice with targeted liver-specific PIP4K deletions (Lundquist et al., 2018) depend on the ability of these enzymes to convert PI(5)P to PI(4,5)P2. However, our present study revealed that the increased insulin sensitivity previously observed in Pip4k2b−/− mice (Lamia et al., 2004) is due to the loss of a non-catalytic function of PIP4K2B in suppressing the ability of PIP5Ks to produce PI(4,5)P2 to be used as the substrate for PI 3-kinase. One confounding issue could be the increase in PI(5)P observed when PIP4Ks are depleted. Previous studies characterizing Ipgd, a bacterial 4-phosphatase of PI(4,5)P2, have demonstrated that increasing PI(5)P levels prolongs Akt signaling (Carricaburu et al., 2003; Niebuhr et al., 2002; Pendaries et al., 2006). The model presented by Carricaburu et al. (2003) describes how increased cellular PI(5)P inhibits PI(3,4,5)P3 -specific phosphatase, thereby leading to an increase in PI(3,4,5)P3 levels. This suggests that in certain settings, the catalytic activity of PIP4K modulates Akt signaling through the control of PI(5)P levels. However, our data show that reconstitution with kinase-dead PIP4K fully rescues PI(3,4,5)P3 levels and Akt phosphorylation in response to insulin signaling, despite not reducing PI(5)P, arguing that the model proposed above cannot account for our observations.
We show that PIP4Ks and PIP5Ks directly interact and that mutating five conserved aas of PIP4K2C can abrogate this interaction and prevent PIP5K suppression. A role in regulating PIP5K activity may explain why PIP4Ks are highly abundant in cells, in excess of PIP5Ks, which are thought to produce most of the PI(4,5)P2. Because both PIP4Ks and PIP5Ks are bound to negatively charged membranes, these enzymes could also alter local membrane structures or recruit other enzymes to modify PIP5Ks to further modulate their activities. Future studies will address these possibilities.
Our results indicate that the design of drugs to promote the degradation of PIP4Ks may mitigate insulin resistance and possibly retard the progression of type 2 diabetes. Recently emerging technologies raise the possibility that one could degrade specific PIP4K proteins by linking them to E3 ligases (Bondeson and Crews, 2017). We initially had reservations about the development of such drugs due to the embryonic lethality of germline co-deletion of PIP4K2B and PIP4K2C or PIP4K2A and PIP4K 2B (Emerling et al., 2013; Shim et al., 2016). In addition, Emerling et al. (2013) and Lundquist et al. (2018) reported the reduced viability of MEFs upon the loss of PIP4K2A and/or PIP4K2B in cell lines with deficient p53 signaling. However, our results in a panel of cancer lines suggest that most cell lines do not require PIP4K isoforms for viability when grown in tissue culture with full nutrients (Figure S2). Embryonic lethality from the co-deletion of murine PIP4K isoforms may be attributed to the developmental defects required for mammalian development or nutrient stress in the perinatal period (Emerling et al., 2013; Shim et al., 2016), and partial and/or tissue-specific targeted depletion may be well tolerated.
We note that each tissue has a different ratio of PIP4K2A: PIP4K2B:PIP4K2C, and therefore it remains to be seen whether targeting a subset of the PIP4K family in humans will enhance systemic insulin sensitivity. The PIP4K2B knockout mice exhibit increased insulin sensitivity that is not seen in PIP4K2A and PIP4K2C knockout mice. This is likely because PIP4K2B is the predominant isoform in skeletal muscle, a primary site of insulin-mediated glucose uptake. It will be interesting to see whether other insulin-responsive tissues, such as liver and adipose tissue, will become sensitized to insulin if multiple PIP4K isoforms are depleted.
PIP4K family members repress the conversion of PI(4)P to PI(4,5)P2 and subsequently to PI(3,4,5)P3 to limit PI3K pathway signaling. This occurs by a mechanism that does not require the catalytic activity of these enzymes, while they directly catalyze the conversion of PI(5)P to PI(4,5)P2 to promote autophagy. How did these two disparate functions come to reside in the same protein? One possibility is to ensure that the levels of PI(4)P remain high at local sites of autophagosome-lysosome fusion. Several recent studies indicate that PI(4)P is required for autophagosome-lysosome fusion and that in some cases this PI(4)P is derived from an inositol 5-phosphatase acting on local pools of PI(4,5)P2 (De Leo et al., 2016; Wang et al., 2015). Thus, the PIP4Ks may be both producing the local PI(4,5)P2 at the autophagosome-lysosome junction and ensuring that upon conversion to PI(4)P, it is not converted back to PI(4,5)P2 by a PIP5K. This model would ensure a unidirectional conversion of PI(5)P to PI(4)P and efficient autophagosome-lysosome fusion. The partial localization of PIP4K2C to the Golgi (Clarke et al., 2008) could also suppress the conversion of Golgi PI(4)P to PI(4,5)P2 to maintain the high local concentration of the PI(4)P that is needed for vesicle trafficking at this location.
Given the global increase in PI(4,5)P2 observed, there are undoubtedly multiple PI(4,5)P2 mediated functions that will be affected. It will be interesting to characterize how the accumulation of PI(4,5)P2 in PIP4K-depleted cells affects cellular processes beyond growth factor signaling, such as cell adhesion, migration, and/or calcium signaling. Furthermore, it will be essential to examine how this change affects the localization and levels of other phosphoinositides within cells.
Our findings highlight an unexpected and important separation of catalytic and non-catalytic functions in PIP4K family enzymes and indicate new avenues for intervention in enhancing insulin signaling.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lewis C. Cantley (LCantley@med.cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines, authentication
Cell lines were purchased from ATCC and/or fingerprinted with the University of Arizona genetics core. Cells were tested to be mycoplasma free with Lonza Mycoalert.
Cell culture conditions
293T, HeLa, PaTu 8988t, and BJ cells were cultured using DMEM media supplemented with 10% FBS, glutamine and pyruvate. H1299 and H1975 cells were cultured in RPMI media.
METHOD DETAILS
Cell lysis and immunoblotting
Cells were lysed in RIPA buffer supplemented with 1 tablet of protease and phosphatase inhibitor. After incubation on ice for 20 minutes, lysates were cleared by centrifugation at 14,000 × g and supernatant was quantified using BCA assay. Lysates were subjected to SDS-PAGE using Novex NuPAGE system. Proteins were separated on 4%–12% Bis Tris Pre-Cast Gels 10% Bis Tris gels using MOPS buffer. Proteins were transferred to 0.45 μm nitrocellulose membranes at 350 mA for 1h. Membranes were blocked in 5% non-fat milk in TBST and incubated with primary antibody overnight: For chemi-luminescently detect antibody binding, membranes were blotted with HRP conjugated secondary antibodies. Membranes were developed using ECL solution, and exposed to film. For insulin signaling westerns, protocol was modified such that cells were lysed in triton buffer, IRDye secondary was used for Li-Cor Odyssey detection with quantification using Image Studio Lite software.
Generation of lentivirus, viral transduction
293T cells were used to generate lentivirus. Once cells were at 90%–95% confluence in a 10cm dish, transfection was performed with Opti-mem, Lipofectamine 2000, lentiviral vector, and accessory plasmids VSVG and Δ8.2. Virus supernatant was harvested 2 days and 3 days post transfection, then filtered and concentrated in an ultracentrifuge at 25,000 rpm for 120 minutes at 4°C.
Generation of cell lines with CRISPR knockout of PIP4K
CRISPR guides in pX458 were transfected in 293T cells. At 48-96 hours post transfection, GFP positive cells were single-cell sorted in 96-well plates using the Influx sorter at the WCMC Flow Cytometry Core. Two weeks later, wells were scored to contain single cell colony and expanded to screen for successful PIP4K2A/PIP4K2B/PIP4K2C knockout. Validation was performed by western blotting as well as PCR around each cut site.
Generation of cell lines with miR-E knockdown of PIP4K
LT3GEPIR vectors containing desired miR-E shRNA(s) were double digested downstream of existing shRNA(s) with EcoRI-HF/ MluI-HF and PCR purified (Fellmann et al., 2013). The miR-E sequence to be added was PCR amplified with Multi-sh-F and Multi-sh-R. PCR products were purified, double digested with BbsI/MluI, and PCR purified once more. Ligations were performed with PCR product and open LT3GEPIR vectors using T4 ligase. Colonies were screened using miR-E-F. Primers listed in Table S3
Generation of mammalian and bacterial expression vectors
To generate vectors to express hairpin-resistant coding sequences for PIP4K isoforms, we cloned wild-type PIP4K2A and PIP4K2C into a lentiviral backbone with a PGK promoter. Next, we generated mutations in PIP4K cDNA using QuikChange to make kinase-dead variants and silent mutations in wobble positions for hairpin-resistant cDNA. Primers listed in Table S3. No wobble mutations were needed for PIP4K2C cDNA since all hairpins targeted the 3′ UTR. To generate PIP4K2C N-terminal mutants, Quikchange kit from Agilent (220521) was used and listed in Table S3.
Measurement of phosphoinositides with high performance liquid chromatography
Cellular phosphoinositides were metabolically labeled for 48 hours in inositol-free DMEM supplemented with glutamine,10% dialyzed FBS, and 10 μCi/ mL 3H myo-inositol. Cells were washed with PBS and then transferred on ice. Cells were killed and then harvested by scraping using 1.5 mL ice-cold aqueous solution (1M HCl, 5 mM Tetrabutylammonium bisulfate, 25 mM EDTA). 2 mL of ice cold MeOH and 4mL of CHCl3 were added to each sample. After ensuring each vial is tightly capped, samples were vortexed and then centrifuged at 1000 rpm for 5 min. If a significant intermediate layer was visible, sample were gently agitated and spun again, until there were predominately two clear layers. The organic layer (lower) was cleaned using theoretical upper, while the aqueous layer was cleaned using theoretical lower (theoretical upper and lower made by combining CHCl3: MeOH: aqueous solution in 8:4:3 v/v ratio). Organic phases were collected and dried under nitrogen gas. Lipids were deacylated using monomethylamine solution (47% Methanol, 36% of 40% Methylamine, 9% butanol, and 8% H2O, by volume). Samples were incubated at 55° for 1 hour and subsequently dried under nitrogen gas. To the dried vials, 1 mL of theoretical upper and 1.5 mL of theoretical lower were added (theoretical upper and lower made by combining CHCl3:MeOH:H2O in 8:4:3 v/v ratio). Samples were vortexed and spun at 1000rpm. The aqueous phase (upper) was collected and dried under nitrogen gas. Samples were resuspended in 150 μL Buffer A (1mM EDTA), filtered and transferred to Agilent polypropylene tubes. Samples were analyzed by anion-exchange HPLC using Partisphere SAX column. The compounds were eluted with a gradient starting at 100% Buffer A (1mM EDTA) and increasing Buffer B (1mM EDTA, 1M NaH2PO4) overtime: 0-1 min 100% Buffer A, 1-30 min 98% Buffer A/2% Buffer B, 30-31 min 86% Buffer A/14% Buffer B, 31-60 min 70% Buffer A/30% Buffer B, 60-80 min 34% Buffer A/ 66% Buffer B, 80-85 min 100% Buffer B, 85-120 min 100% Buffer A. Buffers were pumped at 1 mL/min through column. Eluate from the HPLC column flowed into an on-line continuous flow scintillation detector for isotope detection. The detector was set to observe events between 10 minutes and 85 minutes, with scintillation fluid flowing at 4 mL/min.
Measurement of phosphatidic acid using LCMS
Cellular lipids were extracted using same method described for phosphoinositide analysis. Lipids were dried and diluted with 113 μL of chloroform:methanol:water (73:23:3) mixture and filtered (0.45 μm) before analysis on an Agilent 6230 electrospray ionization–time-of-flight (ESI-TOF) MS coupled to an Agilent 1260 HPLC equipped with a Phenomenex Luna silica 3 μm 100 Å 5 cm × 2.0 mm column. LCMS analysis was performed using normal phase HPLC with a binary gradient elution system where solvent A was chloroform:methanol:ammonium hydroxide (85:15:0.5) and solvent B was chloroform:methanol:water:ammonium hydroxide (60:34:5:0.5). Separation was achieved using a linear gradient from 100% A to 100% B over 9 min. Phospholipid species were detected using a dual ESI source operating in positive mode, acquiring in extended dynamic range from m/z 100–1700 at one spectrum per second; gas temperature: 325°C; drying gas 10 L/min; nebulizer: 20 psig; fragmentor 300 V
Immunoprecipitation
293T cell lines expressing 3xHA empty vector or 3xHA-PIP5K1A were fixed for 10 minutes in 4% PFA, washed 3x with PBS, and lysed in lysis buffer (10mM Tris 7.4, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP40) using probe sonicator for 2 minutes. Lysates were pre-cleared with control magnetic beads for 1 hour and 4° and Pierce magnetic anti-HA beads added for overnight incubation at 4°. Beads were washed 3x with lysis buffer and aliquots were added to sample buffer loading dye with beta mercaptoethanol, boiled, and run on SDS-page for immunoblotting.
Protein Purification
BL21 bacterial cells were transfected with pGEX vectors with PIP4K isoform coding sequence variants. Cells were grown in 1L of TB at 37° at 200rpm until 0.8 OD, at which point 500 uL of 1M IPTG was added. Cultures were shaken overnight at room temp for protein induction, and pelleted at 5000 g for 15 minutes. Cells were lysed (50 mM Tris pH 7.5, 500 mM NaCl, 10 mM MgCl2, 10% glycerol, DTT, lysozyme, protein/phosphatase inhibitors), sonicated for 1 minute (output 4, continuous duty cycle) and spun down at 10,000 g for 1 hour. Supernatant was kept for further purification and cleavage according to manufacturer’s protocol using glutathione Sepharose beads.
In-vitro kinase assays
Cells were trypsinized and normalized for cell number. Cell pellets, or E.Coli purified proteins were resuspended in HNE buffer (20 mM HEPES pH 7.4, 100mM NaCl, 0.5mM EGTA), and sonicated in the presence of 32P-γ-ATP, and liposomes (10ug PS, 1ug PI(4)P in 30mM HEPES pH 7.4, 1mM EGTA). Liposome lipids were purchased from Avanti. Reactions were stopped by addition of 50uL of 4N HCL. To extract lipids, 100 μL of MeOH:CHCl3 (1:1) was added and samples were vortexed 2× 30 s. Samples were spun down at top speed for 2 min and the organic phase containing phosphatidylinositol lipids (bottom) were separated using thin-layer chromatography (TLC) using 1-propanol: 2N acetic acid (65:35 v/v). TLC plates were prepared ahead of time by coating with 1% Potassium Oxalate. Phosphorylated lipids were visualized by autoradiography on a GE Typhoon FLA 7000 and quantified using ImageQuant TL software.
GST Pulldown assays
GST tagged proteins were isolated as described above except that after washing, aliquots of beads with bound protein were added to 293T cell lysates generated by lysis in IP buffer (50 mM Tris-Cl pH 8.0, 150 mM, 1% NP-40, complete PI/Pis with EDTA) and spinning out DNAat 22,000×g ×10min. These lysates and purified protein were incubated together at 4 degrees for 1 hour before the beads were again pelleted, washed with IP buffer three times, eluted with 2M NaCl, and analyzed by SDS-PAGE and western blot.
Fluorescence microscopy
293T cells were grown on glass coverslips pre-treated with poly-d-lysine. Adherent cell lines were rinsed with phosphate-buffered saline, pH 7.4 (PBS) and subsequently fixed/permeabilized in −20 MeOH for 20 minutes. After fixation, the cells were blocked for 30 minutes in blocking buffer (PBS with 3% BSA) and labeled with primary antibodies in blocking buffer for 1 hour at room temperature. Coverslips were washed three times with blocking buffer and incubated with Alexa Fluor-conjugated goat secondary antibodies in blocking buffer for 1 hour at room temperature. After incubation with secondary antibodies, coverslips were washed three times with PBS, once with water, and then mounted on a glass microscope slide with Prolong Gold with DAPI. The following primary antibodies were used: LAMP1. Alexa Fluor-conjugated secondary antibodies were used at 1:1000 (Thermo Scientific). Fluorescent and phase contrast images were acquired on a Nikon Eclipse Ti microscope equipped with an Andor Zyla sCMOS camera. Within each experiment, exposure times were kept constant and in the linear range throughout. When using the 60x oil immersion objectives, stacks of images were taken and deconvoluted using AutoQuant.
qRT-PCR
Total RNA was prepared using RNeasy. cDNA was synthesized using Superscript Vilo and qRT-PCR performed utilizing Fast SYBR green and the Realplex Mastercycler. For a list of primers used see oligos in Table S3. Isolation of mRNA and qPCR was performed as follows. 200,000 cells were plated in 6-well plastic dishes. 24 hours later, the RNA in the lysates was extracted using the RNeasy protocol. The RNA was resuspended in 50 μL H2O at a concentration of 1 μg/μL. cDNA was transcribed using the SuperScript Vilo. The sequences of the oligonucleotides used as primers in the PCR reactions are given in Table S3. The genes that were quantified here were previously shown to be regulated by TFEB
QUANTIFIICATION AND STATISTICAL ANALYSIS
Experiments were repeated with at least three biological replicates with the following exceptions: 1) Experiments in Figure S3 were performed once, and panels with error bars were performed with technical triplicates; 2) Experiments in Figures S4H-S4J were performed once. No samples were excluded from analysis. When comparing two groups, a two-tailed t test was used. P values are indicated in figure legends. When comparing greater than two groups, significance was calculated using ANOVA and Holm-Sidack post hoc test. Figure legends indicate which comparisons are significant, with respective p values.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PIP4K2A | Cell Signaling Technologies | Cat# 5527; RRID:AB_2722636 |
| PIP4K2B | Cell Signaling Technologies | Cat# 9694; RRID:AB_2164572 |
| PIP4K2C | Proteintech | Cat# 17077-1-AP; RRID:AB_2715526 |
| PIP5K1A | Cell Signaling Technologies | Cat# 9693; RRID:AB_2164698 |
| PIP5K1C | Abcam | Cat# Ab109192; RRID:AB_10862359 |
| Rac1/2/3 | Cell Signaling Technologies | Cat# 2465; RRID:AB_2176152 |
| Lamp1 | Cell Signaling Technologies | Cat# 15665; RRID:AB_2798750 |
| pAKT-473 | Cell Signaling Technologies | Cat# 4060; RRID:AB_2315049 |
| pAKT-308 | Cell Signaling Technologies | Cat# 13038; RRID:AB_2629447 |
| AKT | Cell Signaling Technologies | Cat# 2920; RRID:AB_1147620 |
| pPRAS40 | Cell Signaling Technologies | Cat# 13175; RRID:AB_2798140 |
| PRAS40 | Cell Signaling Technologies | Cat# 2610; RRID:AB_916206 |
| Beta-Actin | Abcam | Cat# Ab6276; RRID:AB_2223210 |
| Tubulin | Abcam | Cat# Ab7291; RRID:AB_2241126 |
| IRDye-secondary mouse | Li-Cor | Cat# 926-68070; RRID:AB_10956588 |
| IRDye-secondary rabbit | Li-Cor | Cat# 926-32213;RRID:AB_621848 |
| HRP-secondary mouse | Thermo Fisher Scientific | Cat# 45000679 |
| HRP-secondary rabbit | Thermo Fisher Scientific | Cat# NA934-1ML |
| Alexa Fluor anti-mouse 568 | Thermo Fisher Scientific | Cat# A10037 |
| Alexa Fluor anti-rabbit 488 | Thermo Fisher Scientific | Cat# A21206 |
| Bacterial and Virus Strains | ||
| Stbl3 | Life Technologies | Cat# C737303 |
| BL21 | Life Technologies | Cat# C600003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RIPA buffer | Sigma | Cat# R0278-500mL |
| Protease and phosphatase inhibitor | Thermo Fisher, Pierce | Cat# 88668 |
| ECL substrate | Thermo Fisher, Pierce | Cat# 32106 |
| Triton Lysis buffer | Cell Signaling Technologies | Cat# 9803 |
| BCA Assay | Thermo Fisher | Cat# 23225 |
| Precast gels | Life Technologies | Cat# NP0336BOX |
| DMEM | Corning | Cat# 10-013-CV |
| RPMI | Corning | Cat# 10-040-CV |
| Lipofectamine 2000 | Life Technologies | Cat# 11668500 |
| Opti-Mem | Life Technologies | Cat# 31985070 |
| 3H inositol | Perkin Elmer | Cat# NET114A005MC |
| 32ATP | Perkin Elmer | Cat# BLU002A001MC |
| Dialyzed FBS | Life Technologies | Cat# 26400044 |
| Inositol free DMEM | Thermo Fisher | Cat# ICN1642954 |
| Glutamine | Life Technologies | Cat# 51985034 |
| Dialyzed FBS | Life Technologies | Cat# 26400044 |
| Inositol free DMEM | Thermo Fisher | Cat# ICN1642954 |
| Glutamine | Life Technologies | Cat# 51985034 |
| Ultima flo scintillation fluid | Perkin Elmer | Cat# 6013599 |
| Anti-HA magnetic beads | Pierce | Cat# 88836 |
| Control magnetic beads | Cell signaling Technologies | Cat# 8726 |
| Phosphatidylserine | Avanti | 840032C |
| Phosphatidylinositol-4-phosphate | Avanti | Cat# 840151 |
| Phosphatidylinositol-5-phosphate | Avanti | Cat# 850152P |
| IPTG | Technova | Cat# I3431 |
| Glutathione Sepharose | Thermo Fisher | Cat# 45000285 |
| TEV protease | Sigma | Cat# T4455-10KU |
| Fast SYBER green | Applied Biosystems | Cat# C0110820 |
| Critical Commercial Assays | ||
| RNEasy | QIAGEN | Cat# 74106 |
| SuperScript VILO Mastermix | Life Technologies | Cat# 11755050 |
| QuikChange | Agilent | Cat# 200522 |
| BCA protein assay | Thermo Fisher | Cat# 23225 |
| Mycoalert | Lonza | Cat# LT07318 |
| Experimental Models: Cell Lines | ||
| 293T | Clontech | Cat# 632180 |
| HeLa | ATCC | Cat# CCL-2 |
| PaTu 8988t | DSMZ | Cat# ACC162 |
| H1299 | ATCC | Cat# CRL-5803 |
| H1975 | ATCC | Cat# CRL-5908 |
| BJ | ATCC | Cat# CRL-2522 |
| Oligonucleotides | ||
| Table S3 | This paper | N/A |
| Recombinant DNA | ||
| LG3GEPIR | Fellmann et al., 2013 | N/A |
| SGEP | Fellmann et al., 2013 | N/A |
| SG3REPIR | This paper | N/A |
| SREN | This paper | N/A |
| pPIG-3xHA-empty | This paper | N/A |
| pPIG-3xHA-PIP4K2A | This paper | N/A |
| pPIG-3xHA-PIP4K2A-KD | This paper | N/A |
| pPIG-3xHA-PIP4K2C | This paper | N/A |
| pPIG 3xHA-PIP4K2C-VD | This paper | N/A |
| Software and Algorithms | ||
| AutoQuant | Media Cybernetics | N/A |
| ImageQuant TL v8.1 | GE | N/A |
| Prism | Graphpad | N/A |
| Accuri | BD Biosciences | N/A |
| Prism 7 | Graphphad Prism Software | N/A |
| Other | ||
| HiChrom Partisphere SAX HPLC 5 micron | VWR | Cat# 101946-560 |
| Typhoon FLA 7000 | GE | N/A |
Highlights.
PIP4Ks suppress PIP5K activity and insulin-stimulated production of PI(3,4,5)P3
PIP5K inhibition is independent of PIP4K catalytic activity
PIP5K inhibition is dependent on the N-terminal motif VMLLPDD on PIP4K
PIP4Ks have distinct catalytic and structural roles regulating metabolism
ACKNOWLEDGMENTS
We thank all of the members of the Cantley laboratory for feedback. We thank Jared Johnson for chemistry and biochemistry consultations. We thank Yuxiang Zheng for sharing expertise on the HPLC analysis of phosphoinositides. We thank Hyeseok Shim for insight on PIP4K2C biology. This work was supported by grants from the NIH (R35 CA197588 and U54 U54CA210184) and the Lustgarten Foundation and by grants to J.M.B., including the NIH Pathway to Independence (R00 GM110121), the National Science Foundation (NSF) CAREER (CHE-1749919), and the Beckman Young Investigator (Arnold and Mabel Beckman Foundation) to L.C.C. L.E.D. was supported by a K22 Career Development Award from the National Cancer Institute (NCI)/NIH (CA 181280-01). D.G.W. was supported by a Medical Scientist Training Program grant from the NIH (T32GM007739) to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program, and T.W.B. was supported by an NSF graduate research fellowship (DGE-1650441). This research was funded in part by Petra Pharmaceuticals. Cell sorting was partially supported by an NIH S10 award (1S10 OD019986-01). The laboratory of L.C.C. also receives funding from Petra Pharmaceuticals for research on phosphoinositide kinases.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.Org/10.1016/j.celrep.2019.04.070.
DECLARATION OF INTERESTS
L.C.C. is a founder and member of the SAB and holds equity in Agios Pharmaceuticals and Petra Pharmaceuticals, companies developing drugs for treating cancer. L.E.D. is a consultant and an advisory board member for Mirimus, which has licensed some of the shRNA technology used in this study. The laboratory of L.C.C. holds patents on targeting phosphoinositide kinases for cancers and insulin resistance.
REFERENCES
- Balla T (2013). Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev 93, 1019–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, and Crews CM (2017). Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol 57, 107–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA (2015). PIP2Clustering: from model membranes to cells. Chem. Phys. Lipids 192, 33–40. [DOI] [PubMed] [Google Scholar]
- Bultsma Y, Keune WJ, and Divecha N (2010). PIP4Kbeta interacts with and modulates nuclear localization of the high-activity PtdIns5P-4-kinase isoform PIP4Kalpha. Biochem. J. 430, 223–235. [DOI] [PubMed] [Google Scholar]
- Carricaburu V, Lamia KA, Lo E, Favereaux L, Payrastre B, Cantley LC, and Rameh LE (2003). The phosphatidylinositol (PI)-5-phosphate 4-kinase type II enzyme controls insulin signaling by regulating PI-3,4,5-trisphosphate degradation. Proc. Natl. Acad. Sci. USA 100, 9867–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Thapa N, Tan X, Hedman AC, and Anderson RA (2015). PIP kinases define PI4,5P2signaling specificity by association with effectors. Biochim. Biophys. Acta 1851, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Hedman AC, Sayedyahossein S, Thapa N, Sacks DB, and Anderson RA (2016). Agonist-stimulated phosphatidylinositol-3,4,5-trisphosphate generation by scaffolded phosphoinositide kinases. Nat. Cell Biol. 18, 1324–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JH, and Irvine RF (2013). Evolutionarily conserved structural changes in phosphatidylinositol 5-phosphate 4-kinase (PI5P4K) isoforms are responsible for differences in enzyme activity and localization. Biochem. J. 454, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JH, Emson PC, and Irvine RF (2008). Localization of phosphatidylinositol phosphate kinase IIgamma in kidney to a membrane trafficking compartment within specialized cells of the nephron. Am. J. Physiol. Renal Physiol 295, F1422–F1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leo MG, Staiano L, Vicinanza M, Luciani A, Carissimo A, Mutarelli M, Di Campli A, Polishchuk E, Di Tullio G, Morra V, et al. (2016). Autopha-gosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 18, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM, Bell EL, Shim HS, Lamia KA, Rameh LE, et al. (2013). Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell 155, 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Zuber J, McJunkin K, Chang K, Malone CD, Dickins RA, Xu Q, Hengartner MO, Elledge SJ, Hannon GJ, and Lowe SW (2011). Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol. Cell 41, 733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. (2013). An optimized micro-RNA backbone for effective single-copy RNAi. Cell Rep. 5, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, and Abraham RT (2017). The PI3K Pathway in Human Disease. Cell 170, 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Wenk MR, Pellegrini L, Onofri F, Benfenati F, and De Camilli P (2003). Phosphatidylinositol 4-kinase type IIalpha is responsible for the phosphatidylinositol 4-kinase activity associated with synaptic vesicles. Proc. Natl. Acad. Sci. USA 100, 3995–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Toscano S, Trivedi D, Jones DR, Mathre S, Clarke JH, Divecha N, and Raghu P (2013). Phosphatidylinositol 5-phosphate 4-kinase (PIP4K) regulates TOR signaling and cell growth during Drosophila development. Proc. Natl. Acad. Sci. USA 110, 5963–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchliffe KA, Ciruela A, Letcher AJ, Divecha N, and Irvine RF (1999). Regulation of type IIalpha phosphatidylinositol phosphate kinase localisation by the protein kinase CK2. Curr. Biol 9, 983–986. [DOI] [PubMed] [Google Scholar]
- Hinchliffe KA, Giudici ML, Letcher AJ, and Irvine RF (2002). Type IIal-pha phosphatidylinositol phosphate kinase associates with the plasma membrane via interaction with type I isoforms. Biochem. J 363, 563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma K, Terui S, Minemura M, Qadota H, Anraku Y, Kanaho Y, and Ohya Y (1998). Phosphatidylinositol-4-phosphate 5-kinase localized on the plasma membrane is essential for yeast cell morphogenesis. J. Biol. Chem 273, 15779–15786. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Ijuin T, and Takenawa T (1998). A novel phosphatidylinositol-5-phosphate 4-kinase (phosphatidylinositol-phosphate kinase IIgamma) is phosphorylated in the endoplasmic reticulum in response to mitogenic signals. J. Biol. Chem 273, 20292–20299. [DOI] [PubMed] [Google Scholar]
- Itzhak DN, Tyanova S, Cox J, and Borner GH (2016). Global, quantitative and dynamic mapping of protein subcellular localization. eLife 5, e16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GH, Fisette PL, and Anderson RA (1994). Type I phosphatidylinositol 4-phosphate 5-kinase isoforms are specifically stimulated by phosphatidic acid. J. Biol. Chem 269, 11547–11554. [PubMed] [Google Scholar]
- Jones DR, Bultsma Y, Keune WJ, Halstead JR, Elouarrat D, Mohammed S, Heck AJ, D’Santos CS, and Divecha N (2006). Nuclear PtdIns5P as a transducer of stress signaling: an in vivo role for PIP4Kbeta. Mol. Cell 23, 685–695. [DOI] [PubMed] [Google Scholar]
- Kunz J, Wilson MP, Kisseleva M, Hurley JH, Majerus PW, and Anderson RA (2000). The activation loop of phosphatidylinositol phosphate kinases determines signaling specificity. Mol. Cell 5, 1–11. [DOI] [PubMed] [Google Scholar]
- Kunz J, Fuelling A, Kolbe L, and Anderson RA (2002). Stereo-specific substrate recognition by phosphatidylinositol phosphate kinases is swapped by changing a single amino acid residue. J. Biol. Chem 277, 5611–5619. [DOI] [PubMed] [Google Scholar]
- Lamia KA, Peroni OD, Kim YB, Rameh LE, Kahn BB, and Cantley LC (2004). Increased insulin sensitivity and reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase beta−/− mice. Mol. Cell. Biol 24, 5080–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist MR, Goncalves MD, Loughran RM, Possik E, Vijayaraghavan T, Yang A, Pauli C, Ravi A, Verma A, Yang Z, et al. (2018). Phosphatidylinositol-5-Phosphate 4-Kinases Regulate Cellular Lipid Metabolism By Facilitating Autophagy. Mol. Cell 70, 531–544.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey AM, Sarkes DA, Bettencourt I, Asara JM, and Rameh LE (2014). PIP4kγ is a substrate for mTORC1 that maintains basal mTORC1 signaling during starvation. Sci. Signal. 7, ra104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, and Toker A (2017). AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu Y, Xue Y, Gao X, Wu D, and Ha Y (2016). Mechanism of substrate specificity of phosphatidylinositol phosphate kinases. Proc. Natl. Acad. Sci. USA 113, 8711–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu F, Baskin JM, Chung J, Tanner LB, Shui G, Lee SY, Pirruc-cello M, Hao M, Ingolia NT, Wenk MR, and De Camilli P (2012). PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol. 199, 1003–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, and Payrastre B (2002). Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J. 21, 5069–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelossof R, Fairchild L, Huang CH, Widmer C, Sreedharan VT, Sinha N, Lai DY, Guan Y, Premsrirut PK, Tschaharganeh DF, et al. (2017). Prediction of potent shRNAs with a sequential classification algorithm. Nat. Biotechnol 35, 350–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendaries C, Tronchère H, Arbibe L, Mounier J, Gozani O, Cantley L, Fry MJ, Gaits-Iacovoni F, Sansonetti PJ, and Payrastre B (2006). PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 25, 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameh LE, Tolias KF, Duckworth BC, and Cantley LC (1997). A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature 390, 192–196. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Tolias KF, Saci A, Koon HB, Humphries LA, Scharenberg A, Rawlings DJ, Kinet JP, and Carpenter CL (2003). BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19, 669–678. [DOI] [PubMed] [Google Scholar]
- Sharma S, Mathre S, Ramya V, Shinde D, and Raghu P (2019). Phosphatidylinositol 5 Phosphate 4-Kinase Regulates Plasma-Membrane PIP3 Turnover and Insulin Signaling. Cell Rep. 27, this issue, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Wu C, Ramsamooj S, Bosch KN, Chen Z, Emerling BM, Yun J, Liu H, Choo-Wing R, Yang Z, et al. (2016). Deletion of the gene Pip4k2c, a novel phosphatidylinositol kinase, results in hyperactivation of the immune system. Proc. Natl. Acad. Sci. USA 113, 7596–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Thapa N, Hedman AC, and Anderson RA (2013). Phosphatidylinositol 4,5-bisphosphate: targeted production and signaling. BioEssays 35, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bout I, and Divecha N (2009). PIP5K-driven PtdIns(4,5)P2 synthesis: regulation and cellular functions. J. Cell Sci 122, 3837–3850. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun H-Q, Zhu X, Zhang L, Albanesi J, Levine B, and Yin H (2015). GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc. Natl. Acad. Sci. USA 112, 7015–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weernink PA, Meletiadis K, Hommeltenberg S, Hinz M, Ishihara H, Schmidt M, and Jakobs KH (2004). Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J. Biol. Chem. 279, 7840–7849. [DOI] [PubMed] [Google Scholar]
- Wilcox A, and Hinchliffe KA (2008). Regulation of extranuclear PtdIns5P production by phosphatidylinositol phosphate 4-kinase 2alpha. FEBS Lett. 582, 1391–1394. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Hein MY, Cox J, and Mann M (2014).A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics 13, 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Chang SM, Pennypacker SD, Liao EY, and Bikle DD (2009). Phosphatidylinositol-4-phosphate 5-kinase 1alpha mediates extracellular calcium-induced keratinocyte differentiation. Mol. Biol. Cell 20, 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.