

Abstract
The modulation of synaptic plasticity by NMDA receptor (NMDAR)-mediated processes is essential for many forms of learning and memory. Activation of NMDARs by glutamate requires the binding of a coagonist to a regulatory site of the receptor. In many forebrain regions, this coagonist is d-serine. Here, we show that experimental epilepsy in rats is associated with a reduction in the CNS levels of d-serine, which leads to a desaturation of the coagonist binding site of synaptic and extrasynaptic NMDARs. In addition, the subunit composition of synaptic NMDARs changes in chronic epilepsy. The desaturation of NMDARs causes a deficit in hippocampal long-term potentiation, which can be rescued with exogenously supplied d-serine. Importantly, exogenous d-serine improves spatial learning in epileptic animals. These results strongly suggest that d-serine deficiency is important in the amnestic symptoms of temporal lobe epilepsy. Our results point to a possible clinical utility of d-serine to alleviate these disease manifestations.
Introduction
NMDA receptors (NMDARs) are of central importance in excitatory synaptic transmission. The activation of these receptors by synaptically released glutamate is required for different forms of synaptic long-term plasticity in the brain, and is thought to be important for intact learning and memory processes. However, full activation of the NMDAR complex requires not only binding of the agonist glutamate but also binding of either d-serine or glycine to an allosteric coagonist binding site (Johnson and Ascher, 1987; Kemp and Leeson, 1993). This has led to the idea that coagonist binding to the NMDAR complex modulates the induction of NMDAR-dependent synaptic plasticity. Indeed, applying antagonists for the NMDAR coagonist binding site blocks induction of long-term potentiation (LTP) both in vivo (Mizutani et al., 1991; Thiels et al., 1992) and in vitro (Bashir et al., 1990; Yang et al., 2003). More importantly, decreasing the levels of endogenously released d-serine by application of the d-serine degrading enzyme d-amino acid oxidase (DAO) suppressed NMDAR activity and the induction of LTP in cell culture, as well as to a lesser extent in acute hippocampal slices (Yang et al., 2003). Indeed, several studies revealed that d-serine is released from astroglia in a Ca2+-dependent manner and regulates activation of NMDARs (Schell et al., 1995; Mothet et al., 2000; Yang et al., 2003; Panatier et al., 2006). A study has shown that, in intact brain tissue, astrocytes control the levels of d-serine within the domain they occupy. Their d-serine release appears to be controlled by neuronal and astrocytic activity and modulates NMDARs and LTP (Henneberger et al., 2010). Recently, experiments using selective degrading enzymes for d-serine and glycine have revealed that d-serine modulates synaptic NMDARs, whereas glycine gates extrasynaptic NMDARs, matching the preferential affinity of synaptic and extrasynaptic NMDARs for d-serine and glycine, respectively (Fossat et al., 2012; Papouin et al., 2012). Furthermore, NMDAR-dependent synaptic plasticity in the normal hippocampus seems to require synaptic NMDARs only (Fossat et al., 2012; Papouin et al., 2012). These studies collectively suggest that precisely controlled d-serine release from individual astrocytes gates synaptic NMDARs, thereby controlling synaptic plasticity.
The central importance of d-serine-mediated cotransmission begs the question whether d-serine levels in the brain might be altered in neurological disorders. Indeed, several studies have shown that CSF and serum levels of d-serine are reduced in patients with schizophrenia (Hashimoto et al., 2003). Moreover, genetic variants in the genes encoding the major enzyme synthesizing d-serine, serine racemase (Srr), and the d-serine degrading enzyme DAO have been shown to be associated with schizophrenia or schizophrenia-like symptoms in mice (Labrie et al., 2009, 2010; for review, see Boks et al., 2007). However, how NMDAR cotransmission, and as a consequence NMDAR function and synaptic plasticity, is altered is not known for any neurological disorders. Temporal lobe epilepsy is associated with manifold cognitive changes, but their underlying mechanisms are unknown. Here, we show that d-serine levels are reduced in experimental temporal lobe epilepsy, causing desaturation of synaptic and extrasynaptic NMDARs and thereby deficits in hippocampal LTP. Exogenous d-serine rescues hippocampal LTP and ameliorates behavioral deficits in epileptic animals.
Materials and Methods
Pilocarpine-induced temporal lobe epilepsy.
A sustained status epilepticus (SE) was induced in young adult male Wistar rats (∼30 d, 140–150 g; Charles River) by administration of the muscarinic agonist pilocarpine according to protocols described previously (Turski et al., 1983; Cavalheiro et al., 1987). Methyl scopolamine nitrate (1 mg/kg, i.p. or s.c.; Sigma) was preapplied 30 min before pilocarpine injection to reduce peripheral cholinergic effects. Then animals of the experimental group received pilocarpine hydrochloride, freshly dissolved in 0.9% saline (340 mg/kg, i.p. or s.c.; Sigma). Control rats received the same dose of methyl scopolamine nitrate and 30 min later sterile saline. In most rats, SE developed within 30–60 min and was stopped after 40 min by application of diazepam (4 mg/kg, i.p. or s.c.), which was occasionally repeated when necessary. The remaining animals received a second injection of pilocarpine after 60 min (170 mg/kg, i.p. or s.c.). Overall, 60–90% of treated animals developed SE. After recovery of 7–10 d, rats were monitored for 2–6 weeks (8 h/d) to observe behavioral limbic seizures. Only rats experiencing more than three seizures were included in this study. Control rats were held in the same observation facility to exclude housing effects. Experiments were performed at the age of 50–100 d (300–500 g). All animal experiments were done in accordance with institutional and national guidelines for animal welfare.
Total RNA preparation and RT-PCR.
Rat brain tissues from pilocarpine and control rats were rapidly dissected, snap frozen in isopentane, and stored at −80°C. Total RNA was prepared by the Trizol method (Invitrogen). Five micrograms of RNA and 0.5 μg oligo-dT(20) primers (Invitrogen) were heated at 70°C for 4 min, chilled on ice, and reverse transcribed in a total volume of 20 μl containing 4 μl first-strand buffer (Invitrogen), 2 μl of 0.1 m DTT, 1 μl of 10 mm dNTPs, 0.5 μl of RNase OUT (Invitrogen), and 200 U of Superscript II reverse transcriptase (Invitrogen) at 42°C for 50 min.
Taqman analyses.
RT-qPCR was performed using an ABI 7900 sequence detector (PerkinElmer Life and Analytical Sciences) on cDNA samples, at 50°C for 2 min, 95°C for 10 min followed by 40 cycles of 95°C for 15 s, and then 60°C for 1 min using the Universal PCR Master Mix (PerkinElmer Life and Analytical Sciences). Taqman primer and probe sets were from Applied Biosystems: Rn01648369_m1 for Srr, Rn01480801_m1 for DAO, Rn00586891_m1 for sodium-independent alanine-serine-cysteine transporter 1 (Asc-1), and Rn01528258_m1 synaptophysin. Synaptophysin was used to normalize for the amount of sample in a given reaction (Chen et al., 2001). Results are expressed as relative expression to the control gene (ΔCt).
HPLC analyses.
Tissue samples were homogenized in 10 vol of 5% trichloroacetic acid (TCA), and the homogenates were centrifuged at 18,000 × g at 4°C for 30 min. To remove TCA, the supernatants were washed three times with water-saturated diethyl ether. The resultant samples were used for HPLC analysis. Amino acid enantiomers were separated by HPLC using a carbon 18 reverse-phase column (250 mm) (Knauer; Advanced Scientific Instruments) with fluorimetric detection after derivatization with N-isobutyryl-l-cysteine and o-phthalaldehyde (OPA). The N-isobutyryl-l-cysteine/OPA derivatives were immediately applied to the HPLC system (Knauer; Advanced Scientific Instruments). Mobile phase was 11% MeCN in 0.1 m sodium acetate buffer, pH 6.0. Amino acids were separated isocratically for 37 min, and then the column was eluted with 50% H2O/50% MeCN for 5 min. After that, the column was re-equilibrated for 15 min under the initial conditions. The flow rate was 0.125 ml/min. Fluorescence detection of each amino acid derivative was performed at 443 nm with excitation at 344 nm according to Grant et al., 2006. We calculated the d-serine ratio compared with total serine content [d-serine/(d-serine + l-serine)] according to Hikida et al., 2008 to minimize variability. From each chromatogram, d-serine ratio compared with total serine was calculated.
Protein extraction and Western blot.
Hippocampal samples from pilocarpine-treated and control rats were extracted and snap frozen in liquid nitrogen. Protein extracts were generated using ultrasonication in RIPA buffer (10 mm Tris-HCl, pH 8, 150 mm NaCl, 5 mm EDTA, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) and protease inhibitors. After centrifugation, 15 μg of supernatant was used for Western blot analysis. Protein concentration was determined using the BCA Protein Assay (Pierce). The Western blot was conducted using NuPAGE 4–12% Bis-Tris gels from Invitrogen. After blotting, the membranes were blocked with 5% milky powder overnight and immunostained with a rabbit-derived serine racemase antibody (ab45543, 1:500, 2 h; Abcam) and enhanced chemiluminescence (Pierce) followed by goat α-rabbit heavy/light (H/L) peroxidase (1:3000, 1 h; The Jackson Laboratory). After stripping, the blot was labeled with a rabbit-derived synaptophysin antibody (ab52636, 1:5000, 2 h; Abcam) as a reference and enhanced chemiluminescence (Pierce), followed by goat α-rabbit H/L peroxidase development (1:3000, 1 h; The Jackson Laboratory). The blots were analyzed using the ChemiDoc MP Imagine System from Bio-Rad and the ImageLab 4.01 software.
Slice preparation.
For LTP experiments, animals were deeply anesthetized with diethylether and decapitated, and the brains were dissected out quickly. Transversal hippocampal slices (400 μm) were prepared using a vibratome (Leica 1000S; Integraslice 7550 mm from Campden Instruments) in ice-cold dissection fluid containing the following (in mm): 125 NaCl, 25 NaHCO3, 3 KCl, 1.25 NaH2PO4, 0.2 CaCl2, 5 MgSO4, and 13 d-glucose. All procedures were performed according to the guidelines laid down by the Animal Care and Use Committees at the Universities of Bonn and Rostock, respectively. Slices were then transferred into an interface chamber heated within 30–40 min from room temperature to 35°C and superfused (1.8–2.0 ml/min) with bath solution containing the following (in mm): 125 NaCl, 26 NaHCO3, 3 KCl, 1.25 NaH2PO4, 2.5 CaCl2, 1.3 MgCl2, and 13 d-glucose. This solution was continuously bubbled with carbogen (95% O2, 5% CO2) to maintain the pH of 7.4. Sucrose was used to adjust the osmolality to 306–308 mOsm/kg. Slices were allowed to recover at least 60 min before recordings were performed. For patch-clamp recordings, rats were deeply anesthetized with a mixture of 0.7 ml of Ketavet injection solution and 0.3 ml of Rompun solution injected into the abdomen of the rat. To achieve rapid cooling of brain tissue already during preparation, which maximizes cell preservation, heart perfusion was performed with ice-cold ACSF slicing solution containing the following (in mm): 125 NaCl, 25 NaHCO3, 3 KCl, 1.25 NaH2PO4, 0.2 CaCl2, 5 MgSO4, and 13 d-glucose (306 mOsm osmolality). Hippocampal slices (300 μm) were obtained with a vibratome (Microm HM 650V) in the same solution and transferred to ACSF containing the following (in mm): 125 NaCl, 3.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2.5 MgCl2, and 15 d-glucose (306 mOsm osmolality). Brain slice were then incubated at 35°C for 35 min and were allowed to recover at least 60 min before recordings were performed. All solutions were continuously bubbled with 95% O2/5%CO2.
Field EPSP recording.
Lateral (LPP) and medial (MPP) perforant path fibers were selectively and alternately stimulated at a frequency of 0.033 Hz via two bipolar platinum wire electrodes (0.1–0.3 mA, 100 μs, paired-pulse stimulation, 40 ms apart) placed in both the outer (OML) and middle (MML) molecular layers of the dentate gyrus (DG). Field EPSPs (fEPSPs) were simultaneously recorded in the OML and MML via glass pipettes (borosilicate glass capillaries, ∼4–6 MΩ) filled with bath solution. All recordings were performed at 35°C. The stimulation strength was adjusted to yield half-maximal fEPSP amplitudes. LTP was induced by high-frequency patterned stimulation (10 trains of 20 impulses at 100 Hz, 1 s apart). Analog data were digitized (Digidata 1322 from Molecular Devices; Micro1401 from Cambridge Electronic Design) and stored for offline analysis (pClamp software from Molecular Devices; Signal2 from Cambridge Electronic Design). The group III metabotropic glutamate receptor agonist l-2-amino-4-phosphonobutyrate (l-AP-4) was obtained from Tocris Cookson. All other chemicals were purchased from Sigma.
LPP stimulation evoked negative fEPSPs in the OML and large positivities in the MML. In contrast, MPP stimulation resulted in negative MML potentials and OML positivities as described previously (Dietrich et al., 1997).
Patch-clamp recordings.
Slices (300 μm) were placed in a submerged chamber on the stage of an upright microscope (Carl Zeiss Axioskop) and superfused with ACSF containing the following (in mm): 125 NaCl, 3.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, and 15 d-glucose. Micropipettes were fabricated with a Flaming/Brown-type micropipette puller (model P-97, 3–5 MΩ; Sutter Instruments) and filled with intracellular solution containing the following (in mm): 100 CsMS, 20 TEA-Cl, 0.05 CaCl2, 10 HEPES acid, 0.5 EGTA, 2 Mg-ATP, and 1 Na2-ATP. Whole-cell recordings were obtained from visually identified granule cells using either an Axopatch 200B or a Multiclamp 700B amplifier (Molecular Devices). To improve the voltage control, capacitance and series resistance was compensated (50–70%). Current signals were sampled at 10 kHz, digitized, and stored on hard disk for later analysis using the pClamp9 acquisition software (Molecular Devices). A glass stimulation electrode filled with ACSF with a tip diameter of ∼7 μm was positioned in the OML of the DG for LPP stimulation.
Behavioral analyses.
Learning behavior was tested using the hidden platform task in the Morris water maze (110 cm maze diameter and 50 cm water depth, 15 cm platform diameter located 25 cm from the maze rim, four large black-and-white cues). For habituation purposes, all animals were allowed to explore the water maze without platform on the day before the experiments started (day 0, two trials for 60 s each). On day 1, the platform was inserted 1–2 cm below the water level within a randomly chosen quadrant. From day 1 through day 7, each animal was allowed six consecutive trials to reach the platform from one of eight random, evenly spaced starting points at the water maze rim. When the animal failed to reach the platform within 60 s, it was manually placed on the platform. Regardless of whether the animal had located the platform or not, the animal was allowed to stay for another 30 s on the platform, before it was placed back into its cage. After 60 s recovering time in the cage, the next trial was started. On the final day of the experiment (i.e., day 8), a probe trial was performed in which the platform was omitted and the animals were allowed to swim for 60 s. Animal location was tracked using the Ethovision Color software (Noldus). A subgroup of animals was orally treated with d-serine. To this end, each animal was given an individual d-serine dosage in the drinking water. First, the drinking volume and the body weight were recorded daily during a baseline phase of 1 week. Based on these values, the individual d-serine content in the drinking water was calculated to reach 32 mg/kg body weight. During the treatment phase, the drinking volume and the body weight were recorded every 2–3 d to adjust the d-serine content in the drinking water. After 1 week of d-serine supply, the behavioral experiments were started, and the d-serine treatment was further maintained. After completing the Morris water maze learning tasks, all animals were decapitated under anesthesia, and the hippocampi were dissected and subsequently frozen in liquid nitrogen for HPLC-based d-serine measurements.
Seizure frequency analyses.
Rats were video monitored for at least five 12-hour periods on consecutive days for the occurrence of spontaneous seizures. All spontaneous seizures were classified as follows: stage I, immobility, rigid posture; stage II, facial clonic movements, head bobbing; stage III, additional clonus of the forelimbs; stage IV, severe seizures with rearing; and stage V, severe seizures with rearing and falling/loss of righting ability.
Data analyses and statistics.
Data analysis was performed in Igor analysis software. A mathematical fit function of the following form was used to define kinetic parameters of the recorded EPSCs:
 |
The rise time was determined by the slope between the data points at 20 and 80% of the maximal EPSC amplitude. The time course from the data point at 80% of EPSC amplitude value after EPSC maximum until 10% of the amplitude value were reached described the decay of the EPSC. Half-width was defined as the width of the EPSC (in milliseconds) at the half-maximal amplitude value. NMDA/AMPA ratios were determined by dividing the average of five consecutive baseline NMDA EPSCs by the average of five consecutive baseline AMPDA EPSCs. The NMDA/AMPA ratio of 16 cells was used to calculate a reasonable average of the performed EPSC recordings. The success rate of LTP induction was quantitatively analyzed by setting a threshold of >5% potentiation after 1 h of post-tetanus recording over the baseline values.
All data are expressed as the mean ± SEM. Effects within one group were tested using the paired Student's t test. Differences in the effects between groups were tested using the nonparametric Mann–Whitney test, unpaired Student's t test, or ANOVA as indicated. p values <0.05 were considered statistically significant and marked with a single asterisk, whereas p values <0.01 were marked with two asterisks.
Results
Altered synaptic NMDAR-mediated EPSCs in epileptic animals
We first studied the properties of synaptic NMDARs in control and epileptic animals at LPP synapses formed onto hippocampal dentate granule neurons, which are easily discriminated based on morphological and functional criteria (see Materials and Methods). The NMDA-mediated component of LPP EPSCs was isolated from the total glutamatergic EPSC by application of 10 μm CNQX (Fig. 1A, black and dark gray traces). The additional application of 25 μm d-APV completely abolished the EPSC (Fig. 1A, light gray traces). There was no significant difference in the NMDA/AMPA ratio when comparing control and chronically epileptic animals (sham, 0.29 ± 0.05 and post-SE, 0.22 ± 0.05, n = 8 for both groups; p = 0.68). However, the kinetics of the pharmacologically isolated NMDA EPSCs appeared substantially slower in epileptic rats (Fig. 1B, black traces indicate control, gray traces indicate SE). Indeed, the time course of activation, as well as the decay time course were slowed and the half-width increased (Fig. 1C, phalf-width = 0.003, pdecay = 0.03, prise time = 0.01, n = 16 for both groups, unpaired t test). To confirm the identity of LPP EPSCs, the paired-pulse ratio of the responses was determined (two stimulations, 40 ms apart, paired pulse ratio defined as the ratio of the second amplitude to the first) and found to be 1.4 ± 0.05 for NMDA and 1.2 ± 0.07 for AMPA EPSCs, as expected for LPP responses (Dietrich et al. 1997). Application of l-AP-4 similarly reduced the NMDAR-mediated EPSCs in both control and epileptic animals (by 32 ± 6 and 34 ± 7%, respectively, n = 6 for both groups, Mann–Whitney test, p = 0.90), similar to values described previously (Dietrich et al. 1997).
Figure 1.
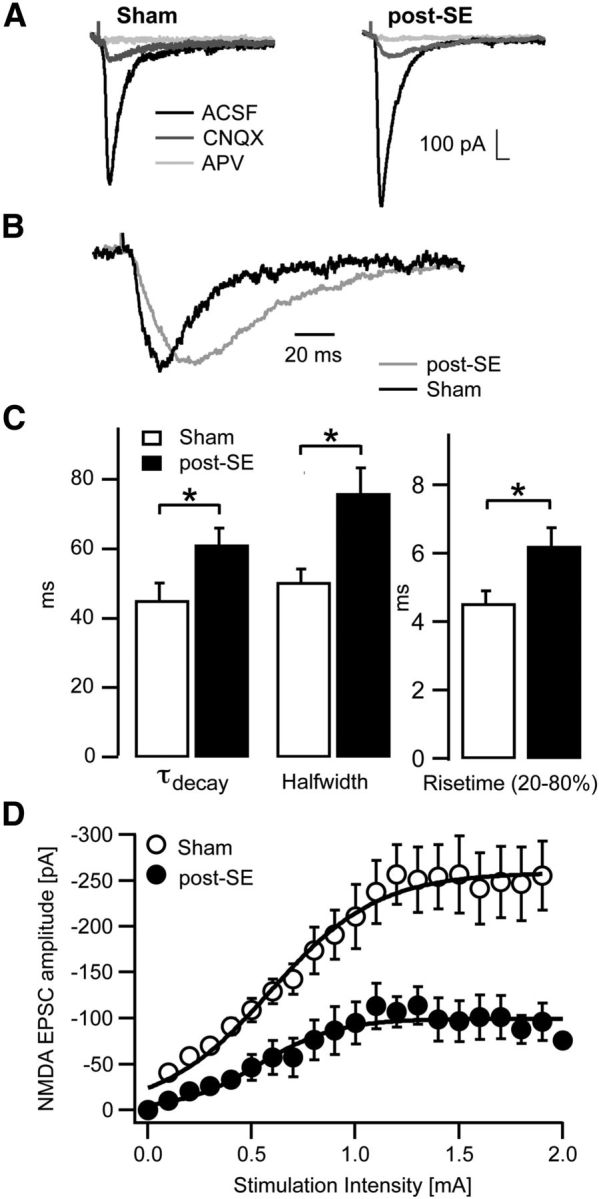
Altered synaptic NMDAR-mediated EPSCs in epileptic animals. A, Representative synaptic EPSCs in sham-control and epileptic (post-SE) animals at LPP synapses in ACSF, followed by application of 10 μm CNQX (gray) and additional application of 25 μm d-APV (light gray). B, Representative NMDA EPSCs showing the slower kinetics in epileptic rats. C, Quantification of the decay time course (decay time constant τdecay), the half-width, and the 20–80% rise time of NMDA EPSCs. D, Input–output curve of NMDA EPSCs in sham-control (open symbols) and epileptic animals (filled symbols), stimulating the LPP and recording from DGCs. Lines represent average curves of the two groups fitted with sigmoidal functions. *p < 0.05, **p < 0.01.
Input–output curves of NMDA EPSCs in control and epileptic animals showed a significant difference in the maximal EPSC amplitude (sham, −261 ± 26 pA, n = 6; post-SE, 141 ± 22 pA, n = 9, p = 0.009, unpaired t test of the maximum of a sigmoidal curve fitted to the data points; Fig. 1D), whereas the stimulation intensity required for the half-maximal EPSC amplitude is not significantly different between the two groups (sham, 0.60 ± 0.03 mA, n = 6; post-SE, 0.81 ± 0.09 mA, n = 9, p = 0.13). This suggests a reduction in NMDAR-mediated synaptic currents in epileptic animals.
The slowing of NMDAR kinetics might indicate altered subunit composition, i.e., as a result of a higher contribution of NR2B-containing receptors in chronic epilepsy. Therefore, we recorded synaptic NMDA EPSCs and applied the NR2A, NR2B, and NR2C/D selective antagonists NVP-AAM007 ([(R)-[(S)-1-(4-bromo- phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydro-quinoxalin-5-yl)-methyl]phosphonic acid), Ro25-6981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol], and PPDA [(2S*, 3R*)-1-(phenanthren-2-carbonyl)piperazine-2,3-dicarboxylic acid], respectively (Fig. 2A–C). In control animals, Ro25-6981 (0.5 μm) reduced NMDA EPSCs by 17 ± 5% (paired t test, p = 0.020, n = 12; Fig. 2A). PPDA (1 μm; Fig. 2A) and NVP-AAM077 (50 nm; Fig. 2C) also caused a significant reduction of the NMDA EPSCs by 27 ± 5% (paired t test p = 0.003, n = 8) or 22 ± 3% (paired t test p = 0.008, n = 6), respectively (summary in Fig. 2D, white bars). In pilocarpine-treated animals, Ro25-6981 reduced NMDA EPSCs by 39 ± 4% (paired t test p = 0.009, n = 9; Fig. 2B), whereas PPDA did not cause significant reductions (by 10 ± 5%, paired t test, p = 0.08, n = 7). The difference between the fractions blocked by Ro25-6981 and PPDA in control and epileptic animals was significant (Fig. 2D, §p = 0.046 and §§p = 0.005, Mann–Whitney test). The application of the NR2A-preferring blocker NVP-AAM077 caused blocking effects similar to those in control animals (31 ± 5%, paired t test, p = 0.001, n = 6; Fig. 2C, summary in D). These data demonstrate an increase in the proportion of synaptic NR2B-containing receptors in chronic epilepsy, with a concomitant decrease in NR2C/D-containing receptors.
Figure 2.

Altered subunit composition of NMDARs. A–D, Examination of NMDAR subunit composition using the NR2A-, NR2B-, and NR2C/D-specific antagonists NVP-AAM007 (50 nm), Ro25-6981 (0.5 μm), and PPDA (1 μm), respectively. A and B show representative experiments with consecutive application of Ro25-6981 and PPDA in sham-control (A) and epileptic animals (B). C, Experiments, either with application of NVP-AAM007 (top traces) or Ro25-6981 (middle traces) in sham-control and epileptic animals. Also in these experiments, subsequent application of d-APV blocked the EPSC completely (bottom traces). D, Quantification of the fraction of the NMDA peak EPSC blocked by the various subtype-selective antagonists in sham-control and epileptic rats. These data demonstrate an increase in the proportion of synaptic NR2B-containing receptors in chronic epilepsy. Error bars represent SEM. *p < 0.05, **p < 0.01, §p = 0.046, §§p = 0.005.
Reduction of the NMDAR coagonist d-serine in chronic epilepsy
The properties of synaptic NMDARs are powerfully modulated by the endogenous coagonist d-serine (Fossat et al., 2012; Papouin et al., 2012). To examine whether a reduction in d-serine content of the hippocampus could contribute to reduced NMDA EPSCs (Fig. 1D), we examined the ratio of d-serine/l-serine in the hippocampus of normal and epileptic rats. Using HPLC, we found that the d-serine/l-serine ratios are significantly reduced in the hippocampus of epileptic rats (Fig. 3A; two-way ANOVA treatment effect, F(1,22) = 16.30, p = 0.0006; brain region effect, two-way ANOVA, F(1,22) = 4.51, p = 0.0451; no interaction), consistent with a reduction of hippocampal d-serine levels. We then examined whether pilocarpine treatment of rats affects the expression of genes involved in generation (Srr), degradation (DAO), or transport (Asc-1) of d-serine using quantitative real-time RT-PCR. We analyzed gene expression in different hippocampal subfields and the cerebral cortex. The expression of serine racemase was unaffected by SE in DG, with a small but significant downregulation in the CA1 subfield (Fig. 3B, p = 0.0344). In contrast, the expression of the enzyme DAO was significantly elevated in all subfields of the hippocampus and the cerebral cortex (Fig. 3C; CA1, p = 0.0007; CA3, p = 0.0001; DG, p = 0.0202; cerebral cortex, p = 0.0246, Mann–Whitney test), but it should be noted that the levels of expression overall were low. The expression of the high-affinity d- and l-serine transporter Asc-1, which is localized in dendrites and somata of neurons and clears d-serine from synaptic sites, was unaltered in CA3 and DG, with a significant downregulation only in the CA1 region (Fig. 3D; CA1, p = 0.0025, Mann–Whitney test). We investigated whether the observed changes in Srr and DAO mRNA result in altered hippocampal protein levels. For Srr, Western blot analyses revealed a significant reduction of relative Srr protein levels normalized to synaptophysin levels in whole hippocampal homogenates (Fig. 3E; sham, 0.28 ± 0.007, n = 6; post-SE, 0.21 ± 0.02, n = 6, p = 0.0043, Mann–Whitney test). DAO protein levels were undetectable using Western blots in our hands.
Figure 3.
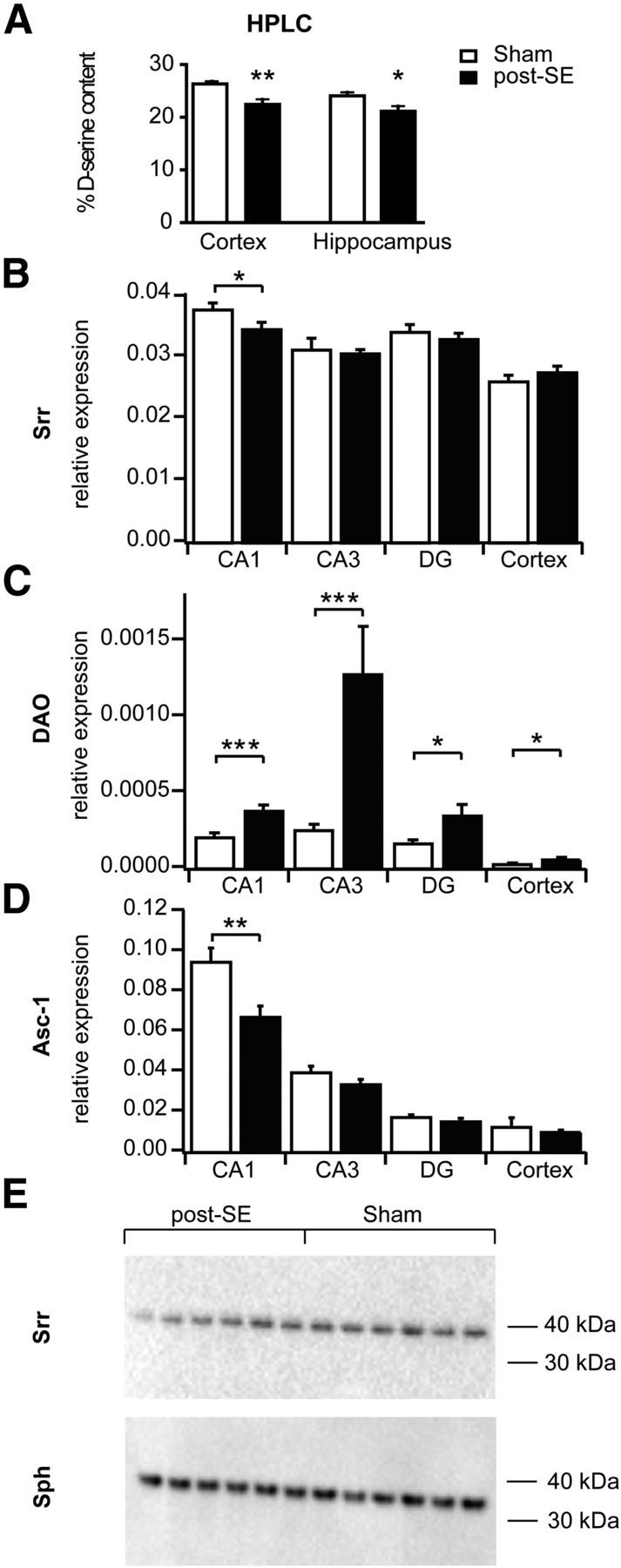
Reduction of the NMDAR coagonist d-serine in chronic epilepsy. A, Analysis of the d-serine content in the hippocampus of normal and epileptic rats using HPLC (see Materials and Methods). Levels of d-serine are significantly reduced in the hippocampus of epileptic rats. B–D, Quantitative real-time RT-PCR quantification of the expression of Srr (B), DAO (C), and the amino acid transporter Asc-1 (D), which transports d-serine, in different hippocampal subfields and the cerebral cortex as indicated. Relative expression units using synaptophysin as control gene were calculated (ΔCt). Note the downregulation of Srr in the CA1 subfield, as well as the significantly elevated expression of DAO in all subfields of the hippocampus and the cerebral cortex (*p < 0.05, **p < 0.01, ***p < 0.001). E, Western blot analyses of the Srr hippocampal protein levels normalized to synaptophysin (Sph) levels in whole hippocampal homogenates. Levels of Srr are significantly reduced in epileptic animals.
Together, these results show that hippocampal d-serine levels are reduced in chronic epilepsy and suggest that this may be attributable to changes in the enzymes that synthesize and degrade d-serine.
Decreased occupancy of the glycine-binding site of synaptic NMDARs
Decreased levels of d-serine predict that a decreased occupancy of the coagonist binding site might contribute to the reduction of synaptic NMDA currents in pilocarpine-treated animals. We examined this hypothesis by applying saturating concentrations of 10 μm d-serine after baseline NMDA EPSC recording (Fig. 4A,B). We stimulated either with a single stimulation (open symbols) or burst stimulation (five stimuli at 100 Hz; Fig. 4A,B, filled symbols). Application of d-serine caused a significant increase of single and burst stimulation-evoked NMDA EPSC amplitudes in both control and epileptic animals (Fig. 4C; single, p = 0.0002; burst, p = 0.002; D, single, p = 0.003; burst, p = 0.004, t test). However, the magnitude of the d-serine-mediated increase in NMDA EPSCs was larger in pilocarpine-treated animals compared with sham animals for both single and burst stimulation (summary in Fig. 4E; single stimulation: increase by 47 ± 7%, n = 8 and 88 ± 19%, n = 9, in control and epileptic tissue, respectively, p = 0.026; burst stimulation: increase of NMDA EPSCs by 65 ± 12%, n = 8 and 134 ± 31%, n = 9 for control and epileptic animals, respectively, p = 0.04, Mann–Whitney test). The charge transferred during NMDA EPSCs was similarly augmented for single and burst stimulation in sham and epileptic animals (Fig. 4F,G), with a significantly larger increase observed for the epileptic animals (summary in Fig. 4H). This suggests that the decreased d-serine levels in the epileptic brain cause a decrease in the occupancy of the coagonist binding site of synaptic NMDARs, resulting in reduced NMDAR-mediated currents and charge transfer. In these experiments, we frequently observed a prominent sustained inward current after burst stimulation, in particular in epileptic animals after d-serine application (Fig. 4I). We quantified the sustained inward current 217–222 ms after the onset of the synaptic burst stimulation (Fig. 4I, gray markers) and found that it is significantly augmented by d-serine in control and epileptic animals (p = 0.03 and 0.008, respectively, paired t test; Fig. 4J). In epilepsy, the augmentation was significantly larger (p = 0.003, Mann–Whitney test; Fig. 4K).
Figure 4.
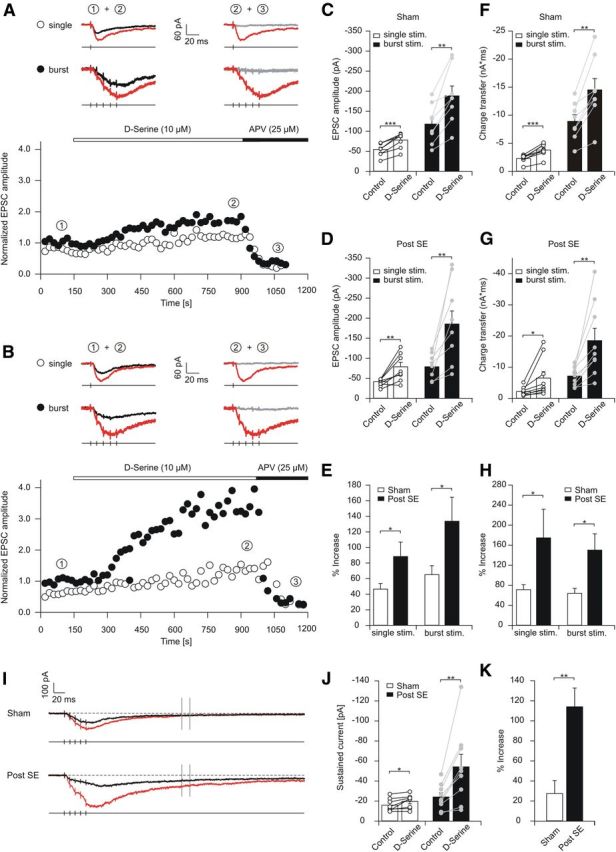
Decreased levels of d-serine cause desaturation of the allosteric modulatory site of synaptic NMDARs. A, B, Representative experiments showing application of saturating concentrations (10 μm) of d-serine after baseline NMDA EPSC recording in sham-control (A) and epileptic (B) rats. Open and filled symbols indicate peak magnitude of NMDA EPSCs elicited with single stimulation and burst stimulation, respectively. C, D, Increase in EPSC magnitude with application of d-serine for single stimulations (white bars) and burst stimulation (black bars) in control (C) and epileptic (D) animals. E, Summary of the degree of NMDA EPSC potentiation by d-serine for single and burst stimulation in control and epileptic rats. F, G, Analysis of the charge transfer increase with application of d-serine for single stimulations (white bars) and burst stimulation (black bars) for control (F) and epileptic (G) animals. H, Degree of charge transfer increase by d-serine for single and burst stimulation in control and epileptic rats. I, Long example traces of NMDA EPSCs elicited by burst stimulation before and after d-serine application (black and red traces, respectively). Note the slow NMDA EPSC component observed, in particular after d-serine application in epileptic rats. Vertical gray lines indicate the time range at which the slow current was quantified. J, The slow current increased significantly both in sham and epileptic rats. K, The fraction of increase was much larger in epileptic animals. Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Effects of d-serine on perisynaptic NMDARs in control and epileptic animals
This effect could be explained by activation of extrasynaptic NMDARs (Lozovaya et al., 2004), which can be activated by spillover of synaptically released glutamate during bursts of high-frequency activity (Kullmann and Asztely, 1998). Therefore, we addressed more directly whether there are epilepsy-associated changes in these receptors and whether the saturation of their coagonist binding site changes. To isolate the contribution of perisynaptic receptors, we evoked NMDA EPSCs with either single or burst stimulation (either a single stimulus or five stimulations at 100 Hz, repeated every 10 s; example traces in Fig. 5A indicated with 1). Application of the use-dependent blocker MK-801 while stimulating with a single stimulus caused a progressive and irreversible block of the resulting synaptic NMDA responses (Fig. 5B). After washout of MK-801, burst stimulation was initiated and evoked a small compound NMDA EPSC (Fig. 5A, example traces indicated with 2, B). This compound NMDA EPSC was attributable to the recruitment of perisynaptic NMDARs remained unblocked by MK-801 (−16.5 ± 1.3 pA, n = 6). As for synaptic NMDARs, we tested whether the coagonist binding sites of perisynaptic NMDARs are saturated or not. We added 10 μm exogenous d-serine, which did not significantly increase the magnitude of perisynaptic NMDA EPSCs (change by 16 ± 18%, p = 0.4, paired t test, n = 6; Fig. 5B, d-serine application shown at higher magnification in inset), implying that the coagonist binding sites of perisynaptic NMDARs are saturated under our recording conditions in control animals. In epileptic animals, a compound perisynaptic EPSC to burst stimulation could also be elicited after blocking synaptic NMDARs (−22.5 ± 5.5 pA, n = 6; example traces in Fig. 5D indicated with 2, E). The magnitude of perisynaptic EPSCs was not different when comparing control and pilocarpine-treated animals. However, the application of d-serine caused a potent increase in the magnitude of the perisynaptic EPSC in epileptic rats by 67.5 ± 13.0% (paired t test, p = 0.0004; example traces in Fig. 5D indicated with 2 and 3, d-serine applications shown at higher magnification in inset). The potentiation of perisynaptic NMDA EPSCs was significantly greater in pilocarpine-treated compared with control animals (Fig. 5E; Mann–Whitney test, p = 0.02). Additionally, the perisynaptic EPSCs in the presence of d-serine, expressed as fraction of the synaptic EPSC, was significantly larger in epileptic animals (perisynaptic EPSC, 10 ± 3 vs 22 ± 4% of initial synaptic EPSCs, respectively, Mann–Whitney test, p = 0.013). These data indicate that perisynaptic NMDARs are not saturated under epileptic conditions and are capable of mediating larger EPSC components than in control animals when saturated by exogenous d-serine.
Figure 5.
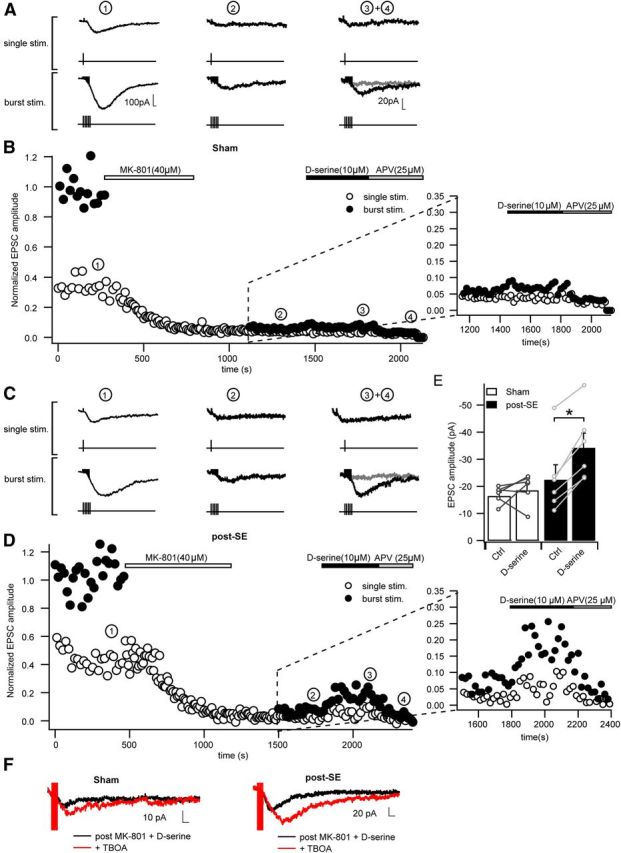
The allosteric modulatory site of perisynaptic NMDARs is not fully occupied in epileptic tissue. A, B, Time course of a representative experiment designed to analyze perisynaptic NMDARs. A shows sample traces of EPSCs in control animals elicited at the time points indicated by the numbers in B. Baseline EPSCs were recorded using a protocol that is alternating between a single stimulus and burst stimulation (5 stimulations at 100 Hz), applied every 10 s. During application of MK-801, single stimulation only was performed to block synaptic NMDARs (B). After washout of MK-801, perisynaptic EPSCs could be elicited with the burst stimulation (example traces in A, 2). Application of d-serine (10 μm) did not significantly increase the burst elicited NMDA EPSCs in control animals (2 and 3 in A and B). Subsequent application of d-APV completely blocked the perisynaptic EPSC (4 in A and B). In B, NMDA EPSC amplitudes are normalized to the baseline of the burst EPSCs recorded under baseline conditions. C, D, Experiments performed as in A and B in epileptic animals. Note the significant increase in perisynaptic EPSCs during d-serine application visible in C (traces 2 and 3), D, and the inset of D at larger magnification. E, Changes in amplitudes of perisynaptic NMDA EPSCs induced by d-serine in sham and post-SE animals. Data points indicate individual experiments. Application of d-serine induces no significant change of perisynaptic NMDA EPSC amplitude in the sham group, whereas NMDA EPSC amplitudes are significantly increased in the post-SE group (*p < 0.05). F, Representative sample traces showing the effect of the glutamate transporter inhibitor TBOA on perisynaptic NMDAR-mediated EPSCs under saturating glycine-binding site conditions. Black traces indicate NMDA EPSCs after stimulation in the presence of MK-801 and in the continuous presence of d-serine. Red traces indicate NMDA EPSCs after additional application of TBOA. Application of TBOA increased perisynaptic NMDA EPSCs to the same extent in sham and post-SE groups.
The maximal perisynaptic NMDA EPSCs with saturating concentrations of exogenous d-serine were larger in epileptic compared with control animals (Fig. 5E). This could be attributable to an increase in the number of perisynaptic NMDAs or a more efficient activation of these receptors by synaptically released glutamate, i.e., via a downregulation of glutamate uptake transporters (Diamond, 2001; Arnth-Jensen et al., 2002). We addressed the latter possibility by evaluating the effects of the glutamate uptake blocker dl-threo-β-benzyloxyaspartic acid (TBOA) (30 μm) on perisynaptic EPSCs. Perisynaptic EPSCs were isolated as described in Figure 5, B and D, with application of MK-801 but with saturating concentrations of d-serine present during the whole experiment. TBOA caused potentiation of perisynaptic EPSCs in both control and pilocarpine-treated animals to the same extent (sham. 58.2 ± 9.3%; post-SE, 47.7 ± 14%; example traces in Fig. 5F, n = 5 and n = 7 respectively, NS). Moreover, perisynaptic EPSCs were still significantly larger in pilocarpine-treated compared with control animals in the presence of TBOA (sham, −17.2 ± 1.2 vs post-SE, −39.3 ± 2.9 pA, p = 0.0001, ratio of perisynaptic burst EPSC and initial burst EPSC: sham, 0.15 ± 0.02 vs post-SE, 0.34 ± 0.05, p = 0.006, Mann–Whitney test).
These results indicate that a higher density of extrasynaptic or perisynaptic NMDARs is present in epileptic animals. To further support this idea, we recorded tonic NMDA currents from granule cells by clamping neurons to +40 mV. This allowed us to isolate the tonic NMDA current via application of d-APV in control versus epileptic rats (Fig. 6). The magnitude of the tonic NMDA current proved larger in epileptic compared with sham-control rats (sham, 26.5 ± 6.3 pA, n = 6; post-SE, 67.0 ± 13.6 pA, n = 5, p = 0.018, unpaired t test).
Figure 6.

Increased tonic NMDA currents in granule cells from epileptic animals. Tonic NMDA currents recorded in epileptic animals by clamping neurons to +40 mV and applying the NMDAR blocker d-AP-5. The magnitude of the tonic NMDA current proved larger in epileptic compared with sham-control rats.
The small magnitude of perisynaptic NMDA EPSCs in control animals precluded an analysis of the contribution of different NMDAR subtypes. In epileptic animals, in which perisynaptic NMDA EPSCs were larger, we examined the contribution of NR2B-containing subunits by applying Ro25-6981 (0.5 μm). The experiments were done as in Figure 4, but d-serine was included from the onset in all recording solutions. After washout of MK-801 and another 5 min baseline recording period, Ro25-6981 was applied and caused a reduction of the perisynaptic EPSC amplitude by 40 ± 5% (n = 5), a fraction similar to the synaptic NMDARs in epileptic animals.
Collectively, these data support the notion that extrasynaptic or perisynaptic NMDA EPSCs are larger in epileptic animals, most likely as a result of an increased density of these receptors. Moreover, the coagonist binding site of these receptors seems to be unsaturated as evidenced by the facilitating effects of exogenously applied coagonists in epileptic animals.
Impaired LTP in the epileptic DG is rescued by exogenous d-serine
The results so far indicate that synaptic NMDAR coagonist binding sites are less occupied in epileptic animals, most likely as a result of a deficit in d-serine caused by DAO upregulation. Because coactivation of the NMDAR coagonist site by glial release of d-serine is obligatory in the induction of LTP (Henneberger et al., 2010), we asked whether synaptic plasticity is impaired at LPP synapses. In this pathway, tetanic stimulation of slices caused stable LTP in control animals (131 ± 6%, n = 10; Fig. 7A, open symbols, p = 0.005, Wilcoxon's signed-rank test vs baseline, LTP success rate of 90%) but failed to elicit stable synaptic changes in epileptic animals [102 ± 5%, n = 14 (filled symbols), p = 0.925, Wilcoxon's signed-rank test vs baseline, comparison of LTP levels with control animals, p = 0.001, unpaired t test; Fig. 7A, see summary in F, LTP success rate of 43%]. Similarly, the post-tetanic potentiation was strongly reduced in epileptic animals (123 ± 4 vs 171 ± 9%, p = 0.003, unpaired t test). Unlike the LPP, LTP in the MPP was not significantly affected (146 ± 10%, n = 14 in pilocarpine-treated rats vs 175 ± 17%, n = 13 in control rats, p = 0.168; data not shown). Input–output curves did not reveal any significant differences between control animals (n = 7) and animals that had experienced SE (n = 10), in either the LPP or MPP (LPP: maximum fEPSP, p = 0.188; stimulation intensity required for half maximal fEPSP amplitudes (xhalf), p = 0.483; MPP: maximum fEPSP, p = 0.421; xhalf, p = 0.333, unpaired t test of curve parameters obtained by fitting with a sigmoidal function; data not shown).
Figure 7.

Impaired LTP in the epileptic DG is rescued by exogenous d-serine. A, Tetanic stimulation of the LPP with a high-frequency patterned stimulation (HFS) causes stable LTP in control animals (open symbols) but not in epileptic animals (filled symbols). B, Short-term plasticity of LPP and MPP synapses during stimulation trains of 10 stimuli at different frequencies. C, LTP at the LPP is restored by exogenous d-serine (100 μm). D, Low concentrations of the glycine site antagonist CGP78608 (0.2 μm) cause a reversal of the rescue effect without affecting LTP in control animals. E, Blocking NR2B-containing NMDARs blocks rescued LTP in epileptic animals. F, Summary of levels of potentiation under native conditions, followed by application of d-serine (D-ser) and after combined application of d-serine and CGP78608. Asterisks indicate significant differences in the post hoc Tukey's test. G, Summary of the effects of subunit-selective NMDAR blockers on LTP under conditions of saturating concentrations of the coagonist d-serine, in control and epileptic animals (white and black bars, respectively). **p < 0.01.
Using an interstimulus interval of 40 ms, the LPP responses exhibited a typical paired-pulse facilitation (paired-pulse ratio: 136 ± 4%, n = 24), whereas paired-pulse depression was always observed in the MPP (paired-pulse ratio: 84 ± 4%, n = 15). In both portions of the perforant path, this form of short-term plasticity remained unchanged after SE (paired-pulse ratios of 127 ± 5%, n = 15 and 88 ± 3%, n = 14 in the LPP and MPP, respectively). As described previously (Bushell et al., 1995; Dietrich et al., 1997), the metabotropic glutamate receptor group III selective agonist l-AP-4 could be used to further discriminate the lateral and medial aspects of the perforant path in control animals. l-AP-4 (10 μm) selectively inhibited LPP-evoked fEPSPs (to 46 ± 5% in LPP, n = 6 vs 114 ± 8% in MPP, n = 5, p < 0.01). Unlike NMDA EPSCs, fEPSPs were insensitive to l-AP-4 after SE (fEPSP amplitude, 101 ± 11% of control in LPP vs 117 ± 2% in MPP). One explanation for this discrepancy may be that the stronger stimulation incurred with bipolar stimulation electrodes leads to a pronounced glutamate release in epileptic animals that saturates presynaptic mGluRs. In contrast to LTP, short-term plasticity of both LPP and MPP synapses during brief stimulation trains at various frequencies (10 stimulations at 0.1, 1, 5, 10, and 20 Hz) was unaltered (Fig. 7B). Thus, SE causes a long-lasting loss of NMDA-dependent long-term, but not short-term, plasticity at LPP synapses.
If loss of LTP is caused by a deficit in d-serine in epileptic hippocampus, addition of exogenous d-serine during LTP induction should restore LTP in epileptic rats. Saturating concentrations of d-serine (100 μm) were applied via the bath solution, and equilibration was allowed to take place for 1–2 h. LTP levels were strongly impaired in pilocarpine animals in the absence of d-serine, as described above, whereas in the presence of d-serine, LTP levels in sham and epileptic animals did not seem different (sham, 120 ± 4%, n = 14, LTP success rate of 79%; post-SE, 122 ± 7%, n = 13, LTP success rate of 77%; Fig. 7C). In the presence of both d-serine and low concentrations of the glycine site antagonist CGP78608 ([(1S)-1-[[(7-Bromo-1,2,3,4-tetrahydro-2,3-dioxo-5-quinoxalinyl)methyl]amino]ethyl]phosphonic acid hydrochloride; 0.2 μm), significant differences between sham and epileptic animals could again be observed (Fig. 7D), similar to the native condition. ANOVA revealed a significant effect of pilocarpine treatment (F(1,67) = 17.24, p < 0.0001), no effect of drug treatment (F(2,67) = 0.33, p = 0.72), but a significant interaction between these factors (F(2,67) = 5.89, p = 0.004). Tukey's post hoc tests to uncover differences between individual groups revealed significant differences between sham and post-SE groups in the absence of d-serine (p = 0.005) and in the presence of both d-serine and CGP78608 (p = 0.003) but not in the presence of d-serine only (p = 0.99, indicated by asterisks in Fig. 7F). This suggests that an action on the glycine modulatory site is relevant for the actions of d-serine. So far, these experiments are consistent with the idea that lack of d-serine causes a deficient activation of NMDARs during LTP induction and that this causes impaired LTP.
Mechanism of LTP rescue in epileptic animals
We next tested which types of NMDARs are important for the LTP rescue effect in epileptic animals, i.e., in the continued presence of 100 μm d-serine (Fig. 7G, leftmost bars indicate potentiation in the presence of 100 μm d-serine for the sake of comparison). We applied the selective NR2B antagonist Ro25-6981 (0.5 μm), the selective NR2C/D antagonist PPDA (0.3 μm), or the NR2A-preferring blocker NVP-AAM077 (50 nm; Fig. 7G). In sham-control animals, significant potentiation versus baseline was obtained, regardless of which NMDAR antagonist was present (potentiation of 125 ± 9%, n = 9; 114 ± 4%, n = 13; and 123 ± 5%, n = 12, respectively; p = 0.015, 0.019, and 0.002, respectively, Wilcoxon's signed-rank test vs baseline). In pilocarpine-treated animals, no significant potentiation was observed in the presence of Ro25-6981 (96 ± 9%, n = 10, p = 0.646) or PPDA (110 ± 13%, n = 9, p = 0.767). In the presence of NVP-AAM077, significant potentiation was observed (120 ± 5%, n = 7, p = 0.046, Wilcoxon's signed-rank test vs baseline). The loss of significant LTP in the presence of Ro25-6981 is consistent with the relative upregulation of synaptic NR2B-containing receptors described in Figure 2. However, it should be noted that ANOVA did not reveal significant effects of treatment over all compounds tested.
Impaired spatial memory after SE is reversed by dietary d-serine
Although the contribution of different NMDAR subunit combinations could not be well resolved, synaptic plasticity in pilocarpine-treated animals was rescued to the level of control animals by d-serine application. Therefore, we tested whether d-serine application in epileptic rats is associated with a rescue of cognitive dysfunction observed in these animals. We analyzed spatial memory in control and pilocarpine-treated rats, with and without oral d-serine supplementation (see Materials and Methods). Feeding the rats with 32 mg/kg resulted in enhanced hippocampal d-serine content in both control and epileptic animals (from 25.6 ± 0.6 to 31.1 ± 0.8% in control animals; from 22.7 ± 0.4 to 28.0 ± 0.6% in epileptic animals). ANOVA revealed a significant effect of chronic epilepsy (F(1,30) = 12.3, p = 0.001), as well as a significant effect of d-serine treatment (F(1,30) = 28.2, p < 0.0001), with no interaction between these two factors. Importantly, the supplementation was able to rescue d-serine levels in epileptic animals to a magnitude that was not significantly different from control animals.
Pilocarpine-treated rats displayed a severe learning deficit in the Morris water maze. We first tested the acquisition of platform position on the first six consecutive trials on the first day of the behavioral experiment. In these trials, sham-control animals acquire the platform location well, whereas epileptic animals perform much worse, regardless of whether they were treated with d-serine or not (Fig. 8A, open vs filled symbols). The difference between the average distance traveled during trials 1 and 2 versus trials 5 and 6 was accordingly much larger in sham compared with epileptic animals (ANOVA, F(3,36) = 4.08, p = 0.014, significant effect of pilocarpine treatment, F(1,36) = 12.0, p = 0.001, no significant effect of d-serine treatment and no significant interaction between these factors). Individual comparisons with a Tukey's post hoc test revealed significant differences between the sham versus post-SE groups (p = 0.016) and the sham d-serine versus post-SE d-serine groups (p = 0.041, indicated by asterisks in Fig. 8B). This indicates a strong deficit in memory acquisition during these consecutive trials in epileptic animals that does not seem to be affected by d-serine.
Figure 8.

Impaired spatial memory after SE is reversed by dietary d-serine. A, The distance moved to reach the platform on the first day (6 consecutive trials) shows a significant working memory deficit in post-SE animals compared with sham control. However, there was no treatment effect of d-serine. B, The difference of the average distance traveled during trials 1 and 2 and trials 5 and 6 was significantly different between sham and epileptic animals during consecutive trials of the first day. This indicates impaired learning in epileptic animals that does not seem to be affected by d-serine. C, The average distance traveled during the first trial of each day until reaching the platform, quantified during 7 consecutive days of training. Note the significant deficit in acquiring the spatial information in post-SE animals and a significant improvement by oral d-serine supplementation in epileptic rats on days 6 and 7. D, Representative traces of the probe trial on day 8. The platform was removed, but for the sake of clarity, the platform position during the days 1–7 is indicated in gray. Data are depicted such that the target quadrant is in the top right corner for all animals. E, Average occupancy of the target and nontarget quadrants during the probe trial. Chance level is at 25% (indicated by red line). Sham-control animals prefer the target quadrant regardless of whether they are treated with d-serine, demonstrating intact spatial memory. The performance of post-SE animals in the probe trial is at chance level, whereas d-serine-treated post-SE animals show some recovery of target quadrant preference. *p < 0.05, **p < 0.01, ***p < 0.001.
Subsequently, we examined the acquisition of platform position on the consecutive 7 d of the acquisition period. Although sham-control animals invariably showed stable acquisition of the platform position (Fig. 8C, open symbols), pilocarpine-treated animals were strongly impaired (Fig. 8C, filled circles). d-Serine treatment of pilocarpine-treated animals appeared to cause a marked improvement in acquisition (Fig. 8C, filled squares). When analyzing the average performance on the last 2 d of the acquisition period, ANOVA revealed a significant effect of pilocarpine treatment (F(1,36) = 42.1, p < 0.0001), a significant effect of d-serine treatment (F(1,36) = 7.23, p = 0.011), and a significant interaction between the two factors (F(1,36) = 9.11, p = 0.005). Individual comparisons with a Tukey's post hoc test revealed that the post-SE group acquired the task significantly worse than either sham-control group (p < 0.0001) and that d-serine treatment significantly improved performance in epileptic animals (post-SE vs post-SE d-serine, p < 0.001). We then performed a probe trial in the absence of the platform on the day immediately after the 7 d acquisition period (Fig. 8D). Sham-control animals showed stable retention of the platform position, evidenced by a preferential occupancy of the target quadrant. Epileptic animals without d-serine treatment searched equally in target and nontarget quadrant. d-Serine application in epileptic animals appeared to reconstitute preference of the target quadrant (Fig. 8E). ANOVA revealed a significant effect of pilocarpine treatment (F(1,36) = 5.44, p = 0.025) but not of d-serine treatment (F(1,36) = 2,22, p = 0.145) and no interaction between these two factors.
Epileptic rats showed a significantly higher swimming velocity compared with control animals, whereas there was no significant effect of treatment with d-serine (post-SE, 23.4 ± 0.2 cm/s; post-SE d-serine, 23.3 ± 0.3 cm/s; sham, 21.1 ± 0.5 cm/s; sham d-serine, 20.4 ± 0.3 cm/s; ANOVA on ranks; Tukey's post hoc test: post-SE vs sham, p < 0.05; post-SE d-serine vs post-SE, p > 0.05; sham d-serine vs sham, p > 0.05). We also analyzed thigmotaxis by determining the fraction of time spent close to the rim (within 10 cm). Epileptic rats showed significantly higher thigmotaxis compared with control animals (post-SE, 58.5 ± 3.6%; post-SE d-serine, 50.3 ± 4.0%; sham, 28.1 ± 3.1%; sham d-serine, 28.3 ± 4.0%; ANOVA, Holm–Sidak post hoc test: post-SE vs sham, p < 0.001; post-SE d-serine vs post-SE, p = 0.133; sham d-serine vs sham, p = 0.965), but there was no significant effect of d-serine on thigmotaxis in both experimental groups. This suggests that the d-serine treatment effects seen in memory acquisition are not likely to be caused by changes in motor function or altered thigmotaxis.
Effects on seizure frequency after dietary d-serine administration
These results demonstrate that epileptic animals show a severe spatial learning deficit. Furthermore, reconstituting CNS d-serine levels significantly reversed deficits in the acquisition of spatial memories. Together, these results suggest that reversing deficient NMDAR function and synaptic plasticity with exogenous d-serine leads to improvement in cognitive deficits associated with chronic epilepsy. However, increasing NMDAR function in the epileptic brain might also have unwanted effects, for instance, an increase in seizure frequency. Therefore, we examined seizure frequencies by video monitoring in epileptic animals receiving dietary d-serine and compared them with epileptic animals without d-serine supplementation (see Materials and Methods). These experiments revealed average seizure frequencies of 0.6 ± 0.2 versus 0.9 ± 0.2 generalized stage 4/5 seizures per day in animals without and with d-serine treatment, respectively (p = 0.43, unpaired t test, n = 15 for both groups). Likewise, the severity of the seizures when classified as described in Materials and Methods was not significantly different (average severity score, 2.6 ± 0.7 and 3.5 ± 0.9, p = 0.30, Mann–Whitney test).
Discussion
NMDARs are central for induction of most forms of synaptic long-term plasticity and for learning and memory. The activation of the NMDAR complex and the resulting synaptic plasticity requires not only binding of the neurotransmitter glutamate but also occupancy of a coagonist binding site on the NR1 subunit (Schell et al., 1995; Mothet et al., 2000; Panatier et al., 2006; Henneberger et al., 2010). In this study, we show marked changes in excitatory synaptic transmission in epilepsy. First, we show a change in NMDAR subunit composition, with synaptic NR2B-containing receptors being more prevalent in epileptic neurons. Second, we show that levels of the endogenous NMDAR coagonist d-serine are strongly reduced in the epileptic brain. This leads to desaturation of synaptic NMDARs, causing a reduction in synaptic NMDA currents. The modulatory site of perisynaptic receptors is also desaturated, with a compensatory increase in the number of perisynaptic NMDARs. Finally, we demonstrate that the deficit in NMDAR-dependent synaptic transmission is associated with deficient synaptic long-term plasticity and that plasticity as well as learning and memory are reinstated by exogenous application of the coagonist d-serine in vitro and in vivo.
Changes in d-serine mediated cotransmission in epilepsy
d-Serine is considered to be the predominant physiological ligand of the NMDAR coagonist site in forebrain regions (Panatier et al., 2006) and has been shown to be more effective in potentiating the NMDAR than glycine (Matsui et al., 1995; Stevens et al., 2010). Our data suggest that the coagonist binding sites of synaptic NMDARs in the hippocampus are not saturated, consistent with previous studies (Yang et al., 2003; Junjaud et al., 2006; Henneberger et al., 2010). Our results further suggest that epilepsy results in a significant decrease of d-serine, which is associated with a desaturation of the coagonist binding site of synaptic and extrasynaptic NMDARs. We found that the mRNA expression of the d-serine synthesizing enzyme serine racemase is decreased in the CA1 region, with a concomitant significant decrease in hippocampal protein levels. This finding is similar to the changes observed in the aging brain, in which decreased d-serine content, desaturation of NMDARs, and impaired LTP were linked to an decreased expression of serine racemase (Junjaud et al., 2006; Mothet et al., 2006). In addition, we found a significant increase in the expression of the d-serine degrading enzyme DAO from very low levels normally present in forebrain but were unable to verify this finding with Western blot analyses because of the still very low levels of this protein. Thus, although DAO could contribute to decreased d-serine content, the significance of this finding is unclear. These mechanisms do not exclude the contribution of additional mechanisms. For instance, protein kinase C regulates d-serine availability (Vargas-Lopes et al., 2011), and the expression of PKC isoforms is upregulated in some epilepsy models (Guglielmetti et al., 1997). EphrinB powerfully regulates d-serine synthesis and release in astrocytes, but how this molecule is regulated in epilepsy is not known (Zhuang et al., 2010).
Mechanism of d-serine-mediated restoration of synaptic plasticity
We found that supplying exogenous d-serine to compensate for the loss of endogenous d-serine was sufficient to restore impaired LTP in the epileptic DG. We have shown that d-serine application is sufficient to rescue impaired LTP at the LPP to dentate granule cell (DGC) synapse. What is the mechanism of this rescue? Our data show that saturating d-serine concentrations caused a potentiation of synaptic NMDARs but also in particular perisynaptic NMDA EPSCs in chronic epilepsy. Our experiments cannot discriminate whether the d-serine action on synaptic or perisynaptic sites is more important in the rescue phenomenon. Most likely, both are involved. For instance, perisynaptically located NMDARs have been shown to be involved in the induction of LTP (Harney et al., 2008). Furthermore, Henneberger et al. (2010) showed that LTP induction at the Schaffer collateral to CA1 synapse depends on d-serine release from astrocytes. It is likely that this reflects, at least in part, activation of extrasynaptic NMDARs by astrocytic glutamate. An intriguing corollary of our finding is that, at least in the DG, downstream signaling cascades for the expression of LTP seem to be intact, suggesting that substitution of exogenous d-serine might indeed be a successful strategy to restitute deficient LTP and cognition in vivo.
Changes in NMDAR subunit composition
A changed NMDAR subunit composition was observed at the LPP to DGC in chronic epilepsy. Although functional expression of NR2B-containing receptors was significantly increased, functional expression of NR2C/D-containing receptors was reduced. This change in subunit composition was associated with significantly slower NMDA EPSC kinetics, consistent with the slower kinetics of NR2B-containing receptors (Erreger et al., 2005; Rauner and Köhr, 2011). Increases in NR2B expression have been observed in various forms of epilepsy models, as well as human epilepsy. An increased NR2B expression in dysplastic human neocortex was correlated with the expression of in situ epileptogenesis in human cortical dysplasia, which is often associated with pharmacoresistant epilepsy (Möddel et al., 2005). Furthermore, a recent study of short-term effects of pilocarpine in primary cultures of hippocampal neurons showed an increased expression of NR2B subunits (Sun et al., 2009; Di Maio et al., 2011, Müller et al., 2013; but see Auzmendi et al., 2009). Our findings rely on the use of subunit-specific antagonists and therefore are contingent on their specificity. Based on IC50 values for NR2B, the selectivity of Ro25-6981 for NR2B- over NR2A-containing NMDARs is considered to be ∼5000-fold (Fischer et al., 1997) and was used in very low concentrations (0.5 μm) to further ensure specificity. The NR2A-selective antagonist NVP-AAM077 is 100-fold selective for NR2A- over NR2B-containing receptors (Auberson et al., 2002) at low concentrations equivalent to those used in this study (Feng et al., 2004) and has been used extensively to characterize native NR2A subunit function (Liu et al., 2004; Berberich et al., 2005). It should be noted that the NR2C/D-selective antagonist PPDA at the concentration range used is less selective, with a threefold to sixfold higher affinity for NR1A/NR2D receptors compared with NR1/NR2A or NR1/NR2B (Feng et al., 2004). Nevertheless, this compound has also been used to pharmacologically dissect the role of NR2D-containing receptors in synaptic plasticity (Hrabetova et al., 2000; Harney et al., 2008).
d-Serine and novel therapies in epilepsy
Our results suggest that exogenous d-serine cannot only restitute NMDAR function and synaptic plasticity but can also improve cognitive function in epileptic animals. Indeed, evidence is accumulating that d-serine might be useful in treating cognitive dysfunction. d-Serine improves performance in recognition memory and working memory tasks in mice (Bado et al., 2011), as well as social memory in rats (Shimazaki et al., 2010). In schizophrenia, a reduction in NMDAR function may account for a substantial portion of the dysfunction seen in this disorder (Coyle, 2006). Blocking NMDARs (Krystal et al., 2002) or genetically reducing NMDAR function in interneurons causes schizophrenia-like symptoms (Belforte et al., 2010). Interestingly, d-serine was found to improve positive, negative, and cognitive symptoms in patients with schizophrenia (Tsai et al., 1998, 1999). Collectively, this suggests that augmenting NMDA-mediated transmission through the NMDA coagonist binding site is promising for the pharmacotherapy of schizophrenia but perhaps also of epilepsy. One advantage of d-serine may be the allosteric nature of NMDAR augmentation, as well as the fact that it affects no other known neurotransmitter system, including the strychnine-sensitive inhibitory glycine receptor. An alternate strategy to increase d-serine levels has been the development of DAO inhibitors for the treatment of schizophrenia, which are currently being examined for clinical efficacy (Smith et al., 2009, 2010). Because DAO expression was elevated in the DG in chronic epilepsy, at least on the mRNA level, such compounds might be candidates for treating disturbed cognition in chronic epilepsy. DAO inhibitors might be safer than d-serine, because DAO catabolism of high doses of d-serine can have cytotoxic effects in astrocytes (Park et al., 2006; Chung et al., 2010). The fact that disturbed metabolism of d-serine is a feature of both epilepsy and psychosis and that d-serine treatment alleviates symptoms in both disorders is also intriguing in view of the fact that there is a clinical comorbidity of both disorders (Johnson and Shorvon, 2011; Clarke et al., 2012; Hesdorffer et al., 2012).
One critical question in using d-serine as a treatment option in epilepsy is whether the increase in NMDAR function that benefits cognition might cause increased seizures attributable to augmented NMDAR-dependent excitation. We found no significant effect on seizure frequencies measured by video monitoring in epileptic animals receiving dietary d-serine. It should be noted that video monitoring may not detect all partial seizures and that a definitive determination of partial seizure frequencies would require long-term telemetric analysis. Nevertheless, these results indicate that there are no deleterious effects of d-serine on generalized seizure frequency.
Together, our results have uncovered a deficiency in d-serine as a cause for deficient NMDAR activation and synaptic plasticity, as well as cognitive function in chronic epilepsy, and suggest novel avenues to treat cognitive dysfunction in epilepsy patients.
Footnotes
This work was supported by the German Research Foundation (Collaborative Research Centre TR3), Functional Genomics and Neurobiology of Epilepsy: a Basis for New Therapeutic Strategies, European Research Area ERA-NET Neuron “EpiNet,” the program of the Federal Ministry of Education and Research/Project Management Agency of the National Aeronautics and Space Research Center 01GW0511, the National Genome Research Net, and the Intramural Research Program of the Medical Faculty of the University of Bonn. We thank T. Ivanova for contributing electrophysiological recordings.
References
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphonomethylqui-noxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/S0960-894X(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Auzmendi J, González N, Girardi E. The NMDAR subunit NR2B expression is modified in hippocampus after repetitive seizures. Neurochem Res. 2009;34:819–826. doi: 10.1007/s11064-008-9828-0. [DOI] [PubMed] [Google Scholar]
- Bado P, Madeira C, Vargas-Lopes C, Moulin TC, Wasilewska-Sampaio AP, Maretti L, de Oliveira RV, Amaral OB, Panizzutti R. Effects of low-dose d-serine on recognition and working memory in mice. Psychopharmacology (Berl) 2011;218:461–470. doi: 10.1007/s00213-011-2330-4. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Tam B, Collingridge GL. Activation of the glycine site in the NMDA receptor is necessary for the induction of LTP. Neurosci Lett. 1990;108:261–266. doi: 10.1016/0304-3940(90)90651-O. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boks MP, Rietkerk T, van de Beek MH, Sommer IE, de Koning TJ, Kahn RS. Reviewing the role of the genes G72 and DAO in glutamate neurotransmission in schizophrenia. Eur Neuropsychopharmacol. 2007;17:567–572. doi: 10.1016/j.euroneuro.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Bushell TJ, Jane DE, Tse HW, Watkins JC, Davies CH, Garthwaite J, Collingridge GL. Rapid communication. Neuropharmacoloy. 1995;34:239–241. doi: 10.1016/0028-3908(95)00012-U. [DOI] [PubMed] [Google Scholar]
- Cavalheiro EA, Silva DF, Turski WA, Calderazzo-Filho LS, Bortolotto ZA, Turski L. The susceptibility of rats to pilocarpine-induced seizures is age-dependet. Brain Res. 1987;465:43–58. doi: 10.1016/0165-3806(87)90227-6. [DOI] [PubMed] [Google Scholar]
- Chen J, Sochivko D, Beck H, Marechal D, Wiestler OD, Becker AJ. Activity-induced expression of common reference genes in individual CNS neurons. Lab Invest. 2001;81:913–916. doi: 10.1038/labinvest.3780300. [DOI] [PubMed] [Google Scholar]
- Chung SP, Sogabe K, Park HK, Song Y, Ono K, Abou El-Magd RM, Shishido Y, Yorita K, Sakai T, Fukui K. Potential cytotoxic effect of hydroxypyruvate produced from d-serine by astroglial d-amino acid oxidase. J Biochem. 2010;148:743–753. doi: 10.1093/jb/mvq112. [DOI] [PubMed] [Google Scholar]
- Clarke MC, Tanskanen A, Huttunen MO, Clancy M, Cotter DR, Cannon M. Evidence for shared susceptibility to epilepsy and psychosis: a population-based family study. Biol Psychiatry. 2012;71:836–839. doi: 10.1016/j.biopsych.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26:365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Mastroberardino PG, Hu X, Montero L, Greenamyre JT. Pilocapine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK1/2 mechanisms. Neurobiol Dis. 2011;42:482–495. doi: 10.1016/j.nbd.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Dietrich D, Beck H, Kral T, Clusmann H, Elger CE, Schramm J. Metabotropic glutamate receptors modulate synaptic transmission in the perforant path: pharmacology and localization of two distinct receptors. Brain Res. 1997;767:220–227. doi: 10.1016/S0006-8993(97)00579-9. [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Tse HW, Skifter DA, Morley R, Jane DE, Monaghan DT. Structure-activity analysis of a novel NR2C//NR2D-preferring NMDA receptor antagonist: 1-(phenanthrene-2-carbonyl) piperazine-2,3-dicarboxylic acid. Br J Pharmacol. 2004;141:508–516. doi: 10.1038/sj.bjp.0705644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25–6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vivo. J Pharmacol Exp Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Fossat P, Turpin FR, Sacchi S, Dulong J, Shi T, Rivet JM, Sweedler JV, Pollegioni L, Millan MJ, Oliet SH, Mothet JP. Glial d-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cereb Cortex. 2012;22:595–606. doi: 10.1093/cercor/bhr130. [DOI] [PubMed] [Google Scholar]
- Grant SL, Shulman Y, Tibbo P, Hampson DR, Baker GB. Determination of d-serine and related neuroactive amino acids in human plasma by high-performance liquid chromatography with fluorimetric detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;844:278–282. doi: 10.1016/j.jchromb.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Guglielmetti F, Rattray M, Baldessari S, Butelli E, Samanin R, Bendotti C. Selective up-regulation of protein kinase C epsilon in granule cells after kainic acid-induced seizures in rat. Brain Res Mol Brain Res. 1997;49:188–196. doi: 10.1016/S0169-328X(97)00142-3. [DOI] [PubMed] [Google Scholar]
- Harney SC, Jane DE, Anwyl R. Extrasynaptic NR2D-containing NMDARs are recruited to the synapse during LTP of NMDAR-EPSCs. J Neurosci. 2008;28:11685–11694. doi: 10.1523/JNEUROSCI.3035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, Nakazato M, Kumakiri C, Okada S, Hasegawa H, Imai K, Iyo M. Decreased serum levels of d-serine in patients with schizophrenia: evidence in support of the N-methyl-d-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572–576. doi: 10.1001/archpsyc.60.6.572. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of d-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesdorffer DC, Ishihara L, Mynepalli L, Webb DJ, Weil J, Hauser WA. Epilepsy, suicidality, and psychiatric disorders: a bidirectional association. Ann Neurol. 2012;72:184–191. doi: 10.1002/ana.23601. [DOI] [PubMed] [Google Scholar]
- Hikida T, Mustafa AK, Maeda K, Fujii K, Barrow RK, Saleh M, Huganir RL, Snyder SH, Hashimoto K, Sawa A. Modulation of d-serine levels in brains of mice lacking PICK1. Biol Psychiatry. 2008;63:997–1000. doi: 10.1016/j.biopsych.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabetova S, Serrano P, Blace N, Tse HW, Skifter DA, Jane DE, Monaghan DT, Sacktor TC. Distinct NMDA receptor subpopulations contribute to long-term potentiation and long-term depression induction. J Neurosci. 2000;20:RC81. doi: 10.1523/JNEUROSCI.20-12-j0002.2000. (1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Johnson MR, Shorvon SD. Heredity in epilepsy: neurodevelopment, comorbidity, and the neurological trait. Epilepsy Behav. 2011;22:421–427. doi: 10.1016/j.yebeh.2011.07.031. [DOI] [PubMed] [Google Scholar]
- Junjaud G, Rouaud E, Turpin F, Mothet JP, Billard JM. Age-related effects of the neuromodulator d-serine on neurotransmission and synaptic potentiation in the CA1 hippocampal area of the rat. J Neurochem. 2006;98:1159–1166. doi: 10.1111/j.1471-4159.2006.03944.x. [DOI] [PubMed] [Google Scholar]
- Kemp JA, Leeson PD. The glycine site of the NMDA receptor—five years on. Trends Pharmacol Sci. 1993;14:20–25. doi: 10.1016/0165-6147(93)90108-V. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Anand A, Moghaddam B. Effects of NMDA receptor antagonists: implications for the pathophysiology of schizophrenia. Arch Gen Psychiatry. 2002;59:663–664. doi: 10.1001/archpsyc.59.7.663. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci. 1998;21:8–14. doi: 10.1016/S0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- Labrie V, Fukumura R, Rastogi A, Fick LJ, Wang W, Boutros PC, Kennedy JL, Semeralul MO, Lee FH, Baker GB, Belsham DD, Barger SW, Gondo Y, Wong AH, Roder JC. Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum Mol Genet. 2009;18:3227–3243. doi: 10.1093/hmg/ddp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie V, Wang W, Barger SW, Baker GB, Roder JC. Genetic loss of d-amino acid oxidase activity reverses schizophrenia-like phenotypes in mice. Genes Brain Behav. 2010;9:11–25. doi: 10.1111/j.1601-183X.2009.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lozovaya NA, Grebenyuk SE, Tsintsadze TSh, Feng B, Monaghan DT, Krishtal OA. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape “superslow” afterburst EPSC in rat hippocampus. J Physiol. 2004;558:451–463. doi: 10.1113/jphysiol.2004.063792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Sekiguchi M, Hashimoto A, Tomita U, Nishikawa T, Wada K. Functional comparison of d-serine and glycine in rodents: the effect on cloned NMDA receptors and the extracellular concentration. J Neurochem. 1995;65:454–458. doi: 10.1046/j.1471-4159.1995.65010454.x. [DOI] [PubMed] [Google Scholar]
- Mizutani A, Saito H, Abe K. Evidence for involvement of endogenous glycine in the induction of long-term potentiation in the dentate gyrus of anesthetized rats. Eur J Pharmacol. 1991;205:303–305. doi: 10.1016/0014-2999(91)90914-C. [DOI] [PubMed] [Google Scholar]
- Möddel G, Jacobson B, Ying Z, Janigro D, Bingaman W, González-Martínez J, Kellinghaus C, Prayson RA, Najm IM. The NMDA receptor NR2B subunit contributes to epileptogenesis in human cortical dysplasia. Brain Res. 2005;1046:10–23. doi: 10.1016/j.brainres.2005.03.042. [DOI] [PubMed] [Google Scholar]
- Mothet JP, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. d-serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc Natl Acad Sci U S A. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Rouaud E, Sinet PM, Potier B, Jouvenceau A, Dutar P, Videau C, Epelbaum J, Billard JM. A critical role for the glial-derived neuromodulator d-serine in the age-related deficits of cellular mechanisms of learning and memory. Aging Cell. 2006;5:267–274. doi: 10.1111/j.1474-9726.2006.00216.x. [DOI] [PubMed] [Google Scholar]
- Müller L, Tokay T, Porath K, Köhling R, Kirschstein T. Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol Dis. 2013;54:183–193. doi: 10.1016/j.nbd.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Park HK, Shishido Y, Ichise-Shishido S, Kawazoe T, Ono K, Iwana S, Tomita Y, Yorita K, Sakai T, Fukui K. Potential role for astroglial d-amino acid oxidase in extracellular d-serine metabolism and cytotoxicity. J Biochem. 2006;139:295–304. doi: 10.1093/jb/mvj036. [DOI] [PubMed] [Google Scholar]
- Rauner C, Köhr G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-d-aspartate receptor population in adult hippocampal synapses. J Biol Chem. 2011;286:7558–7566. doi: 10.1074/jbc.M110.182600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell MJ, Molliver ME, Snyder SH. d-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci U S A. 1995;92:3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki T, Kaku A, Chaki S. d-Serine and a glycine transporter-1 inhibitor enhance social memory in rats. Psychopharmacology (Berl) 2010;209:263–270. doi: 10.1007/s00213-010-1794-y. [DOI] [PubMed] [Google Scholar]
- Smith SM, Uslaner JM, Yao L, Mullins CM, Surles NO, Huszar SL, McNaughton CH, Pascarella DM, Kandebo M, Hinchliffe RM, Sparey T, Brandon NJ, Jones B, Venkatraman S, Young MB, Sachs N, Jacobson MA, Hutson PH. The behavioral and neurochemical effects of a novel d-amino acid oxidase inhibitor compound 8 [4H-thieno [3,2-b]pyrrole-5-carboxylic acid] and d-serine. J Pharmacol Exp Ther. 2009;328:921–930. doi: 10.1124/jpet.108.147884. [DOI] [PubMed] [Google Scholar]
- Smith SM, Uslaner JM, Hutson PH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med Chem J. 2010;4:3–9. doi: 10.2174/1874104501004020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens ER, Gustafson EC, Miller RF. Glycine transport accounts for the differential role of glycine vs. d-serine at NMDA receptor coagonist sites in the salamander retina. Eur J Neurosci. 2010;31:808–816. doi: 10.1111/j.1460-9568.2010.07135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QJ, Duan RS, Wang AH, Shang W, Zhang T, Zhang XQ, Chi ZF. Alterations of NR2B and PSD-95 expression in hippocampus of kainic acid-exposed rats with behavioural deficits. Behav Brain Res. 2009;201:292–299. doi: 10.1016/j.bbr.2009.02.027. [DOI] [PubMed] [Google Scholar]
- Thiels E, Weisz DJ, Berger TW. In vivo modulation of N-methyl-d-aspartate receptor-dependent long-term potentiation by the glycine modulatory site. Neuroscience. 1992;46:501–509. doi: 10.1016/0306-4522(92)90139-S. [DOI] [PubMed] [Google Scholar]
- Tsai GE, Yang P, Chung LC, Tsai IC, Tsai CW, Coyle JT. d-serine added to clozapine for the treatment of schizophrenia. Am J Psychiatry. 1999;156:1822–1825. doi: 10.1176/ajp.156.11.1822. [DOI] [PubMed] [Google Scholar]
- Tsai G, Yang P, Chung LC, Lange N, Coyle JT. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. 1998;44:1081–1089. doi: 10.1016/S0006-3223(98)00279-0. [DOI] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- Vargas-Lopes C, Madeira C, Kahn SA, Albino do Couto I, Bado P, Houzel JC, De Miranda J, de Freitas MS, Ferreira ST, Panizzutti R. Protein kinase C activity regulates d-serine availability in the brain. J Neurochem. 2011;116:281–290. doi: 10.1111/j.1471-4159.2010.07102.x. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S. Contribution of astrocytes to hippocampal long-term potentiation through release of d-serine. Proc Natl Acad Sci U S A. 2003;100:15194–15199. doi: 10.1073/pnas.2431073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Z, Yang B, Theus MH, Sick JT, Bethea JR, Sick TJ, Liebl DJ. EphrinBs regulate d-serine synthesis and release in astrocytes. J Neurosci. 2010;30:16015–16024. doi: 10.1523/JNEUROSCI.0481-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]