

Abstract
Memory consolidation requires gene expression regulation by transcription factors, which eventually may induce chromatin modifications as histone acetylation. This mechanism is regulated by histone acetylases and deacetylases. It is not yet clear whether memory consolidation always recruits histone acetylation or it is only engaged in more persistent memories. To address this question, we used different strength of training for novel object recognition task in mice. Only strong training induced a long-lasting memory and an increase in hippocampal histone H3 acetylation. Histone acetylase inhibition in the hippocampus during consolidation impaired memory persistence, whereas histone deacetylase inhibition caused weak memory to persist. Nuclear factor κB (NF-κB) transcription factor inhibition impaired memory persistence and, concomitantly, reduced the general level of H3 acetylation. Accordingly, we found an important increase in H3 acetylation at a specific NF-κB-regulated promoter region of the Camk2d gene, which was reversed by NF-kB inhibition. These results show for the first time that histone acetylation is a specific molecular signature of enduring memories.
Introduction
A classical tenet in the field of the neurobiology of memory is that, to persist, memory storage requires changes in gene expression patterns (Alberini, 2009). However, persistent forms of memory require specific mechanisms that are differentially induced by particular learning conditions that warrant remote memory stabilization. Such mechanisms should not be required in less lasting forms of memories. Recent research has identified a specific time window, present ∼12 h after training, in which BDNF action is required for memories to persist. Such BDNF signals are considered to be part of the recurrent rounds of consolidation-like events that take place in the hippocampus for the maintenance of the memory trace (Bekinschtein et al., 2007; Rossato et al., 2009). However, beyond these mechanisms of memory maintenance, specific mechanisms activated during consolidation may make some memories more persistent than others, according to either the strength of training or the level of attention or motivation during learning.
Epigenetic mechanisms are postulated to provide potentially stable marks in the genome to regulate the expression of specific genes continuously during memory formation (Levenson and Sweatt, 2006; Barrett and Wood, 2008). Chromatin structure and function can be affected by various posttranslational modifications of nucleosomal histones (Strahl and Allis, 2000; Kouzarides, 2007). Lysine acetylation is one of the best-characterized histone modifications. Chromatin-modifying enzymes, such as histone acetyl transferases (HATs) and histone deacetylases (HDACs), perform protein acetylation and deacetylation, respectively (Sterner and Berger, 2000). Histone acetylation induces transcriptional activation by increasing the accessibility of the DNA to the transcriptional machinery (Vettese-Dadey et al., 1996). Recruitment of epigenetic mechanisms, such as histone acetylation, methylation, and phosphorylation, has already been implicated in long-term plasticity and memory consolidation (Guan et al., 2002; Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005; Gupta et al., 2010; Gräff et al., 2012). In agreement with such findings, HDAC inhibitor administration has been shown to improve long-term memory (Levenson et al., 2004; Yeh et al., 2004; Fischer et al., 2007; Vecsey et al., 2007) and to revert memory deficits induced by genetic manipulations (Alarcón et al., 2004; Korzus et al., 2004; Guan et al., 2009).
Some studies have evidenced the participation of epigenetic mechanisms in novel object recognition (NOR) memory. The activity of the HAT CREB-binding protein (CBP) and p300 during recognition memory consolidation was studied in genetically modified mice (Bourtchouladze et al., 2003; Barrett et al., 2011; Oliveira et al., 2011; Zhao et al., 2012). To our knowledge, epigenetic mechanisms in general and chromatin acetylation in particular have not been specifically ascribed to memory persistence. The aim of this study was to determine the involvement of histone acetylation in different strengths of memory, with the hypothesis that this mechanism is a molecular feature of the formation of enduring memories. In previous work in invertebrates, we obtained data supporting this hypothesis (Federman et al., 2009; Federman et al., 2012), although no specific genes were studied in that model, and no evidence was obtained regarding the persistence of strong memories.
Materials and Methods
Animals.
C57BL/6J male mice, 6–9 weeks of age and weighing ∼25–30 g were used. These mice were provided by the animal facility of the Universidad Nacional de La Plata, Argentina. In our laboratory, these animals were housed in boxes in groups of four individuals; they were later housed individually after cannulation. They were provided with water and food ad libitum and were kept under a 12 h light/dark cycle (lights on at 8:00 A.M.) at a temperature of 21–22°C. Experiments were performed in accordance with Facultad de Ciencias Exactas y Naturales (Universidad de Buenos Aires) regulations and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH publication 80–23/96). All efforts were made to minimize animal suffering and to reduce the number of animals used.
Training-testing apparatus and behavioral procedure.
The experimental chamber consisted of a wooden box with a transparent lid and white walls (30 × 23 × 21.5 cm); the floor was covered with shavings. The novel object recognition task consisted of 2 d of handling for 3 min once a day and 3 d of habituation that involved the placement of each animal in the experimental box for 5 min once a day with no object presentation. The following day, mice were trained in the experimental chamber with two identical objects. The objects used were 100 ml transparent beakers or blue blocks (Rasti toys), both of similar size. They were allowed to explore the objects for 3 min (weak training), 10 min (standard training), or 15 min (strong training), depending on the experiment. Between trials, the objects were scrubbed with a tissue soaked in 96% alcohol (Sanicol) and then rinsed with bi-distilled water to ensure that no olfactory cues were present. Memory retention was assessed either 24 h or 1 week after training. The testing session involved introducing the animals to the experimental chamber and allowing them to explore two different objects (a beaker and a block) for 5 min. One of the objects was identical to those explored during the training session (familiar object) and the other was a different object (novel object). The objects were exposed in the same locations of the chamber as they were in the training session. The location of the novel object was exchanged between the left and the right for different animals to avoid place preference during the evaluation session. The training and testing sessions were filmed with a web camera (Tessar 2.0/3.7 2MP autofocus; Logitech), which was connected to a computer using the program Quicktime (Logitech). The camera was placed 30 cm above the experimental chamber for later analysis. All manipulations were performed with the investigator wearing gloves.
Behavioral data analysis.
The time that mice spent exploring the objects was established as the time during which the animal oriented its head toward an object with his nose 1 cm or less around it. The time that the animal spent above the object was not considered. During training, the total time of exploration and the percentage of time that the animal spent exploring the left and right objects (with respect to the initial position of the mouse) were determined. During the testing session, the total exploration time for each object was determined and the relative time of novel object exploration was calculated as the index of discrimination (ID%) calculated as follows: (t novel − t familiar)/(t novel + t familiar) × 100%. The mean ID% value was calculated for the different groups of animals. Depending of the number of groups, Student's t test or one-way ANOVA was used with a per comparison error rate (α) of 0.05 for statistical comparisons between groups. The total times of exploration for each group in training and testing sessions were compared in all of the experiments to verify that there were no differences between groups in this parameter. Animals showing low exploration times were excluded from the experiments.
Surgery and drug infusion.
Under deep anesthesia (70 μl of ketamine 5% w/v and 20 μl of xylazine at 20 mg/ml), mice were implanted with 23-gauge guide cannulae 1 mm dorsal to their dorsal hippocampi at coordinates of anterior, −1.9; lateral, +/−1.2; and ventral, 1.2, in accordance with the atlas of Franklin and Paxinos (2001). Guide cannulae were fixed to the skulls with dental acrylic containing calcium hydroxide. Experiments were performed 3 d after surgery to ensure animal recovery. The injection device consisted of a 30-gauge cannula connected to a 5 μl Hamilton syringe with tubing. Initially, the infusion device was filled with distilled water and a small air bubble was sucked into the injection cannula, followed by the injection solution. The air bubble allowed for visual inspection of the injection progress. The injection cannula was inserted into the guide cannula with its tip extending beyond the guide by 1 mm so as to reach the dorsal hippocampus. The injections were administered by hand across a 30 s time period. The injection cannula was removed after 60 s to avoid reflux and to allow the diffusion of drugs. The volume of each intrahippocampal infusion was 0.5 μl/side. Different injection devices were used for drug and vehicle. The injections were performed immediately after the training session. After behavioral procedures, the animals were injected with black ink and decapitated. The brains were placed in 4% paraformaldehyde for 1 d, followed by 30% sucrose for an additional 24 h. To verify cannulae placement, frozen brains were sliced using a cryostat and analyzed with a magnifying glass. The deepest position of the needle was superimposed on serial coronal maps. Only data from animals with cannulae located in the intended sites were included in the analysis.
Drugs.
The κB-decoy (double-stranded DNA oligonucleotide 5′-GAGGGGACTTTCCCA-3′; consensus sequence in bold) and the m-decoy (5′-GAGGCGACTTTCCCA-3′; base changed underlined) (Albensi and Mattson, 2000) were dissolved in sodium chloride-Tris-EDTA solution. κB-decoy or m-decoy was used at a concentration of 0.47 μg/μl and administered bilaterally, delivering 0.26 pmol per side (Freudenthal et al., 2005). The HDAC inhibitor sodium butyrate (NaB; Sigma-Aldrich) was dissolved in saline solution and used at a concentration of 0.1 μg/μl. The HDAC inhibitor trichostatin A (TSA, Sigma-Aldrich) was dissolved in dimethylsulfoxide (DMSO) and used at 6 μg/μl (3.5 μg/g). The HAT inhibitor c646 (Sigma-Aldrich) was dissolved in DMSO and used at 2 μg/μl. Drug concentrations used were chosen from previous work (NaB from Korzus et al., 2004; Federman et al., 2009; TSA from Hawk et al., 2011; and c646 from Marek et al., 2011).
Histone-enriched extracts and Western blot assay.
The mice were killed by cervical dislocation immediately after training (see Results). The brains were rapidly removed, and both hippocampi were dissected according to the method described by Glowinski and Iversen (1966). All procedures were performed at 4°C. After dissection, the hippocampus was immediately placed in buffer A (10 mm HEPES, pH 7.8, 1.5 mm MgCl2, 10 mm KCl, 1 mm DDT, 5 mm sodium butyrate, 1 μg/ml pepstatin A, 10 μg/ml leupeptin, 0.5 mm PMSF, and 10 μg/ml aprotinin). Each hippocampus was then homogenized in buffer A with 12 strokes of a Teflon-glass homogenizer, rotating the pestle with a motor at 1800 rpm. The homogenate was centrifuged at 1000 × g for 15 min. The pellet was completely resuspended in buffer A, and H2SO4 was then added to a concentration of 0.4N. The homogenate was then incubated for 30 min at 4°C to extract the histones from the nuclear fraction. The solution was centrifuged for 5 min at 14,000 × g. The supernatant was transferred to a new tube and proteins were precipitated in 10 volumes of acetone at −20°C overnight. The nuclear basic-protein precipitate was collected by centrifugation at 14,000 × g for 15 min. The pellet was resuspended in distilled water. For Western blots, loading buffer was added and samples were incubated at 100°C for 5 min and then immediately placed on ice. Ten micrograms of protein from each sample were resolved on 15% SDS-PAGE. Proteins were electrotransferred to a PVDF membrane for detection by specific antibody binding. Acetyl-specific histone H3 antibody (acetylated K9 and K14), concentration 1:5000, and total anti-histone H3 antibody, concentration 1:25000 (Millipore) were used following the manufacturer's protocol. The detection was performed using a luminol chemiluminescence kit (Santa Cruz Biotechnology) following the manufacturer's protocol, and signals were digitized using an imaging device (Fuji Intelligent Dark Box II) and LAS-1000 software. The relative optic density was estimated using NIH ImageJ 1.29x software. The intensity of the bands was compared using the integrated density value, which represents the sum of gray values of the selected area minus background. A profile of the intensity values of gray for each band was made and the area of the histogram was measured to determine the optical density. Each membrane was analyzed with the two antibodies and the proportions of acetylated/total H3 were determined. The Student's t test or one-way ANOVA was used with a per comparison error rate (α) of 0.05 for statistical comparisons between groups.
Chromatin immunoprecipitation assay.
For the chromatin immunoprecipitation (ChIP) assay, each hippocampus was dissected, placed in 1.5 ml of 1.8% formaldehyde in PBS, and tissue was fixed for 20 min at room temperature. The reaction was stopped with glycine (final concentration 0.125 m) for 5 min at room temperature. After washing with PBS, tissue was homogenized in cell lysis buffer (5 mm PIPES, pH 8, 85 mm KCl, 0.5% NP40) supplemented with proteinase inhibitors (1 μg/ml pepstatin A, 10 μg/ml leupeptin, 0.5 mm PMSF, and 10 μg/ml aprotinin) with 12 strokes in a Dounce homogenizer, type B pestle. The homogenate was centrifuged for 5 min at 3000 × g. The pellet was resuspended in sonication lysis buffer (1% SDS, 1% Triton X-100, 10 mm EDTA, and 50 mm Tris-HCl pH 8.1) supplemented with proteinase inhibitors and incubated for 30 min on ice. Four hippocampi were pooled together and sonicated to generate DNA fragments with an average length of 600–700 bp, which were centrifuged for 10 min at 14,000 × g at 4°C. Supernatants were precleared with Protein G-Agarose (Invitrogen) adsorbed with sheared salmon sperm DNA (sSS-DNA; Eppendorf) and bovine serum albumin (Sigma-Aldrich). At this step, a portion of the precleared sample (∼5%) was stored and considered the “input” sample. Immunoprecipitation of acetyl H3-DNA or NF-κB-DNA complexes was achieved by incubating the precleared samples overnight at 4°C with 10 μg of acetyl-specific histone H3 antibody, acetylated K9 and K14 (Millipore), or 10 μg of NF-κB p65 (c-20) antibody (sc-372; Santa Cruz Biotechnology), respectively. A “non-antibody” immunoprecipitation was performed as a negative control. Samples were added to Protein G-Agarose/sSS-DNA and incubated for 2 h at 4°C. All samples were eluted with 1% SDS and 0.1 m NaHCO3, and the cross-links were reverted by overnight incubation with 0.2 m NaCl at 65°C. Proteins were digested with Proteinase K (Roche) and DNA was purified by phenol/chloroform extraction. The DNA was subjected to real-time PCR in 25 μl of reaction buffer containing 0.8 mm dNTPs, 2.5 units of TaqDNA polymerase (Invitrogen), and 10 μm concentrations of the primers specific for mouse Zif268 or the Camk2d promoter, respectively (κB site included), designed as follows: Zif268 forward: 5′-AACCCCGGCCTGACTCGCCCTC-3′, Zif268 reverse: 5′-CTGAGCGCGCGCGGGAGT-3′; Camk2d forward: 5′-GCACTTTTGGGTTTCATTATGTTAG-3′, Camk2d reverse: 5′-CGTCTTCGCCCTTCTCTCC-3′. For both promoters, PCRs were performed at 95°C for 15 s, 64°C for 20 s, and 72°C for 20 s, for a total of 39 cycles. The regulatory region of the Camk2d gene (−1000 to +1 bp) was screened using the TRANSFAC method (TRANSFAC 7.0 Public 2005; Matys et al., 2003) to identify a putative NF-κB-binding sequence. One target region, containing a κB consensus sequence at ∼−400 bp from the initiation site, was identified and used to make the primers for real-time PCR. The cumulative fluorescence for each amplicon was normalized to input amplification.
RNA extraction and gene expression analysis.
The mice were killed by cervical dislocation 3 h after training (see Results). The brains were rapidly removed, and both hippocampi were dissected. Total RNA was extracted using TRIzol LS reagent (Invitrogen). Each hippocampus was placed in 1 ml of TRIzol and tissue was homogenized with 15 strokes in a glass Dounce homogenizer, type A pestle. The samples were incubated for 5 min at room temperature and then 0.2 ml of chloroform was added. Tubes were shaken for 15 s, incubated for 3 min at room temperature, and then centrifuged at 12,000 × g for 15 min at 4°C. The aqueous phase from each sample was obtained and the RNA was precipitated by adding 0.5 ml of isopropyl alcohol. Samples were incubated at room temperature for 10 min and then centrifuged at 12,000 × g for 10 min at 4°C. RNA was washed, air-dried, and dissolved in RNase-free water and 5 μg of RNA was used for the retrotranscription reaction. The cDNA obtained was subjected to real-time PCR in 25 μl of reaction buffer containing 0.8 mm dNTPs, 2.5 units of TaqDNA polymerase (Invitrogen) and 10 μm concentrations of the primers specific for mouse βactin or Camk2d mRNA, designed as follows: βactin RNA forward: 5′-TCCTTCCTGCCTATGGAATC-3′, βactin RNA reverse: 5′-ACTCATCGTACTCCTGCTTG-3′, Camk2d RNA forward: 5′-TTCGGACACGGAAAGTGAGG-3′, Camk2d RNA reverse: 5′-TTCTCACCACTGAGAACGCC-3′. For both genes, PCRs were performed at 95°C for 15 s, 60°C for 20 s, and 72°C for 20 s for a total of 39 cycles. The cumulative fluorescence for each amplicon of each group of animals was normalized to the control habituation (HAB) group.
Results
Strong training induces a persistent recognition memory
NOR is a widely investigated form of declarative memory. It refers to the ability to determine whether a newly found item or object has been previously encountered (Squire et al., 2007). Initially, we assessed whether different training intensities could induce different memory persistence in an NOR task. In the first experiment, 3 groups of animals were formed: 1 group received a weak training of 3 min (TR3), another group received a standard training of 10 min (TR10), and the last group received a strong training of 15 min (TR15). All 3 groups were tested 24 h later. As shown in Figure 1a, 3 min of exploration yielded no memory retention, whereas 10 min induced memory retention. These results coincide with previous findings (Stefanko et al., 2009). The TR15 group had a similar discrimination index (DI%) as the TR10 group; this DI% was significantly higher than that of the TR3 group (one-way ANOVA, F(2,27) = 13.88, p < 0.01; Duncan test TR3 vs TR10, p < 0.01; TR3 vs TR15, p < 0.01; TR10 vs TR15, ns). Therefore, 10 min and 15 min of exploration rendered similar levels of retention 24 h after training. The percentage of time spent exploring the objects during training did not differ significantly between training conditions (percentage of time spent with left object: TR3, 50.5% ± 1.4; TR10, 52.4% ± 3.1; TR15, 49.4% ± 1.9; right object: TR3: 49.5 ± 1.4; TR10, 47.6 ± 3.1; TR15, 50.6 ± 1.9). Furthermore, the total times of exploration during testing were similar for all groups (average of total exploration time: TR3, 28.4 ± 3.3; TR10, 21.9 ± 5.1; TR15, 23.5 ± 3.7). Similarly, no differences in these parameters were noted in the other experiments (data not shown).
Figure 1.

Strong training induces a persistent recognition memory and histone acetylation increment during consolidation. a, Diagram outlining the experimental design. TR3 (n = 10), TR10 (n = 9), and TR15 (n = 9) groups received 3, 10, or 15 min of training, respectively. b, Graph representing the mean ± SEM of DI% for each group. **p < 0.01 in a one-way ANOVA, followed by the Duncan post hoc test. c, Diagram outlining the experimental design. TR3 (n = 11), TR10 (n = 11), and TR15 (n = 9) groups as in a and b, plus a nontrained HAB group (n = 9). Animals were killed 1 h after training. Graph represents the mean ± SEM of acetyl H3 levels in the hippocampus estimated by Western blot normalized to total H3 levels. *p < 0.05, one-way ANOVA, followed by the Duncan post hoc test.
In the second experiment, the same groups were formed, but they were tested 1 week after training. As shown in Figure 1b, the DI% of the TR3 and TR10 groups were similarly low and both differed significantly from the DI% of the TR15 group (one-way ANOVA, F(2,26) = 5.596, p < 0.01; Duncan test TR3 vs TR15, p < 0.01; TR10 vs TR15, p < 0.01; TR3 vs TR10, ns). These results indicated that the memories induced by standard and strong trainings differed in their persistence.
Only the consolidation of strong memory involves changes in the levels of hippocampal histone H3 acetylation
Next we performed an experiment aimed at investigating whether the different training conditions recruited histone acetylation mechanisms during memory consolidation. We determined the levels of acetylated histone H3 in the hippocampus 1 h after training. The experiment consisted of 3 groups trained with 3, 10, or 15 min of exploration (TR3, TR10, and TR15, respectively) and a fourth group that was placed in the experimental chamber without objects for 10 min, as in the 2 previous sessions of HAB. At 1 h after training, animals were killed, the hippocampi were dissected, and histone-enriched extracts were obtained. The levels of acetylated H3 histone relative to total H3 were assessed by Western blot. As shown in Figure 1c, only the TR15 group showed a significantly higher level of acetylated H3 with respect to the HAB group (one-way ANOVA F(3,38) = 2.852, p < 0.05; Duncan test p < 0.05). This result indicates that strong training induced a significant increase in hippocampal H3 acetylation 1 h after training. Conversely, such an increase was not induced after weak or standard training.
Administration of a HAT inhibitor in the hippocampus impairs long-lasting recognition memory
The HAT p300 plays a critical role in regulating gene expression, and its activity has been found to be required for memory formation (Oliveira et al., 2007; 2011; Marek et al., 2011). In the following experiments, we evaluated whether the inhibition of p300 in the hippocampus immediately after strong training impairs memory persistence. One group of mice was trained with 15 min of exploration and then injected with 2 μg of the p300 inhibitor c646 in each hippocampus (TR15+HATi). Another group received the same training and was injected with the vehicle DMSO (TR15+Veh). Both groups were tested 24 h after training. As shown in Figure 2a, left, no differences were identified between the groups (Student's t test; t = 0.83; p = 0.41). A second experiment was performed with the same groups, but the animals were tested 1 week after training. As shown in Figure 2a, right, significant differences were found when the DI% of the groups were compared (Student's t test; t = 3.85; p < 0.01). To evaluate the effect of c646 on histone acetylation, we performed an experiment with the same TR15+HATi and TR15+Veh groups that were trained and injected as in the previous experiments, but the animals were killed 1 h after training, the hippocampi were dissected, and histone-enriched extracts were obtained. The levels of acetylated H3 histone relative to the total H3 were assessed by Western blot. The results are shown in Figure 2b. The TR15+Veh group showed a significantly higher level of acetylated H3 than the TR15+HATi group. These results indicate that HATi administration impaired the increment of H3 acetylation in the hippocampus during the consolidation of strong training (Student's t test; t = −2.32; p < 0.05).
Figure 2.

Administration of a HAT inhibitor in the hippocampus impairs long-lasting recognition memory and the histone acetylation increment during consolidation. a, Diagram outlining the experimental design. Left: Mean ± SEM of DI% for the 24 h test of the TR15+HATi (n = 11) and TR15+Veh (n = 6) groups. Right: Mean ± SEM of DI% for the 7 d test of the TR15+HATi (n = 8) and TR15+Veh groups (n = 9). **p < 0.01, Student's t test. b, Diagram outlining the experimental design. Animals were killed 1 h after training (n = 6 per group). Graph represents the mean ± SEM of acetyl H3 levels in hippocampus estimated by Western blot normalized to total H3 levels. *p < 0.05 Student's t test. c, Example of cannula position (cresyl violet stained).
Administration of HDAC inhibitors in the hippocampus enhanced recognition memory
The results obtained in the first experiment and in previous studies using the NOR task showed that a training session of 3 min did not induce long-term memory. However, the posttraining injection of HDAC inhibitors enhanced this memory (Stefanko et al., 2009; Haettig et al., 2011). Nevertheless, in these studies, drug administration was intraperitoneal and no information regarding the sites and areas involved in memory enhancement was obtained. In the following experiment, we analyzed the effect of HDAC inhibition in the hippocampus on recognition memory. All animals were trained for 3 min, followed immediately by direct hippocampal injection of either the HDAC inhibitor sodium butyrate (TR3+NaB group) or saline (TR3+Veh group). Testing was performed 24 h later. Comparison of the DI% of the groups revealed significant differences (Fig. 3a, Student's t test; t = 3.895; p < 0.05). Therefore, the inhibition of HDAC in the hippocampus immediately after training enhanced object recognition memory. Together with the results of the previous section, this fact supports the notion that the hippocampus is a functional region for the consolidation of recognition memory.
Figure 3.

Administration of HDAC inhibitors in the hippocampus enhanced memory persistence by the induction of histone acetylation. a–c, Diagrams outlining the experimental design. a, Graph representing the mean ± SEM of DI% for the TR3+NaB (n = 7) and TR3+Veh (n = 8) groups. *p < 0.05, Student's t test. b, Graph representing the mean ± SEM of DI% for the TR3+TSA (n = 11) and TR3+Veh (n = 10) groups. **p < 0.01, Student's t test. c, Graph representing the mean ± SEM of DI% for the TR3+NaB (n = 7), TR3+Veh (n = 7), and TR15+Veh (n = 8) groups. *p < 0.05, one-way ANOVA, followed by a Duncan post hoc test. d, Graph representing the mean ± SEM of acetyl H3 levels in hippocampus estimated by Western blot normalized to total H3 levels. Animals were killed 1 h after training. *p < 0.05, Student's t test. n = 10.
In addition to its effect on the HDAC enzyme, NaB is also a putative inhibitor of chloride channels and phosphodiesterases (Resta-Lenert et al., 2001). Therefore, we evaluated the effect of another HDAC inhibitor, trichostatin A (TSA), which is structurally unrelated to NaB. A similar experiment was performed, but the hippocampi were injected with either TSA (TR3+TSA group) or the vehicle DMSO (TR3+Veh group) immediately after training. In agreement with the findings obtained with NaB, significant differences in DI% were found between the TSA and Veh groups (Student's t test; t = 3.68; p < 0.01; Fig. 3b). The results of the last two experiments support the idea that the hippocampal inhibition of HDAC enzymes enhances recognition memory after 24 h.
Weak memory enhanced by administration of an HDAC inhibitor persists as a strong-training memory
The following experiment investigated whether HDAC inhibition induced a persistent memory similar to the memory induced by strong training. The following 3 groups were established: 2 groups of animals were trained for 3 min, after which 1 group was injected with NaB (TR3+NaB group) and the other was injected with vehicle (TR3+NaB group). The third group of animals was trained for 15 min and injected with vehicle (TR15+Veh group). All of the injections were performed at the end of training and the mice were tested 1 week after training. As Figure 3c shows, the DI% of the Veh group differed significantly from those of both the TR3+NaB and the TR15+Veh groups (one-way ANOVA F(2,19) = 5.164, p < 0.05; Duncan test, p < 0.05). These results indicate that NaB-enhanced memory was as long lasting as the memory induced by strong training.
Consolidation of a weak memory facilitated by HDAC inhibition involves an increase in histone H3 acetylation
To obtain further evidence of the effect of HDAC inhibition on weak memory consolidation, we assessed whether NaB injection would induce an increase in histone H3 acetylation. Two groups were established: a weak-trained group injected with vehicle (TR3+Veh) and a weak-trained group injected with NaB (TR3+NaB). All animals were killed 1 h after training, the hippocampi were dissected, and histone-enriched extracts were obtained. Histone H3 acetylation was measured by Western blots and the results of these experiments are shown in Figure 3d. A significantly higher level of H3 acetylation was observed in the TR3+NaB group relative to the TR3+Veh group (Student's t test; t = 2.27; p < 0.05). This result suggests that HDAC inhibition enhances memory by increasing histone H3 acetylation during consolidation.
Hippocampal NF-κB activity is required for consolidation and persistence of object recognition memory
The regulation of chromatin structure is closely related to the function of transcription factor (TFs; Norton et al., 1989; Vettese-Dadey et al., 1996; Kouzarides, 2007). Specifically, the recruitment of HAT and HDAC enzymes is associated with the presence of TFs in the regulatory regions of target genes. NF-κB is a TF that plays a key role in neural plasticity and memory in both vertebrates and invertebrates (Romano et al., 2006; Alberini, 2009) and is an important regulator of synaptogenesis during development and in excitatory synapses of mature neurons (Boersma et al., 2011; Gutierrez and Davies, 2011). We studied the participation of NF-κB in the process of acetylation during NOR memory formation. We used a highly specific inhibitor of NF-κB, the κB-decoy, which was previously used in the in vivo inhibition of this TF in passive avoidance and fear conditioning tasks in mice (Freudenthal et al., 2005; Boccia et al., 2007; de la Fuente et al., 2011). The κB-decoy consists of a double-stranded DNA oligonucleotide of 15 bp that contains the NF-κB binding consensus sequence. The κB-decoy oligonucleotide binds to NF-κB and impedes its normal action at the genome sites. Delivery of the κB-decoy to the dorsal hippocampus at the concentration used here induced an inhibition of 60% of the basal NF-κB activity and blocked its training-induced activation (Boccia et al., 2007). A mutated κB-decoy (m-decoy) was used as a control. In this oligonucleotide, a G base is replaced by a C base, with the consequent replacement of C to G in the complementary strand. With this mutation, the oligonucleotide is not recognized by NF-κB, controlling the effect of DNA administration in the brain while maintaining the same overall composition of bases.
This experiment consisted of two groups of animals that received training for 15 min. After the training session, one group received the κB-decoy (TR15+Dec), whereas the other group was injected with m-decoy (TR15+mDec). Testing took place 24 h after training and the results are shown in Figure 4a. The group of animals injected with the κB-decoy had a DI% that was significantly lower than that of the control group injected with m-decoy (Student's t test; t = −2.7; p < 0.05). A similar experiment was then performed, but the mice were tested 7 d after training. As expected, the κB-decoy group exhibited significantly lower levels of discrimination than observed in the control group (Fig. 4b, Student's t test; t = 6.1; p < 0.05). These results suggest that the TF NF-κB is necessary in the hippocampus for NOR memory consolidation.
Figure 4.
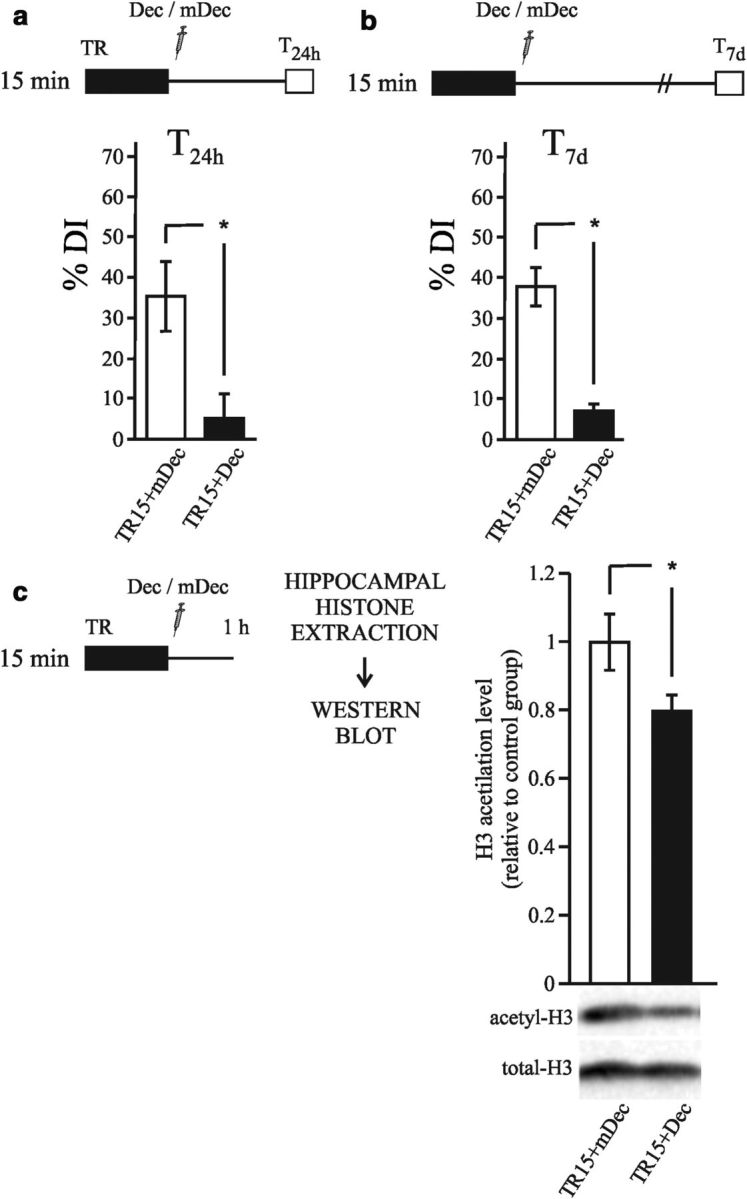
Hippocampal NF-κB activity is required for the histone acetylation increment during consolidation of object recognition memory. a–c, Diagrams outlining the experimental design. a, Graph representing the mean ± SEM of DI% for the TR15+Dec (n = 11) and TR15+mDec (n = 10) groups. *p < 0.05, Student's t test. b, Graph representing the mean ± SEM of DI% for the TR15+Dec (n = 7) and TR15+mDec (n = 7) groups. *p < 0.05). c, Graph representing the mean ± SEM of acetyl H3 levels in hippocampus estimated by Western blot normalized to total H3 levels. Animals were killed 1 h after training. *p < 0.05, Student's t test.
Inhibition of NF-κB reduces the levels of the histone H3 acetylation after strong training
In other cell types, activated NF-κB recruits CBP/p300 acetylases to the regulatory regions of different genes, contributing to the increase in histone acetylation (Vo and Goodman, 2001; Teferedegne et al., 2006; Hayden and Ghosh, 2008). In this experiment, we evaluated whether the inhibition of NF-κB reduces the increase in H3 acetylation observed in the hippocampus after strong training. Two groups of animals received 15 min of training and were then injected with either κB-decoy (TR15+Dec) or m-decoy (TR15+mDec). All animals were killed 1 h after training, the hippocampi were dissected, and histone-enriched extracts were obtained. Histone H3 acetylation levels were estimated by Western blots and the results are shown in Figure 4c. A significantly lower level of H3 acetylation was found in the TR15+Dec group compared with the TR15+mDec group (Student's t test; t = −2.32; p < 0.05). This result suggests that NF-κB contributes to the increase in the level of acetylation that is induced by the strong training, likely via CBP/p300 recruitment to specific gene promoters.
Acetylation of a promoter region that included an NF-κB consensus sequence is specifically increased after strong training
In the previous experiments, the general level of hippocampal histone H3 acetylation was analyzed using chromatin-enriched extracts. With this method, we identified increases in acetylation that were specifically related to the formation of long-lasting memory. In the last two sections, we obtained evidence supporting the role of NF-κB in this process. Next, to determine the level of histone acetylation in specific genome locations, we studied promoter regions of particular genes that are associated with neural plasticity and memory. We studied two genes that codify important proteins involved in memory formation: Zif268 and CaMKII. Zif268 (also known as Egr-1, Ngfi-A, Krox 24, Tis 8, and Zenk) is an immediate-early gene (Davis et al., 2003; Soulé et al., 2008) and calcium/calmodulin kinase II δ (Camk2d) is a late gene (Sirri et al., 2010; Lucchesi et al., 2011). In particular, we focused the analysis on promoter regions containing NF-κB-regulatory elements. Previous studies have reported that NF-κB regulates the Zif268 promoter (Carayol et al., 2006; Lubin and Sweatt, 2007). We analyzed the regulatory regions of the Camk2d gene and identified a bona fide NF-κB target region containing a κB consensus sequence at ∼−400 bp from the initiation site (see experimental procedures section; Fig. 5). This experiment consisted of three groups: two of the groups were trained with either 10 or 15 min of exploration (TR10 and TR15, respectively), whereas another group was placed in the experimental chamber without objects for the same length of time as the two previous sessions of HAB. One hour after training, animals were killed, and the hippocampi were then dissected. A ChIP assay was performed using the acetyl-specific H3 antibody combined with quantitative real-time PCR. No changes were identified at the level of acetylation at the Zif268 promoter for the TR10 and TR15 groups (Fig. 5a, one-way ANOVA ns, F(2,3) = 2.686, p = 0.21). Conversely, a 2.5-fold increase in H3 acetylation levels was found at the Camk2d promoter in the TR15 group relative to the levels observed in the HAB and the TR10 groups (one-way ANOVA F(2,3) = 19.257, p < 0.05; Duncan test HAB vs TR15, p < 0.05; TR10 vs TR15, p < 0.05). In the same promoter region, no changes in H3 acetylation were found for the TR10 group. Therefore, strong training triggered a significant increase in histone H3 acetylation levels in the NF-κB-regulatory region of the Camk2d promoter.
Figure 5.
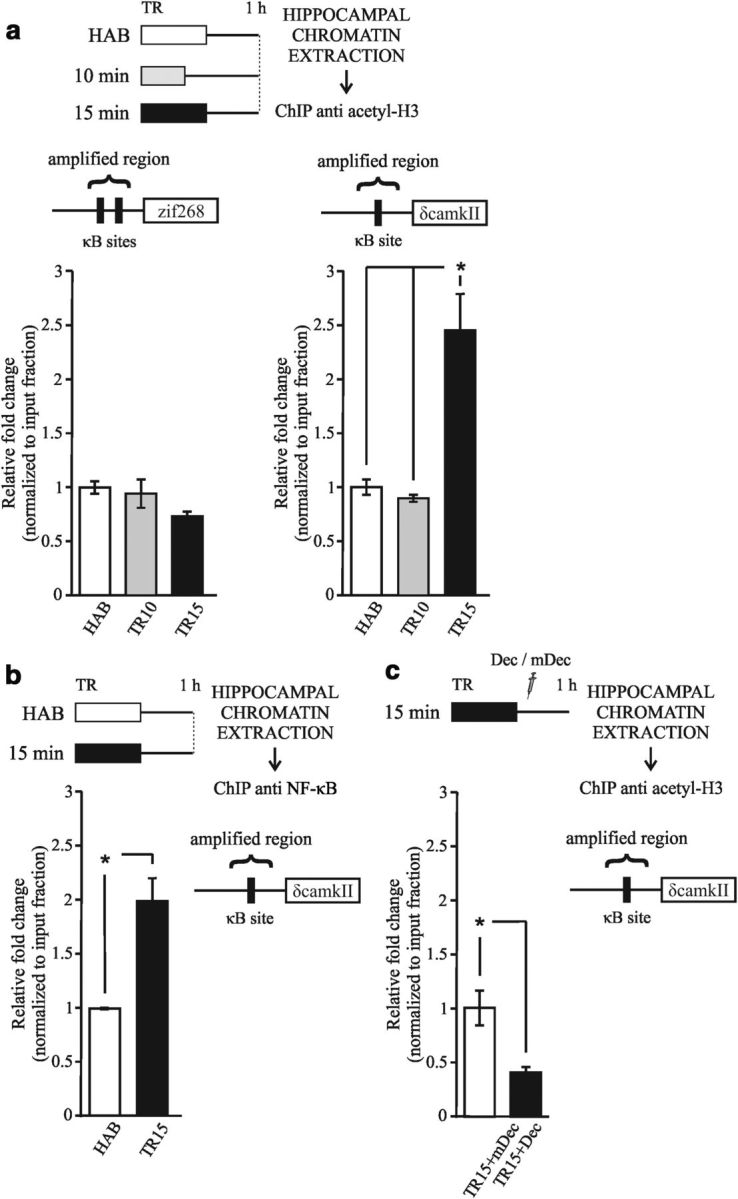
Acetylation of a promoter region that included an NF-κB consensus sequence is specifically increased after strong training. a, Diagram outlining the experimental design. Left upper diagram: NF-kB-binding sites identified within 1 kbp promoter upstream sequences of the Zif268 gene (GenBank Gene ID: 13653). Left graph: mean ± SEM of the fold change relative to input fraction of histone H3 acetylation at the Zif268 promoter in the hippocampus 1 h after training obtained from a ChIP assay. Right upper diagram: Bona fide NF-κB-binding site identified within 1 kbp promoter upstream sequences of the Camk2d gene (GenBank Gene ID: 108058). Right graph: Mean ± SEM of the fold change relative to input fraction of histone H3 acetylation at the Camk2d promoter in the hippocampus 1 h after training obtained from a ChIP assay. *p < 0.05, one-way ANOVA, followed by a Duncan post hoc test. For this study, 2 independent experiments were performed, with 4 animals per group. b, Diagram outlining the experimental design. Graph represents the mean ± SEM of the fold change relative to input fraction of p65 levels at the Camk2d promoter in the hippocampus 1 h after training obtained from ChIP assay. *p < 0.05, Student's t test. For this study, 2 independent experiments were performed with 4 animals per group. c, Diagram outlining the experimental design. Graph represents the mean ± SEM of the fold change relative to input fraction of H3 acetyl levels at the Camk2d promoter in the hippocampus 1 h after training and decoy injection obtained from ChIP assay. *p < 0.05, Student's t test. For this study, 2 independent experiments were performed with 4 animals per group.
To investigate whether NF-kB is in fact bound to Camk2d promoter during memory consolidation, we performed a new experiment using an antibody for the p65 protein instead of an acetyl-specific H3 antibody. P65 is one of the components of the NF-κB TF dimer. This assay revealed that p65 was present in this promoter region in the TR15 group but not in the HAB group (Fig. 5b, Student's t test; t = −4.80; p < 0.05). These findings suggest that the Camk2d gene promoter is regulated by NF-κB in this region and that NF-kB increases its interaction with the promoter after strong training. These results are also consistent with those of the previous experiments showing memory impairment with κB-decoy administration and suggest that NF-κB is implicated in histone acetylation during strong memory consolidation.
Inhibition of NF-κB reduces the level of histone acetylation at Camk2d promoter after strong training
We also investigated whether NF-κB inhibition affects histone H3 acetylation levels at the Camk2d gene promoter. Two groups of animals received 15 min of training and were then injected with either κB-decoy (TR15+Dec) or m-decoy (TR15+mDec) (Fig. 5c). All animals were killed 1 h after training and the hippocampi were dissected. A ChIP assay was performed using the acetyl-specific H3 antibody combined with quantitative real-time PCR. The results show a decrease in the histone H3 acetylation level at Camk2d promoter after decoy administration (TR15+Dec group) during strong memory consolidation (Student's t test; t = 3.534; p < 0.05) compared with the control group (TR15+mDec group). This result supports that NF-κB-dependent histone acetylation is specifically involved in persistent forms of memory.
Camk2d expression is specifically induced after strong training
CaMKII is a multifunctional serine/threonine kinase with a broad range of substrates. In mammals, CaMKII comprises multiple isoforms that are the product of four closely related genes: α, β, γ, and δ (Gaertner et al., 2004). The α and β isoforms are primarily localized in the brain, whereas γ and δ isoforms are ubiquitously expressed. αCaMKII is highly expressed in the forebrain, where it constitutes the major postsynaptic density protein (Cheng et al., 2006), and this isoform has been studied extensively in relation to memory formation (Elgersma et al., 2004; Irvine et al., 2006). However, little is known about the function of βCaMKII in the brain and virtually nothing is known about the γ and δ isoforms. Therefore, we next investigated whether δCaMKII expression is in fact induced during strong memory consolidation. The experiments consisted of three groups: two of the groups were trained with either 10 or 15 min of exploration (TR10 and TR15, respectively), whereas another group was placed in the experimental chamber without objects for the same length of time as the two previous sessions of HAB. Three hours after training, animals were killed, the hippocampi were dissected, and mRNA extraction and gene expression analysis were performed. As shown in Figure 6, we found a significant increment in the δCaMKII mRNA level 3 h after strong training compared with the TR10 and control groups (one-way ANOVA F(2,19) = 4.394, p < 0.05; Duncan test HAB vs TR15, p < 0.05; TR10 vs TR15, p < 0.05). This result shows that δCaMKII expression is only induced during consolidation of a persistent form of memory.
Figure 6.

Camk2d expression is specifically induced after strong training. Diagram outlines the experimental design. Graphs represent the mean ± SEM of gene expression levels in hippocampus of Camk2d (left) and βactin (right) measured by real-time PCR for each group (HAB, TR10, and TR15). Animals were killed 3 h after training. *p < 0.05, one-way ANOVA, followed by a Duncan post hoc test.
Discussion
Covalent modifications at the regulatory tail of histones, such as the acetylation of lysine residues, have been implicated in the formation of long-term memory (Levenson and Sweatt, 2006; Barrett and Wood, 2008; Roth and Sweatt, 2009). In this work, we obtained evidence from the NOR task indicating that histone acetylation plays a specific role in the formation of more persistent forms of memory. Initially, we found that the general level of H3 acetylation was increased in the hippocampus only during the consolidation of a memory that is patently present 1 week after training. In contrast, no increases in acetylation were detected after training conditions that rendered no retention after 7 d. We then found that the increases in H3 acetylation were reversed by the inhibition of NF-κB, a TF that plays a key role in memory formation (Meffert and Baltimore, 2005; Romano et al., 2006; Alberini, 2009). The involvement of this TF in the process of histone acetylation was further evidenced by analyzing a regulatory region of the Camk2d gene promoter. This promoter region is a bona fide NF-κB site and we detected the presence of p65 in this region during memory consolidation using ChIP assays. In agreement with the observed increases in the general acetylation levels, H3 acetylation increased in this particular region only during the consolidation of a more persistent memory. Furthermore, we observed that the inhibition of NF-kB reverted the acetylation state of this promoter. The last finding provides a direct evidence of NF-kB-dependent epigenetic regulation specifically associated with memory persistence. Accordingly, we found that Camk2d mRNA levels are specifically induced after strong training. These data support that Camk2d gene is specifically expressed in more persistent forms of memory.
The manipulation of HAT and HDAC activities in the hippocampus by means of specific inhibitors elicited additional supportive results. HAT inhibition impaired memory persistence after strong training but did not impair retention after 24 h. In agreement with this finding, the administration of the drug blocked the general increase in acetylation that was induced by the strong training. Conversely, the inhibition of HDAC activity in the hippocampus allowed a weak memory to persist for at least 1 week. This behavioral finding is consistent with the concomitant increase in H3 acetylation that was induced by the HDAC inhibitors. These results suggest that a shift in the regulatory balance that favors acetylation is sufficient to render a memory more persistent.
Treatment with HDAC inhibitors not only induced memory enhancement after 7 d, but also caused memory enhancement 24 h after training. Pharmacological manipulation could not only increase the acetylation of histones, but also increased the acetylation of other target proteins such as TFs. Specifically, NF-κB is a substrate for these enzymes (Sterner and Berger, 2000) and its acetylation has been shown to regulate the transcriptional activity of this TF (Chen and Greene, 2004). The inhibition of HDACs has been shown to increase the activity of NF-κB during memory enhancement by increasing its acetylated status (Yeh et al., 2004). The action on these alternative targets could explain the fact that memory facilitation by HDAC inhibition is not restricted to long-lasting memory, but also enhances 1 d memory.
The effects of the experimental manipulations used in this work, such as the varying intensities/durations of training and the actions of the administered drugs, can be described in terms of the level and quality of transcription attained during consolidation in each case. Weak training is unable to induce a sufficient amount of gene transcription, resulting in the production of a weak memory. The enhancement of gene transcription induced by HDAC inhibitors may enable a sufficient amount of mRNA synthesis to occur, transforming a weak memory trace into a strong one. An intermediate level of training may induce the transcription of target genes that does not require histone acetylation. Zif268, an immediate-early gene, is possibly an example of a gene that is ready for transcription in these training conditions. We did not find changes in the level of acetylation in the Zif268 promoter after strong training. Such transcriptional activity should be enough to induce long-term memory that is present 1 d after training but that decays in the following days. However, this does not dismiss the role of Zif268 in NOR memory consolidation, as described previously (Jones et al., 2001). In fact, preliminary results of our group shows an increase in Zif268 expression at the level of mRNA and protein after NOR training, which is dependent on NF-kB activity (Zalcman et al., manuscript in preparation). In contrast, strong training is able to recruit epigenetic mechanisms during consolidation, allowing for the expression of genes that are not directly available for transcription. Camk2d is possibly an example of a late gene that is normally less accessible for transcription and requires epigenetic mechanisms to reach the necessary level of expression for the formation of an enduring memory. Similarly, in other cellular systems, two forms of NF-κB transcriptional regulation were found in gene promoters of macrophage cells stimulated by LPS (Saccani et al., 2001). One of these forms is caused by the recruitment of NF-κB to immediately accessible genes and the other corresponds to the recruitment of NF-κB to a set of promoters that require histone acetylation to make them accessible to the transcriptional machinery. Consistent with the interpretation that the level and quality of transcription determine the persistence of memory, we found that the inhibition of HAT or NF-κB during consolidation indeed impaired NOR memory persistence.
It is important to note that our study was focused on epigenetic mechanisms involved in gene expression during consolidation around the first hour after memory acquisition. The quality and quantity of transcription during memory consolidation defines, at least in part, memory persistence. The kinetics of the general acetylation state of the chromatin and the acetylation of the particular promoter studied here was not analyzed in the present work. However, on the basis of previous studies, we expected that the increments in chromatin acetylation should be transient, returning to the basal levels at the end of the consolidation process (Chwang et al., 2006; Federman et al., 2009). Our findings do not rule out further processes that could be involved in memory persistence, such as subsequent recurrent reactivations of the trace that involve BDNF synthesis and releasing (Bekinschtein et al., 2007; Rossato et al., 2009).
The experiments in Figure 1 and Figure 2 showed that increased histone acetylation is necessary for memory to persist 7 d, but is not necessary for 1 d memory trace. This increment in acetylation is dependent, at least in part, on NF-kB activity (Fig. 4 and Fig. 5). However, the inhibition of NF-kB induced memory impairment at 24 h (Fig. 4). The last result is indicative that NF-kB plays a role in memory formation beyond the regulation of chromatin acetylation. In fact, we found that a NF-kB-regulated promoter region of Zif268 gene was not acetylated even after a strong training. As mentioned before, some genes, such as Zif268, would be readily available for transcription even in training conditions that do not induce histone acetylation. Such acetylation-independent transcriptional activity of NF-kB in these genes may be necessary to warrant a memory that is present 1 d after training but that decays before 1 week.
CaMKII is an abundant synaptic signaling molecule that is essential for both memory formation and for the induction of synaptic potentiation. In addition, CaMKII plays a prominent role in synaptic tagging and metaplasticity (Lucchesi et al., 2011). In mammals, CaMKII exists in multiple isoforms that are the product of four closely related genes: α, β, γ, and δ. Little information is known about the role of the δ CaMKII. The gene was expressed in a late manner, with an increase in its messenger RNA in mouse hippocampus 6 h after trace fear conditioning (Sirri et al., 2010). In addition, the protein exhibited higher levels of expression in the amygdala of a transgenic mouse with Zif268 overexpression (Baumgärtel et al., 2009). In this study, we found that histone acetylation occurred in a promoter region of the Camk2d gene only after strong training, and that only high levels of training were able to recruit the epigenetic mechanisms necessary for the expression of this gene. Ongoing experiments are aimed at determining the particular role of δ CaMKII isoform in memory persistence.
Learned information is stored in the nervous system, at least in part through changes in synaptic connections. The TF NF-κB has been involved in the upregulation of the structural plasticity of dendritic spines and of synaptic density in both developing (Gutierrez and Davies, 2011) and matured neurons after neuronal excitability and activation of modulatory pathways (Boersma et al., 2011). Accordingly, it was found that sustained HDAC inhibition induced the sprouting of dendrites and an increased number of synapses; it also reinstated learning capacity and promoted retrieval of long-term memory in mice, even after massive neuronal loss (Fischer et al., 2007). In contrast, the neuron-specific overexpression of HDAC2 decreased dendritic spine density, synapse numbers, synaptic plasticity, and memory formation (Guan et al., 2009). Consistent with these previous data, this study found that NF-κB inhibition significantly reduced the increase in histone acetylation after strong training. This finding suggests a possible link between epigenetic mechanisms and the induction of synaptogenesis and synaptic reinforcement. In this way, histone acetylation may promote the stability of synaptic changes during long-lasting memory consolidation.
Some studies initially suggested that the hippocampus does not participate in the NOR task, but other areas of the temporal lobe do (e.g., the insular cortex; Brown and Xiang, 1998; Eldridge et al., 2000). More recent studies are controversial, with some reports supporting the notion that the hippocampus is not involved (Bermudez-Rattoni et al., 2005; Balderas et al., 2008) and others supporting the idea that the hippocampus is indeed a functional area for this task (Kelly et al., 2003; Rossato et al., 2007; Squire et al., 2007; Broadbent et al., 2009; Clarke et al., 2010). The experimental results presented here indicate that the hippocampus participates in object recognition memory. The inhibition of HAT and NF-κB in the hippocampus immediately after learning prevented the formation of long-term recognition memory, whereas the inhibition of HDAC in the hippocampus after weak training enhanced recognition memory. We also found that strong training induced an increase in the levels of histone H3 acetylation in the hippocampus 1 h after training. These results support that the hippocampus participates in both the encoding and storage of recognition memory.
These data coincide with results obtained by our group in an invertebrate, the crab Chasmagnathus (Federman et al., 2009; Federman et al., 2012), in which we found that histone acetylation is necessary for the formation of stronger memories. All of this evidence suggests that the molecular mechanisms described here are not specific to the rodent brain, but are conserved mechanisms in both vertebrates and invertebrates.
According to some reports, there is a constant proportion of neurons recruited to a memory trace, regardless of the training intensity used (Repa et al., 2001; Jin-Hee Han et al., 2007). Providing that a similar process is induced in the NOR task, the increase in the level of acetylation found in this study would not be due to an increase in the number of neurons recruited to the memory trace, but to the recruitment of HAT activity in the same neurons.
The process of histone acetylation is a regulatory mechanism for transcription induction that exhibits slow dynamics. This mechanism would be able to more accurately control the quality and quantity of gene expression than other faster and less-specific mechanisms. Adequate training conditions or the action of modulatory pathways can induce the acetylation of promoter regions of memory-related genes, a property that might define the persistence of memory over time.
Footnotes
This work was supported by FONCYT PICT 2049, FONCYT PICT 1482, Argentina; UBACYT X198, Universidad de Buenos Aires; and PIP 5466, Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina. We thank Marcelo Wood for help in some experiments and helpful discussions, Angel Vidal for technical assistance, and Ruth Barrett and Ignacio Schor for help in some of the experiments.
The authors declare no competing financial interests.
References
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Alberini CM. Transcription factors in long term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balderas I, Rodriguez-Ortiz CJ, Salgado-Tonda P, Chavez-Hurtado J, McGaugh JL, Bermudez-Rattoni F. The consolidation of object and context recognition memory involves different regions of the temporal lobe. Learn Mem. 2008;15:618–624. doi: 10.1101/lm.1028008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15:460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RM, Malvaez M, Kramar E, Matheos DP, Arrizon A, Cabrera SM, Lynch G, Greene RW, Wood MA. Hippocampal focal knockout of CBP affects specific histone modifications, long-term potentiation, and long-term memory. Neuropsychopharmacology. 2011;36:1545–1556. doi: 10.1038/npp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgärtel K, Tweedie-Cullen RY, Grossmann J, Gehrig P, Livingstone-Zatchej M, Mansuy IM. Changes in the proteome after neuronal zif268 overexpression. J Proteome Res. 2009;8:3298–3316. doi: 10.1021/pr801000r. [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, Medina JH. Persistence of long-term memory storage requires a late protein synthesis- and BDNF- dependent phase in the hippocampus. Neuron. 2007;53:261–277. doi: 10.1016/j.neuron.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Bermudez-Rattoni F, Okuda S, Roozendaal B, McGaugh JL. Insular cortex is involved in consolidation of object recognition memory. Learn Mem. 2005;12:447–449. doi: 10.1101/lm.97605. [DOI] [PubMed] [Google Scholar]
- Boccia M, Freudenthal R, Blake M, de la Fuente V, Acosta G, Baratti C, Romano A. Activation of hippocampal nuclear factor-kappa B by retrieval is required for memory reconsolidation. J Neurosci. 2007;27:13436–13445. doi: 10.1523/JNEUROSCI.4430-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK. A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis. J Neurosci. 2011;201:5414–5425. doi: 10.1523/JNEUROSCI.2456-10.2011. 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T. A mouse model of Rubinstein-Taybi syndrome: Defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci U S A. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent NJ, Gaskin S, Squire LR, Clark RE. Object recognition memory and the rodent hippocampus. Learn Mem. 2009;17:5–11. doi: 10.1101/lm.1650110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MW, Xiang JZ. Recognition memory: neuronal substrates of the judgement of prior occurrence. Prog Neurobiol. 1998;55:149–189. doi: 10.1016/S0301-0082(98)00002-1. [DOI] [PubMed] [Google Scholar]
- Carayol N, Chen J, Yang F, Jin T, Jin L, States D, Wang CY. A dominant function of IKK/NF-kappaB signaling in global lipopolysaccharide-induced gene expression. J Biol Chem. 2006;281:31142–31151. doi: 10.1074/jbc.M603417200. [DOI] [PubMed] [Google Scholar]
- Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Mol Cell Biol Rev. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, Sheng M, Peng J. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics. 2006;5:1158–1170. doi: 10.1074/mcp.D500009-MCP200. [DOI] [PubMed] [Google Scholar]
- Chwang WB, O'Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JR, Cammarota M, Gruart A, Izquierdo I, Delgado-García JM. Plastic modifications induced by object recognition memory processing. Proc Natl Acad Sci U S A. 2010;107:2652–2657. doi: 10.1073/pnas.0915059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Bozon B, Laroche S. How necessary is the activation of the immediate early gene zif268 in synaptic plasticity and learning? Behav Brain Res. 2003;142:17–30. doi: 10.1016/S0166-4328(02)00421-7. [DOI] [PubMed] [Google Scholar]
- de la Fuente V, Freudenthal R, Romano A. Reconsolidation or extinction: transcription factor switch in the determination of memory course after retrieval. J Neurosci. 2011;31:5562–5573. doi: 10.1523/JNEUROSCI.6066-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge LL, Knowlton BJ, Furmanski CS, Bookheimer SY, Engel SA. Remembering episodes: a selective role for the hippocampus during retrieval. Nat Neurosci. 2000;3:1149–1152. doi: 10.1038/80671. [DOI] [PubMed] [Google Scholar]
- Elgersma Y, Sweatt JD, Giese KP. Mouse genetic approaches to investigating calcium/calmodulin-dependent protein kinase II function in plasticity and cognition. J Neurosci. 2004;24:8410–8415. doi: 10.1523/JNEUROSCI.3622-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federman N, Fustiñana MS, Romano A. Histone acetylation is recruited in consolidation as a molecular feature of stronger memories. Learn Mem. 2009;16:600–606. doi: 10.1101/lm.1537009. [DOI] [PubMed] [Google Scholar]
- Federman N, Fustiñana MS, Romano A. Reconsolidation involves histone acetylation depending on the strength of the memory. Neuroscience. 2012;219:145–156. doi: 10.1016/j.neuroscience.2012.05.057. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. London: Academic; 2001. [Google Scholar]
- Freudenthal R, Boccia MM, Acosta GB, Blake MG, Merlo E, Baratti CM, Romano A. NF-kappaB transcription factor is required for inhibitory avoidance long-term memory in mice. Eur J Neurosci. 2005;21:2845–2852. doi: 10.1111/j.1460-9568.2005.04126.x. [DOI] [PubMed] [Google Scholar]
- Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, Waxham MN. Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:12484–12494. doi: 10.1074/jbc.M313597200. [DOI] [PubMed] [Google Scholar]
- Glowinski J, Iversen LL. Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J Neurochem. 1966;13:655–669. doi: 10.1111/j.1471-4159.1966.tb09873.x. [DOI] [PubMed] [Google Scholar]
- Gräff J, Woldemichael BT, Berchtold D, Dewarrat G, Mansuy IM. Dynamic histone marks in the hippocampus and cortex facilitate memory consolidation. Nat Commun. 2012;3:991. doi: 10.1038/ncomms1997. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–63. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long term memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/S0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez H, Davies AM. Regulation of neural process growth, elaboration and structural plasticity by NF-κB. Trends Neurosci. 2011;34:316–325. doi: 10.1016/j.tins.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haettig J, Stefanko DP, Multani ML, Figueroa DX, McQuown SC, Wood MA. HDAC inhibition modulates hippocampus-dependent long-term memory for object location in a CBP-dependent manner. Learn Mem. 2011;18:71–79. doi: 10.1101/lm.1986911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, Josselyn SA. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- Hawk JD, Florian C, Abel T. Post-training intrahippocampal inhibition of class I histone deacetylases enhances long-term object-location memory. Learn Mem. 2011;18:367–370. doi: 10.1101/lm.2097411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Irvine EE, von Hertzen LS, Plattner F, Giese KP. alphaCaMKII autophosphorylation: a fast track to memory. Trends Neurosci. 2006;29:459–465. doi: 10.1016/j.tins.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;3:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- Kelly A, Laroche S, Davis S. Activation of mitogen activated protein kinase/extracellular signal regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J Neurosci. 2003;12:5354–5360. doi: 10.1523/JNEUROSCI.23-12-05354.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their functions. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms: a common theme in vertebrate and invertebrate memory formation. Cell Mol Life Sci. 2006;63:1009–1016. doi: 10.1007/s00018-006-6026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Sweatt JD. The IkB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. 2007;55:942–957. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi W, Mizuno K, Giese KP. Novel insights into CaMKII function and regulation during memory formation. Brain Res Bull. 2011;85:2–8. doi: 10.1016/j.brainresbull.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Marek R, Coelho CM, Sullivan RK, Baker-Andresen D, Li X, Ratnu V, Dudley KJ, Meyers D, Mukherjee C, Cole PA, Sah P, Bredy TW. Paradoxical enhancement of fear extinction memory and synaptic plasticity by inhibition of the histone acetyltransferase p300. J Neurosci. 2011;31:7486–7491. doi: 10.1523/JNEUROSCI.0133-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Münch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Norton VG, Imai BS, Yau P, Bradbury EM. Histone acetylation reduce nucleosome core particle linking number change. Cell. 1989;108:449–457. doi: 10.1016/0092-8674(89)90920-3. [DOI] [PubMed] [Google Scholar]
- Oliveira AM, Wood MA, McDonough CB, Abel T. Transgenic mice expressing an inhibitory truncated form of p300 exhibit long-term memory deficits. Learn Mem. 2007;14:564–572. doi: 10.1101/lm.656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AM, Estévez MA, Hawk JD, Grimes S, Brindle PK, Abel T. Subregion-specific p300 conditional knock-out mice exhibit long-term memory impairments. Learn Mem. 2011;18:161–169. doi: 10.1101/lm.1939811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE. Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci. 2001;4:724–731. doi: 10.1038/89512. [DOI] [PubMed] [Google Scholar]
- Resta-Lenert S, Truong F, Barrett KE, Eckmann L. Inhibition of epithelial chloride secretion by butyrate: role of reduced adenylyl cyclase expression and activity. Am J Physiol Cell Physiol. 2001;281:C1837–1849. doi: 10.1152/ajpcell.2001.281.6.C1837. [DOI] [PubMed] [Google Scholar]
- Romano A, Freudenthal R, Merlo E, Routtenberg A. Evolutionarily-conserved role of the NF-kappaB transcription factor in neural plasticity and memory. Eur J Neurosci. 2006;24:1507–1516. doi: 10.1111/j.1460-9568.2006.05022.x. [DOI] [PubMed] [Google Scholar]
- Rossato JI, Bevilaqua LR, Myskiw JC, Medina JH, Izquierdo I, Cammarota M. On the role of hippocampal protein synthesis in the consolidation and reconsolidation of object recognition memory. Learn Mem. 2007;14:36–46. doi: 10.1101/lm.422607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossato JI, Bevilaqua LR, Izquierdo I, Medina JH, Cammarota M. Dopamine controls persistence of long-term memory storage. Science. 2009;325:1017–1020. doi: 10.1126/science.1172545. [DOI] [PubMed] [Google Scholar]
- Roth TL, Sweatt JD. Regulation of chromatin structure in memory formation. Curr Opin Neurobiol. 2009;19:336–342. doi: 10.1016/j.conb.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G. Two waves of nuclear factor kappaB recruitment to target promoters. J Exp Med. 2001;193:1351–1359. doi: 10.1084/jem.193.12.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirri A, Bianchi V, Pelizzola M, Mayhaus M, Ricciardi-Castagnoli P, Toniolo D, D'Adamo P. Temporal gene expression profile of the hippocampus following trace fear conditioning. Brain Res. 2010;1308:14–23. doi: 10.1016/j.brainres.2009.10.049. [DOI] [PubMed] [Google Scholar]
- Soulé J, Penke Z, Kanhema T, Alme MN, Laroche S, Bramham CR. Object-place recognition learning triggers rapid induction of plasticity-related immediate early genes and synaptic proteins in the rat dentate gyrus. Neural Plast. 2008:269097. doi: 10.1155/2008/269097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, Wixted JT, Clark RE. Recognition memory and the medial temporal lobe: a new perspective. Nat Rev Neurosci. 2007;8:872–883. doi: 10.1038/nrn2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA. Modulation of long term memory for object recognition via HDAC inhibition. Proc Natl Acad Sci U S A. 2009;106:9447–9452. doi: 10.1073/pnas.0903964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/MMBR.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Teferedegne B, Green MR, Guo Z, Boss JM. Mechanism of action of a distal NF-kappaB-dependent enhancer. Mol Cell Biol. 2006;26:5759–5770. doi: 10.1128/MCB.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vettese-Dadey M, Grant PA, Hebbes TR, Crane-Robinson C, Allis CD, Workman JL. Acetylation of histone H4 plays a primary role in enhancing transcrption factor binding to nucleosomal DNA in vitro. EMBO J. 1996;15:2508–2518. [PMC free article] [PubMed] [Google Scholar]
- Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SH, Lin CH, Gean PW. Acetylation of nuclear factor-kB in rat amygdala improves long-term but not short-term retention of fear memory. Mol Pharmacol. 2004;65:1286–1292. doi: 10.1124/mol.65.5.1286. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Fan L, Fortress AM, Boulware MI, Frick KM. Hippocampal histone acetylation regulates object recognition and the estradiol-induced enhancement of object recognition. J Neurosci. 2012;32:2344–2351. doi: 10.1523/JNEUROSCI.5819-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]