

Abstract
During mammalian cortical development, division of progenitor cells occurs at the apical ventricular zone. Apical complex proteins and adherens junctions regulate the different modes of division. Here, we have identified the membrane-associated guanylate kinase protein membrane palmitoylated protein 3 (MPP3) as an essential protein for the maintenance of these complexes. MPP3 localizes at the apical membrane in which it shows partial colocalization with adherens junction proteins and apical proteins. We generated Mpp3 conditional knock-out mice and specifically ablated Mpp3 expression in cortical progenitor cells. Conditional deletion of Mpp3 during cortical development resulted in a gradual loss of the apical complex proteins and disrupted adherens junctions. Although there is cellular disorganization in the ventricular zone, gross morphology of the cortex was unaffected during loss of MPP3. However, in the ventricular zone, removal of MPP3 resulted in randomization of spindle orientation and ectopically localized mitotic cells. Loss of MPP3 in the developing cortex resulted in delayed migration of progenitor cells, whereas the rate of cell division and exit from the cell cycle was not affected. This resulted in defects in cortical stratification and ectopically localized layer II–IV pyramidal neurons and interneurons. These data show that MPP3 is required for maintenance of the apical protein complex and adherens junctions and for stratification and proper migration of neurons during the development of the cortex.
Introduction
Mammalian cortical development is a highly regulated process, in which cell division occurs at the apical side in the ventricular zone (Lehtinen and Walsh, 2011). Progenitor cells undergo symmetric division, after which the daughter cells stay as progenitors at the ventricular zone, or asymmetric division, after which one of the daughter cells relocates to the basal surface along radial glia and differentiates into a specific type of neuron (Zhong and Chia, 2008). There is a delicate balance between maintaining the progenitor pool via self-renewal and movement toward the basal surface and differentiation. Already subtle disturbances in this balance can have a dramatic impact resulting in aberrant cortex formation, defects in cortical layering, and cortical connectivity and affected behavior.
At the ventricular zone, adherens junctions are vital to maintain cellular integrity and play an important role in determining the mode of division and interkinetic nuclear migration, and their disruption affects cell migration and layering patterns (Cappello et al., 2006; Imai et al., 2006; Kadowaki et al., 2007). Furthermore, adherens junction proteins are involved in determining proliferation and pool size of progenitor cells (Costa et al., 2008; Sottocornola et al., 2010). Besides adherens junction proteins, there is another protein complex important in cortical development, the apical protein complex (Lehtinen and Walsh, 2011). The core protein in this complex, PALS1, is essential in proper cortical development. Conditional deletion of Pals1 resulted in disrupted apical protein complex and adherens junctions. These mice show a near total lack of the neocortex as a result of premature exit of cells from the cell cycle (Kim et al., 2010). Thus, disruption of these complexes clearly affects proper cortical development.
PALS1, also known as MPP5, is a membrane-associated guanylate kinase protein (MAGUK), belonging to the membrane palmitoylated protein (MPP) family, consisting of seven members. All MPP proteins have a guanylate kinase, postsynaptic density-95/Discs large/zona occludens-1, and Src homology 3 domain and, except for MPP1, contain a tandem L27 domain (Gosens et al., 2008). These MPP proteins are scaffolding proteins that can interact with several other proteins, including interactions with family members. In the retina, PALS1 interacts with MPP1, MPP3, and MPP4 and is the core protein of the apical Crumbs complex (Kantardzhieva et al., 2005,2006; Gosens et al., 2007; Park et al., 2011). In addition to interaction with PALS1, MPP3 also interacts with the Nectin1 adherens junction protein (Dudak et al., 2011).
Here, we show that MPP3 is expressed during cortical development and partially colocalized with PALS1 and with adherens junction proteins. To further address the role of MPP3 in the developing cortex, we removed MPP3 specifically in the developing cortex. Removal of MPP3 results in a loss of the apical protein complex and a disruption of adherens junctions. Furthermore, we show that, although cell division is not affected, loss of MPP3 results in delayed migration of progenitor cells and ectopically localized late-born neurons in adult cortex. As such, we hypothesize that MPP3 plays an important role as a link between the apical protein complex and adherens junctions in the developing cortex.
Materials and Methods
Generation of Mpp3 conditional knock-out mice.
To generate Mpp3 conditional knock-out (cKO) mice, a targeting construct was generated with an flippase recognition target (FRT) flanked Neomycin cassette positioned between exons 6 and 7 of the Mpp3 gene and flanked by 10 and 4 kb 5′ and 3′ homology arms, respectively. The FRT–Neomycin cassette also contained a loxP site, and a loxP site was introduced in the 5′ homology arm 2 kb upstream of the third exon of Mpp3, which is the exon containing the start codon. The targeting vector was linearized with NotI and NruI and electroporated in 129Ola embryonic day 14 (E14) embryonic stem cells. Neomycin-resistant embryonic stem cells were tested with Southern blotting to confirm homologous recombination. The 5′ and 3′ probes were generated using primer pairs 5′-GCTCATTGACACTTCACTAGAGACC-3′, 5′-GTAGGCACACACATATATACGC-3′ and 5′-CCCAGAACCCATGTAAAAACC-3′, 5′-CAGGTATACAGATATATGGGTACACGG-3′, respectively. For Southern blotting with the 5′ and 3′ probes, genomic DNA was digested with EcoRV or AseI for the 5′ and 3′ probes, respectively. A positive embryonic stem cell clone was used for blastocyst injections, and this resulted in two male chimeric mice that gave germ-line transmission of the targeted allele. The Neomycin cassette was successfully removed by crossing the Mpp3F/+ mice with a transgenic mouse that expressed flippase recombinase (FLP) in the germ line [129S4/SvJaeSor–t(ROSA)26Sortm1(FLP1)Dym/J mice; The Jackson Laboratory]. All mice were maintained as a cross of C57BL/6JOlaHsd and 129/Ola (50%/50%). To conditionally delete Mpp3 in the developing cortex, Emx1Cre mice, maintained as a cross of C57BL/6JOlaHsd and 129/Ola (50%/50%), were used for Cre-mediated recombination. Genotyping of Mpp3 mice was done by PCR on genomic DNA. For genotyping, the following primers were used: 5′-ATGCAAGTGCAGGCATCTGCTG-3′ and 5′-AATGTCTCAACTGTGGGGCTGG-3′. Genotyping of Emx1Cre mice was performed as described previously (Guo et al., 2000a). Mpp3F/F Emx1–CreTg/+ (Mpp3 cKO) mice were used as experimental animals, and Mpp3F/+ Emx1–CreTg/+ (Mpp3 control) mice were used as control animals. All animals were kept at a 12 h light/dark cycle (100 lux) with food and water provided ad libitum. All animal experiments performed were approved by the experimental animal committee of the Royal Netherlands Academy of Arts and Sciences.
Tissue collection.
To obtain embryonic mice, pregnant female mice from timed matings were used, in which detection of a plug was considered E0.5. To obtain the embryos, pregnant female mice were killed with CO2/O2, after which a cesarean section was made to obtain the embryos. Embryos were incubated in 4% PFA in PBS, pH 7.4, overnight at 4°C. For immunohistochemistry on postnatal day 7 (P7), pups were killed with CO2/O2 or decapitation, and the brains were removed from the skull and incubated in 4% PFA in PBS overnight at 4°C. Subsequently, brains were incubated overnight in PBS–15% sucrose and PBS–30% sucrose. After incubation in sucrose, brains were embedded in Tissue Tek (Sakura Finetek) and stored at −20°C.
Immunohistochemistry.
On a cryostat, 10 μm sections were cut and stored at −20°C until use for immunohistochemistry. First, sections were incubated in PBS, pH 7.4, supplemented with 0.3% Triton X-100 (PBS–TX100) and 10% normal goat serum (Dako) for 1 h. Next, sections were incubated in the primary antibodies in PBS–TX100 for 2 h at room temperature or overnight at 4°C. After incubation in primary antibodies, sections were washed with PBS and incubated in secondary antibodies diluted in PBS for 1 h. Finally, sections were washed in PBS and embedded in Dabco-Mowiol 4–88 (Sigma-Aldrich). All reactions were performed at room temperature unless otherwise stated.
The following primary antibodies were used: MPP3 CPH8 (1:100) (Kantardzhieva et al., 2006), MPP3 (1:250; Proteintech Group), MPP5/PALS1 SN47 (1:100), PALS1 (1:1000; Proteintech Group), CRB2 SK11 (1:700; obtained from Dr. Rashbass, University of Sheffield, Sheffield, UK), MUPP1 (1:200; BD Biosciences), PATJ (1:400; obtained from Dr. Le Bivic, Developmental Biology Institute of Marseille Luminy, Marseille, France), Nectin1 (1:500; MBL), β-catenin (1:100; BD Biosciences), p120 catenin (1:100; BD Biosciences), N-cadherin (1:500; BD Biosciences), M-cadherin (1:500; BD Biosciences), PAR3 (1:200; Millipore), phospho-Histone H3 (pH3; 1:500; Millipore), BrdU (1:250, Abcam), Ki67 (1:100; BD Biosciences), CUX1 (1:200; Santa Cruz Biotechnology), CTIP2 (1:100; Abcam), Calretinin (1:500; Millipore Bioscience Research Reagents), Sox2 (1:300; Millipore Bioscience Research Reagents), Tbr1 (1:200; Abcam), Tbr2 (1:200; Abcam), and Tuj1 (1:200; Covance).
Fluorescent-labeled secondary antibodies were rabbit anti-chicken, goat anti-mouse, goat anti-rabbit, or goat anti-rat IgGs conjugated to Cy3, Alexa Fluor 488, or Alexa Fluor 555 (1:500; Jackson ImmunoResearch or Invitrogen). Nuclei were counterstained with TO-PRO-3 iodine (Invitrogen) at 1 μm. Sections were analyzed using a Carl Zeiss CLSM 510 confocal microscope. Image analysis was performed with Carl Zeiss LSM Image Browser 3.5 and Adobe Photoshop 7.0 (Adobe Systems) software.
Protein chemistry.
Cortices from embryonic mice were dissected and homogenized in ice-cold lysis buffer containing protease inhibitors. After overnight incubation at 4°C, samples were centrifuged, and protein concentration in the supernatant was measured by Bradford's assay. Cortical lysate was loaded on a gradient SDS-PAGE gel (NuPAGE gels; Invitrogen) and subsequently transferred onto PVDF membranes. After transfer, membranes were blocked by TBS (50 mm Tris, pH 7.5, 150 mm NaCl) supplemented with 1% BSA and 1% nonfat milk and incubated with primary antibodies (anti-MPP3 CPH8 and anti-actin; Millipore Bioscience Research Reagents) for 1 h in T-TBS (TBS supplemented with 0.05% Tween 20). Blots were washed with T-TBS and incubated in secondary antibodies (streptavidin IRDye conjugated; Rockland Immunochemicals) in TBS for 1 h. After washing, bands were visualized using the Odyssey Infrared Imaging system (Li-Cor Biosciences) and quantified by NIH ImageJ or Adobe Photoshop 7.0.
BrdU experiments.
For BrdU cell labeling experiments, pregnant female mice obtained from timed matings were injected intraperitoneally at E12.5 with 200 μl (1 mg/ml) of BrdU in physiological salt solution, the mice were killed 30 min or 24 h after BrdU injections, and the embryos were obtained. For injections at E16.5, mice were injected intraperitoneally with 200 μl (1 mg/ml) of BrdU, and pups were killed at P7. Tissue was collected as described above, and for immunohistochemistry, 10 μm cryostat sections were treated with 2 m HCl for 30 min at 37°C, after which sections were incubated three times for 5 min in 10 mm sodium citrate, pH 7.5, supplemented with 0.05% Tween 20. Subsequently, immunohistochemistry was performed using anti-BrdU as described above. For Ki67 labeling, sections were heated in the microwave in 10 mm sodium citrate and further treated as described above.
Quantification and data analyses.
For determining spindle orientation, the angle of the spindle in pH3-positive (pH3+) mitotic cells was quantified using the apical membrane as the reference line, and spindle orientation was divided in three bins: 0–30° (horizontal), 31–60° (oblique), and 61–90° (vertical), respectively. For quantifying distribution and number of mitotic or BrdU+ cells, pH3+ or BrdU+ labeled cells were manually counted in E12.5 cortex in 100 μm bins. Mitotic or BrdU+ cells within three cell nuclei from the apical membrane were considered as mitotic or BrdU+ cells in the ventricular zone in E12.5 cortex, whereas all other cells were quantified as ectopic pH3+ or BrdU+ cells. For determining percentage of cells exiting the cell cycle in E13.5 cortex, BrdU+/Ki67− cells were counted in 100 μm bins and divided by the total number of BrdU+ cells. For quantification of ectopic CUX1+ and Calretinin+ cells in P7 cortex, the number of CUX1+ cells in layers V–VI and number of Calretinin cells in layer VI were manually counted per image. To quantify the BrdU+ cell distribution at P7, the cortex was divided in five equal bins, and the number of BrdU+ cells in each bin per image manually counted. Quantification of CUX1+ and Calretinin+ distribution in E16.5 and E17.5 cortex, respectively, was performed by manually counting the cells in the different layers per 100 μm cortex. Quantification of Tbr2+ cells at E16.5 was performed by manually counting the Tbr2+ cells per 100 μm cortex. For statistical analysis, Student's t tests or one-way ANOVA with Bonferroni's correction were used. Data are shown as average ± SEM.
Results
Localization of MPP3 in the developing cortex
In the retina, MPP3 and PALS1 have a direct interaction and colocalize at the subapical region adjacent to adherens junctions (Kantardzhieva et al., 2006). In the developing cortex, apical proteins including PALS1 are localized at the apical membrane. To address whether during development of the cortex MPP3 colocalized with the apical complex protein PALS1, we investigated the localization of MPP3 in the developing cortex. This showed that MPP3 is expressed in E12.5 dorsal telencephalon, in which MPP3 is enriched at the apical membrane (Fig. 1A,B). In the mouse brain, apical proteins such as PALS1 have the highest level of expression during early neurogenesis, at approximately E12.5–E14.5 (Kim et al., 2010). Studying the localization of MPP3 during development showed that MPP3 localized at the apical membrane at E12.5 and E14.5, but at E18.5, the level of MPP3 at the apical membrane is clearly diminished (Fig. 1C–E). MPP3 partly colocalized with PALS1, but in contrast to the restricted localization of PALS1 at the most apical side of the developing cortex, MPP3 had a more broad localization (Fig. 1F–H). Because at the ventricular zone apical complex proteins localize adjacent to adherens junctions, we performed immunohistochemistry for the adherens junction protein Nectin1 and showed partial colocalization with MPP3 (Fig. 1I–K). Thus, in the developing cortex, MPP3 is enriched at the apical membrane and colocalized with apical proteins and adherens junction proteins.
Figure 1.
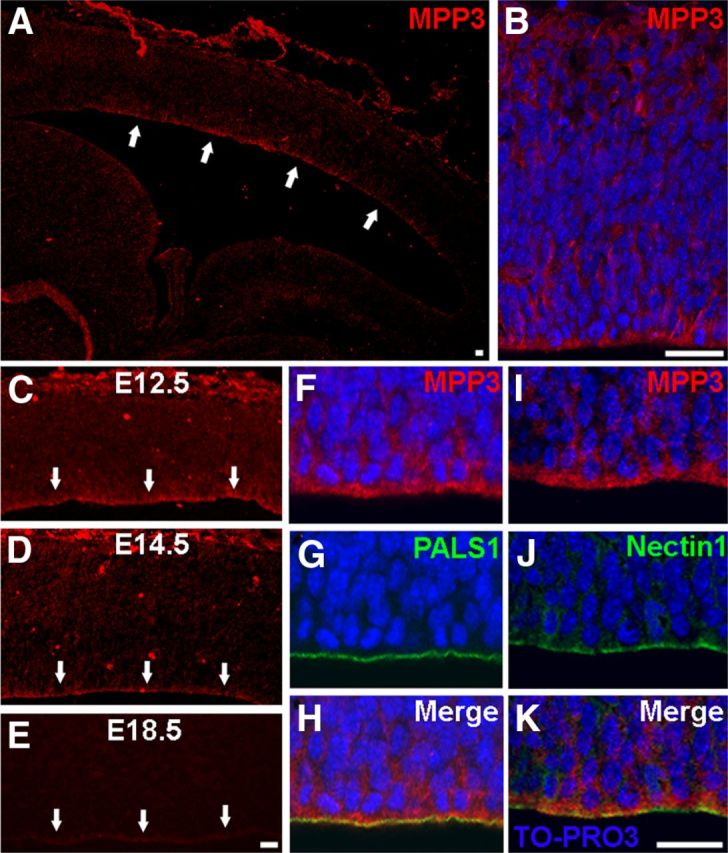
MPP3 localization in the developing cortex. A, B, In E12.5 dorsal telencephalon, MPP3 is enriched at the apical membrane at the ventricular zone. C–E, Expression of MPP3 at the apical membrane is highest at E12.5 (C), decreased at E14.5 (D), and clearly diminished at E18.5 (E). Arrows in A and C–E indicate apical membrane. F–H, Partial colocalization (H) of MPP3 (F) with the apical protein PALS1 (G). I–K, Partial colocalization (K) of MPP3 (I) with adherens junction protein Nectin1 (J). Scale bars, 20 μm.
Generation of Mpp3 cKO mice
The murine Mpp3 gene has 19 exons with the start codon in the third exon (Lin et al., 1998). To generate Mpp3 cKO mice, loxP sites were positioned around exons 3–6, which includes the start codon in exon 3 (Fig. 2A). After initial PCR screening, candidate embryonic stem cell clones were checked by Southern blotting to verify the proper homologous recombination of the targeting construct (Fig. 2B,C). Blastocyst injections resulted in two male chimeric mice, which gave germ-line transmission of the targeted gene. Genotyping of mutant mice resulted in a PCR product of ∼200 bp attributable to the addition of the loxP site (Fig. 2D).
Figure 2.

Conditional deletion of Mpp3 in the developing cortex. A, A targeting construct was designed with loxP sites flanking exons 3–6 and with a FRT site flanked Neomycin cassette for positive selection. The 5′ and 3′ probes used for Southern blot analysis of homologous recombination are indicated together with the fragment lengths resulting from an EcoRV or AseI digestion, respectively. Bottom line shows modified cKO Mpp3 gene, as well as PCR screening strategy over the 5′ loxP site. B, C, Southern blot analysis showing additional 12 and 11 kb restriction fragments attributable to the addition of EcoRV and AseI restriction sites, respectively. D, Genotyping PCR of the cKO locus, in which wild-type (+/+) locus gives rise to an ∼175 bp product, whereas the Mpp3 cKO locus (F/F) leads to a fragment of ∼200 bp. E, F, In E12.5 control cortex (E), MPP3 localized at the apical membrane whereas in Mpp3 cKO cortex, the level of MPP3 at the apical membrane is decreased (F). G, H, More prominent reduction of MPP3 in E16.5 Mpp3 cKO cortex (H) compared with control cortex (G). I, At E18.5, the level of MPP3 at the ventricular zone was decreased compared with E12.5 and E16.5 cortex. J, At E18.5, the level of MPP3 in Mpp3 cKO cortex is clearly reduced, with only some puncta of MPP3 left. K, Western blotting for MPP3 in cortex lysate shows that, in E12.5 cKO, there is only a limited reduction of MPP3 compared with control, whereas in E16.5 cKO cortex lysate, there is a clear reduction in levels of MPP3 compared with control. Scale bar, 20 μm.
To address the role of MPP3 in cortical neurogenesis, we crossed the Mpp3 cKO mice with Emx1Cre mice, which express Cre recombinase starting at E9.5 in cortical progenitor cells (Guo et al., 2000b; Jin et al., 2000; Gorski et al., 2002). In E12.5 cKO cortex, levels of MPP3 at the apical membrane are reduced compared with control cortex (Fig. 2E,F,K). In E16.5 Mpp3 cKO cortex, there is a more prominent and widespread reduction of MPP3 (Fig. 2G,H,K). Finally, as shown in Figure 1, in E18.5 control cortex, MPP3 levels at the apical membrane are clearly reduced compared with earlier time points (Fig. 2I) and still further decreased in E18.5 cKO cortex (Fig. 2J).
Loss of MPP3 results in cellular disorganization at the ventricular zone
Interestingly, in E12.5 cKO cortex, we observed a mild cellular disorganization at the ventricular zone (Fig. 2F). In E16.5 cortex, there was a more prominent and broad cellular disorganization at the ventricular zone (Figs. 2H, 3A–D). Importantly, at the level of the third ventricle, in which Cre recombinase is not expressed, MPP3 expression and localization and cellular organization was indistinguishable from control (Fig. 3E,F). However, analysis of gross cortical morphology at E16.5 did not show a defect on either cortical thickness or lamination (Fig. 3G,H). Thus, loss of MPP3 in the cortex results in a cellular disorganization in the ventricular zone, but gross cortical morphology and lamination is unaffected.
Figure 3.

Cellular disorganization at the ventricular zone in cortex lacking MPP3. A, C, At E16.5, control cortex progenitor cells at the ventricular zone are orderly organized. B, D, At E16.5, the Mpp3 cKO cortex showed cellular disorganization at the ventricular zone with cells detaching from the apical membrane. C and D are magnifications of the red boxes in A and B, respectively. E, F, As a control, MPP3 level and localization and cellular organization are indistinguishable between control (E) and Mpp3 cKO (F) in the third ventricle in which Cre recombinase is not expressed. G, H, Although there is cellular disorganization at the ventricular zone, the gross morphology, thickness, and organization of the layers is not different between Mpp3 cKO (H) and control (G). VZ, Ventricular zone; IZ, intermediate zone; CP, cortical plate. Scale bars, 20 μm.
Loss of apical protein complex precedes the loss of adherens junctions in Mpp3 cKO cortex
Because loss of MPP3 resulted in cellular disorganization, we were interested whether apical proteins and adherens junction proteins still localized properly in the absence of MPP3. Already at E12.5, there was a clear decrease in PALS1 immunostaining intensity in Mpp3 cKO cortex compared with control cortex (Fig. 4A–F). At areas in which MPP3 levels were still comparable with control, also PALS1 was unaffected, suggesting that there is a direct relationship between loss of MPP3 and loss of PALS1 (Fig. 4G). In addition to PALS1, also the localization of the other apical proteins CRB2, MUPP1, and PATJ was impaired in Mpp3 cKO cortex (Fig. 4H–M). Interestingly, PATJ-labeled progenitor cells are clearly disorganized in Mpp3 cKO cortex compared with control, similar to that observed in retina with a disrupted apical protein complex attributable to removal of CRB2 (Fig. 4L,M) (Alves et al., 2013). Thus, loss of MPP3 results in a disruption of the apical protein complex and a disorganization of progenitor cells in the ventricular zone. Previous research showed that loss of the apical protein complex resulted in disruption of adherens junctions (Kim et al., 2010). Therefore, we next assessed the effect of removal of Mpp3 on adherens junctions. In E12.5 Mpp3 cKO cortex, the level and localization of the adherens junction markers p120 catenin, Nectin1, N-cadherin, and β-catenin and cellular organization was still essentially unaffected (Fig. 5A,B,D and data not shown). However, at areas in which there was already loss of adherens junction proteins, we observed cellular disorganization at the ventricular zone (Fig. 5C,E). Next, we investigated the distribution of adherens junction proteins in E16.5 cortex. At the level of the third ventricle, in which Cre is not expressed, levels and localization of β-catenin was indistinguishable between control and Mpp3 cKO (Fig. 5F,G). In contrast, the pattern of β-catenin expression was clearly disrupted in the Mpp3 cKO cortex, with both fewer and mislocalized β-catenin puncta, suggesting a loss and disruption of adherens junctions (Fig. 5H,I). Transmembrane adherens junction proteins, including Catenins, Cadherins, and Nectins, are required for proper adherens junction integrity. In contrast to control cortex, in which these proteins showed a punctate pattern, in Mpp3 cKO cortex, there is a partial loss and dyslocalization of p120 catenin, N-cadherin, M-cadherin, and Nectin1 (Fig. 5J–Q). Immunohistochemistry for another apical complex protein, PAR3, showed that removal of MPP3 also resulted in a loss of PAR3 in E16.5 cortex (Fig. 5R,S). These data suggest that loss of MPP3 first results in loss of the PALS1 apical protein complex at E12.5, followed by a loss of the PAR apical protein complex and disruption of adherens junctions at the apical membrane at E16.5.
Figure 4.
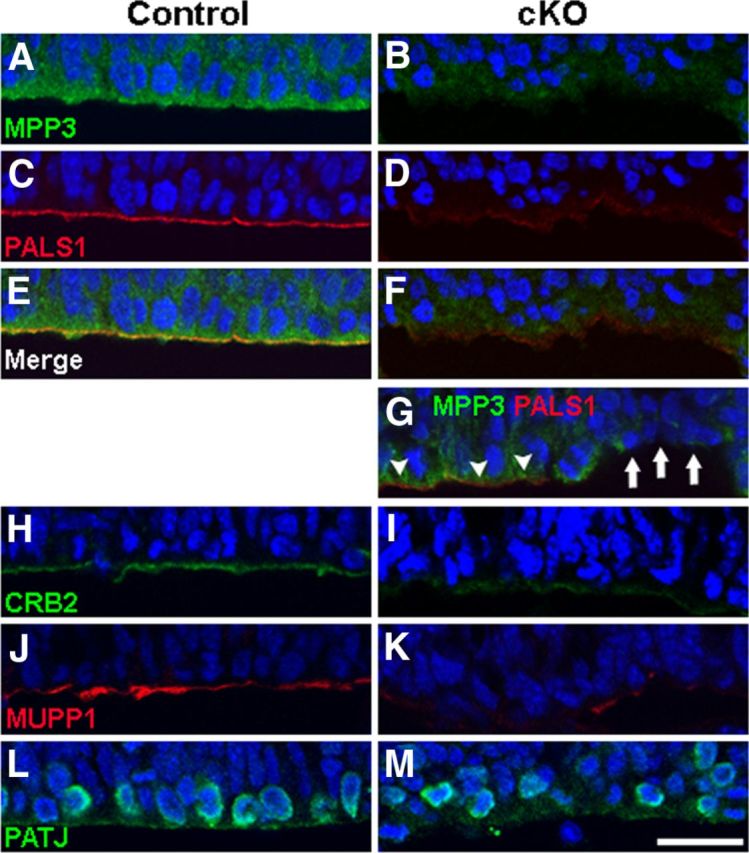
Loss of apical protein complex in Mpp3 cKO cortex. A–F, At E12.5, in control cortex (A), PALS1 is localized at the apical membrane (C, E), whereas in Mpp3 cKO cortex (B), the levels of PALS1 at the apical membrane are reduced (D, F). G, Interestingly, at areas with remaining MPP3, PALS1 levels are unchanged (arrowheads), whereas at areas with reduced MPP3, PALS1 levels are decreased (arrows). H–M, In control cortex, the apical complex proteins CRB2 (H), MUPP1 (J), and PATJ (L) are localized at the apical membrane. In Mpp3 cKO cortex, there is a clear loss of CRB2 (I), MUPP1 (K), and PATJ (M) from the apical membrane. Note that there is a disorganization of the cells labeled by PATJ in Mpp3 cKO cortex compared with control. Scale bar, 20 μm.
Figure 5.

Disruption of adherens junctions in Mpp3 cKO cortex. A–C, p120 catenin levels and apical membrane localization are essentially unaffected in E12.5 Mpp3 cKO cortex. At areas with a loss of p120 catenin, there is cellular disorganization in the ventricular zone (C). B and C are magnification of boxes 1 and 2 in A, respectively. D, E, Likewise, at areas in which Nectin1 is unaffected, cellular organization is indistinguishable from control cortex, but at areas with a loss of Nectin1, a clear disruption of cellular organization was observed (E). F, G, No difference in level and localization of adherens junction protein β-catenin between control (F) and Mpp3 cKO (G) at the level of the third ventricle in which Cre recombinase is not expressed. H, I, Whereas β-catenin displays a punctate pattern at the apical membrane in control cortex (H), in the Mpp3 cKO, there is a loss and delocalization of β-catenin (I). J–O, The same differences were observed between control and Mpp3 cKO for adherens junction proteins p120 catenin (J, K), N-cadherin (L, M), and M-cadherin (N, O). P, Q, Clear loss of Nectin1, which directly interacts with MPP3, during loss of MPP3. R, S, The PAR complex member PAR3 has a punctate distribution in the control cortex (R), but in the Mpp3 cKO cortex, there is a prominent loss of PAR3 from the apical membrane (S). Scale bars, 20 μm.
Loss of MPP3 affects spindle orientation and results in increased number of basally localized mitotic cells
Integrity of adherens junctions and apical complex proteins is required for proper spindle orientation in apical progenitor cells and is involved in determining the mode of cell division (Siller and Doe, 2009; Morin and Bellaiche, 2011). Because adherens junctions and apical protein complexes are disrupted in cortex lacking MPP3, we investigated the spindle orientation in Mpp3 cKO mice. To this end, we quantified the spindle orientation in apically localized phospho-pH3+ mitotic progenitor cells at the ventricular zone in E12.5 cortex. In control cortex, the majority of pH3+ progenitor cells have either a horizontal (0–30°, 38.3 ± 4.5%) or a vertical (61–90°, 47.3 ± 4.1%) spindle orientation, with only a minority of cells with oblique spindle orientation (31–60°, 14.5 ± 3.6%; Fig. 6A–C,G). In Mpp3 cKO cortex, the percentage of pH3+ progenitor cells with vertical spindle orientation was not different compared with control (control, 47.3 ± 4.1%; cKO, 41.2 ± 1.7%; n = 2–3 embryos per group, 6–9 images per embryo), but in contrast to control cortex, there was a dramatic increase in percentage of cells with an oblique spindle orientation, suggesting a shift from horizontal to oblique spindle orientation (horizontal: control, 38.3 ± 4.5%; cKO, 22.7 ± 2.0%, p < 0.00448; oblique: control, 14.5 ± 3.6%; cKO, 36.2 ± 2.5%, p < 0.000014; n = 2–3 embryos per group, 6–9 images per embryo; Fig. 6D–G). It is hypothesized that cells with an oblique spindle orientation give rise to two daughter cells with more basal localization. To further resolve this issue, we determined the distribution and number of pH3+ cells in E12.5 cortex. In control cortex, the majority of mitotic cells were localized at the ventricular zone at the apical membrane, with only few mitotic cells at more basal locations (Fig. 6H). In Mpp3 cKO cortex, several mitotic cells were localized at the ventricular zone, but there were also several mitotic cells with a more basal distribution (Fig. 6I). Quantification of the total number of pH3+ mitotic cells in E12.5 cortex showed an increased number of mitotic cells in cortex lacking MPP3 (control, 4.71 ± 0.17; cKO, 6.20 ± 0.18 mitotic cells/100 μm, p < 0.00001, n = 3 embryos per group, 5–13 images per embryo; Fig. 6J). Remarkably, the number of apically localized mitotic cells was not different between control and cKO (control, 3.83 ± 0.14; cKO, 4.07 ± 0.15 mitotic cells/100 μm; n = 3 embryos per group, 5–13 images per embryo; Fig. 6J). Instead, the increase in the number of basally localized mitotic cells in cortex lacking MPP3 was attributable for the increase in the total number of mitotic cells (control, 0.88 ± 0.08; cKO, 2.13 ± 0.13 mitotic cells/100 μm, p < 0.00001, n = 3 embryos per group, 5–13 images per embryo; Fig. 6J). This suggests that the shift toward more mitotic cells with oblique spindle orientation results in an increased number of basally localized pH3+ cells in cortex lacking MPP3.
Figure 6.

Removal of MPP3 affects spindle orientation, results in ectopically localized mitotic cells, and affects migration. A–F, Determination of mitotic spindle orientation in pH3+ cells in E12.5 control (A–C) and Mpp3 cKO (D–F) cortex. G, Quantification of mitotic spindle orientation revealed that, in Mpp3 cKO cortex, there is a shift from horizontal to oblique spindle orientation. H, In E12.5 control cortex, the majority of pH3+ cells are localized in the ventricular zone at the apical membrane, with some ectopic more basally localized pH3+ cells (arrowheads). I, In contrast, in Mpp3 cKO, there are several ectopic basally localized pH3+ cells (arrowheads). J, Quantification of pH3+ cells in E12.5 cortex showed no difference in number of pH3+ cells in VZ but an increase in total number of pH3+ cells attributable to an increased number of basally localized pH3+ cells in Mpp3 cKO cortex. K, L, Distribution of BrdU+ cells in E12.5 cortex exposed to BrdU for 30 min showed no difference in total number of BrdU+ cells between control (K) and Mpp3 cKO (L) cortex but an increased number of apically localized BrdU+ cells (arrowheads). M–T, Exposure of BrdU for 24 h showed no difference in number of BrdU+ cells or a difference in percentage of cells exiting the cell cycle between control and Mpp3 cKO cortex. Cells that are BrdU+ and Ki67− have exited the cell cycle. h, Horizontal (0–30°); o, oblique (31–60°); v, vertical (61–90°); VZ, ventricular zone. *p < 0.05, **p < 0.001. Scale bars, 20 μm.
Loss of MPP3 does not affect proliferation but does affect migration
To address whether loss of MPP3 affects cell division, we performed BrdU incorporation experiments and colabeling with BrdU and Ki67 to determine the rate of cell division and percentage of cells exiting the cell cycle. BrdU incorporation occurs during the S-phase of the cell cycle, whereas Ki67 is a marker for dividing cells in all phases of the cell cycle. First, we quantified the number of BrdU+ cells after a short 30 min pulse of BrdU in E12.5 cortex. This revealed that the number of BrdU+ cells was not different between control and Mpp3 cKO in E12.5 cortex (control, 71.7 ± 2.2 BrdU+ cells/100 μm cortex; cKO, 70.3 ± 2.1 BrdU+ cells/100 μm cortex, n = 3 embryos per group, 3–4 images per embryo; Fig. 6K,L). In control cortex, the majority of BrdU+ cells were positioned in the subventricular zone, with only few BrdU+ cells outside this zone (Fig. 6K). Strikingly, in Mpp3 cKO cortex, there were BrdU+ cells that were apically localized from the subventricular zone (Fig. 6L). Quantification of the number of BrdU+ cells within three cell nuclei from the apical membrane showed a twofold increase in apically localized BrdU+ cells in Mpp3 cKO cortex, suggesting either affected interkinetic nuclear movement or neuronal migration (control, 1.1 ± 0.18 BrdU+ cells/100 μm cortex; cKO, 2.35 ± 0.37 BrdU+ cells/100 μm cortex, p < 0.00151, n = 3 embryos per group, 3–4 images per embryo). Next, we determined the percentage of cells exiting from the cell cycle in E13.5 cortex exposed to 24 h of BrdU. We determined this by quantifying the BrdU+/Ki67− cells as a percentage of the total pool of BrdU+ cells. This showed that the number of BrdU+ cells was indistinguishable between control and cKO (control, 90.8 ± 3.9 BrdU+ cells/100 μm cortex; cKO, 97.5 ± 9.3 BrdU+ cells/100 μm cortex, n = 2 embryos per group, 4–6 images per embryo; Fig. 6M,Q), as well as the percentage of cells exiting the cell cycle (control, 17.3 ± 1.6%; cKO, 17.1 ± 2.0%; Fig. 6N–P,R–T). Interestingly, whereas in control cortex the majority of BrdU+ and Ki67+ cells were outside the subventricular zone, in cKO cortex, several BrdU+ and Ki67+ cells still resided in the subventricular zone (Fig. 6M–T). Indeed, quantification of the percentage of BrdU+ cells within three nuclei of the apical membrane revealed a twofold increase in Mpp3 cKO cortex (control, 10.8 ± 1.0%; cKO, 19.2 ± 1.5%, p < 0.00003, n = 2 embryos per group, 4–6 images per embryo). Therefore, these data suggest that MPP3 is not required for regulating cell cycle length or exit from the cell cycle but that loss of MPP3 affects migration. Next, we investigated whether loss of MPP3 resulted in a change in cell fate. Immunohistochemistry for Sox2, which marks the progenitor pool of cells, did not reveal a difference between control and Mpp3 cKO cortex (Fig. 7A,B). Additionally, we did not observe a difference in Tbr1+ early-born neurons and Tuj1+ neurons (Fig. 7C–F). Finally, we investigated the distribution of Tbr2+ cells, which are the intermediate progenitor pool of cells. This showed that the distribution and number of Tbr2+ cells was not different between control and Mpp3 cKO (control, 48.9 ± 2.8 Tbr2+ cells/100 μm; cKO, 45.7 ± 2.4 Tbr2+ cells/100 μm, n = 2–3 embryos per group, 4–6 images per embryo; Fig. 7G,H).
Figure 7.

Loss of MPP3 does not result in a change in cell fate. A, B, No difference in Sox2+ progenitor pool between control (A) and Mpp3 cKO (B) at E12.5. C, D, Also, no difference in early-born Tbr1+ neurons between control (C) and Mpp3 cKO (D). E, F, Labeling for Tuj1, which labels the neuronal pool, shows no difference between control (E) and Mpp3 cKO (F) at E14.5. G, H, Number and distribution of Tbr2+ intermediate progenitors at E16.5 was not different between control (G) and Mpp3 cKO (H). Scale bars, 20 μm.
Ectopic localization of late-born neurons attributable to a delay in migration in Mpp3 cKO cortex
Next, we investigated whether loss of MPP3 affected cellular stratification in the cortex. To this end, we performed immunohistochemistry on P7 cortex for CTIP2 and CUX1, markers for early-born (E12.5–E14.5) layer V–VI and late-born (E14.5–E16.5) layer II–IV neurons, respectively. Although gross distribution of CTIP2 neurons in lower layers was not affected, stratification was affected during loss of MPP3 because we observed some CTIP2 neurons in upper layers (Fig. 8A–D). Analysis of CUX1+ neuron distribution in control cortex showed that the majority of CUX1+ cells were localized in layers II–IV, with only few ectopic cells in deeper cortical layers (Fig. 8B,E). In contrast, although in Mpp3 cKO the majority of CUX1+ cells occupied layer II–IV, an increased number of ectopic CUX1+ cells in layer V–VI were observed (control, 32 ± 7.1 ectopic CUX1+ cells/image; cKO, 71 ± 8.8 ectopic CUX1+ cells/image, p < 0.00227, n = 2–3 mice per group, 2–4 images per mouse; Fig. 8E,F). This suggests that loss of MPP3 results in a delayed migration of late-born neurons. Next we focused on the distribution of inhibitory interneurons, which are early-born neurons generated in the caudal and medial ganglionic eminences. Interneurons migrate toward the ventricular zone in the dorsal telencephalon and from there migrate toward upper cortical layers during late-developmental stages (Nery et al., 2002; Fishell, 2007). Investigating the distribution of Calretinin interneurons showed an increased number of Calretinin+ interneurons in layer VI of Mpp3 cKO cortex (control, 57.4 ± 8.5 Calretinin+ neurons in layer VI/image; cKO, 130.1 ± 13.4 Calretinin+ neurons in layer VI/image, p < 0.00034, n = 3 mice per group, 2–4 images per mouse; Fig. 8G,H). This again suggests a delayed migration of neurons during late developmental stages. To further delineate the delayed migration of neurons during late developmental stages and to exclude a change in cell fate, we performed BrdU injections in E16.5 embryos and checked the distribution of BrdU cells in P7 cortex. Quantifying the distribution of BrdU+ cells in the cortex, subdivided into five bins (Fig. 8I,J), showed that less BrdU+ cells occupied the superficial layers in Mpp3 cKO cortex (Bin1 control, 43.3 ± 6.1%; cKO, 27.8 ± 2.1%, p < 0.00763; Bin2 control, 27 ± 3.1%; cKO, 18.7 ± 2.1%, p < 0.04644, n = 2–3 mice per group, 2–4 images per mouse; Fig. 8I–K) and that more BrdU+ cells occupied deeper layers (Bin4 control, 6.5 ± 2%; cKO, 18.8 ± 2.2%, p < 0.00446, n = 2–3 mice per group, 2–4 images per mouse; Fig. 8I–K).
Figure 8.

Delayed migration and ectopic localization of late-born neurons in Mpp3 cKO cortex. A–C, In both control (A) and Mpp3 cKO (C) cortex, CTIP2 neurons are localized in lower layers. However, in Mpp3 cKO cortex, there are some ectopically localized CTIP2 neurons in upper layers (arrowheads in C). C, D, Removal of MPP3 results in stratification defects with ectopic CUX1 (arrows in D) and CTIP2 (arrowheads in D) neurons. E, Distribution of CUX1+ cells in control P7 cortex showed that the majority of CUX1+ cells reside in layer II–IV with some ectopic CUX1+ cells in layer V–VI. F, Also in Mpp3 cKO cortex, the majority of CUX1+ cells occupied layers II–IV, but there is an increase in number of ectopically localized CUX1+ cells in deeper layers. G, H, Distribution of Calretinin interneurons in control (G) and Mpp3 cKO (H) cortex shows an increased number of interneurons in layer VI. I, J, BrdU injection at E16.5 and analysis of distribution of BrdU+ cells in P7 control (I) and Mpp3 cKO (J) cortex. K, Quantification of the distribution of the BrdU+ cells shows that, in Mpp3 cKO cortex, fewer cells occupied superficial layers and there are an increased number of BrdU+ cells in deeper layers. L–N, Analyzing the distribution of CUX1+ cells between E16.5 control (L) and Mpp3 cKO (M) cortex showed that the total number of CUX1+ cells was not different. However, the number of CUX1+ cells still residing in the ventricular zone was increased, whereas the number of CUX1+ cells occupying the cortical plate was decreased (N). O–S, Distribution of Calretinin+ interneurons in E17.5 control (O, P) and Mpp3 cKO (Q, R) cortex. P and R are magnifications of Calretinin interneurons in the ventricular zone from O and Q, respectively. There is no difference in the number of Calretinin+ interneurons between control and Mpp3 cKO cortex, but the distribution is altered during removal of Mpp3, with a lower percentage of Calretinin+ interneurons in the cortical plate, and an increased percentage in the ventricular zone (S). VZ, Ventricular zone; IZ, intermediate zone; CP, cortical plate. *p < 0.05, **p < 0.001. Scale bars, 50 μm.
Finally, we investigated the distribution of CUX1 and Calretinin cells in the developing cortex. CUX1 upper layer neurons are born at approximately E16; therefore, at E16.5, CUX1 neurons start to migrate to layer II–IV. First of all, quantification of the number of CUX1+ cells at E16.5 revealed that no difference was observed between control and cKO (control, 28.4 ± 1.4 CUX1+ cells/100 μm cortex; cKO, 30.6 ± 1.7 CUX1+ cells/100 μm cortex, n = 3–4 embryos per group, 7–17 images per embryo; Fig. 8L,M). However, quantification of the percentage of CUX1+ cells in the ventricular and intermediate zone and in the cortical plate showed that, although the percentage of CUX1+ cells in the intermediate zone was not different (control, 44.6 ± 1.3%; cKO, 41.3 ± 1.3%), in Mpp3 cKO cortex, a smaller percentage of CUX1+ cells had reached the cortical plate (control, 22.4 ± 1.2%; cKO, 14 ± 0.9%, p < 0.00017, n = 3–4 embryos per group, 7–17 images per embryo; Fig. 8L–N). Concomitantly, in Mpp3 cKO cortex, a larger percentage of CUX1+ cells still resided in the ventricular zone (control, 33 ± 1.1%; cKO, 44.7 ± 1.3%, p < 0.00001, n = 3–4 embryos per group, 7–17 images per embryo; Fig. 8L–N). Additionally, we also investigated the distribution of Calretinin interneurons in E17.5 cortex. This showed that the total number of Calretinin interneurons was not different between control and Mpp3 cKO (control, 27.5 ± 1.4 Calretinin+ cells/100 μm cortex; cKO, 24.2 ± 1.4 Calretinin+ cells/100 μm cortex, n = 2 embryos per group, 2–4 images per embryo; Fig. 8O–R). However, like the distribution of CUX1+ cells, in Mpp3 cKO cortex, a smaller percentage of Calretinin+ interneurons occupied the cortical plate (control, 41.2 ± 2.2%; cKO, 23.9 ± 2.4%, p < 0.00001) and a larger percentage of interneurons was still in the ventricular zone (control, 30.8 ± 1.7%; cKO, 48.1 ± 2.8%, p < 0.00001), whereas the percentage of interneurons in the intermediate zone was not different between control and Mpp3 cKO (control, 28 ± 1.3%; cKO, 28 ± 2.3%, n = 2 embryos per group, 2–4 images per embryo; Fig. 8O–S). These data suggest that removal of MPP3 in the cortex results in delayed migration of late-born neurons and ectopically localized pyramidal CUX1 neurons and Calretinin interneurons in postnatal mice.
Discussion
In this study, we showed that loss of the MAGUK protein MPP3 in the developing cortex results in a disruption of apical complex proteins and adherens junctions, resulting in cellular disorganization at the ventricular zone. Furthermore, although removal of MPP3 does not affect cell division, it does affect spindle orientation and localization of mitotic cells. Finally, we showed that loss of MPP3 results in ectopic localization of late-born neurons attributable to a delay in migration.
We showed that MPP3 is required for maintenance of PALS1 at the apical membrane. There is a striking difference between the Mpp3 and the Pals1 cKO phenotype in the cortex. Gross morphology of Mpp3 cKO cortex is indistinguishable from control cortex, whereas conditional deletion of Pals1 in the cortex resulted in a virtually absent cortex attributable to premature exit from the cell cycle (Kim et al., 2010). An explanation for this could be that remaining levels of PALS1 are still sufficient to maintain proper cell division. However, one could expect that partial loss of PALS1 would result in an intermediate phenotype. Alternatively, we hypothesize that, apart from its role as being a core component of the apical protein complex, PALS1 has another autonomous function in cell division that is independent from MPP3. Finally, it might be that depletion of PALS1 in the Mpp3 cKO cortex is only reached after PALS1 has fulfilled its most important role in cell division and maintenance of the pool of progenitor cells.
Like in the Pals1 cKO, there is a disruption of adherens junctions during removal of MPP3 in the cortex. Maintenance of adherens junction integrity is vital for proper cell division and cortical organization. For example, loss of the adherens junction protein N-cadherin resulted in basally localized mitotic cells and disrupted cortical lamination (Kadowaki et al., 2007). Furthermore, loss of Reelin, which regulates the function of Cadherin, results in aberrant neuron migration and cortical lamination (Franco et al., 2011). Additionally, loss of Numb and Numbl, proteins interacting with the Cadherin–Catenin adhesion complex, disrupts adherens junctions and affects progenitor cell polarity in the cortex (Rasin et al., 2007). However, the phenotype observed in the Mpp3 mutant cortex is not as prominent as in the N-cadherin, Reelin, and Numb/Numbl mutant mice. Additionally, the cellular disorganization observed is not as prominent as would be expected after a complete loss of adherens junctions. Therefore, we conclude the disruption and loss of adherens junctions is only partial in our cKO. Indeed, we showed that, in Mpp3 cKO cortex, there is a partial loss and dyslocalization of adherens junction markers. This might be explained by the fact that there is probably functional redundancy with other MAGUK protein scaffolding adherens junction proteins.
Another complex important for proper development of the cortex is the PAR complex (Costa et al., 2008). Deletion of CDC42, a protein that interacts with the PAR complex, in the cortex results in gradual loss of the PAR complex and adherens junctions, accompanied by an affected interkinetic nuclear migration and a thicker and disorganized cortex (Cappello et al., 2006). Likewise, removal of ASPP2, which is required for proper localization of PAR3, results in aberrant neural progenitor proliferation and affected interkinetic nuclear migration and lamination (Sottocornola et al., 2010). We showed that removal of MPP3 leads to a loss of PAR3 from the apical membrane. However, this is possibly a secondary effect from the loss of other apical complex proteins, and therefore the observed phenotype in the Mpp3 cKO might be more attributable to a loss of apical proteins and adherens junctions rather than a loss of PAR3, an issue that remains difficult to segregate.
In addition to affected migration, the Mpp3 cKO mutant mice display ectopic CUX1+ layer II–IV pyramidal neurons in layer V–VI and ectopic interneurons. Interestingly, the delayed migration and ectopic localization of late-born CUX1+ neurons is strikingly similar to the phenotype observed after removal of CNTNAP2, a protein that interacts with several MPP proteins, including MPP3 (Horresh et al., 2008; Peñagarikano et al., 2011). Additionally, these CNTNAP2 KO mice have a reduced number of interneurons and display autism-related behavior and suffer from epileptic seizures (Peñagarikano et al., 2011). In contrast to the CNTNAP2 KO, the Mpp3 cKO does not have a reduction in the number of interneurons but rather has an altered distribution of interneurons. These interneurons are generated in the ganglionic eminences, after which they migrate toward the dorsal telencephalon in which they migrate to the upper layers of the cortex (Nery et al., 2002; Fishell, 2007). Because Cre recombinase in the Emx1Cre mice is not expressed in the ganglionic eminences (Gorski et al., 2002; Cappello et al., 2006; Liang et al., 2012), generation or initial migration of interneurons is likely not affected, but the final migration from the subventricular zone toward superficial layers might be. Additionally, it cannot be excluded that the affected distribution of interneurons in Mpp3 cKO cortex is a secondary effect attributable to the ectopic CUX1+ pyramidal neurons in layers V–VI. This delayed migration in Mpp3 cKO cortex could be attributable to affected radial glia-mediated migration of progenitor cells and neurons. Indeed, we showed that not only neuronal migration but also migration of progenitor cells was affected. Mutant mice, such as MARCKS and PTB that show a gradual loss of adherens junctions in the cortex (Weimer et al., 2009; Shibasaki et al., 2012), also have affected migration of neural progenitor cells, suggesting that the disruption of adherens junctions in Mpp3 cKO cortex leads to this aberrant migration.
Importantly, aberrant migration or connectivity of interneurons is associated with a number of neurodevelopmental disorders, such as autism, schizophrenia, and mental retardation (Levitt, 2005; Di Cristo, 2007; Rossignol, 2011). Although we found that the gross morphology of Mpp3 cKO cortex was unaffected, there is a clear stratification defect in Mpp3 cKO cortex and ectopically localized pyramidal neurons and interneurons. Therefore, it is tempting to speculate that MPP3 is a new candidate gene for neurodevelopmental disorders.
Footnotes
This work was supported by the Royal Netherlands Academy of Arts and Sciences, European Commission Grant HEALTH-F2-200234, Stichting Blindenhulp, and Rotterdamse Stichting Blindenbelangen. We thank Robert Hoek, Lucie Pellissier, and Henrique Alves for comments that contributed to the final version of this manuscript, Marian Verhage for blastocyst injections, and Inge Versteeg for excellent technical assistance. We thank Pen Rashbass for anti-CRB2 and André le Bivic for anti-PATJ.
The authors declare no competing financial interests
References
- Alves CH, Sanz AS, Park B, Pellissier LP, Tanimoto N, Beck SC, Huber G, Murtaza M, Richard F, Sridevi Gurubaran I, Garcia Garrido M, Levelt CN, Rashbass P, Le Bivic A, Seeliger MW, Wijnholds J. Loss of CRB2 in the mouse retina mimics human retinitis pigmentosa due to mutations in the CRB1 gene. Hum Mol Genet. 2013;22:35–50. doi: 10.1093/hmg/dds398. [DOI] [PubMed] [Google Scholar]
- Cappello S, Attardo A, Wu X, Iwasato T, Itohara S, Wilsch-Bräuninger M, Eilken HM, Rieger MA, Schroeder TT, Huttner WB, Brakebusch C, Götz M. The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat Neurosci. 2006;9:1099–1107. doi: 10.1038/nn1744. [DOI] [PubMed] [Google Scholar]
- Costa MR, Wen G, Lepier A, Schroeder T, Götz M. Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development. 2008;135:11–22. doi: 10.1242/dev.009951. [DOI] [PubMed] [Google Scholar]
- Di Cristo G. Development of cortical GABAergic circuits and its implications for neurodevelopmental disorders. Clin Genet. 2007;72:1–8. doi: 10.1111/j.1399-0004.2007.00822.x. [DOI] [PubMed] [Google Scholar]
- Dudak A, Kim J, Cheong B, Federoff HJ, Lim ST. Membrane palmitoylated proteins regulate trafficking and processing of nectins. Eur J Cell Biol. 2011;90:365–375. doi: 10.1016/j.ejcb.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishell G. Perspectives on the developmental origins of cortical interneuron diversity. Novartis Found Symp. 2007;288:21–35. discussion 35–44:96–98. [PubMed] [Google Scholar]
- Franco SJ, Martinez-Garay I, Gil-Sanz C, Harkins-Perry SR, Müller U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011;69:482–497. doi: 10.1016/j.neuron.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosens I, van Wijk E, Kersten FF, Krieger E, van der Zwaag B, Märker T, Letteboer SJ, Dusseljee S, Peters T, Spierenburg HA, Punte IM, Wolfrum U, Cremers FP, Kremer H, Roepman R. MPP1 links the Usher protein network and the Crumbs protein complex in the retina. Hum Mol Genet. 2007;16:1993–2003. doi: 10.1093/hmg/ddm147. [DOI] [PubMed] [Google Scholar]
- Gosens I, den Hollander AI, Cremers FP, Roepman R. Composition and function of the Crumbs protein complex in the mammalian retina. Exp Eye Res. 2008;86:713–726. doi: 10.1016/j.exer.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Guo H, Mao C, Jin XL, Wang H, Tu YT, Avasthi PP, Li Y. Cre-mediated cerebellum- and hippocampus-restricted gene mutation in mouse brain. Biochem Biophys Res Commun. 2000a;269:149–154. doi: 10.1006/bbrc.2000.2263. [DOI] [PubMed] [Google Scholar]
- Guo H, Hong S, Jin XL, Chen RS, Avasthi PP, Tu YT, Ivanco TL, Li Y. Specificity and efficiency of Cre-mediated recombination in Emx1-Cre knock-in mice. Biochem Biophys Res Commun. 2000b;273:661–665. doi: 10.1006/bbrc.2000.2870. [DOI] [PubMed] [Google Scholar]
- Horresh I, Poliak S, Grant S, Bredt D, Rasband MN, Peles E. Multiple molecular interactions determine the clustering of Caspr2 and Kv1 channels in myelinated axons. J Neurosci. 2008;28:14213–14222. doi: 10.1523/JNEUROSCI.3398-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai F, Hirai S, Akimoto K, Koyama H, Miyata T, Ogawa M, Noguchi S, Sasaoka T, Noda T, Ohno S. Inactivation of aPKClambda results in the loss of adherens junctions in neuroepithelial cells without affecting neurogenesis in mouse neocortex. Development. 2006;133:1735–1744. doi: 10.1242/dev.02330. [DOI] [PubMed] [Google Scholar]
- Jin XL, Guo H, Mao C, Atkins N, Wang H, Avasthi PP, Tu YT, Li Y. Emx1-specific expression of foreign genes using “knock-in” approach. Biochem Biophys Res Commun. 2000;270:978–982. doi: 10.1006/bbrc.2000.2532. [DOI] [PubMed] [Google Scholar]
- Kadowaki M, Nakamura S, Machon O, Krauss S, Radice GL, Takeichi M. N-cadherin mediates cortical organization in the mouse brain. Dev Biol. 2007;304:22–33. doi: 10.1016/j.ydbio.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Kantardzhieva A, Gosens I, Alexeeva S, Punte IM, Versteeg I, Krieger E, Neefjes-Mol CA, den Hollander AI, Letteboer SJ, Klooster J, Cremers FP, Roepman R, Wijnholds J. MPP5 recruits MPP4 to the CRB1 complex in photoreceptors. Invest Ophthalmol Vis Sci. 2005;46:2192–2201. doi: 10.1167/iovs.04-1417. [DOI] [PubMed] [Google Scholar]
- Kantardzhieva A, Alexeeva S, Versteeg I, Wijnholds J. MPP3 is recruited to the MPP5 protein scaffold at the retinal outer limiting membrane. FEBS J. 2006;273:1152–1165. doi: 10.1111/j.1742-4658.2006.05140.x. [DOI] [PubMed] [Google Scholar]
- Kim S, Lehtinen MK, Sessa A, Zappaterra MW, Cho SH, Gonzalez D, Boggan B, Austin CA, Wijnholds J, Gambello MJ, Malicki J, LaMantia AS, Broccoli V, Walsh CA. The apical complex couples cell fate and cell survival to cerebral cortical development. Neuron. 2010;66:69–84. doi: 10.1016/j.neuron.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Walsh CA. Neurogenesis at the brain-cerebrospinal fluid interface. Annu Rev Cell Dev Biol. 2011;27:653–679. doi: 10.1146/annurev-cellbio-092910-154026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P. Disruption of interneuron development. Epilepsia. 2005;46(Suppl 7):22–28. doi: 10.1111/j.1528-1167.2005.00305.x. [DOI] [PubMed] [Google Scholar]
- Liang H, Hippenmeyer S, Ghashghaei HT. A Nestin-cre transgenic mouse is insufficient for recombination in early embryonic neural progenitors. Biol Open. 2012;1:1200–1203. doi: 10.1242/bio.20122287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Peters LL, Ciciotte SL, Chishti AH. cDNA sequence and chromosomal localization of mouse Dlgh3 gene adjacent to the BRCA1 tumor suppressor locus. Biochim Biophys Acta. 1998;1443:211–216. doi: 10.1016/S0167-4781(98)00209-7. [DOI] [PubMed] [Google Scholar]
- Morin X, Bellaïche Y. Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev Cell. 2011;21:102–119. doi: 10.1016/j.devcel.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Nery S, Fishell G, Corbin JG. The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat Neurosci. 2002;5:1279–1287. doi: 10.1038/nn971. [DOI] [PubMed] [Google Scholar]
- Park B, Alves CH, Lundvig DM, Tanimoto N, Beck SC, Huber G, Richard F, Klooster J, Andlauer TF, Swindell EC, Jamrich M, Le Bivic A, Seeliger MW, Wijnholds J. PALS1 is essential for retinal pigment epithelium structure and neural retina stratification. J Neurosci. 2011;31:17230–17241. doi: 10.1523/JNEUROSCI.4430-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasin MR, Gazula VR, Breunig JJ, Kwan KY, Johnson MB, Liu-Chen S, Li HS, Jan LY, Jan YN, Rakic P, Sestan N. Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nat Neurosci. 2007;10:819–827. doi: 10.1038/nn1924. [DOI] [PubMed] [Google Scholar]
- Rossignol E. Genetics and function of neocortical GABAergic interneurons in neurodevelopmental disorders. Neural Plast. 2011;2011:649325. doi: 10.1155/2011/649325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki T, Tokunaga A, Sakamoto R, Sagara H, Noguchi S, Sasaoka T, Yoshida N. PTB deficiency causes the loss of adherens junctions in the dorsal telencephalon and leads to lethal hydrocephalus. Cereb Cortex. 2012 doi: 10.1093/cercor/bhs161. doi: 10.1093/cercor/bhs161. Advance online publication. Retrieved April 12, 2013. [DOI] [PubMed] [Google Scholar]
- Siller KH, Doe CQ. Spindle orientation during asymmetric cell division. Nat Cell Biol. 2009;11:365–374. doi: 10.1038/ncb0409-365. [DOI] [PubMed] [Google Scholar]
- Sottocornola R, Royer C, Vives V, Tordella L, Zhong S, Wang Y, Ratnayaka I, Shipman M, Cheung A, Gaston-Massuet C, Ferretti P, Molnár Z, Lu X. ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev Cell. 2010;19:126–137. doi: 10.1016/j.devcel.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Weimer JM, Yokota Y, Stanco A, Stumpo DJ, Blackshear PJ, Anton ES. MARCKS modulates radial progenitor placement, proliferation and organization in the developing cerebral cortex. Development. 2009;136:2965–2975. doi: 10.1242/dev.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Chia W. Neurogenesis and asymmetric cell division. Curr Opin Neurobiol. 2008;18:4–11. doi: 10.1016/j.conb.2008.05.002. [DOI] [PubMed] [Google Scholar]