

Abstract
In Huntington's disease (HD), cognitive symptoms and cellular dysfunction precede the onset of classical motor symptoms and neuronal death in the striatum and cortex by almost a decade. This suggests that the early cognitive deficits may be due to a cellular dysfunction rather than being a consequence of neuronal loss. Abnormalities in dendritic spines are described in HD patients and in HD animal models. Available evidence indicates that altered spine and synaptic plasticity could underlie the motor as well as cognitive symptoms in HD. However, the exact kinetics of spine alterations and plasticity in HD remain unknown. We used long-term two-photon imaging through a cranial window, to track individual dendritic spines in a mouse model of HD (R6/2) as the disease progressed. In vivo imaging over a period of 6 weeks revealed a steady decrease in the density and survival of dendritic spines on cortical neurons of R6/2 mice compared with control littermates. Interestingly, we also observed increased spine formation in R6/2 mice throughout the disease. However, the probability that newly formed spines stabilized and transformed into persistent spines was greatly reduced compared with controls. In cultured neurons we found that mutant huntingtin causes a loss, in particular of mature spines. Furthermore, in R6/2 mice, aggregates of mutant huntingtin associate with dendritic spines. Alterations in dendritic spine dynamics, survival, and density in R6/2 mice were evident before the onset of motor symptoms, suggesting that decreased stability of the cortical synaptic circuitry underlies the early symptoms in HD.
Introduction
Huntington's disease (HD) is caused by expansion of CAG triplets in the gene huntingtin (Htt) (The Huntington's Disease Collaborative Research Group, 1993). HD patients exhibit abnormal movement “chorea” and cognitive and psychiatric disturbances caused by degeneration of striatal medium spiny neurons and cortical pyramidal neurons (The Huntington's Disease Collaborative Research Group, 1993). Evidence demonstrates that the cognitive disturbances and cellular dysfunction precede overt motor symptoms and cell death by many years in humans and occur long before, or in the absence of, cell death in animal models. This suggests that early deficits in cognition in HD are likely caused by synaptic dysfunction rather than as a consequence of neuronal cell death (Marder et al., 2000; Milnerwood and Raymond, 2007; Verny et al., 2007; Schippling et al., 2009; Orth et al., 2010).
Several neurodegenerative disorders are associated with pathology in dendritic spines (the site of excitatory synapses) and in most cases, spine alterations underlie deficits in cognition, learning, and memory (Fiala et al., 2002). Postmortem HD patients show both proliferative and degenerative changes in dendritic spines whereas HD animal models mostly show lower spine densities on cortical and striatal neurons (Graveland et al., 1985; Ferrante et al., 1991; Sotrel et al., 1993; Guidetti et al., 2001; Spires et al., 2004; Heck et al., 2012). Further, animal model studies suggest that cognitive impairment is due to altered synaptic plasticity (Murphy et al., 2000; Mazarakis et al., 2005; Lynch et al., 2007; Milnerwood and Raymond, 2007). Although these findings suggest a relationship between declined spine densities and HD, the techniques used in previous studies (Golgi–Cox staining and postmortem materials) do not provide insight into exact kinetics, the nature, and temporal course of spine alterations in HD. Furthermore, since these studies have mostly reported spine alterations in an advanced stage of HD, it remains unclear whether spine loss is an epiphenomenon of neurodegeneration or whether it is a leading event in the disease. To better understand progression of dendritic spine pathology in HD, we imaged cortical dendrites in the R6/2 mouse model of HD in vivo, using two-photon laser scanning microscopy through a cranial window (Holtmaat et al., 2009). This allowed us to track the fate and lifetime of individual spines over a period of 6 weeks. We confirm decreased spine density in R6/2 mice compared with wild-type controls. Interestingly, we observed a dramatically increased turnover and a lower survival rate of dendritic spines. Additionally, R6/2 mice fail to generate long-lasting connections on cortical neurons due to an impaired stabilization of newly formed spines. As a result, cortical neurons of R6/2 mice contain a significantly lower fraction of stable spines and a larger population of transient spines. Additionally, we found that mutant huntingtin causes a loss of dendritic spines, especially mature spines, in cultured neurons. Our findings suggest that synapses are highly unstable in R6/2 mice. Spine alterations in R6/2 mice occur well before the onset of the motor symptomatic phase, suggesting that synapse pathology may be causal to the early cognitive deficits observed in HD.
Materials and Methods
Transgenic mice.
All animal procedures were performed in accordance with the protocols approved by the Animal Care Welfare Committee of Lund University (Sweden). All mice were kept in a normal 12 h light/dark cycle and had access to food and water ad libitum. A colony of R6/2 mice was maintained by crossing R6/2+/− males with F1 of C57BL/6 × CBA hybrid females in the animal facility at Lund University. The colony of Thy1-GFP-M was maintained by crossing Thy1-GFP-M males or females with C57BL/6 females or males. The colony was also maintained on F1 background. Heterozygous female Thy1-GFP-M mice expressing eGFP under the control of Thy-1 promoter (line M) were crossed with heterozygous R6/2 male mice (F1 hybrids of R6/2+/− males × hybrid of C57BL/6 × CBA females) to produce double transgenic reporter mice, i.e., R6/2+/−-Thy1-GFP-M+/−, for time-lapse imaging. Throughout the text we will refer to these double transgenic reporter mice as R6/2 mice, and single transgenic reporter mice, i.e., R6/2−/−-Thy1-GFP-M+/−, as wild-type control mice. Thy1-GFP-M is a transgenic reporter line in which eGFP is expressed in a subset of layer II/III and V cortical neurons (Feng et al., 2000). The R6/2 mice represent a well characterized model of HD. R6/2 mice express exon 1 and promoter sequences of the human HD gene inserted into the mouse genome and carry 141–157 CAG repeats (Mangiarini et al., 1996). The CAG repeat lengths were verified for each mouse by sequencing. In our R6/2 mice, the repeat length ranged between 255 and 280 CAGs.
Surgery and time-lapse in vivo imaging.
We implanted a cranial window on 1-month-old isoflurane anesthetized (1.5% (v/v) maintenance) single (R6/2−/−Thy1-GFP-M+/−) and double (R6/2+/−-Thy1-GFP-M+/−) transgenic reporter mice as previously described (Holtmaat et al., 2009). The skull overlying the right somatosensory cortex (bregma, anteroposterior −1.5 mm; lateral −3.5 mm laterally) was removed and replaced with a small coverslip (5 mm in diameter), keeping the dura intact (see Fig. 1B). Seven to 10 d after the surgery, imaging was performed using a two-photon upright (Zeiss LSM 7 MP, AxioCam MRM) microscope equipped with a tunable Ti:sapphire ultrafast oscillating laser (Mai Tai; Spectra Physics) and a 12 W solid-state pump laser. For imaging, the laser was tuned to 900 nm to excite eGFP. During chronic experiments, mice were anesthetized with isoflurane (1.5% (v/v) maintenance) for every imaging session. Imaging sessions were 45–50 min in duration. In each animal, apical dendritic tufts of pyramidal neurons with soma position in layers II/III and V were imaged over a period of 6 weeks. Apical dendritic tufts of these neurons are located in L1 and L2. The unique vascular pattern was captured with a Canon PowerShot A650 camera and was used to relocate the same dendritic segments over subsequent imaging sessions (Fig. 1C). Imaging was performed every eighth day starting from postnatal day (PND) 41 to PND81 (P41–P81). Images were acquired using a 20× objective (NA 1.0). Lower resolution image stacks (512 × 512 pixels, × 1.0 zoom, 1.0 μm step size) were acquired to visualize the dendritic branching pattern and the position of the soma (Fig. 1D). For higher magnification spine imaging, 5–15 random apical dendritic tufts (per mouse) each 42.51 × 42.51 μm, up to 300 μm below the pial surfaces, were imaged. For spines, image stacks with a digital zoom of × 10, 512 × 512 pixels (0.08 μm/pixel) taken at 0.6 μm increments were collected. Care was taken to ensure similar fluorescence across imaging sessions. To avoid phototoxicity, the lowest laser power that could discern all spines was used. Scanning and image acquisitions were controlled by Zen 2009 software from Zeiss.
Figure 1.

Progression of dendritic spine pathology in HD. A, Experimental time line for in vivo imaging. B, Schematic of a cranial window on the right somatosensory cortex of mice where neurons were imaged. C, The vascular pattern as seen through the cranial window. The picture is captured on the first imaging day and is used to find the same dendrites in the subsequent imaging sessions. D, Low-magnification image of an apical dendritic tuft of layer II/III pyramidal neuron in control-Thy1-GFP-M mice. E, Mean spine density in week 3 (PND22) and week 4 (PND30) control and R6/2 mice. At both ages R6/2 mice display tendencies toward lower spine densities compared with controls (n = 4 mice/group; unpaired t test). F, Time-lapse image of a dendritic branch from a R6/2 mouse showing decreased spine density as the disease progressed. Fewer spines were evident on the dendritic segment of R6/2 mice at the late phase PND81 as compared with the first view (PND41). Arrows mark lost spines. G, Mean spine density of 5–15 randomly selected apical dendritic branches of layers II/III and V pyramidal neurons in control (Thy1-GFP-M) and R6/2 mice (n = 13 mice/group). Already, before the onset of motor symptoms (5 ½ weeks), R6/2 mice display a significant reduction in dendritic spine density that continued to decrease steadily as the disease progressed (two-way repeated-measure ANOVA). H, Survival function of spines in control and R6/2 mice examined over 6 weeks (n = 6 mice/group). Note that R6/2 mice show decreased survival of dendritic spines before the onset of motor symptoms, and as the disease progressed. Spine survival over 6 weeks in control mice was >80% whereas in R6/2 only 59% of spines survived by the end of the examination period, which represents the late phase of disease onset in R6/2. Data are presented as mean ± SEM. Significant differences between groups are indicated by asterisks. Data were considered to be statistically significant if p < 0.05. Scale bar, D, 100 μm.
Immunofluorescence on tissues.
R6/2 and control mice were perfused using 4% paraformaldehyde (PFA). Coronal sections (35 μm in thickness) were cut using a frozen microtome (Leica). Free-floating sections were rinsed (3 × 5 min) in 0.1 m PBS containing 0.1% Triton X-100, then blocked for 1 h in blocking buffer containing 0.3% bovine albumin serum (BSA), 5% normal goat serum in 0.1 m PBS, incubated overnight with mouse primary anti-human huntingtin antibody (MAB 5374-EM48; Millipore), 1:200 in blocking buffer containing 0.3% BSA/1× PBS. After washing in 0.1% Triton X-100/1× PBS (3 × 5 min), the sections were incubated for 1 h with goat anti-mouse Cy3 secondary antibody (1:1000, Jackson ImmunoResearch). Tissue sections were mounted with polyvinyl alcohol containing DABCO (PVA-DABCO; Sigma-Aldrich). Fluorescent images were acquired with a confocal laser scanning system equipped with Zen 2009 software at 63× objective and 5× digital zoom, 0.056 μm/pixel (Zeiss, Meta 510). Care was taken to ensure similar fluorescence across images.
Neuronal cultures and constructs.
Wild-type C57BL/6 males were crossed with wild-type C57BL/6 females to generate the embryos. Primary neuronal cultures were prepared from cortices and hippocampi of embryonic day (E15) C57BL/6 mice as described by Tampellini et al. (2007, 2009). In this study, we combined cortical/hippocampal neuronal cultures for several reasons. First, because both cortex and hippocampus are involved in regulating cognition, learning, and memory and both these areas are affected in HD (Rosas et al., 2003). Second, we combined these cultures to facilitate the possibility of having more neurons for in vitro spine analysis. At 7 days in vitro (DIV), the neurons were transfected with plasmids containing a hemagglutinin-tagged exon 1 human huntingtin with either 17 (short-Q huntingtin) or 69 (long-Q huntingtin) glutamine residues under the control of a cytomegalovirus (CMV) promoter. Cells were cotransfected with actin-GFP (under the control of CMV promoter) using Lipofectamine for 5 h. Since the Thy1-GFPM mice that we used for our in vivo two-photon imaging does not express GFP in utero (GFP expression detected only postnatally), we cotransfected cortical/hippocampal neurons with actin-GFP to label neurons and to visualize dendritic spines. Studies have shown that GFP-tagged actin effectively labels the actin in dendritic spines without altering their morphology (Kaech et al., 1997; Fischer et al., 1998). At 14 DIV, cortical/hippocampal neurons were fixed in 4% PFA for 20 min at room temperature, permeabilized, and rinsed (3 × 5 min each) in 0.3% Triton X-100/1× PBS, blocked for 1 h in 10% normal donkey serum in 0.3% Triton X-100/1× PBS, incubated overnight at 4°C with primary antibodies (polyclonal rabbit anti-synaptophysin, 1:200; rabbit anti-PSD-95, 1:400; Abcam). After washing in 0.3% Triton X-100/1× PBS (3 × 5 min), the sections were incubated for 1 h with donkey anti-rabbit Cy3 secondary antibody, 1:500 (Jackson ImmunoResearch). After washing (3 × 5 min) in 0.3% Triton X-100/1× PBS sections were mounted in Vectastain (Vector Laboratories). Fluorescent images were acquired with a confocal laser scanning system equipped with Zen 2009 software at 63× objective and 5× digital zoom, 0.056 μm/pixel (Zeiss Meta 510). Care was taken to ensure similar fluorescence across images.
Behavioral tests.
Motor coordination and balance of R6/2 and C57BL/6 control mice were tested using a rotarod apparatus (Rotamex 4/8; Columbus Instruments) at 4, 6, 8, 10, and 12 weeks. First, all the mice were placed on the rotarod for a 30 s training period. Fifteen minutes after the first trial, each mouse was placed on a rotarod at an accelerating speed (4–40 rpm) for three trials, each lasting for a maximum of 300 s (Carter et al., 1999, 2001). The mean latency to fall off the rotarod (for the three trials) was recorded and used in subsequent analysis. At the end of the third trial the body weight of each mouse was measured.
Image processing and data analysis.
Image processing and data analysis were performed using the National Institutes of Health (Bethesda, MD)-based public domain software ImageJ, according to published methods (Holtmaat et al., 2009). All images in the study are 3D projected Z-stacks. Before projections, images were loss pass and median filtered. In some figures, distracting fluorescent processes were removed. Dendritic spines were counted manually by scrolling through the Z-stacks of subsequent time points of the same position. All clear protrusions, regardless of their shape (stubby, mushroom, thin spines), emanating laterally from the dendrite not above or below the dendrite were measured. Analysis was done blindly, with the analyzer unaware of the experimental conditions. Only frames with good signal-to-noise ratio, where spines on all parts of the dendrite were clearly visible, were used for analysis; dim frames were discarded. Spines were considered the same between two views on the basis of their spatial relationship to adjacent landmarks and their relative position to immediately adjacent spines. Spines were considered different if they were >0.5 μm away from their previous position. Spine length was measured by manually drawing a line from the base of the spine to the tip. Spines were considered as lost if they consisted of <4 pixels and gained if they consisted of >4 pixels in length protruding from the dendrite. Because the two-photon microscope has a relatively poor resolution in the z-axis, only protrusions emanating in the x-y directions were included. Spines that survived for 8 d or more, i.e., >8 d, were considered as “persistent spines,” while spines that did not survive for 8 d, i.e., <8 d, were defined as “transient spines.” For each imaging session, the following parameters were calculated: fraction of persistent spines (Fp), fraction of transient spines (Ft), fraction of gained spines (Fgained) and fraction of lost spines (Flost) as previously described (Holtmaat et al., 2005). Fp = Npersistent/Ntotal × 100; Ft = Ntransient/Ntotal × 100 where Npersistent and Ntransient are the number of spines that were classified as persistent and transient for the analyzed session or day (t), whereas Ntotal is the number of all spines present on imaging session (t). Fgained = Ngained/Ntotal; Flost = Nlost/Npresent where Ngained and Nlost represent the number of spines gained and lost from one imaging session to the next, Ntotal is the number of all spines present on imaging session (t), and Npresent is (t − 8), i.e., the number of spines present at the previous imaging session. Similarly, turnover ratios (TOR) representing gained and lost spines between imaging sessions (i.e., one week to the next) were measured. TOR = (Ngained/Nlost)/(2 × Ntotal). The time-dependent survival function was calculated as SF(t) = N(t)/N0, where N0 is the number of spines at t = 0, and N(t) is the number of spines of the original set surviving after time t. New persistent spines (NP) were defined as spines that were not present at the first time point (PND41), but were gained during subsequent imaging sessions, i.e., PND49, 57, 65, and 73 and were present for at least 8 d or more, >8 d, i.e., the later time points (see Fig. 2A). Total new (TN) include new spines that were gained throughout the imaging sessions. Spine density was measured by dividing the total number of spines on the dendritic branch by its length, which was 35–40 μm per frame. For cell culture experiments, N = 37–51 random neuronal segments (at high magnification) which were derived from 13 neurons obtained from 4 × technical replicates for each genotype (3–5 neurons each genotype). All kinds of spines, regardless of their shape (mushroom, stubby, or thin spines) were included in the analysis given that they protruded laterally (x-y-axis) from the dendrite and not above or below (z-axis) the dendrite.
Figure 2.
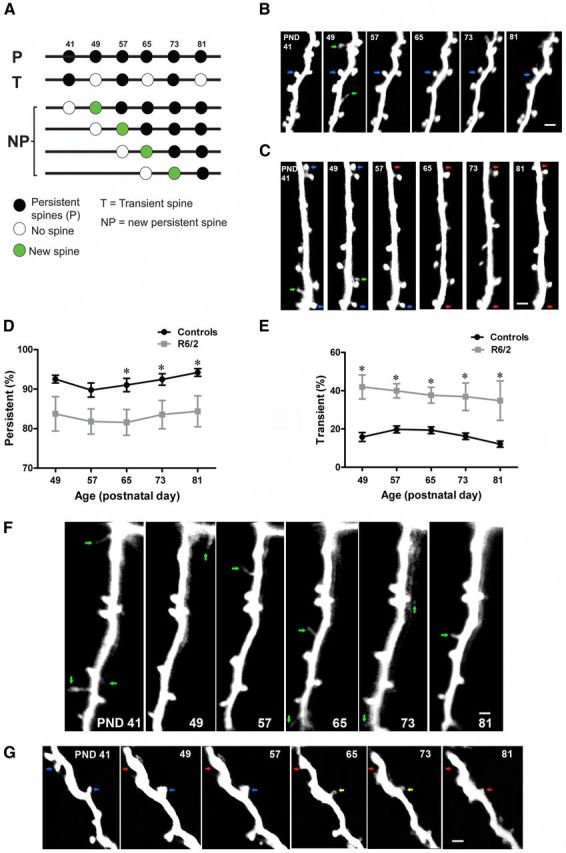
Loss of persistent spines in HD. A, Time line for imaging and definition of spine categories. Imaging days are represented by 41, 49, 57, 65, 73, and 81. B, Time-lapse images of a dendritic branch, from a control Thy1-GFP-M mouse. Notice the presence of two types of spines, persistent spines (blue arrows) and transient spines (green arrows). Persistent spines are larger and contain a head, which is clearly separated from the dendrite by a thin neck. The transient spines are thin, filopodia-like, and do not survive until the next imaging session (green arrows). C, Time-lapse image of a dendritic branch from R6/2 mice. Blue arrows indicate persistent spines, red arrows indicate lost persistent spines, and green arrows mark transient spines. D, Average fraction of persistent spines in control and R6/2 mice. Throughout the examination period, R6/2 mice show lower fraction of persistent spines compared with controls. E, Average fraction of transient spines in control and R6/2 mice, which is greater than twofold in R6/2 mice (∼38%) compared with controls (∼16%). F, Time-lapse imaging of a dendritic branch from R6/2 mice. Green arrows indicate transient spines. Note the presence of several transient spines that appear and disappear throughout the entire imaging period in the R6/2 mice. In the control mice (B, green arrows) very few transient spines appear during the imaging period. G, Changes in brightness of persistent spines before their complete loss in R6/2 mice. Yellow arrow indicates loss of fluorescence in the persistent spine. Note that over the course of the imaging experiment, this dendritic branch lost most of its spines. Significant differences between groups are indicated by asterisks. Data are presented as mean ± SEM. Means were considered to be statistically significant if p < 0.05. N = 13 mice/group. Scale bar, 2 μm.
Statistics.
A two-way repeated-measure ANOVA (genotype × time) was performed to analyze significant difference in dendritic spine density, survival, and turnover. Post hoc comparisons were made using the Bonferroni test. All data are presented as mean ± SEM. N = 4–13 mice per group. Student's t test (two-tailed) was used to test significant differences in dendritic spine density between 3-week-old (PND22) and 4-week-old (PND30) R6/2 mice compared with wild-type controls and between PND30 and PND41 R6/2 mice. In addition, Student's t test (two-tailed) was performed to test significant difference in spine number in cultured neurons overexpressing Q17 or Q69 huntingtin and Actin-GFP. A two-way repeated-measure ANOVA was performed to test differences in motor behavior between R6/2 and wild-type control mice at age of 4, 6, 8, 10, and 12 weeks (N = 12 mice/group at each time point). Differences were considered to be statistically significant if p < 0.05. All statistical analyses were performed with GraphPad Prism (GraphPad Software).
Results
Decreased density and survival of dendritic spines on cortical neurons in a model of HD
In many neurodegenerative conditions, such as in tauopathies, alterations in synapses and dendritic spines are some of the earliest manifestations of the disease (Yoshiyama et al., 2007). The HD mouse model, R6/2, is an excellent model to study progressive early synaptic dysfunctions, because this model mimics the cognitive as well as motor phenotype of HD in the absence of any clear neuronal loss. In R6/2 mice, neuronal loss is not evident until the very end stage of the disease (Turmaine et al., 2000). To study the structural plasticity of dendritic spines in HD, we applied long-term in vivo two-photon imaging through a cranial window to follow individual spines in the cortex of R6/2 mice as the disease progressed. Starting from PND41 (i.e., week 5 ½), which represents the period before the onset of symptomatic phase (motor symptoms) in our R6/2 mice, we repeatedly imaged 5–15 apical dendrites of layers II/III and V pyramidal neurons in the right somatosensory cortex of R6/2 and age-matched wild-type (Thy1-GFP-M) mice (Fig. 1A; experimental time line). We followed 1503 and 1129 spines in wild-type and R6/2 mice, respectively, for 6 weeks with a 7 d interval. First, we compared the spine density of wild-type and R6/2 mice at PND41 (the presymptomatic phase) to the density at PND81 (which represents the advanced phase of the disease). Two-way repeated-measure ANOVA revealed a significant effect of genotype, time, and a significant interaction between genotype and time on dendritic spine density. R6/2 mice show decreased spine density on apical dendrites of layers II/III and V pyramidal neurons compared with wild-type controls at both time points (Fig. 1F,G; F(1,24) = 7.34; p = 0.0122; n = 13 animals/group). Already at the presymptomatic phase (PND41), R6/2 mice display reduced spine density compared with controls, and as the disease progresses, R6/2 mice exhibit a steady decrease in spine density (Fig. 1G; F(5,120) = 14.93; p = <0.0001; two-way repeated-measure ANOVA). At the last imaging session (PND81), spine density was less than half of that observed in wild-type controls (Fig. 1G). To examine at which time point during the presymptomatic phase R6/2 mice start losing spines on the cortical neurons, we performed acute imaging on 3-week-old (PND22) and 4-week-old (PND30) R6/2 mice and compared spine density of R6/2 mice at both ages with that of age-matched wild-type mice. Young R6/2 mice did not show a significant difference in spine density compared with controls (Fig. 1E; R6/2 PND22: 0.36 ± 0.03; controls: 0.41 ± 0.05; n = 4 mice/group; p = 0.46; R6/2 PND30: 0.33 ± 0.03; controls: 0.40 ± 0.01; n = 4 mice/group; p = 0.07; unpaired t test). However, at PND41 (the day when time-lapse imaging started) spine density was significantly lower than at PND30 (R6/2, PND41: 0.26 ± 0.022; PND30: 0.33 ± 0.026; n = 4–7 mice/group; p = 0.045), suggesting that spines are progressively lost before the onset and during the progression of the symptomatic phase. To determine the underlying mechanisms of reduced spine density in R6/2 mice, we tracked the fates of individual spines observed in the first imaging session (PND41) and calculated the fraction of spines surviving (survival fraction) as a function of time (SF[t]). We found that R6/2 mice display a significant reduction in dendritic spine survival compared with controls (Fig. 1H; F(1,10) = 46.36; p = <0.0001; n = 6 mice per group; two-way repeated-measure ANOVA). Already, before the onset of motor symptoms, R6/2 mice show ∼20% lower spine survival compared with wild-type controls (Fig. 1H). Furthermore, R6/2 mice display a steady decrease in dendritic spine survival as the disease progresses (Fig. 1H; F(5,50) = 221.87; p = <0.0001; two-way repeated-measure ANOVA). At the end of the examination period (PND81), only ∼59% of the spines that were observed at the start (PND41) had survived in the R6/2 mice, whereas the controls displayed a survival rate of ∼81%. These data show that in R6/2 mice, dendritic spine survival is affected during the early phase, as well as throughout the symptomatic phase, which could be a plausible explanation for the reduced spine density and/or numbers reported in HD patients and HD animal models.
Loss of persistent spines and a high fraction of transient spines in HD
In vivo studies have revealed the existence of at least two types of dendritic spines in the neocortex, persistent and transient spines (Holtmaat et al., 2005). In naive mice, persistent spines are typically large, mushroom-like, and stable for weeks or months. Transient spines are usually thin and live shorter than a few days (Holtmaat et al., 2005). In contrast to transient spines, persistent spines always bear synapses (Knott et al., 2006). Most transient spines do not make a synapse. However, those that stabilize and become persistent are capable of rapid functional maturation (Holtmaat et al., 2006; Knott et al., 2006). During development, the fraction of persistent spines increases, while the fraction of transient spines decreases (Holtmaat et al., 2005). The pathological decrease in spine survival and density in R6/2 mice prompted us to measure the fractions of persistent and transient spines before and during the symptomatic phase. We classified persistent and transient spines (Fig. 2A) as previously described (see Material and Methods; Holtmaat et al., 2005). Two-way repeated-measure ANOVA revealed a significant effect of genotype and time in which persistent spines were reduced in R6/2 mice compared with controls (Fig. 2B–D; F(1,24) = 8.83; p = 0.0066; n = 13 animals/group). Comparing the temporal course of persistent spine loss in R6/2, we found that R6/2 mice display a reduction in the persistent spine fraction at the initial phase of the disease, which is maintained throughout the course of the disease (Fig. 2D; F(5,120) = 13.86; p = <0.0001; two-way repeated-measure ANOVA). A significant effect of genotype was also observed on the transient spine fraction. R6/2 mice display a significant increase in the fraction of “transient-type” spines compared with controls (Fig. 2E,F; F(1,24) = 13.99; p = 0.0010; N = 13 animals/group; two-way repeated-measure ANOVA). We found that most of the spines that appeared and disappeared between imaging sessions were thin, filopodia-like or stubby spines, but rarely mushroom-type mature spines (Fig. 2F). We further observed that in the R6/2 mice, spine loss was not due to the degeneration of and/or a loss of fluorescence in the neurons under study, since all neurons that were imaged remained present, and dendritic shafts contained the same level of fluorescence throughout the whole imaging period (see Fig. 2B,C,F,G). In addition, small filopodia-like structures remained to be seen, indicating that small protrusions could still be resolved on those neurons. Neurons and dendrites remained present until the end of the imaging experiment. Only spines were lost (Fig. 2G).
Increased turnover and impaired stabilization of newly formed spines in HD
There is considerable evidence indicating that spine appearance and disappearance in response to neural activity and experience involves synapse formation and elimination (Holtmaat et al., 2006). To further analyze dendritic spine instability in detail, we quantified dTOR, which represents the fraction of daily gained and lost spines. We quantified the weekly spine TORs in R6/2 and age-matched wild-type control mice. To our surprise, R6/2 mice exhibited significantly increased spine turnover (TOR) compared with controls (Fig. 3A; F(1,24) = 13.45; p = 0.0012; n = 13 animals/group; effect of genotype; two-way repeated-measure ANOVA). Increased spine turnover in R6/2 mice was evident as early as at the presymptomatic phase and remained elevated throughout disease progression (Fig. 3A). To examine whether the increased TORs observed in R6/2 mice were not only due to increased spine loss, we examined the gain and loss of spines independently of one another in both R6/2 and wild-type controls. As expected, R6/2 mice display a significant increase in the fraction of lost spines throughout the disease. Interestingly, the peak in spine loss was evident right before the onset of the symptomatic phase (Fig. 3B; lost spines; F(1,24) = 21.55; p = 0.0001; n = 13 animals/group; effect of genotype; two-way repeated-measure ANOVA). In addition to a higher spine loss, R6/2 mice also gain more spines as compared with wild-type controls (Fig. 3C,D) (gained spines; F(1,24) = 6.09; p = 0.0211; n = 13 animals/group; effect of genotype; two-way repeated-measure ANOVA). Comparing the temporal course of spine gain in R6/2, we found that the gain is higher at the symptomatic phase of the disease than toward the advanced phase of the disease (Fig. 3D; F(4,96) = 2.80; p = 0.0301; effect of time; two-way repeated-measure ANOVA). To investigate why the increased gain of spines in R6/2 mice does not result in an apparent increase in the fraction of persistent spines, we tracked how many of those newly formed spines became persistent over time in R6/2 as compared with control mice. We counted the total new spines (TN) and the new persistent spines (NP) in both R6/2 and controls (for definition of spine categories see Materials and Methods and Fig. 2A). The density of new persistent spines (NP density) was significantly reduced in R6/2 mice compared with controls (NP mm−1, controls: 13.55 ± 1.50; R6/2: 2.73 ± 1.11; n = 6 mice/group; p = 0.0002; unpaired t test; Fig. 3E). Furthermore, in the R6/2 mice, the probability that new spines became persistent (NP/TN) was significantly lower compared with controls (NP/TN, controls: 0.114 ± 0.02; R6/2: 0.017 ± 0.007; n = 6 mice/group; p = 0.0027; unpaired t test; Fig. 3F). These data imply that new spine stabilization is impaired in R6/2 mice.
Figure 3.

Increased turnover and impaired stabilization of newly formed spines in HD. A, Average weekly spine TOR in control (Thy1-GFP-M) and R6/2 mice, which is ∼8% in controls and ∼19% in R6/2 mice. Compared with controls, R6/2 mice display a significant increase in spine turnover that remained elevated as the disease progressed. B, Average lost spine fraction per week of controls and R6/2 mice. Throughout the examination period, R6/2 mice lose more spines per week (∼21%) compared with controls (∼8%). C, Time-lapse images of a dendritic branch showing newly formed spines. Newly formed spines indicated by arrows are mostly thin, filopodia-like, and do not survive very long. D, Average gain spine fraction per week of control and R6/2 mice. R6/2 mice gain more spines (∼12%) compared with controls (∼8%) as the disease progresses. E, Quantification of new persistent spine density (NP). F, The probability (NP/TN) that new spines became persistent. Every circle and square represents a mouse. Significant differences between groups are indicated by asterisks. Horizontal bars represent the group averages. Data are presented as mean ± SEM. N = 7–13 mice/group. Scale bar, C, 2 μm.
Characterization of motor deficits in R6/2 mice
Studies have suggested that expanded polyglutamine stretch (CAG > 300) paradoxically delays the onset of motor symptoms and increases the survival duration in R6/2 mice (Morton et al., 2009; Cummings et al., 2012). Since the CAG repeat length has expanded in our colony, we performed a rotarod test to determine whether the symptomatic phase (motor deficit) in our R6/2 mice is also delayed. We subjected the same mice repeatedly to rotarod starting from the age of 4–12 weeks. We found that the R6/2 mice with expanded CAG repeats did not display any motor deficits at week 4, nor at week 6. These mice had no difficulty maintaining balance on the rotarod and the latency to fall off the rotarod was not significantly different between the R6/2 mice and wild-type controls at this age (mean latency to fall; 4 weeks: R6/2 – 76 ± 7.5; controls: 82 ± 6.8; 6 weeks: R6/2 100 ± 6.8; controls: 111 ± 7.6; Fig. 4). However, with increasing age, R6/2 mice displayed severe deficits in motor coordination and balance (F(2,66) = 18.52; p < 0.0001; N = 12 mice/group; effect of time, two-way repeated-measure ANOVA). R6/2 mice at 8, 10, and 12 weeks displayed decreased latency to fall off the rotarod compared with wild-type controls (F(1,66) = 4.41; p = 0.039; N = 12 mice/group; effect of genotype, two-way repeated-measure ANOVA). (Mean latency to fall: 8 weeks R6/2 − 88 ± 3.4; controls: 158 ± 4.4; 10 weeks: R6/2 81 ± 6.16; controls: 170 ± 9.5; 12 weeks: R6/2 62 ± 5.7; controls: 203 ± 14; Fig. 4). A significant interaction of genotype and time was observed in the motor behavior of R6/2 mice compared with controls (F(2,66) = 3.36; p = 0.040; two-way repeated-measure ANOVA). Wild-type mice, as expected, demonstrated enhanced motor learning with repeated exposure to rotarod in contrast to R6/2 mice that performed worse on rotarod with repetitive exposure at weeks 4, 6, 8, 10, and 12. These data suggest that motor learning is impaired in R6/2 mice.
Figure 4.

Characterization of motor-learning deficit in R6/2 mice compared with wild-type controls at five different ages. Young R6/2 mice (weeks 4 and 6) spent the same amount of time running on an accelerating rotarod as the wild-type controls, indicating that they did have any motor deficit. Further, with repeated exposure to rotarod (weeks 4 to 12), wild-type mice improve their performance indicating motor learning in wild-type mice. R6/2 mice, on the other hand, display diminished performance after repetitive exposure to rotarod, indicating impaired motor learning in the R6/2 mice compared with wild types. Older R6/2 mice (8, 10, and 12 weeks) displayed decreased latency to fall off the rotarod compared with wild types indicating that R6/2 mice at these ages have developed a motor phenotype. Two-way repeated-measure ANOVA was used to test the significant effect of genotype and age on motor behavior. N = 12 mice/group at each time point. Data are presented as mean ± SEM. Note that the error bars for R6/2 mice are difficult to see because they are small. Significant differences are indicated by asterisks.
Mutant huntingtin causes loss of mature spines in cortical/hippocampal neurons in vitro
To investigate the direct effect of mutant huntingtin on dendritic spines, we first transfected cultured cortical and hippocampal neurons from wild-type C57BL/6 mice either with short-Q huntingtin (huntingtin with 17 glutamine residues; Q17) or with long-Q huntingtin (huntingtin with 69 glutamine residues; Q69). Cells were cotransfected with actin-GFP to visualize dendritic spines. We then immunostained actin-GFP-labeled neurons with markers of presynaptic and postsynaptic compartments such as synaptophysin and PSD-95 to determine whether the cultured neurons resemble neurons in vivo in that they express these markers or whether the actin-GFP protrusions on these neurons represent true spines. Our results show that cortical/hippocampal neurons overexpressing Q69 huntingtin exhibit significant decrease in the overall spine number compared with neurons expressing Q17 huntingtin or actin-GFP only (mean spine number/10 μm; Q69: 6.7 ± 0.4; Q17: 10 ± 0.4; actin-GFP: 11 ± 0.4; p < 0.0001; Fig. 5A,C). Moreover, in the neurons over-expressing Q69 huntingtin, we observed a dramatic loss of mature-type spines and a tendency for increased immature spines (mean mature spines/10 μm; Q69: 3 ± 0.27; Q17: 7 ± 0.39; actin-GFP: 7.7 ± 0.34; p < 0.0001; mean immature spines/10 μm; Q69: 4 ± 0.29; Q17: 3 ± 0.21; actin-GFP; 3 ± 0.19; p < 0.07; Fig. 5A,C). The total spine number and the number of mature and immature spines were comparable between neurons overexpressing Q17 huntingtin and actin-GFP only (Fig. 5A,C). Additionally, we were able to show that there is a loss of true dendritic spines in our cortical/hippocampal cultures overexpressing Q69 mutant huntingtin compared with neurons overexpressing Q17 huntingtin or actin-GFP only. In our culture, the actin-GFP-labeled neurons expressed synaptophysin, a presynaptic axonal marker and PSD-95, a postsynaptic dendritic marker. Synaptophysin-positive punctae were found adjacent to dendritic spines and PSD-95-positive punctae were found on dendritic spines or “actin-protrusions” suggesting that these neurons bear true spines (Fig. 6A–D). Furthermore, it is noteworthy, that in our cultured cortical/hippocampal neurons we observed a more robust effect on dendritic spines after overexpression of mutant huntingtin with 69 CAG repeats, which is much shorter than the CAG repeat length in our HD mice R6/2 (CAG 255–280). This robust effect on cultured neurons could be attributed to a higher level of overexpression of mutant huntingtin compared with in vivo situation in the animal model. In summary, the findings suggest that mutant huntingtin might directly affect dendritic spines leading to loss of mature spines in vitro.
Figure 5.
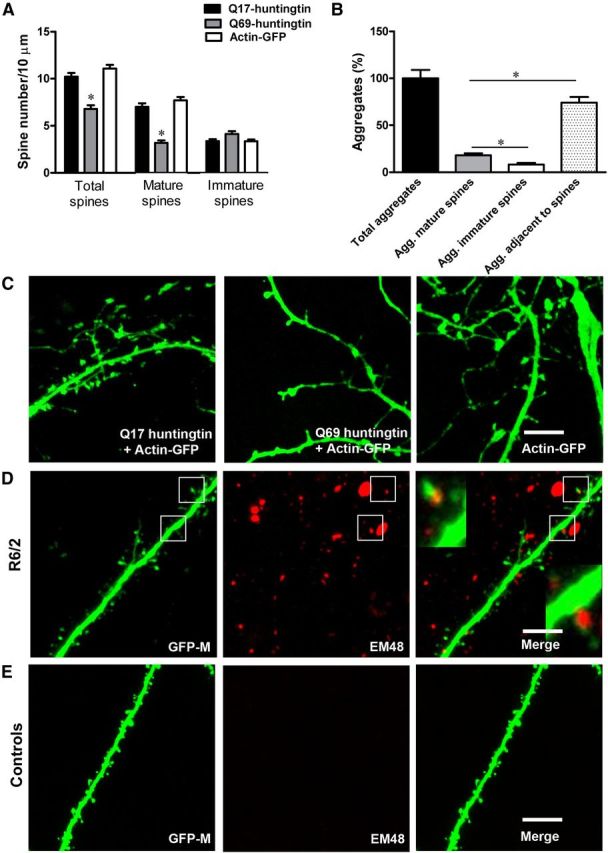
In vitro mutant huntingtin leads to loss of spines, especially of mature-type spines, and in vivo aggregates of mutant huntingtin localize in dendritic spines. A, Mean spine number/10 μm counted on ∼37–51 high-magnification dendritic segments of cortical/hippocampal neurons (13 neurons, 3–5 per genotype 4 × technical replicates) overexpressing short Q17 huntingtin, long Q69 huntingtin, or actin-GFP only. In neurons overexpressing Q69 huntingtin, we observed significant loss, particularly of mature-type spines, compared with neurons overexpressing Q17 huntingtin and actin-GFP only. B, Quantification of aggregates associated with dendritic spines in the R6/2 mice. More huntingtin aggregates were present within mature (mushroom-shaped) spines compared with smaller spines (filopodia-like or stubby spines; Agg, aggregates). However, there are more huntingtin aggregates adjacent to spines than within mature or immature spines. C, Z-projected stack representing total spines on cortical/hippocampal dendrite overexpressing Q17 + actin-GFP, Q69 + actin-GFP, and actin-GFP only. Notice that Q69 huntingtin overexpressing neurons (middle) have fewer spines compared with neurons expressing Q17 huntingtin + actin-GFP (left) and actin-GFP only (right). Also notice the presence of fewer mature spines on neurons overexpressing Q69 huntingtin. D, GFP expressing apical dendrite from double transgenic Thy1-GFP-M × R6/2 mice. In red is mutant huntingtin staining with anti-human huntingtin antibody EM48. Insets in the merged image represent boxed regions. Note the presence of mutant huntingtin aggregates in dendritic spines of R6/2 mice. E, GFP-expressing apical dendrite from the wild-type Thy1-GFP-M mice. No huntingtin aggregates are present in the wild-type mice. In this study, we did not observe any significant difference in shaft diameter between R6/2 and wild-type mice as evident in the figures above. Scale bar, 5 μm.
Figure 6.
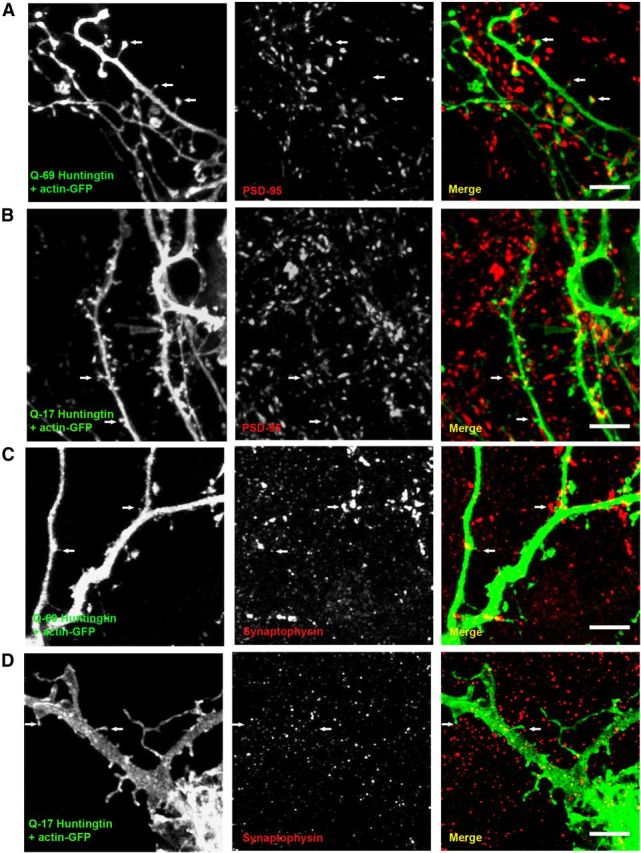
In culture, actin-GFP-labeled cortical/hippocampal neurons coexpress presynaptic and postsynaptic markers. A, Cortical/hippocampal neurons cotransfected with Q69 huntingtin + actin-GFP express PSD-95 in most of the dendritic spines, suggesting that actin-GFP labeled protrusions in these neurons are true spines. B, Q17 huntingtin + actin-GFP overexpressing neurons showing PSD-95 immunopositive punctae in most spines. C, D, Synaptophysin (a presynaptic protein marker)-positive dots were found adjacent to dendritic spines in cortical/hippocampal neurons overexpressing Q69 and Q17 huntingtin. White arrows indicate synaptophysin or PSD-95 immunoreactivity close to or in the dendritic spines. The color of the text (Q-69 huntingtin + actin-GFP, Q17 huntingtin + actin-GFP, PSD-95, synaptophysin) represents the color of the antigen in the corresponding merged section (right). Scale bar, 5 μm.
Mutant huntingtin aggregates are localized in and around dendritic spines in vivo
Mutant huntingtin aggregates are found in human HD patients and in animal models of HD (DiFiglia et al., 1997; Scherzinger et al., 1997). To examine whether mutant huntingtin aggregates localize in dendritic spines in cortical neurons, we performed immunostaining with anti-Htt antibody (EM48) recognizing misfolded human huntingtin and imaged cortical neurons of R6/2 and wild-type control mice using confocal microscopy. We found that some huntingtin-containing aggregates localize within the dendritic spines and some huntingtin aggregates are found around the dendritic spines in R6/2 mice, whereas no huntingtin aggregates are found in the cortical neurons of wild-type control mice (Fig. 5D,E). In the R6/2 mice, we also quantified the total number of huntingtin aggregates on cortical dendrites to determine what fraction of the total huntingtin aggregate associates with dendritic spines (Fig. 5B). We quantified aggregates as follows: (1) total aggregate number, (2) aggregates in mature spines, (3) aggregates in immature spines, and (4) aggregates adjacent to spines. We found that the largest fraction of huntingtin aggregates were adjacent to spines (74%), regardless of their shape (filopodia, stubby, and mature spines). However, mature or mushroom-type spines contained a higher fraction of huntingtin aggregates (18%) compared with smaller spines or filopodia-like spines that contained 8% aggregates (Fig. 5B). In addition, we measured the dendritic shaft diameter of R6/2 and wild-type mice and found no significant difference in shaft diameter between R6/2 mice and wild types. Our data indicate that there are more huntingtin containing aggregates in mature spines compared with immature spines, which might contribute to their loss. These data are in agreement with our in vitro data (Fig. 5A–D).
Discussion
Dendritic spine pathology is observed in several neurodegenerative conditions including HD where spine alterations have been described mostly at the symptomatic and advanced disease stage (Graveland et al., 1985; Ferrante et al., 1991; Sotrel et al., 1993; Guidetti et al., 2001; Klapstein et al., 2001; Fiala et al., 2002; Spires et al., 2004; Nithianantharajah et al., 2009; Lerner et al., 2012). Thus far, studies describing effects on spines in brain tissue have been based on measurements in postmortem material. Therefore, it is not clear how these phenotypes come about. The development of two-photon in vivo imaging techniques allows longitudinal studies of individual spines in live animals to determine how spine phenotypes develop in mouse models of neurodegenerative conditions (Tsai et al., 2004; Fuhrmann et al., 2007; Cruz-Martín et al., 2010).
Here, we used in vivo two-photon imaging to monitor cortical dendritic spine alterations in a mouse model of HD while the disease progresses. Several important conclusions can be drawn from our results. First, our findings are in agreement with previous reports and more importantly, provide new insight into dendritic spine pathogenesis in HD. In accordance with previous findings, we observed reduced spine density in our HD mice (R6/2) that could be due to reduced spine survival on cortical neurons. Importantly, both reduced spine survival and density were evident in the presymptomatic phase during which our R6/2 mice do not show any deficit in motor coordination and balance. This suggests that synaptic dysfunction precedes the motor symptoms in HD. Furthermore, the early spine loss in our model suggests that the decreased spine numbers in HD are not a consequence of neuronal loss, but rather represent loss of excitatory synaptic input. In addition, the fact that spine loss progressed slowly discourages the hypothesis that spines disappear as a consequence of neuronal death and dendritic degeneration. Moreover, previous studies suggest that neuronal loss in R6/2 mice is not evident until the very end stage of the disease (Turmaine et al., 2000). The cause for reduced spine survival in the R6/2 model remains unknown. Reduced spine survival could be due to cellular alterations that occur early in the disease, such as loss of excitatory input due to degeneration of the presynaptic partner, or loss of neurotrophic factors (Zuccato et al., 2008). A study by del Toro et al. (2006) suggests that wild-type huntingtin regulates brain-derived neurotrophic factor (BDNF) trafficking while mutant huntingtin disrupts this transport along the corticostriatal pathway. It is likely that mutant huntingtin, by disrupting vesicular trafficking of BDNF, influences spine survival on cortical neurons. Interestingly, overexpression of BDNF has been shown to rescue defective synaptic plasticity and related mechanisms in animal models of HD (Lynch et al., 2007; Xie et al., 2010).
How does spine loss progress in HD? Particularly, at which phase of the disease do R6/2 mice begin to lose spines? To answer these questions, we analyzed spine density in younger R6/2 mice at PND22 and PND30 and found that R6/2 mice at both ages show tendencies toward lower spine density. Hence, it is possible that subtle alterations in dendritic spines may occur as early as week 3. Significant reduction in spine density became evident at PND41, before the onset of motor symptoms (motor learning deficits) in R6/2 mice. Conversely, we did not detect significant dendritic abnormalities in vivo (R6/2 mice) and in vitro (cultured neurons), indicating that spine loss could be one of the earliest manifestations of the disease.
Second, in this study, we found that persistent spines were preferentially lost in R6/2 mice during the whole imaging period. Selective elimination of persistent synapses that represent intact neuronal circuitry in the brain would mean that previously existing neuronal circuitry has been interrupted in HD. A lost persistent spine might represent the degenerating structure unit underlying early cellular deficits in HD. Interestingly, we also observed a remarkable increase in the pool of transient spines in R6/2 mice. This could be a compensatory mechanism, whereby cortical neurons react to spine and synapse loss by producing more spines. However, we also observed that most of those transient spines had an immature phenotype (filopodia-like, thin spines), which suggests that dendritic spine maturation is affected in HD.
Last, to our surprise, we observed that spine turnover is increased as early as the presymptomatic phase and remains elevated throughout the disease in R6/2 mice compared with controls. This indicates that R6/2 mice exhibit a higher rate of neuronal circuitry rewiring compared with control mice. Interestingly, R6/2 mice also gain more spines compared with controls. Although R6/2 mice display significantly higher newly formed spines, there was significant reduction in persistent spines throughout the disease. Our results show that the reduction in persistent spine numbers is not only due to the loss of spines but also due to an impaired stabilization of newly formed spines. Since only stable spines bear synapses (Knott et al., 2006), our data indicate that the cortical circuitry in R6/2 mice likely fails to generate long-lasting synaptic connections in the brain.
The exact cause of spine loss in HD remains unknown. However, several mechanisms could be implicated. The precise function of the huntingtin protein is unknown. Studies suggest that wild-type huntingtin has a role in vesicular trafficking and axonal transport (Gauthier et al., 2004; Trushina et al., 2004; Smith et al., 2005a; Caviston and Holzbaur, 2009). Mutant huntingtin, however, has a propensity to aggregate in the nucleus and in the cytoplasm, which is reported both in human HD patients and in animal models, in dendrites as well as in dendritic spines (DiFiglia et al., 1997; Scherzinger et al., 1997; Gutekunst et al., 1999; Lunkes et al., 2002). These aggregates could impair synaptic function, distort dendritic morphology, and disrupt trafficking and cell signaling and contribute to the known dendritic pathology that occurs in HD (Li et al., 2003; Smith et al., 2005a). We found that the expression of mutant huntingtin causes a reduction in mature (mushroom type) spine numbers suggesting that mutant huntingtin is directly involved in the loss of mature spines in HD. In addition, we found aggregates of mutant huntingtin within mature spines and also around all types of dendritic spines in the R6/2 mice. Mutant huntingtin aggregates could sequester proteins that are involved in synaptic transmission, such as the scaffolding protein PSD-95, and AMPA receptors among many others and lead to depletion of these proteins at their site of action (Cha et al., 1998; Sun et al., 2001; Luthi-Carter et al., 2003; Smith et al., 2005a,b, 2007; Nithianantharajah and Hannan, 2012). Interestingly, the mRNA and protein levels of PSD-95 and AMPA receptors are reduced in HD patients and in animal models (Cha et al., 1998; Sun et al., 2001; Luthi-Carter et al., 2003). Maintenance and stabilization of dendritic spines are regulated by synaptic activity, as well as by the molecular complex at the postsynaptic density of spines, consisting of neurotransmitter receptors, scaffolding proteins (PSD-95), enzymes, and cytoskeletal proteins (Yoshihara et al., 2009). Close correlation exists between spine head size, PSD size, AMPA receptor number, and spine stability; a phenomenon that possibly shows similarities to LTP (Yoshihara et al., 2009).
Another mechanism by which dendritic spine loss can occur in HD is via defective endocytic recycling and disrupted axonal transport. Impaired endosomal trafficking resulted in loss of mushroom-type, mature spines on hippocampal neurons at the vicinity of huntingtin aggregates (Richards et al., 2011). Hence, huntingtin aggregates, by sequestering molecular components of endosomal trafficking and dendritic spine stability, could contribute to failure of newly formed spines to stabilize and become persistent, eventually causing spine loss, particularly loss of persistent type spines in HD. Furthermore, in HD vesicular transport is altered which could lead to axonal degeneration, eventually causing spine loss (Li and Conforti, 2013). It is widely believed that regulated (de-)polymerization of actin, which forms the spine cytoskeleton, underlies mechanisms of learning and memory, such as long-term potentiation and long-term depression. A study by Munsie et al. (2011) suggested that mutant huntingtin causes defective actin remodeling that could influence the spine appearance, disappearance, and loss observed here. Furthermore, aberrant cortical synaptic plasticity and disturbances in Ca2+ signaling is reported in HD models including R6/2 mice (Murphy et al., 2000; Deckel et al., 2002; Mazarakis et al., 2005; Milnerwood and Raymond, 2007). Interestingly, impaired experience-dependent cortical plasticity in the HD mouse model has been reported to underlie a form of learning, which is regulated by the barrel cortex (Mazarakis et al., 2005). Hence, it is likely that altered synaptic plasticity could have an impact on spine number, stability, and turnover in this cortical area.
In summary, disturbances in the cellular processes described above could contribute to spine pathology in our HD model and this could lead to functional changes in cortical neurons and change their output characteristics. These changes could underlie symptoms such as deficits in cognition, learning, and memory, which constitute early stage pathology in HD. Therapies that stabilize dendritic spines or promote spine maturation may be beneficial for the treatment of early symptoms in HD.
Footnotes
This work received financial support from the Swedish Research Council, BAGADILICO-Excellence in Parkinson and Huntington Research, the ERA-Net Neuron Program (nEUROsyn), and Petrus och Augusta Hedlunds Stifelse, Tore Nilsson Stifelse, and Thorsten och Elsa Segerfalks Stiftelse. We would like to acknowledge Dr. Vanessa Schubert for teaching us the cranial window technique. We also thank Inger Remse for technical support. We would like to acknowledge Dr. Gunnar Gouras and Dr. Davide Tampellini for critical reading and constructive comments and also for providing us the Actin-GFP construct. We thank Andrew C. McCourt for the linguistic revision of this manuscript.
The authors declare no competing financial interests.
References
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Morton J, Dunnett SB. Motor coordination and balance in rodents. Curr Protoc Neurosci Chapter. 2001;8 doi: 10.1002/0471142301.ns0812s15. unit 8.12. [DOI] [PubMed] [Google Scholar]
- Caviston JP, Holzbaur EL. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009;19:147–155. doi: 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci U S A. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Martín A, Crespo M, Portera-Cailliau C. Delayed stabilization of dendritic spines in fragile X mice. J Neurosci. 2010;30:7793–7803. doi: 10.1523/JNEUROSCI.0577-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DM, Alaghband Y, Hickey MA, Joshi PR, Hong SC, Zhu C, Ando TK, André VM, Cepeda C, Watson JB, Levine MS. A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington's disease. J Neurophysiol. 2012;107:677–691. doi: 10.1152/jn.00762.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckel AW, Elder R, Fuhrer G. Biphasic developmental changes in Ca2+/calmodulin-dependent proteins in R6/2 Huntington's disease mice. Neuroreport. 2002;13:707–711. doi: 10.1097/00001756-200204160-00034. [DOI] [PubMed] [Google Scholar]
- del Toro D, Canals JM, Ginés S, Kojima M, Egea G, Alberch J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci. 2006;26:12748–12757. doi: 10.1523/JNEUROSCI.3873-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/S0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/S0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Mitteregger G, Kretzschmar H, Herms J. Dendritic pathology in prion disease starts at the synaptic spine. J Neurosci. 2007;27:6224–6233. doi: 10.1523/JNEUROSCI.5062-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell WO, Jr, Schwarcz R, Tagle DA. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169:340–350. doi: 10.1006/exnr.2000.7626. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck N, Betuing S, Vanhoutte P, Caboche J. A deconvolution method to improve automated 3D-analysis of dendritic spines: application to a mouse model of Huntington's disease. Brain Struct Funct. 2012;217:421–434. doi: 10.1007/s00429-011-0340-y. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441:979–983. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L. Long-term, high resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Fischer M, Doll T, Matus A. Isoform specificity in the relationship of actin to dendritic spines. J Neurosci. 1997;17:9565–9572. doi: 10.1523/JNEUROSCI.17-24-09565.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci. 2006;9:1117–1124. doi: 10.1038/nn1747. [DOI] [PubMed] [Google Scholar]
- Lerner RP, Trejo Martinez Ldel C, Zhu C, Chesselet MF, Hickey MA. Striatal atrophy and dendritic alterations in a knock-in mouse model of Huntington's disease. Brain Res Bull. 2012;87:571–578. doi: 10.1016/j.brainresbull.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wyman T, Yu ZX, Li SH, Li XJ. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet. 2003;12:2021–2030. doi: 10.1093/hmg/ddg218. [DOI] [PubMed] [Google Scholar]
- Li JY, Conforti L. Axonopathy in Huntington's disease. Exp Neurol. 2013;246:62–71. doi: 10.1016/j.expneurol.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haïem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–269. doi: 10.1016/S1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Apostol BL, Dunah AW, DeJohn MM, Farrell LA, Bates GP, Young AB, Standaert DG, Thompson LM, Cha JH. Complex alteration of NMDA receptors in transgenic Huntington's disease mouse brain: analysis of mRNA and protein expression, plasma membrane association, interacting proteins, and phosphorylation. Neurobiol Dis. 2003;14:624–636. doi: 10.1016/j.nbd.2003.08.024. [DOI] [PubMed] [Google Scholar]
- Lynch G, Kramar EA, Rex CS, Jia Y, Chappas D, Gall CM, Simmons DA. Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington's disease. J Neurosci. 2007;27:4424–4434. doi: 10.1523/JNEUROSCI.5113-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Marder K, Zhao H, Myers RH, Cudkowicz M, Kayson E, Kieburtz K, Orme C, Paulsen J, Penney JB, Jr, Siemers E, Shoulson I. Rate of functional decline in Huntington's disease. Huntington Study Group. Neurology. 2000;54:452–458. doi: 10.1212/WNL.54.2.452. [DOI] [PubMed] [Google Scholar]
- Mazarakis NK, Cybulska-Klosowicz A, Grote H, Pang T, Van Dellen A, Kossut M, Blakemore C, Hannan AJ. Deficits in experience-dependent cortical plasticity and sensory-discrimination learning in pre-symptomatic Huntington's disease mice. J Neurosci. 2005;25:3059–3066. doi: 10.1523/JNEUROSCI.4320-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load. J Physiol. 2007;585:817–831. doi: 10.1113/jphysiol.2007.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Glynn D, Leavens W, Zheng Z, Faull RL, Skepper JN, Wight JM. Paradoxical delay in the onset of disease caused by super-long CAG repeat expansion in R6/2 mice. Neurobiol Dis. 2009;33:331–341. doi: 10.1016/j.nbd.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Munsie L, Caron N, Atwal RS, Marsden I, Wild EJ, Bamburg JR, Tabrizi SJ, Truant R. Mutant huntingtin cause defective actin remodeling during stress: defining a new role for transglutaminase 2 in neurodegenerative disease. Hum Mol Genet. 2011;20:1937–1951. doi: 10.1093/hmg/ddr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J Neurosci. 2000;20:5115–5123. doi: 10.1523/JNEUROSCI.20-13-05115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithianantharajah J, Hannan AJ. Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience-dependent mediators of cognitive and psychiatric symptoms in Huntington's disease. Neuroscience 2012. 2012 doi: 10.1016/j.neuroscience.2012.05.043. doi: 10.1016/j.neuroscience.2012.05.043. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Nithianantharajah J, Barkus C, Vijiaratnam N, Clement O, Hannan AJ. Modeling brain reserve: experience-dependent neuronal plasticity in healthy and Huntington's disease transgenic mice. Am J Geriatr Psychiatry. 2009;17:196–209. doi: 10.1097/JGP.0b013e318196a632. [DOI] [PubMed] [Google Scholar]
- Orth M, Schippling S, Schneider SA, Bhatia KP, Talelli P, Tabrizi SJ, Rothwell JC. Abnormal motor cortex plasticity in premanifest and very early manifest Huntington disease. J Neurol Neurosurg Psychiatry. 2010;81:267–270. doi: 10.1136/jnnp.2009.171926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, Glynn P, Cain K, Kyriacou CP, Giorgini F, Nicotera P. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington's disease. Cell Death Differ. 2011;18:191–200. doi: 10.1038/cdd.2010.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM. Evidence for more wide spread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60:1615–1620. doi: 10.1212/01.WNL.0000065888.88988.6E. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/S0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Schippling S, Schneider SA, Bhatia KP, Münchau A, Rothwell JC, Tabrizi SJ, Orth M. Abnormal motor cortex excitability in preclinical and very early Huntington's disease. Biol Psychiatry. 2009;65:959–965. doi: 10.1016/j.biopsych.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Brundin P, Li JY. Synaptic dysfunction in Huntington's disease: a new perspective. Cell Mol Life Sci. 2005a;62:1901–1912. doi: 10.1007/s00018-005-5084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Petersén A, Bates GP, Brundin P, Li JY. Depletion of rabphilin 3A in a transgenic mouse model (R6/1) of Huntington's disease, a possible culprit in synaptic dysfunction. Neurobiol Dis. 2005b;20:673–684. doi: 10.1016/j.nbd.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Smith R, Klein P, Koc-Schmitz Y, Waldvogel HJ, Faull RL, Brundin P, Plomann M, Li JY. Loss of SNAP-25 and rabphilin 3a in sensory-motor cortex in Huntington's disease. J Neurochem. 2007;103:115–123. doi: 10.1111/j.1471-4159.2007.04703.x. [DOI] [PubMed] [Google Scholar]
- Sotrel A, Williams RS, Kaufmann WE, Myers RH. Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington's disease: a quantitative Golgi study. Neurology. 1993;43:2088–2096. doi: 10.1212/WNL.43.10.2088. [DOI] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Garry S, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ. Dendritic spine pathology and deficits in experience-dependent plasticity in R6/1 Huntington's disease transgenic mice. Eur J Neurosci. 2004;19:2799–2807. doi: 10.1111/j.0953-816X.2004.03374.x. [DOI] [PubMed] [Google Scholar]
- Sun Y, Savanenin A, Reddy PH, Liu YF. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem. 2001;276:24713–24718. doi: 10.1074/jbc.M103501200. [DOI] [PubMed] [Google Scholar]
- Tampellini D, Magrané J, Takahashi RH, Li F, Lin MT, Almeida CG, Gouras GK. Internalized antibodies to the Abeta domain of APP reduce neuronal Abeta and protect against synaptic alterations. J Biol Chem. 2007;282:18895–18906. doi: 10.1074/jbc.M700373200. [DOI] [PubMed] [Google Scholar]
- Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, 2nd, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verny C, Allain P, Prudean A, Malinge MC, Gohier B, Scherer C, Bonneau D, Dubas F, Le Gall D. Cognitive changes in asymptomatic carriers of the Huntington disease mutation gene. Eur J Neurol. 2007;14:1344–1350. doi: 10.1111/j.1468-1331.2007.01975.x. [DOI] [PubMed] [Google Scholar]
- Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington's disease phenotypes in YAC128 mice. J Neurosci. 2010;30:14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara Y, De Roo M, Muller D. Dendritic spine formation and stabilization. Curr Opin Neurobiol. 2009;19:146–153. doi: 10.1016/j.conb.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglia activation precede tangles in a P30IS tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Marullo M, Conforti P, MacDonald ME, Tartari M, Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington's disease. Brain Pathol. 2008;18:225–238. doi: 10.1111/j.1750-3639.2007.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]