

Abstract
Large conductance Ca2+- and voltage-activated potassium channels (BKCa) shape neuronal excitability and signal transduction. This reflects the integrated influences of transmembrane voltage and intracellular calcium concentration ([Ca2+]i) that gate the channels. This dual gating has been mainly studied as voltage-triggered gating modulated by defined steady-state [Ca2+]i, a paradigm that does not approximate native conditions. Here we use submillisecond changes of [Ca2+]i to investigate the time course of the Ca2+-triggered gating of BKCa channels expressed in Chinese hamster ovary cells at distinct membrane potentials in the physiological range. The results show that Ca2+ can effectively gate BKCa channels and that Ca2+ gating is largely different from voltage-driven gating. Most prominently, Ca2+ gating displays a pronounced delay in the millisecond range between Ca2+ application and channel opening (pre-onset delay) and exhibits slower kinetics across the entire [Ca2+]i-voltage plane. Both characteristics are selectively altered by co-assembled BKβ4 or an epilepsy-causing mutation that either slows deactivation or speeds activation and reduces the pre-onset delay, respectively. Similarly, co-assembly of the BKCa channels with voltage-activated Ca2+ (Cav) channels, mirroring the native configuration, decreased the pre-onset delay to submillisecond values. In BKCa–Cav complexes, the time course of the hyperpolarizing K+-current response is dictated by the Ca2+ gating of the BKCa channels. Consistent with Cav-mediated Ca2+ influx, gating was fastest at hyperpolarized potentials, but decreased with depolarization of the membrane potential. Our results demonstrate that under experimental paradigms meant to approximate the physiological conditions BKCa channels primarily operate as ligand-activated channels gated by intracellular Ca2+ and that Ca2+ gating is tuned for fast responses in neuronal BKCa–Cav complexes.
Introduction
Large-conductance calcium- and voltage-activated K+ channels (BKCa) are key elements of signal transduction in the CNS that sharpen action potentials (Storm, 1987b; Shao et al., 1999; Edgerton and Reinhart, 2003), generate the fast phase of afterhyperpolarization (Lancaster and Nicoll, 1987; Storm, 1987a; Sah and Faber, 2002; Womack and Khodakhah, 2002), shape Ca2+ spikes in dendrites (Golding et al., 1999; Rancz and Hausser, 2006), and influence synaptic transmission (Robitaille et al., 1993; Pattillo et al., 2001; Raffaelli et al., 2004; Grimes et al., 2009).
All these actions critically depend on the properly timed activation of BKCa channels that is driven by two distinct stimuli, membrane depolarization and elevation in intracellular Ca2+ concentration ([Ca2+]i) (Marty, 1981; Cui et al., 1997; Horrigan and Aldrich, 2002). Both stimuli are captured by distinct structural domains in the channel protein (Xia et al., 2002; Ma et al., 2006; Cui et al., 2009; Wu et al., 2010; Yuan et al., 2010) and trigger distinct conformational changes that converge onto the pore to open the channel (Yuan et al., 2012).
Classically, the allosteric coupling of the two gating stimuli has been documented in experiments where voltage steps were applied to BKCa channels in the presence of defined but constant levels of [Ca2+]i (Marty, 1981; Cox et al., 1997; Cui et al., 1997; Lippiat et al., 2003). In this setting, BKCa channels are primarily gated by voltage and [Ca2+]i acts as a modulator facilitating channel activation. Notwithstanding, a few experiments demonstrated that [Ca2+]i may also act as the gating stimulus at constant transmembrane voltage. Thus, the steady-state open probability of BKCa channels largely increased when [Ca2+]i was raised from 0 to 100 μm at membrane potentials keeping the voltage-sensor(s) at rest (at −120 mV; Horrigan and Aldrich, 2002) or when [Ca2+]i jumps were applied at constant voltages (Markwardt and Isenberg, 1992).
As both, voltage and [Ca2+]i, are able to independently activate BKCa channels the question remains, which stimulus is in command under physiological conditions where both can change in response to cellular activity? In fact, changes in [Ca2+]i usually occur almost instantaneously with the change in voltage, at least with BKCa channels that are located in nano-domains around Cav channels (Neher, 1998; Fakler and Adelman, 2008) or that form stable channel–channel complexes with these channels (Berkefeld et al., 2006; Loane et al., 2007; Berkefeld and Fakler, 2008; Grimes et al., 2009). Accordingly, analysis of such channel–channel complexes showed that their K+-current output is closely controlled by the gating characteristics of the Cav subunit(s): BKCa channels only opened after Ca2+ ions were provided through activated Cav channels and BKCa channels assembled with Cav2.1 (P/Q-type) activated faster and at more negative potentials than those in complex with Cav 1.2 (L-type; Berkefeld and Fakler, 2008). While all these observations demonstrate that voltage-driven elevations in [Ca2+]i are required for BKCa activity under physiological conditions, they cannot answer whether channel gating is predominantly driven by changes in voltage or [Ca2+]i.
The present work pursued this question by detailed analyses of the Ca2+-triggered gating in sole BKCa channels and in BKCa channels co-assembled with Cav2 channels, as they are in many neurons. For this purpose we used a combination of patch-clamp recordings in excised-patch and whole-cell configuration together with rapid piezo-driven changes in [Ca2+]i.
Materials and Methods
Molecular biology.
Ion channels were heterologously reconstituted either in unmodified Chinese hamster ovary (CHO) cells or in CHO cells stably expressing Cav2.2 channels (Cav2.2α1, Cavβ1, α2δ1, γ4; a gift from Novartis). Transfection of cDNAs was done with the JetPEI transfection reagent (Biomol), cells were incubated at 37°C and 5% CO2 and measured 2–3 d after transfection. GenBank accessions of the clones used are ADO63674 (BKα; a gift from Dr. L. Salkoff, Department of Anatomy and Neurobiology, Washington University School of Medicine, St. Louis, MO), NM_021452.1 (BKβ4), AF055477 (Cav2.2α1), NM_017346 (Cavβ1b), AF286488.1 (for transient transfection), and NM_012919 (in stably transfected cells) (α2δ). EGFP was simultaneously added as a transfection control. All cDNAs were verified by sequencing.
Electrophysiology.
Standard whole-cell patch-clamp recordings on CHO cells were done at room temperature (22–24°C) as previously described (Bildl et al., 2004). Briefly, currents were recorded with an Axopatch 200B amplifier, filtered at 3–10 kHz and sampled at 25 kHz; series resistance and capacitance (95%) were compensated.
Recording pipettes were made from quartz glass capillaries and had resistances of 1–5 MΩ when filled with intracellular solution containing the following (in mm): 137.5 KCl, 3.5 MgCl2, 2.5 NaATP, 10 K2EGTA, and 5 HEPES (pH adjusted to 7.3 with HCl). The extracellular solution was composed as follows (in mm): 144 NaCl, 5.8 KCl, 0.9 MgCl2, 1.3 CaCl2, 0.7 NaH2PO4, 5.6 d-glucose, and 10 HEPES (pH adjusted to 7.4 with NaOH).
Rapid solution exchange at excised inside-out patches was performed using a piezo-driven fast application system with a double-barrel application pipette (Schwenk et al., 2009). Speed and exact onset of solution exchange were monitored for each patch at the beginning of the respective experiment with a switch between intracellular solutions containing 150 and 60 mm K+ (K+ equally exchanged for Na+). Patches with exchange times < 1 ms (time constants of exponential functions fitted to the current change ≤ 0.3 ms) were used for evaluation. Solutions applied to the intracellular face of the patches contained the following (in mm): 137 KCl, 1.25 MgCl2, 2 DiBrBAPTA, 5 HEPES, 5 NaCl (pH adjusted to 7.3 with KOH). Free [Ca2+] of 1, 3, 10, and 100 μm were obtained by adding 0.706, 1.232, 1.7, and 2.06 mm CaCl2 (calculated with WEBMAXC 2.22). Free [Ca2+] was verified with a Ca2+-sensitive electrode (Berkefeld et al., 2006). Recording pipette solutions for inside-out patches was identical with the extracellular solution used for whole-cell measurements (see above).
Steady-state activation curves (see Fig. 3A) were determined as follows: for Ca2+ gating, [Ca2+]i steps were applied 20 ms after stepping the membrane potential to constant values between −80 and 110 mV (10 mV increment); for voltage gating voltage steps as above were applied at constant [Ca2+]i of values between 0 and 100 μm.
Figure 3.

Comparison of voltage- and Ca2+-triggered gating: similar steady-states, but distinct time courses. A, Relative steady-state activation of BKCa channels determined in inside-out patches and plotted along [Ca2+]i and transmembrane voltage. Intense colors refer to steady-state activation as obtained with Ca2+-triggered gating, faint colors are steady-state activation resulting from voltage-triggered gating. Data points are mean ± SEM 6–11 (Ca2+ gating) or 5–7 (voltage gating) experiments. Lines are results of fits with a Boltzmann function to the activation curves in both Ca2+ and voltage dimension. B, Time constants obtained for voltage (faint colors) and Ca2+ gating (intense colors) in the experiments in A and plotted against voltage and [Ca2+]i.
Data analysis.
Data analyses were done with Igor Pro 4.05A on a MacBook Pro. Time constants of channel activation (see Figs. 2B, 3, 4) were derived from monoexponential fits to the rising phase of the recorded K+ currents. For the fast application experiments, the delay of the current onset was defined as time between start of effective solution exchange and crossing of the signal threshold (set to 2 SDs from background noise; Figs. 2C, 4).
Figure 2.
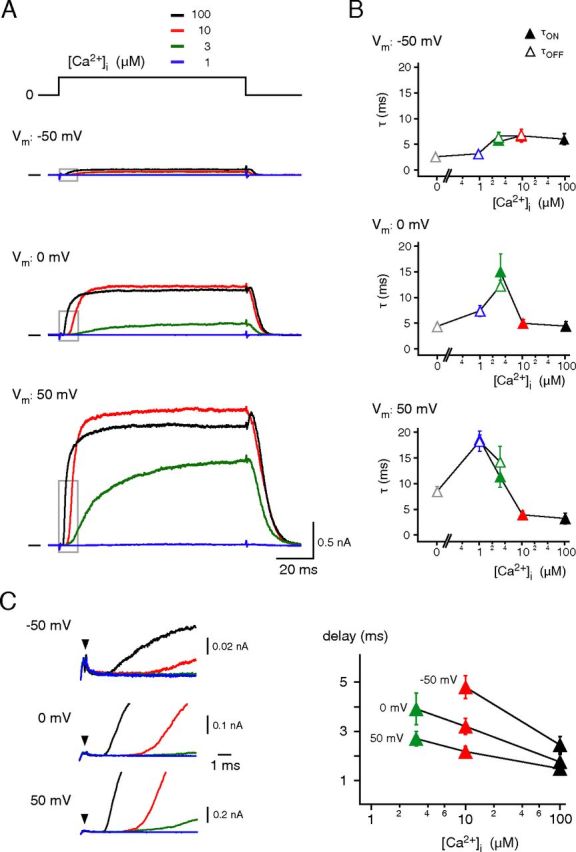
Characterization of Ca2+gating: the time domain. A, Currents through BKCa channels elicited by [Ca2+]i steps from 0 to 1, 3, 10, or 100 μm at membrane potentials of −50 mV, 0 or 50 mV (upper, middle, and bottom). Increase in amplitude upon withdrawal of the 100 μm Ca2+ solution reflects release of the pore block by Ca2+. B, Time constants of Ca2+ gating obtained from experiments as in A. Rising and declining phases of the currents were fitted with a mono-exponential function; closed symbols (τON) represent fits to the rising phase ([Ca2+]i stepped from 0 μm to 1, 3, 10, or 100 μm Ca2+), open symbols (τOFF) are results of fits to the decline of the current ([Ca2+]i stepped from 100 μm to 0, 1, 3, or 10 μm Ca2+). Data points are mean ± SEM for 3–10 (τON) or 3–16 experiments (τOFF). C, Left, Boxed periods of the traces in A at enlarged timescale. Arrows indicate start of effective solution exchange. Right, Pre-onset delay obtained from experiments as on the left and plotted versus the respective [Ca2+]i. Data points are mean ± SEM 7–10 experiments.
Figure 4.
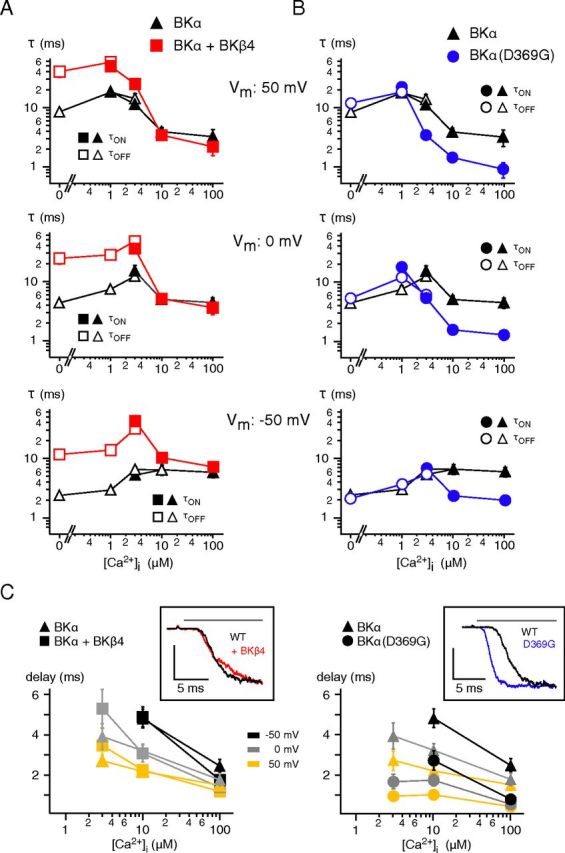
Effects of co-assembled BKβ4 and the BKα(D369G) mutation on Ca2+ gating of BKCa channels. A, B, Time constants of Ca2+ gating measured at the indicated potentials in inside-out patches from CHO cells expressing wild-type BKα either alone or together with BKβ4 (A) or expressing the BKα(D369G) mutant (B). Symbols are as in Figure 2; data points for τON are mean ± SEM 3–16 (BKα), 5–11 (BKα + BKβ4), and 6–9 (BKα(D369G)) experiments; data points for τOFF are mean ± SEM 3–16 (BKα), 6–12 (BKα + BKβ4), and 7–8 (BKα(D369G)). Note the distinct effects of BKβ4 and the D to G mutation on the gating characteristics. C, Pre-onset delay plotted versus [Ca2+]i for BKα and BKα + BKβ4 (left) and BKα and BKα(D369G) (right). Data points are mean ± SEM 7–10 (BKα), 5–9 (BKα + BKβ4), or 3–9 (BKα(D369G)) experiments. Insets, Representative current traces recorded in response to application of Ca2+ (horizontal bar) and normalized to maximum. Note the marked reduction in pre-onset delay observed with the BKα(D369G) mutant channels.
For determination of activation curves (Fig. 3A) steady-state currents obtained through voltage gating as well as Ca2+ gating were analyzed. Along the voltage axis, V1/2, slope factor k and conductance C were calculated with f(V) = (V − Vrev) * C/(1 + exp((V − V1/2)/k)) with a solution-dependent Vrev of −82 mV. First-order Boltzmann curves were deduced from these parameters with I = Ileak + Imax/(1 + exp((V − V1/2)/k)) where Ileak is voltage-independent leak current (set to 0), Imax is the amplitude of the fully activated current (set to 1), V is prepulse voltage, V1/2 is voltage for half-maximal activation, and k is the slope factor. Along the [Ca2+]-axis, V and V1/2 in the Boltzmann function were replaced by [Ca2+]i and [Ca2+]i at half-maximal activation [Ca2+]i,1/2. Data are given as mean ± SEM throughout the manuscript.
The Kolmogorov–Smirnov test was used for testing the pre-onset delays and gating time constants recorded at distinct values of [Ca2+]i and membrane potentials to be different; p values ≤ 0.05 are indicative for different behavior (at a confidence of >99%).
Results
Ca2+-triggered gating of BKCa channels
Classically, the gating properties of BKCa channels have been examined by applying voltage steps in the presence of constant [Ca2+]i, a regime where channels are gated by voltage pulses (“voltage gating”) and [Ca2+]i is considered a modulator (of gating time constants and steady-state open probabilities). Figure 1 shows application of such a paradigm to excised inside-out patches from CHO cells expressing homomeric BKCa channels. Thus, BKCa channels responded instantaneously to a voltage step from −120 to 0 mV with an increase in current whose time course strongly depended on the [Ca2+]i set to defined values between 1 and 100 μm (Fig. 1B, left, middle, and bottom).
Figure 1.
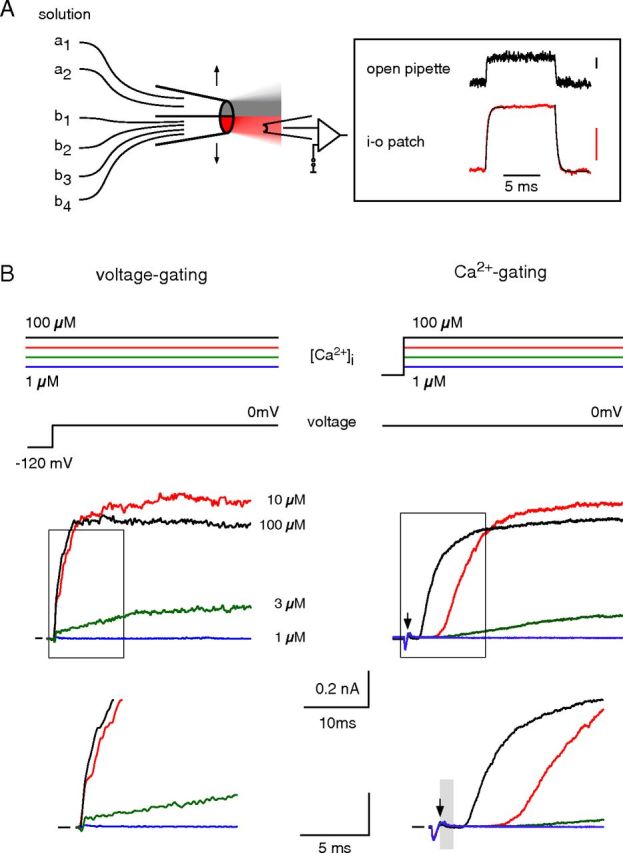
Gating of BKCa channels by voltage and Ca2+ in separation. A, Left, Scheme of the piezo-driven fast application system; each of the two barrels could be perfused with several different solutions. Right, Solution exchange (150 mm K+ vs 60 mm K+, at 10 μm Ca2+) with this system determined either at an open pipette or an excised inside-out patch containing BKCa channels. Exponential fits (black lines) to rise and decline of the currents through inside-out patches yielded time constants of 0.30 and 0.28 ms, respectively. Current scales are 0.1 nA. B, Voltage and Ca2+ gating determined in an inside-out patch excised from a CHO cell expressing BKα. Left, Current response (middle, bottom, and boxed period at expanded timescale) to a voltage step from −120 to 0 mV recorded at the indicated (constant) [Ca2+]i. Note the instantaneous rise in current at the voltage step. Right, Current response to [Ca2+]i steps (indicated by the arrow) from 0 to 1, 3, 10, or 100 μm recorded at a (constant) membrane potential of 0 mV; gray bar denotes three times the value of the exchange time constant (∼0.9 ms). Note the initial delay before the current onset. The reduction in current amplitude at 100 μm Ca2+ reflects pore block by the divalent (Ledoux et al., 2008).
Next, we performed experiments that used concentration steps in [Ca2+]i to trigger channel gating, while the membrane potential was held at constant values (“Ca2+ gating”). Experimentally, [Ca2+]i steps were achieved by a piezo-driven rapid application system that allowed for a complete solution exchange at the cytoplasmic face of inside-out patches within <1 ms (Fig. 1A). This time course was determined from the exchange of high versus low K+ concentrations at open BKCa channels (Fig. 1A, inset; time constants of ≤0.3 ms for switches in either direction) and, due to its critical importance, was repeated at every excised patch before the actual experiment. Similarly, the exchange of Ca2+ was not rate limiting (see below; Figs. 4C, 7). Figure 1B (right) illustrates such a rapid application experiment performed on the same patch as before and held at a membrane potential of 0 mV: [Ca2+]i steps to distinct concentrations between 1 and 100 μm elicited BKCa currents with amplitudes similar to those recorded upon voltage steps, but largely different kinetic properties. Thus, Ca2+-triggered BKCa currents displayed a prominent delay before their onset and rose with a significantly slower time course (Fig. 1B, right). The delay in current onset did not result from the solution exchange as it depended on [Ca2+]i, and exceeded the period required for complete exchange at a saturating [Ca2+]i of 100 μm (Fig. 1B, gray bar, right, bottom). Moreover, it should be noted that [Ca2+]i of ≥1 mm largely abolished BKCa-mediated currents due to the pore block of the divalent (Ledoux et al., 2008; Fig. 2).
Figure 7.

Acceleration of the BKCa channel Ca2+ gating upon complex formation with Cav channels. Time course of BKCa activation recorded either with Cav-free BKCa wild-type and D369G mutant channels upon fast application of 10 and 100 μm Ca2+ (at 50 mV) or in Cav2.2-associated BKCa channels at 40 mV. Data for complexes are from Figure 6, data for free BKCa channels (black traces) are mean ± SEM 8 (10 μm Ca2+) and 4 (100 μm Ca2+) fast application experiments with excised patches. Note the largely decreased pre-onset delay observed for the BKα(D369G) channels and the Cav2.2-associated BKCa channels.
The Ca2+ gating of BKCa channels was further characterized by determining both the pre-onset delay and the gating time constants over broad ranges of physiologically relevant [Ca2+]i and membrane potentials (Figs. 2, 3). The time constants for channel activation (opening, τON) and deactivation (closing, τOFF) were obtained from mono-exponential fits of the rising and falling phases of the currents recorded upon application ([Ca2+]i stepped from 0 to 3, 10, and 100 μm) and withdrawal of Ca2+ ([Ca2+]i stepped from 100 μm to 3, 1, and 0 μm), respectively; the pre-onset delay was determined as the period between the start of effective solution exchange (Figs. 1, 2, arrowheads) and the time point when currents exceeded the background noise. As shown in Figure 2, pre-onset delay and channel gating exhibited marked dependence on [Ca2+]i, with increasing [Ca2+]i promoting faster activation (Fig. 2A,B) and slower deactivation kinetics, together with a shorter pre-onset delay (Fig. 2A–C, left). The gating time constants were distributed in a bell-shaped manner over the [Ca2+]i range investigated (Fig. 2B). They were lowest at either [Ca2+]i ≥10 μm or at [Ca2+]i of 0 and 1 μm where τON and τOFF reflected the mere activation or deactivation kinetics, respectively (Fig. 2B). At 3 μm [Ca2+]i, where current amplitudes were about half-maximal and channel activation and deactivation occurred at balanced levels, the time constants displayed maximal values (Fig. 2B).
These observations were essentially independent of the membrane potential, although Ca2+ gating was modulated by voltage (Fig. 2). Thus, depolarization of the membrane potential (from −50 to 50 mV) accelerated Ca2+-triggered channel opening ∼2-fold, while channel closing was slowed down >3-fold (Fig. 2B). In addition, the pre-onset delay obtained at any [Ca2+]i was significantly decreased by membrane depolarization with values down to ∼1.5 ms at a [Ca2+]i of 100 μm and potentials ≥0 mV (Fig. 2C). Noteworthy, increasing [Ca2+]i from 10 to 100 μm significantly decreased the pre-onset delay at all voltages, while τON remained essentially constant at these [Ca2+]i (Fig. 2B,C).
Comparison of Ca2+- and voltage-triggered gating
These results demonstrated that BKCa channels can be effectively gated by both [Ca2+]i and voltage, consistent with previous studies (Markwardt and Isenberg, 1992; Rothberg and Magleby, 2000; Horrigan and Aldrich, 2002). To directly compare the two gating modes in the same BKCa channels, we determined activation curves for Ca2+ gating ([Ca2+]i steps at constant potentials along the voltage-axis from −80 to 110 mV) and voltage gating (voltage steps at constant [Ca2+]i between 0 and 100 μm) in 5–11 patches and plotted steady-state current amplitudes along the [Ca2+]i and voltage axes. The resulting 3D-plot showed that activation of BKCa channels under steady-state conditions is independent of whether Ca2+ or voltage was used as a gating trigger or, equivalently, that steady-state activation does not discriminate between the two stimuli (Fig. 3A). Different from the steady-state condition, however, the time courses were quite distinct between voltage- and Ca2+-triggered gating (Fig. 3B, Table 1). Thus, only Ca2+ gating exhibited a delay between stimulus and current onset (Fig. 1B), and the time constants determined for Ca2+-triggered gating exceeded those for voltage gating to about the same extent across the entire [Ca2+]i-voltage plane (Fig. 3B, Table 1).
Table 1.
Time constants of Ca2+ and voltage gating obtained for BKCa channels in excised patches
| Voltage gating |
Ca2+ gating |
||
|---|---|---|---|
| [Ca2+]i | Voltage step | Vm | [Ca2+]i step |
| −80 to −50 mV (τON) | −50 mV | 0 to [Ca2+]i (τON) | |
| 100 | 2.20 ± 0.23 (3) | 100 | 5.85 ± 1.08 (7) |
| 10 | 3.10 ± 0.78 (6) | 10 | 6.49 ± 0.50 (10) |
| 3 | 4.18 ± 0.57 (8) | 3 | 5.28 ± 0.72 (3) |
| 100 to −50 mV (τOFF) | 100 μm to [Ca2+]i (τOFF) | ||
| 1 | 0.76 ± 0.08 (4) | 1 | 2.96 ± 0.28 (8) |
| 0 | 0.47 ± 0.02 (3) | 0 | 2.23 ± 0.57 (8) |
| −80 to 0 mV (τON) | 0 mV | 0 to [Ca2+]i (τON) | |
| 100 | 1.71 ± 0.11 (4) | 100 | 4.44 ± 0.88 (7) |
| 10 | 1.72 ± 0.24 (9) | 10 | 5.01 ± 1.90 (9) |
| 3 | 6.13 ± 0.47 (8) | 3 | 15.00 ± 1.09 (8) |
| 100 to 0 mV (τOFF) | 100 μm to [Ca2+]i (τOFF) | ||
| 1 | 3.77 ± 0.46 (10) | 1 | 7.39 ± 1.07 (8) |
| 0 | 0.86 ± 0.17 (8) | 0 | 4.19 ± 1.06 (8) |
| −80 to 50 mV (τON) | 50 mV | 0 to [Ca2+]i (τON) | |
| 100 | 0.68 ± 0.09 (3) | 100 | 3.24 ± 1.03 (7) |
| 10 | 1.09 ± 0.30 (7) | 10 | 3.95 ± 0.68 (10) |
| 3 | 5.56 ± 0.60 (8) | 3 | 11.34 ± 3.05 (8) |
| 1 | 14.57 ± 1.76 (9) | 1 | 18.17 ± 2.01 (3) |
| 100 to 50 mV (τOFF) | 100 μm to [Ca2+]i (τOFF) | ||
| 0 | 3.28 ± 1.01 (4) | 0 | 10.02 ± 3.29 (8) |
All values are mean ± SEM of (n) experiments.
Together these results indicated that while both Ca2+ and voltage converge on the opening mechanism of BKCa channels, their actual coupling is quite distinct. Ca2+-triggered gating likely involves additional steps including Ca2+ binding, which engender the pronounced pre-onset delay.
Selective effects of BKβ4 and an epilepsy-associated mutation in BKα on Ca2+ gating
The time course of Ca2+ gating was further characterized by analyzing alterations induced by co-assembly of BKα with BKβ4, the most abundant auxiliary subunit of brain BKCa channels (Brenner et al., 2000; Berkefeld et al., 2006), or by a point mutation in the cytoplasmic region linking S6 to the “regulator of K+ conductance” (RCK) domain of the BKα protein (D369G; Du et al., 2005). Both have been reported to influence the gating of BKCa channels by shifting steady-state activation, decelerating (BKβ4), or accelerating (D369G) channel opening and by increasing the apparent sensitivity to Ca2+ (D369G) (Ha et al., 2004; Wang et al., 2006; Yang et al., 2010). Figure 4 summarizes the results obtained from analysis of currents recorded upon [Ca2+]i steps at three distinct voltages for channels assembled either from BKα and BKβ4 (Fig. 4A,C) or from BKα(D369G) proteins (Fig. 4B,C). The two alterations exerted quite distinct effects on the dynamics of the Ca2+ gating: while BKβ4 increased the τOFF values (measured at 0 and 1 μm [Ca2+]i) by up to an order of magnitude without significant effects on τON (measured at [Ca2+]i ≥10 μm; p >0.05, Kolmogorov–Smirnov test; Fig. 4A), the D to G exchange in the linker region resulted in marked decrease of τON, but left τOFF virtually unaffected (Fig. 4B). Thus, co-assembly of BKα with BKβ4 predominantly influenced channel deactivation, whereas the (D369G) mutation selectively targeted Ca2+-triggered activation of the channels. In line with this interpretation, the pre-onset delay measured in BKα(D369G) channels was significantly decreased at all activation-triggering [Ca2+]i and at all three membrane potentials (Fig. 4C, right) with values down to <1 ms measured at 100 μm [Ca2+]i (0.77 ± 0.21 ms (−50 mV), 0.53 ± 0.19 ms (0 mV), 0.42 ± 0.18 ms (50 mV), n = 9; Fig. 4C). BKβ4 did not induce any obvious changes at [Ca2+]i ≥10 μm, while a slight increase in the pre-onset delay was observed for [Ca2+]i of 3 μm (p > 0.01, Kolmogorov–Smirnov test; Fig. 4C, left).
Together, these results indicated that BKβ4 and the D369G mutation selectively promote distinct gating processes underlying deactivation and activation of Ca2+-triggered gating, respectively. In fact, the observed decrease in pre-onset delay together with the accelerated channel activation are consistent with the enhanced allosteric coupling proposed for the D369G mutation by Cui et al. (Yang et al., 2010).
Ca2+ gates BKCa channels in BKCa–Cav complexes
Next, we investigated the Ca2+ gating in BKCa channels that are coexpressed with Cav channels. Under these circumstances, the two channel types are co-assembled into macromolecular complexes similar to the association of BKCa and Cav channels in the mammalian brain (Robitaille et al., 1993; Prakriya and Lingle, 2000; Berkefeld et al., 2006; Loane et al., 2007; Berkefeld and Fakler, 2008). This configuration places the BKCa channels within ∼10 nm from their Ca2+ source, effectively delivering [Ca2+]i within microseconds after voltage-dependent pore opening (Neher, 1998; Fakler and Adelman, 2008).
We performed whole-cell patch-clamp recordings under physiological ion conditions on CHO cells that coexpressed BKCa and Cav2.2 channels (N-type channels) and were dialyzed with a high concentration (10 mm) of the Ca2+ buffer EGTA to eliminate currents of Cav-free BKCa channels (Fig. 5A; Berkefeld et al., 2006; Berkefeld and Fakler, 2008). The current-output evoked in BKCa–Cav complexes by voltage commands reflects the combination of K+ and Ca2+ currents (Berkefeld et al., 2006). Therefore, a modified tail-current protocol was used to precisely trigger Ca2+ gating and to selectively focus on the resulting K+-current component (Berkefeld and Fakler, 2008). This tail-current protocol consisted of three voltage steps (Fig. 5B,C, top): (1) a depolarizing prepulse to 70 mV for 10 ms fully activating Cav channels without eliciting Ca2+ currents due to the largely reduced driving force for Ca2+ ions, (2) a conditioning pulse to potentials between −40 and 40 mV promoting Ca2+ influx (increase in driving force for Ca2+ ions of 30 to 110 mV) for a defined period of time (varied between 0.06 and 15.4 ms, details in Figure 5 legend), and (3) a test pulse to 70 mV selectively monitoring the BKCa-mediated K+ currents.
Figure 5.
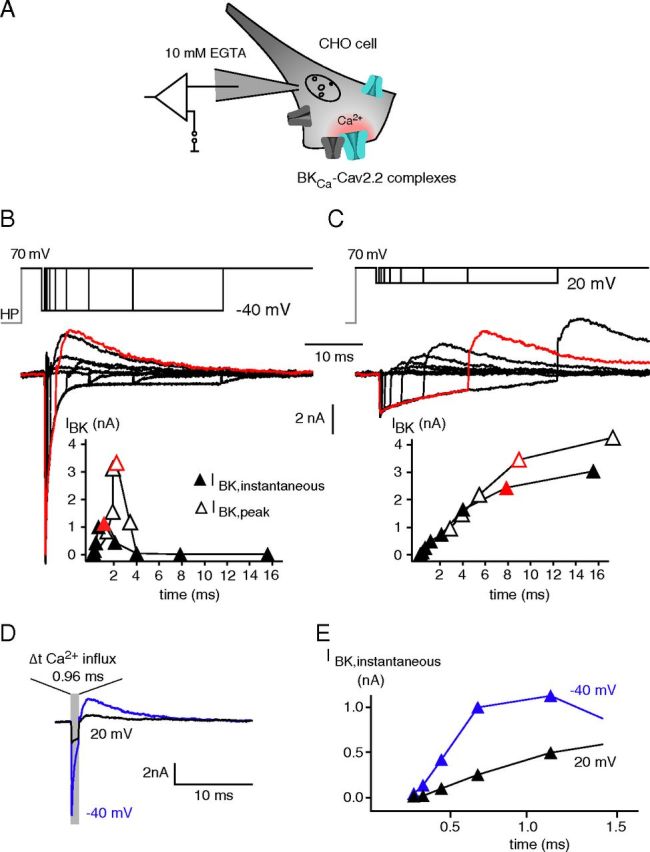
Operation of Cav-associated BKCa channels by their Ca2+ gating. A, Experimental conditions for analysis of the Ca2+ gating in BKCa–Cav2.2 complexes. Channel–channel complexes were reconstituted in CHO cells upon coexpression of BKα, BKβ4, Cav2.2α1, α2δ, and Cavβ1b; whole-cell recordings were done with 10 mm EGTA in the pipette to eliminate currents through Cav-free BKCa channels (Berkefeld et al., 2006). B, C, Representative currents through BKCa–Cav2.2 complexes recorded in whole-cell configuration in response to the indicated “scanning-tail-current protocol” (details in the text). Step to 70 mV activated Cav channels without eliciting Ca2+ influx (effective reversal potential for Ca2+ slightly below this potential), steps to −40 mV (B; termed conditioning pulse), and 20 mV (C) triggered Ca2+ influx (increase in driving force) for defined periods (0.06, 0.12, 0.24, 0.48, 0.96, 1.92, 3.84, 7.68, and 15.36 ms). The final step back to 70 mV switched off the Ca2+-inward currents and elicited K+-outward currents. Traces in red are responses recorded with a conditioning pulse duration of 0.96 ms (B) or 7.68 ms (C); 0 time point is start of the conditioning pulse. Note the similarity in amplitudes of the two BKCa outward currents despite the largely different periods of Ca2+ influx. Bottom, Amplitudes of BKCa currents recorded 0.2 ms after the final pulse step (I BK,instantaneous, filled symbols; “time” is duration of conditioning pulse plus 0.2 ms) or of BKCa peak currents (I BK,ipeak, open symbols) plotted over the period of Ca2+ influx (start of the conditioning pulse). D, BKCa currents from an experiment as in B and C with a conditioning pulse of 0.96 ms to −40 mV (blue) and 20 mV (black). E, IBK,instantaneous-time relation from the experiment in B and C at expanded timescale (time points are conditioning interval plus 0.2 ms). Note that the BKCa current is larger in amplitude at −40 mV than at 20 mV, and that the time course of channel activation is markedly faster at −40 mV than at 20 mV.
Figure 5B–D illustrates representative current traces recorded during such a “scanning tail-current protocol” with two distinct conditioning pulse potentials of −40 and 20 mV. In either case, the Cav-mediated Ca2+ influx effectively activated BKCa channels as visualized by the transient outward K+ currents recorded during the test pulse (see also Figure 5 legend). However, the time course of the BKCa currents was largely different for the two conditioning pulses tightly mirroring the distinct gating kinetics of the Cav channel(s) at the respective potentials (Fig. 5B,C, inset). Thus, BKCa currents rapidly rose and fell at −40 mV as a consequence of the pronounced Cav channel deactivation (time constant of ∼0.5 ms), while they steadily increased over time at 20 mV promoted by the only moderate inactivation of the Cav channels (time constant of ∼5 ms). This distinction was independent of whether BKCa currents were measured as a peak outward current (IBK,peak) or as a tail current at the test step (IBK,instantaneous), which directly monitored the Ca2+-driven gating during the conditioning pulse (Fig. 5B,C).
Closer investigation of IBK (IBK,instantaneous) recorded at conditioning pulses of short length (between 0.06 and 1.2 ms; see also Fig. 5 caption) revealed two further peculiarities of BKCa activation in the BKCa–Cav complexes. First, activation at 20 mV occurred only after an initial time lag reminiscent of the pre-onset delay seen in fast application experiments (Fig. 5E) and, second, activation was markedly faster at −40 mV than at 20 mV (Fig. 5D,E). The latter observation is counterintuitive with voltage-dependent gating, but consistent with channel activation driven by Ca2+ whose delivery at −40 mV largely exceeded that at 20 mV as reflected by the amplitudes of the Ca2+-inward current (Fig. 5B–D). In fact, analysis of the time course of BKCa activation on an extended voltage range from −40 to 40 mV further corroborated this finding: the speed of BKCa activation steadily decreased with depolarization of the membrane potential during the conditioning pulse (Fig. 6A).
Figure 6.

Determination of the K+-current output of BKCa–Cav complexes by the properties of BKCa Ca2+ gating. A–C, IBK,instantaneous-time relations determined for BKCa–Cav complexes of the indicated composition in experiments as in Figure 5 but for the conditioning pulse potentials between −40 and 40 mV given by the inset in A. Currents were normalized to the maximum recorded at a conditioning pulse potential of −40 mV; data points are mean ± SEM 13 (BKα), 10 (BKα+ BKβ4), and 6 (BKα(D369G)) experiments. Note the slowing of BKCa-channel activation with increasing depolarization and the lack of delay obtained with the BKCa(D369G) mutant. D, Overlay of data from A to C recorded at a conditioning pulse potential of 20 mV.
These results strongly suggested that in BKCa–Cav complexes Ca2+ dominates over voltage as the rate-limiting stimulus for activation of the BKCa channels and prompted experiments probing the effect of altered Ca2+ gating on the complexes' K+-current response. For this purpose, the experiments in Figures 5 and 6A were repeated with the BKα(D369G) mutant or with BKβ4 co-assembled with wild-type BKα. As illustrated in Figure 6B–D the K+ currents in either type of macromolecular BKCa–Cav complex increased in amplitude with hyperpolarization of the conditioning pulse; however, while the D to G exchange promoted a significant acceleration of the activation time course together with a reduction in the initial lag time (in particular at voltages ≥0 mV; Fig. 6B,D), the changes induced by the co-assembled BKβ4-subunit were rather moderate (Fig. 6D). Both observations fit the results obtained from the fast application experiments on BKCa channels expressed alone (Fig. 2) and emphasize that activation of Cav-associated BKCa channels showed the hallmark properties of Ca2+ gating. In addition, direct comparison of the activation time courses obtained from Cav-free and Cav-associated BKCa channels revealed a surprising difference (Fig. 7): while the pre-onset delay in isolated BKCa channels were in the range of 1–2 ms (measured for 10–100 μm Ca2+ at 50 mV), it was as short as ∼300 μs for Cav-associated channels (at 40 mV; Fig. 7). This reduction in lag time is very similar to that obtained in the D369G mutant (∼400 μs; Figs. 4C, 7) and thus suggests that it results from altered gating induced by direct interaction between the BKCa and the Cav channel proteins (Berkefeld et al., 2006) rather than from a high local [Ca2+]i.
Together, these results demonstrated that in macromolecular complexes with the Cav channels, BKCa channels are primarily activated by Ca2+ ions suggesting that, under physiological conditions, they are operated as (voltage-sensitive) ligand-gated channels similar to their SKCa counterparts (Xia et al., 1998; Berkefeld et al., 2010).
Discussion
The central finding of the present work is that BKCa channels in macromolecular complexes with Cav channels behave as ligand-operated channels with Ca2+ gating determining channel activation. Accordingly, in this configuration depolarization serves primarily to activate the Cav subunits which, in turn, provide the elevation in [Ca2+]i that drives the Ca2+ gating of the BKCa channels. In addition, the co-assembly with Cav channels appears to allosterically tune BKCa channels for fast responses by markedly reducing the pre-onset delay of their Ca2+ gating.
Ca2+ gating: characteristics and mechanism
We used voltage-clamp recordings on patches excised from CHO cells together with a rapid piezo-driven solution exchange system to obtain precise and fast manipulations of either of the two distinct input stimuli (Fig. 1). In particular, the quality of the [Ca2+]i steps defining the time resolution of Ca2+ gating was ensured by determining the time constants of solution exchange at any inside-out patch before use (Fig. 1A). Respective estimates taken from exchange of K+ concentrations at open BKCa channels yielded values ≤0.3 ms (for either direction; Fig. 1A), and effective Ca2+ exchange was evidenced by the minimal pre-onset delay obtained with BKα(D369G) mutant channels that was as low as 0.4 ms (Figs. 4, 7).
Using this configuration we first showed that BKCa channels can effectively be gated by [Ca2+]i and thus operate as ligand-activated channels (Markwardt and Isenberg, 1992; Horrigan and Aldrich, 2002), analogous to the SK-type Ca2+-activated K+ channels (Xia et al., 1998; Adelman et al., 2012). Different from SKCa channels, however, the Ca2+ gating of BKCa is sensitive to changes in the transmembrane voltage (Fig. 2) as a result of the channels' allosteric coupling of both Ca2+ and voltage gating branches through the pore-forming domains of the channel protein (Cui et al., 1997; Rothberg and Magleby, 2000; Horrigan and Aldrich, 2002). Direct comparison of the two gating branches showed that Ca2+ gating differs from voltage gating in mainly two aspects: (1) a prominent delay between Ca2+ application and pore opening (termed pre-onset delay; Figs. 1, 2) and (2) a slower activation time course to about the same extent over the entire [Ca2+]i-voltage plane (Figs. 2, 3).
These two characteristics suggest that the conformational dynamics responsible for Ca2+ gating are different from those underlying voltage-driven gating. In this sense, the pre-onset delay and its dependence on [Ca2+]i most likely reflect concentration-dependent binding of Ca2+ ions to respective site(s) in all four BKα-subunits and the subsequent structural rearrangements preceding channel opening. Importantly, the delay period measured at gating-saturating [Ca2+]i of 100 μm was ∼1.5 ms and thus considerably longer than the period required for complete solution exchange at the cytoplasmic face of the channels (Figs. 1A, 4C, 7). Indeed, 100 μm is about an order of magnitude higher than what the BKCa channels encounter when co-assembled with Cav channels, as seen by their activation kinetics (Berkefeld et al., 2006; Berkefeld and Fakler, 2008). The slower speed of Ca2+-gated BKCa channel activation may reflect a pore-opening mechanism distinct from the conformational changes underlying voltage-dependent gating. Thus, while voltage gating in BKCa, similar to that in Kv channels (Perozo et al., 1999; Long et al., 2007; Pathak et al., 2007), is envisaged to operate through the moving voltage sensor (S2–4 helices) transferring energy to the pore-forming S5/6 helices via the S4/5 linker, Ca2+ gating implies coupling of the cytoplasmic agonist binding to the pore via the RCK-S6 linker (16 aa in length) and the S6 helix.
In fact, recent crystallographic work on the Ca2+ “gating ring” of BKCa channels in both the apo and Ca2+-bound form put forward a structural model for Ca2+ gating supporting this notion (Wu et al., 2010; Yuan et al., 2010, 2012). Accordingly, binding of Ca2+ to the four RCK2 domains of the gating ring triggers conformational changes in the N-terminal lobes of the adjacent RCK1 domains that, via a petal-like widening, directly enforce the S6 helices to undergo structural rearrangements underlying pore opening (Yuan et al., 2012). Moreover, in the framework of this model, the speed of the force transfer should be directly affected by the structural constraints of the RCK-S6 linker, in line with the strong effect observed for the pre-onset delay with the BKα(D369G) mutation (Figs. 4, 7; Yang et al., 2010).
Ca2+ gates BKCa channels in Cav–BKCa complexes
Experiments in whole-cell configuration examined the gating of BKCa channels in macromolecular BKCa–Cav complexes, reflecting the co-assembly of both channels in the mammalian brain (for review, see Berkefeld et al., 2010). In BKCa–Cav complexes, the K+-current output is tightly controlled by the gating properties of the particular Cav subtype co-assembled with BKCa (Fig. 5B,C; Berkefeld and Fakler, 2008).
Application of a pulse protocol separating voltage-dependent gating of the Cav channels from Ca2+ influx and subsequent increase in [Ca2+]i (Fig. 5B,C) showed that (1) the speed of BKCa activation increased upon hyperpolarization, but decreased with depolarization of the membrane potential (Figs. 5E, 6A–C); (2) BKCa activation displayed an initial delay, in particular at membrane potentials ≥0 mV (Figs. 5E, 7); and (3) both activation speed and initial delay were affected by the D to G mutation in the RCK-S6 linker (Fig. 6D).
All these characteristics are consistent with BKCa channels being activated by [Ca2+]i rather than by voltage and, consequently, Ca2+ gating dictating the time course of the K+-current output in BKCa–Cav complexes. In this context, despite the inherent voltage sensitivity of Ca2+ gating, changes in membrane potential primarily serve as the gating trigger for the Cav channels which, in turn, provide the increase in [Ca2+]i required for effective activation of BKCa. The latter is most effective at highest [Ca2+]i as promoted by hyperpolarization of the membrane potential due to the resulting increase in driving force (Figs. 5, 6).
This “sequential cascade” derived for activation of Cav-associated BKCa channels that reflect the configuration in many CNS neurons should be particularly effective during an action potential (Berkefeld and Fakler, 2008). The depolarizing upstroke initiates activation of the Cav channels, which pass Ca2+ into the cytosol mainly during the repolarization phase of the action potential (Li et al., 2007) when the driving force for Ca2+ is largest. The resulting increase in [Ca2+]i serves as an effective input stimulus to the BKCa Ca2+ gating, which is further speeded by the reduction in the pre-onset delay established through the direct protein–protein interactions between Cav and BKCa channels (Fig. 7). Together, these precisely timed processes should engender effective and rapid activation of the BKCa channels under physiological conditions.
Footnotes
This work was supported by grants of the Deutsche Forschungsgemeinschaft (SFB 746/TP16, DFG Fa 332/5-3) to B.F. We thank J.P. Adelman and the Fakler group for insightful comments and critical reading of this manuscript.
References
- Adelman JP, Maylie J, Sah P. Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol. 2012;74:245–269. doi: 10.1146/annurev-physiol-020911-153336. [DOI] [PubMed] [Google Scholar]
- Berkefeld H, Fakler B. Repolarizing responses of BKCa-Cav complexes are distinctly shaped by their Cav subunits. J Neurosci. 2008;28:8238–8245. doi: 10.1523/JNEUROSCI.2274-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart J-O, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U, Fakler B. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–620. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- Berkefeld H, Fakler B, Schulte U. Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev. 2010;90:1437–1459. doi: 10.1152/physrev.00049.2009. [DOI] [PubMed] [Google Scholar]
- Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B. Protein kinase CK2 is co-assembled with small conductance Ca2+-activated K+ channels and regulates channel gating. Neuron. 2004;43:847–858. doi: 10.1016/j.neuron.2004.08.033. [DOI] [PubMed] [Google Scholar]
- Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem. 2000;275:6453–6461. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol. 1997;110:257–281. doi: 10.1085/jgp.110.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Cox DH, Aldrich RW. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J Gen Physiol. 1997;109:647–673. doi: 10.1085/jgp.109.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Yang H, Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2009;66:852–875. doi: 10.1007/s00018-008-8609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Lüders HO, Shi J, Cui J, Richerson GB, Wang QK. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- Edgerton JR, Reinhart PH. Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol. 2003;548:53–69. doi: 10.1113/jphysiol.2002.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakler B, Adelman JP. Control of KCa channels by calcium nano/microdomains. Neuron. 2008;59:873–881. doi: 10.1016/j.neuron.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Golding NL, Jung HY, Mickus T, Spruston N. Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J Neurosci. 1999;19:8789–8798. doi: 10.1523/JNEUROSCI.19-20-08789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes WN, Li W, Chávez AE, Diamond JS. BK channels modulate pre- and postsynaptic signaling at reciprocal synapses in retina. Nat Neurosci. 2009;12:585–592. doi: 10.1038/nn.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha TS, Heo MS, Park CS. Functional effects of auxiliary beta4-subunit on rat large-conductance Ca2+-activated K+ channel. Biophys J. 2004;86:2871–2882. doi: 10.1016/S0006-3495(04)74339-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120:267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol. 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux J, Bonev AD, Nelson MT. Ca2+-activated K+ channels in murine endothelial cells: block by intracellular calcium and magnesium. J Gen Physiol. 2008;131:125–135. doi: 10.1085/jgp.200709875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bischofberger J, Jonas P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J Neurosci. 2007;27:13420–13429. doi: 10.1523/JNEUROSCI.1709-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippiat JD, Standen NB, Harrow ID, Phillips SC, Davies NW. Properties of BK(Ca) channels formed by bicistronic expression of hSloalpha and beta1–4 subunits in HEK293 cells. J Membr Biol. 2003;192:141–148. doi: 10.1007/s00232-002-1070-0. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Lima PA, Marrion NV. Co-assembly of N-type Ca2+ and BK channels underlies functional coupling in rat brain. J Cell Sci. 2007;120:985–995. doi: 10.1242/jcs.03399. [DOI] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Ma Z, Lou XJ, Horrigan FT. Role of charged residues in the S1–S4 voltage sensor of BK channels. J Gen Physiol. 2006;127:309–328. doi: 10.1085/jgp.200509421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwardt F, Isenberg G. Gating of maxi K+ channels studied by Ca2+ concentration jumps in excised inside-out multi-channel patches (myocytes from guinea pig urinary bladder) J Gen Physiol. 1992;99:841–862. doi: 10.1085/jgp.99.6.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature. 1981;291:497–500. doi: 10.1038/291497a0. [DOI] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/S0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Pathak MM, Yarov-Yarovoy V, Agarwal G, Roux B, Barth P, Kohout S, Tombola F, Isacoff EY. Closing in on the resting state of the Shaker K+ channel. Neuron. 2007;56:124–140. doi: 10.1016/j.neuron.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Pattillo JM, Yazejian B, DiGregorio DA, Vergara JL, Grinnell AD, Meriney SD. Contribution of presynaptic calcium-activated potassium currents to transmitter release regulation in cultured Xenopus nerve-muscle synapses. Neuroscience. 2001;102:229–240. doi: 10.1016/S0306-4522(00)00453-X. [DOI] [PubMed] [Google Scholar]
- Perozo E, Cortes DM, Cuello LG. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lingle CJ. Activation of BK channels in rat chromaffin cells requires summation of Ca2+ influx from multiple Ca2+ channels. J Neurophysiol. 2000;84:1123–1135. doi: 10.1152/jn.2000.84.3.1123. [DOI] [PubMed] [Google Scholar]
- Raffaelli G, Saviane C, Mohajerani MH, Pedarzani P, Cherubini E. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J Physiol. 2004;557:147–157. doi: 10.1113/jphysiol.2004.062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancz EA, Häusser M. Dendritic calcium spikes are tunable triggers of cannabinoid release and short-term synaptic plasticity in cerebellar Purkinje neurons. J Neurosci. 2006;26:5428–5437. doi: 10.1523/JNEUROSCI.5284-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Rothberg BS, Magleby KL. Voltage and Ca2+ activation of single large-conductance Ca2+-activated K+ channels described by a two-tiered allosteric gating mechanism. J Gen Physiol. 2000;116:75–99. doi: 10.1085/jgp.116.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/S0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klöcker N. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323:1313–1319. doi: 10.1126/science.1167852. [DOI] [PubMed] [Google Scholar]
- Shao LR, Halvorsrud R, Borg-Graham L, Storm JF. The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol. 1999;521:135–146. doi: 10.1111/j.1469-7793.1999.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Intracellular injection of a Ca2+ chelator inhibits spike repolarization in hippocampal neurons. Brain Res. 1987a;435:387–392. doi: 10.1016/0006-8993(87)91631-3. [DOI] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987b;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Rothberg BS, Brenner R. Mechanism of beta4 subunit modulation of BK channels. J Gen Physiol. 2006;127:449–465. doi: 10.1085/jgp.200509436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Characterization of large conductance Ca2+-activated K+ channels in cerebellar Purkinje neurons. Eur J Neurosci. 2002;16:1214–1222. doi: 10.1046/j.1460-9568.2002.02171.x. [DOI] [PubMed] [Google Scholar]
- Wu Y, Yang Y, Ye S, Jiang Y. Structure of the gating ring from the human large-conductance Ca2+-gated K+ channel. Nature. 2010;466:393–397. doi: 10.1038/nature09252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- Xia XM, Zeng X, Lingle CJ. Multiple regulatory sites in large-conductance calcium-activated potassium channels. Nature. 2002;418:880–884. doi: 10.1038/nature00956. [DOI] [PubMed] [Google Scholar]
- Yang J, Krishnamoorthy G, Saxena A, Zhang G, Shi J, Yang H, Delaloye K, Sept D, Cui J. An epilepsy/dyskinesia-associated mutation enhances BK channel activation by potentiating Ca2+ sensing. Neuron. 2010;66:871–883. doi: 10.1016/j.neuron.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Leonetti MD, Pico AR, Hsiung Y, MacKinnon R. Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science. 2010;329:182–186. doi: 10.1126/science.1190414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Leonetti MD, Hsiung Y, MacKinnon R. Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature. 2012;481:94–97. doi: 10.1038/nature10670. [DOI] [PMC free article] [PubMed] [Google Scholar]