

Abstract
Alzheimer's disease (AD) chiefly characterizes a progressively neurodegenerative disorder of the brain, and eventually leads to irreversible loss of intellectual abilities. The β-amyloid (Aβ)-induced neurodegeneration is believed to be the main pathological mechanism of AD, and Aβ production inhibition or its clearance promotion is one of the promising therapeutic strategies for anti-AD research. Here, we report that the natural product arctigenin from Arctium lappa (L.) can both inhibit Aβ production by suppressing β-site amyloid precursor protein cleavage enzyme 1 expression and promote Aβ clearance by enhancing autophagy through AKT/mTOR signaling inhibition and AMPK/Raptor pathway activation as investigated in cells and APP/PS1 transgenic AD model mice. Moreover, the results showing that treatment of arctigenin in mice highly decreased Aβ formation and senile plaques and efficiently ameliorated AD mouse memory impairment strongly highlight the potential of arctigenin in anti-AD drug discovery.
Introduction
Alzheimer's disease (AD) is a progressively neurodegenerative disease that eventually leads to the irreversible loss of neurons and intellectual abilities, including cognition and memory. β Amyloid (Aβ), generated from a sequent cleavage of amyloid precursor protein (APP) by β-site APP cleavage enzyme 1 (BACE1) and γ-secretase, contributes much to AD pathology, in that accumulation of this peptide in the hippocampus and cortex causes neuron dysfunction and cognitive deficit (Hardy and Selkoe, 2002). Therefore, targeting Aβ has been considered a promising therapeutic strategy against AD (Yamin et al., 2008; Kurz and Perneczky, 2011).
In the discovery of Aβ production inhibitor, BACE1 as a rate-limiting enzyme for Aβ generation has always received much attention, and most of the anti-AD research has focused on the discovery of BACE1 enzymatic inhibitor (Hong et al., 2000; May et al., 2011). However, none of the inhibitors discovered so far has passed clinical trials yet, an indication that a new tactic besides enzymatic inhibition should be devised against this enzyme (Klaver et al., 2010). Recently, the finding of the elevated protein level of BACE1 in both AD patients and model mice has implied that downregulation of BACE1 expression could be developed as a new approach for anti-AD drug discovery (Fukumoto et al., 2002; Holsinger et al., 2002; Chen et al., 2011). This was further confirmed by the fact that BACE1 expression inhibitors targeting BACE1 transcription or translation effectively improved AD symptoms (Yi et al., 2011).
Apart from Aβ production inhibition, Aβ clearance promotion is also regarded as another valuable strategy in anti-AD research, since failure of Aβ clearance has been confirmed to be strongly associated with AD pathogenesis (Wostyn et al., 2011; Yang et al., 2011). Efforts to target Aβ clearance have resulted in the discovery of some active compounds that could be useful in ameliorating AD symptoms. For example, nilvadipine, an anti-hypertensive drug, highly facilitated Aβ clearance and effectively improved learning abilities and spatial memory in AD mice, and is currently used in Phase 3 clinical trials in Europe (Bachmeier et al., 2011; Kennelly et al., 2011).
Accordingly, based on the above-mentioned facts, we expected that agents with dual-functional activities in both Aβ production inhibition and its clearance promotion might demonstrate more potent anti-AD properties. Considering that natural products are major resources of bioactive agents for their large-scale structural diversity, we thus constructed a “one stone, two birds” platform against our laboratory in-house natural product library. This led to the discovery of arctigenin (Fig. 1A), which could both inhibit Aβ production and promote its clearance. In addition, the mechanisms underlying the arctigenin-mediated anti-AD function were intensively investigated.
Figure 1.
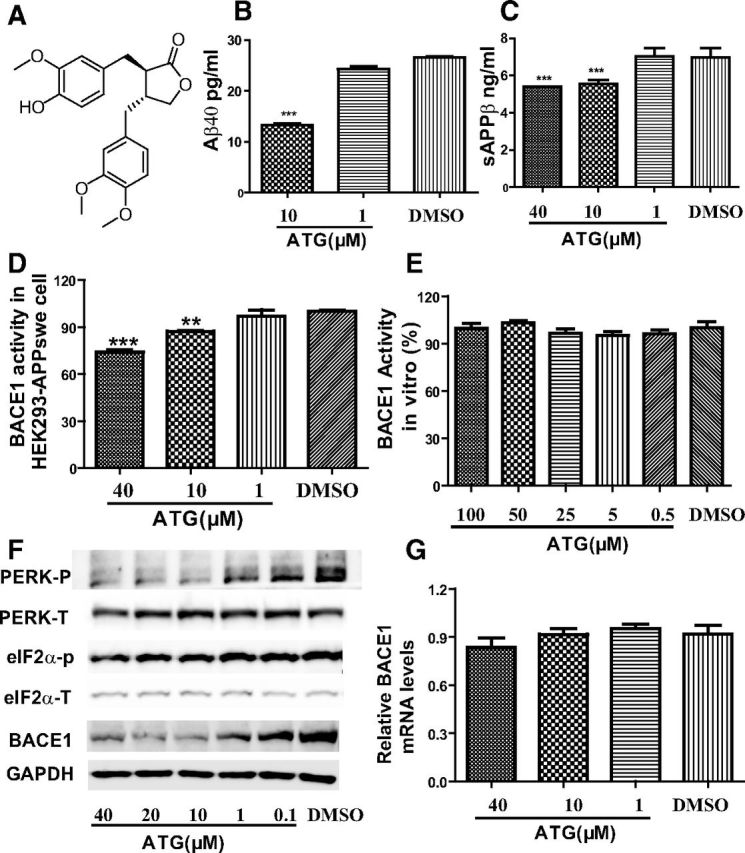
Arctigenin reduced Aβ production with BACE1 translation inhibition in HEK293-APPswe cells. HEK293-APPswe cells were cultured with 0.1% DMSO (as a control) or different concentrations of arctigenin (ATG) for 24 h. A, Chemical structure of arctigenin. B, ELISA assay of arctigenin-decreased Aβ40. C, ELISA assay of arctigenin-reduced sAPPβ. D, Arctigenin inhibited BACE1 activity in cells. E, FRET-based assay demonstrated that arctigenin failed to inhibit BACE1 activity at protein level. F, Western blot assay indicated that arctigenin decreased BACE1 protein level and phosphorylation of eIF2α or PERK. G, RT-PCR assay indicated that arctigenin had no effect on BACE1 transcription. The results shown are representative of three independent experiments. **p < 0.01, ***p < 0.001, n = 3.
We discovered that arctigenin decreased BACE1 translation via dephosphorylation of eIF2α, causing the suppression of Aβ production, and initiated autophagy through both AMPK/Raptor signaling activation and AKT/mTOR pathway inhibition, resulting in enhanced Aβ clearance. Moreover, arctigenin strongly ameliorated the memory impairment of APP/PS1 transgenic model mice. Our results have thereby strongly highlighted the potential of arctigenin in anti-AD drug research.
Materials and Methods
Materials.
Arctigenin was extracted from Fructus arctii, and the purity of arctigenin was determined to be >99% by high-performance liquid chromatography. All cell culture reagents were purchased from Invitrogen. Complete protease inhibitor mixture, penicillin, streptomycin, G418, Tween 80, Congo red, thioflavine S, and DMSO were from Sigma-Aldrich. RNAiso, RT reagent kit, and SYRB Premix Ex-Taq were purchased from TaKaRa Bio. BACE1 antibody was from Sigma-Aldrich. Phospho-double-stranded RNA-dependent protein kinase-like endoplasmic reticulum kinase (phospho-PERK, Thr981) and PERK antibodies were from Santa Cruz Biotechnology. The antibodies of phospho-eIF2α (Ser51), eIF2α, GAPDH, phospho-AMP-activated protein kinase (phospho-AMPK, Thr172), AMPK, phospho-Raptor (Ser792), Raptor, phospho-AKT (Ser473), AKT, phospho-mTOR (Ser2448), mTOR, phospho-ULK1 (Ser757), ULK1, phospho-P70S6K (Thr389), P70S6K, and P62 were from Cell Signaling Technology.
Cell culture.
HEK293 cells expressing APP Swedish mutantK595N/M596L (HEK293-APPswe) were grown in DMEM containing 10% fetal bovine serum (FBS), 100 U/ml penicillin-streptomycin, and 400 μg/ml G418 at 37°C in 5% CO2. BV2 cells were cultured in DMEM containing 10% FBS and 100 U/ml penicillin-streptomycin at 37°C in 5% CO2.
Primary cortical neuron culture.
Embryos (embryonic day 16) were removed from an anesthetized pregnant mouse. Cerebral cortices were dissected from the brain and digested with D-Hanks buffer (5.4 mm KCl, 0.41 mm KH2PO4, 138 mm NaCl, 4.5 mm NaHCO3, 0.22 mm Na2HPO4, pH 7.4) containing 0.125% trypsin and 200 U/ml DNase (Sigma-Aldrich) for 15 min at 37°C, followed by dissociation with pipetting in DMEM with 10% FBS. Dissociated cells were cultured in the neurobasal medium with 2% B27 supplement, 0.5 mm glutamine (Invitrogen), and 50 U/ml penicillin-streptomycin using poly-d-lysine (Sigma-Aldrich)-coated six-well cell culture plates. The plating density was 0.5 × 106 cells/ml. After 10 d, different concentrations of DMSO-dissolved arctigenin were separately incubated in the cultured neurons for 24 h.
Primary cortical astrocytes culture.
Embryos (embryonic day 18) were removed from an anesthetized pregnant mouse, and the isolation procedure of cortical astrocytes was similar to that of neuronal cells. Briefly, dissociated cells were cultured in DMEM/F12 with 10% FBS and 50 U/ml penicillin-streptomycin, using poly-d-lysine-coated 75 cm2 flask by the density of 2 × 105 cells/cm2. After 7 d, the flask was rotated at 220 rpm/min at 37°C overnight. Then, the cells in the medium were removed, leaving behind adhesion cells, which were identified by GFAP and DAPI staining as astrocytes. After 3 d continuing culture, the astrocytes were subcultured in six-well plates, followed by administration with different concentrations of DMSO-dissolved arctigenin for 24 h.
BACE1 activity assay.
The effect of arctigenin on BACE1 activity at protein level was investigated using BACE1 activity kits (Invitrogen) according to the provided kit protocol. Briefly, the BACE1 substrate (5 μm), BACE1 enzyme (550 ng/ml), and varied concentrations of arctigenin (100, 50, 10, 5, 1, 0.5, 0.1 μm) were sequentially incubated for 1 h at 37°C in darkness. Fluorescence intensity was then measured with excitation and emission wavelengths at 420 and 530 nm, respectively.
The cell-based assay of the effect of arctigenin on BACE1 activity was performed with BACE1-specific FRET-based substrate (Invitrogen) according to the provided protocol. In brief, after 24 h incubation of cells with different concentrations of arctigenin, the cells were lysed in RIPA buffer (Thermal), supernatants were collected, and protein concentration was determined by BCA protein assay kit (Thermal). Then the supernatants were diluted to the protein concentration of 10 ng/ml, and incubated with BACE1 substrate and reaction buffer (provided with the purchased kit) at 37°C in darkness. After 1 h incubation, fluorescence test was performed with excitation and emission wavelengths at 420 and 530 nm, respectively.
Western blot.
In cell-based assay, cells were administrated with different concentrations of arctigenin and incubated for 24 h, and then lysed with RIPA buffer (Thermal) containing protease mixture (Sigma-Aldrich). Protein concentration was determined by BCA protein assay kit (Thermal). Proteins were mixed with 2 × SDS-PAGE sample buffer (4% SDS, 62.5 mm Tris-HCl, pH 6.8, 25% glycerol, and 0.1% bromophenol blue) and then boiled for 15 min at 99°C.
In brain tissue-based assay, the cortices of four mice in each group were homogenized with RIPA buffer (Thermal) containing protease mixture (Sigma-Aldrich) by a hand-held motor and kept on ice for 1 h to lyse the cells completely. The homogenates were then centrifuged by 20,000 × g at 4°C for 30 min. The supernatants were collected, and protein concentration was determined using BCA protein assay kit. Equal amounts of lysates (2 mg/ml protein) were mixed with 2 × SDS-PAGE sample buffers, and then boiled for 15 min at 99°C.
All cell and tissue samples were subject to Western blot analyses of BACE1, phospho-eIF2α, eIF2α, phospho-PERK, PERK, P62, GAPDH, phospho-AMPK, AMPK, phospho-Raptor, Raptor, phospho-AKT, AKT, phospho-mTOR, mTOR, phospho-ULK1, ULK1, phospho-P70S6K, and P70S6K.
Intracellular Aβ clearance assay.
Intracellular Aβ clearance in astrocytes, BV2 cells, and primary neuronal cells was assayed according to the Landreth approach (Jiang et al., 2008; Cramer et al., 2012). In brief, astrocytes were sequentially cultured with varied concentrations (40, 10, 1, 0 μm) of arctigenin for 18 h at 37°C. Cells were then treated with 2 μg/ml soluble Aβ40 and varied concentrations (40, 10, 1, 0 μm) of arctigenin for 3 h, followed by lysis in 50 mm Tris buffer containing 1% SDS. Protein concentration was determined by BCA protein assay kit, and intracellular Aβ peptide was evaluated by ELISA and normalized to total protein.
ELISA assay.
In cell-based assay, HEK293-APPswe cells were incubated with series of concentrations of DMSO-dissolved arctigenin for 24 h. The cell culture media were collected, and a complete protease inhibitor mixture was added (Sigma-Aldrich). The culture media were then centrifuged by 20,000 × g at 4°C for 30 min, and supernatants were collected. ELISA kits of Aβ40 (Invitrogen) and sAPPβ (Immuno-Biological Laboratories) were used for evaluation of Aβ40 and sAPPβ contents, respectively.
For the assay in the brains, hippocampus sample was prepared according to the published approach (Zhu et al., 2010). In brief, the hippocampi were homogenized in 5 m guanidine hydrochloride using a hand-held motor, and the homogenates were then centrifuged at 20,000 × g at 4°C for 30 min. The supernatants were collected, and contents of Aβ40 and Aβ42 were tested according to the protocol of Aβ40/Aβ42 ELISA kit.
Real-time quantitative PCR analysis.
The total mRNA was isolated from the arctigenin-treated cells or mice brains using TRIZOL Reagent (TaKaRa Bio) according to the protocols of commercial kits. Complementary DNA synthesis was performed with oligo-dT primers by the instruction of reverse-PCR kit (TaKaRa Bio). Real-time PCR was performed using SYBR green PCR core reagent kit (TaKaRa Bio) for mRNA quantity detection.
PCR primer pairs for amplification of the three genes are as follows: human BACE1: (F) CAT TGG AGG TAT CGA CCA CTC GCT, (R) CCA CAG TCT TCC ATG TCC AAG GTG (human 18sRNA was designed as described previously; Scheid and Woodgett, 2001); mice BACE1: (F) TGGACACCGGAAGCAGTAACTT, (R) AGCTTGATGGCTTGGCCAA; mice β-actin: (F) TCG TGG GCC GCT CTA GGC ACC A, (R) GTT GGC CTT AGG GTT CAG GGG GG.
Animal experiment.
All animal experiments were performed according to the institutional ethical guidelines on animal care. The APP/PS1 mice [B6C3-Tg (APPswe, PS1dE9)], which express chimeric mouse/human Swedish mutant amyloid precursor protein (Mo/HuAPP695swe) and mutant human presenilin 1 protein (PS1-dE9), were purchased from Jackson Laboratory and housed under standard conditions with a 12 h light/dark cycle at room temperature of 22°C. As described previously, APP/PS1 mice exhibited early AD symptoms at 6–7 months of age. These symptoms became more serious after another 3 months or more (Reiserer et al., 2007). In our work, 20 male APP/PS1 mice were divided randomly into two groups and 10 nontransgenic mice were in one group as negative control. Arctigenin was dissolved in 5% Tween 80. The two 6 month transgenic groups were administrated with 3 mg/kg/d arctigenin or vehicle and the 6 month nontransgenic group was administrated with vehicle for 100 d by intraperitoneal injection. After 100 d administration, Morris water maze (MWM) assay was applied to evaluate the cognitive abilities of the mice for 6 d with continuous arctigenin treatment. Upon completion of MWM test, mice were killed, and the brains were then removed and bisected in the midsagittal plane. The right hemispheres were frozen and stored at −80°C, and the left hemispheres were fixed in 4% paraformaldehyde.
MWM test.
The MWM assay was performed as previously described (Zhu et al., 2010). Briefly, over 5 consecutive days with three trials per day, mice were trained to use a variety of visual cues located on the pool wall to find a hidden submerged white platform in a circular pool (120 cm in diameter, 50 cm deep) filled with milk-tinted water. In each trial, each mouse was given 90 s to find the hidden platform. If the mouse found the platform within that time, the animal was allowed to rest on the platform for 20 s. If the mouse failed to find the platform within 90 s, the animal was manually placed on the platform and allowed to remain there for 20 s. On day 6, after training trials, a probe trial was performed by removing the platform, and mice were allowed to swim for 90 s in search of it. All data were collected for mouse performance analysis. For data analysis, the pool was divided into four equal quadrants formed by imaging lines, which intersected in the center of the pool at right angles called north, south, east, and west.
Congo red and thioflavine S staining.
Paraformaldehyde-fixed brain tissues (∼3.5 mm thick) were obtained from the hippocampus and embedded in paraffin. Ten-micrometer-thick coronal sections were for Congo red and thioflavine S staining according to the previous approach (Zhu et al., 2010). For Congo red staining, the sections were deparaffinized, hydrated, and stained with Congo red for 20 min, followed by differentiation in alkaline alcohol solution and counterstaining with Gill's hematoxylin for 30 s, finally mounting with resinous medium. For thioflavine S staining, the sections were stained in a 1% thioflavine S stock for 5 min and differentiated in 70% alcohol for 5 min and mounted in glycerin jelly.
The Congo red-stained plaque numbers and thioflavine S plaque burdens were determined separately in the hippocampus and cortex. One section from each mouse (10 mice per group) was used for plaque quantification with Congo red and thioflavine S staining. Thioflavine S plaque burdens were counted on every five fields throughout the entire hippocampus and cortex (10 mice per group) by Image-Pro Plus (Media Cybernetics). Total number of Congo red-stained plaque in hippocampus and cortex (10 mice per group) was calculated for statistical analysis.
Data analysis.
The statistical difference between multiple treatments and control was analyzed using one-way ANOVA with Dunnett's post-test. p value of <0.05 was considered to be statistically significant.
Results
Arctigenin decreased Aβ content
To screen for natural products able to decrease Aβ content, ELISA assay was performed in HEK293-APPswe cells against the laboratory in-house natural product library. The natural product arctigenin was finally determined. As shown in Figure 1B, arctigenin effectively decreased Aβ content in the cells.
Arctigenin reduced Aβ production
Arctigenin reduced Aβ production by inhibiting BACE1 activity
Given that Aβ level involves a dynamic equilibrium of Aβ production and clearance, we at first investigated the potential effect of arctigenin on Aβ production in HEK293-APPswe cells using ELISA assay by testing the level of sAPPβ, a direct product of APP with BACE1 cleavage, for indication of Aβ production (Zhu et al., 2010). As shown in Figure 1C, sAPPβ level was obviously decreased with arctigenin treatment, suggesting that arctigenin decreased Aβ production.
Next, we determined whether arctigenin inhibited BACE1 activity in HEK293-APPswe cells. In the assay, the cells were cultured in six-well plates and incubated sequentially with varied concentrations of arctigenin for 24 h. Cells were then lysed in RIPA buffer (Thermal), and BACE1 activity of the cell lysates was determined using BACE1-specific substrate (Invitrogen). As indicated in Figure 1D, arctigenin dose-dependently decreased BACE1 activity.
Arctigenin suppressed BACE1 activity via inhibition of BACE1 translation
In view of the arctigenin-induced BACE1 activity inhibition, we used FRET technology-based assay to examine whether arctigenin functioned as a BACE1 enzymatic inhibitor. As indicated in Figure 1E, arctigenin failed to directly inhibit BACE1 activity in vitro, implying that arctigenin possibly downregulated BACE1 activity by decreasing BACE1 protein level.
Accordingly, by Western blot assay, we investigated whether arctigenin decreased BACE1 level in HEK293-APPswe cells. As shown in Figure 1F, arctigenin dose-dependently decreased BACE1 protein level. In addition, by RT-PCR, we investigated the potential influence of arctigenin on BACE1 transcription. The results in Figure 1G suggested that arctigenin exhibited no effects on BACE1 mRNA level. Therefore, these results demonstrated that arctigenin inhibited BACE1 activity by decreasing BACE1 translation.
Considering that BV2 cells could mimic primary microglia with high fidelity and primary neuronal cells function potently in AD pathology (Henn et al., 2009), we further investigated the effect of arctigenin on BACE1 activity in both BV2 and primary neuronal cells. Similar to the cases in HEK293-APPswe cells, in either BV2 or primary neuronal cells arctigenin decreased BACE1 activity through BACE1 protein inhibition (Fig. 2A,C,D,F) without affecting BACE1 mRNA levels (Fig. 2B,E).
Figure 2.
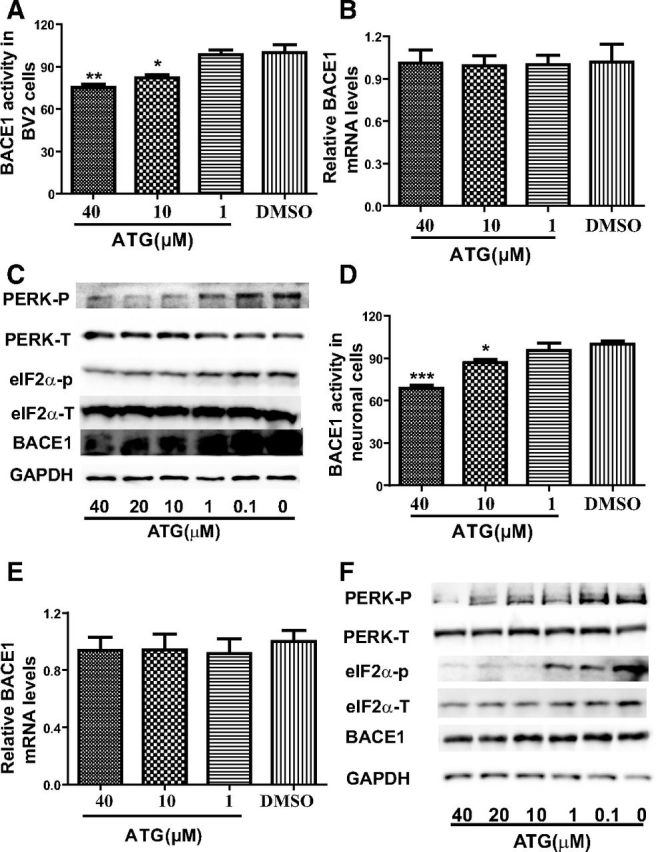
Arctigenin decreased BACE1 activity with BACE1 translation inhibition in BV2 and primary neuronal cells. BV2 and primary cortical neuronal cells were treated with 0.1% DMSO or different concentrations of arctigenin for 24 h. A, Arctigenin inhibited BACE1 activity in BV2 cells. B, RT-PCR assay indicated that arctigenin had no effect on BACE1 transcription in BV2 cells. C, Western blot assay indicated that arctigenin decreased BACE1 protein level and phosphorylation of eIF2α or PERK in BV2 cells. D, Arctigenin inhibited BACE1 activity in primary cortical neuronal cells. E, RT-PCR assay indicated that arctigenin had no effect on BACE1 transcription in primary cortical neuronal cells. F, Western blot assay indicated that arctigenin decreased BACE1 protein level and phosphorylation of eIF2α or PERK in primary cortical neuronal cells. The results shown are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, n = 3.
In addition, we examined whether arctigenin decreased BACE1 protein level by inhibiting BACE1 translation in primary astrocytes. Unexpectedly, arctigenin failed to affect BACE1 activity or protein level (data not shown). This result thus demonstrated the cell-selective character of arctigenin.
PERK/eIF2α-mediated pathway was involved in the arctigenin-reduced BACE1 translation
As reported, transcription factors, such as NFκB, PPARγ, and YY1, could regulate BACE1 protein expression by mediation of BACE1 transcription (Devi and Ohno, 2010; Guglielmotto et al., 2011; Katsouri et al., 2011; Kwak et al., 2011), and phosphorylation of the translation initiation factor eIF2α (eIF2α-P) increased BACE1 translation and further regulated its protein level (O'Connor et al., 2008; Devi and Ohno, 2010). Therefore, based on our above finding that arctigenin failed to induce BACE1 transcription, we used Western blot assay in HEK293-APPswe cells to determine whether arctigenin regulated BACE1 translation via dephosphorylation of eIF2α. As expected, arctigenin could dose-dependently inhibit eIF2α phosphorylation (Fig. 1F). In addition, it was reported that arctigenin blocked the phosphorylation level of phosphorylated PERK in HT-29 cells (Kim et al., 2010). Given that PERK as a kind of eIF2α kinase played an important role in eIF2α phosphorylation (Harding et al., 1999), we detected the potential regulation of arctigenin on PERK phosphorylation in HEK293-APPswe cells. As demonstrated in Figure 1F, arctigenin could dose-dependently inhibit the phosphorylation of PERK. These results thus implied that PERK/eIF2α-mediated pathway was involved in the arctigenin-reduced BACE1 translation.
In addition, the assay in either BV2 or primary neuronal cells further validated the inhibition of arctigenin against PERK/eIF2α pathway (Fig. 2C,F). However, probably due to the cell-selective feature of arctigenin, this natural product rendered no such effect in primary astrocytes (data not shown).
Arctigenin promoted Aβ clearance
In the investigation of the arctigenin-induced promotion of exogenous Aβ clearance, the assays based on primary astrocytes, BV2 cells, and primary neuronal cells were performed according to Landreth's approach (Cramer et al., 2012). The results indicated that arctigenin dose-dependently enhanced exogenous Aβ clearance in primary astrocytes (Fig. 3A), primary neuronal cells (Fig. 3B), and BV2 cells (Fig. 3C).
Figure 3.

Arctigenin promoted Aβ clearance by enhancing autophagy with AKT/mTOR signaling inhibition and AMPK/Raptor pathway activation. A–C, ELISA assay indicated that arctigenin increased Aβ clearance in primary cortical astrocytes (A), primary neuronal cells (B), and BV2 cells (C). D, CQ-based ELISA assay demonstrated that CQ could enhance Aβ levels and partially reverse arctigenin-induced Aβ reduction in HEK293-APPswe cells. Primary cortical astrocytes were treated with 0.1% DMSO or different concentrations of arctigenin for 24 h. E–G, Western blot assay demonstrated that arctigenin reduced P62 protein level and ULK1 or P70S6K phosphorylation (E), decreased AKT or mTOR phosphorylation (F), and increased AMPK or Raptor phosphorylation (G). The results shown are representative of three independent experiments. **p < 0.01, ***p < 0.001, n = 3.
In the subsequent investigation of the potential mechanism underlying the arctigenin-enhanced Aβ clearance, we focused on autophagy-related research, because autophagy as a vital catabolic process in cells is highly responsible for degradation of abnormal and aggregated proteins and dysfunction organelles, and has been confirmed to be a major pathway for Aβ clearance (Klionsky and Emr, 2000). It is noticed that activation of the mammalian target of rapamycin (mTOR), a conserved Ser/Thr protein kinase, effectively inhibited autophagy through phosphorylation of Unc51-like kinase 1 (ULK1), which is an initiator of autophagy process (Chan et al., 2007; Kundu et al., 2008; Kim et al., 2011). In addition, P62 as an autophagic substrate also characterizes the activation of autophagy (Bjørkøy et al., 2005).
Therefore, before we investigated the detailed regulation of arctigenin against the autophagy pathway, the autophagy inhibitor chloroquine (CQ, Kimura et al., 2013) was used for related assays in HEK293-APPswe cells, where the cells were treated with arctigenin (40 μm), vehicle alone, CQ (10 μm), or arctigenin in combination with CQ for 24 h. The cell culture media were then collected and subjected to ELISA testing for Aβ40. As shown in Figure 3D, CQ could enhance Aβ level and partially reverse the arctigenin-induced Aβ reduction in cells. This result thereby indicated that arctigenin decreased Aβ level and load via enhancing autophagy.
Arctigenin effectively actuated the mTOR-mediated autophagy
Given that P70S6K is the direct substrate of mTOR (Kim et al., 2011), we inspected P70S6K phosphorylation to evaluate the arctigenin-regulated mTOR activity. As shown in Figure 3E, arctigenin effectively suppressed the phosphorylation of mTOR substrate P70S6K, thus suggesting that arctigenin inhibited mTOR activity in primary astrocytes.
Moreover, the results showing that arctigenin dose-dependently decreased ULK1 phosphorylation and P62 protein level further indicated the actuation of autophagy (Fig. 3E).
Similarly, arctigenin also suppressed the phospho-P70S6K, phospho-ULK1, and P62 protein level in HEK293-APPswe cells (Fig. 4A), in BV2 cells (Fig. 4G), and in primary neuronal cells (Fig. 4D).
Figure 4.
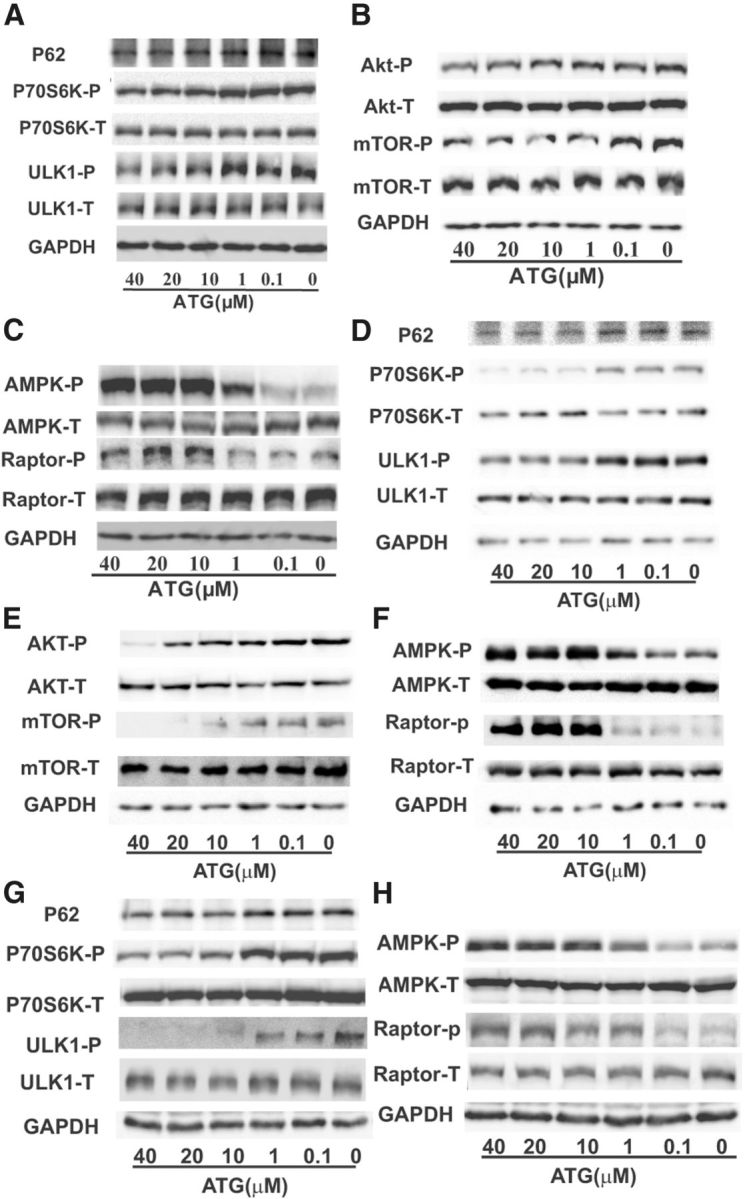
Arctigenin enhanced autophagy with AKT/mTOR signaling inhibition or AMPK/Raptor pathway activation in HEK293-APPswe, primary cortical neuronal, and BV2 cells. HEK293-APPswe, primary cortical neuronal, and BV2 cells were treated with 0.1% DMSO or different concentrations of arctigenin for 24 h. A–C, Western blot assay demonstrated that arctigenin reduced P62 protein level and ULK1 or P70S6K phosphorylation (A), decreased AKT or mTOR phosphorylation (B), and increased AMPK or Raptor phosphorylation (C) in HEK293-APPswe cells. D–F, Western blot assay demonstrated that arctigenin reduced P62 protein level and ULK1 or P70S6K phosphorylation (D), decreased AKT or mTOR phosphorylation (E), and increased AMPK or Raptor phosphorylation (F) in primary cortical neuronal cells. G, H, Western blot assay demonstrated that arctigenin reduced P62 protein level and ULK1 or P70S6K phosphorylation (G) and increased AMPK or Raptor phosphorylation (H) in BV2 cells.
Therefore, all above results demonstrated that arctigenin could enhance autophagy and inhibit mTOR activity.
AKT/mTOR and AMPK/Raptor pathways were both involved in the arctigenin-induced mTOR inhibition
Given that arctigenin has been discovered to effectively regulate mTOR-mediated autophagy, we next explored the potential underlying mechanism for this natural product in the regulation of mTOR pathway.
As indicated in the published results, AKT [protein kinase B (PKB)] is a critical mediator of mTOR, which increases mTOR activity through direct or indirect phosphorylation of mTOR (Nave et al., 1999; Memmott and Dennis, 2009). AMP-activated protein kinase (AMPK) phosphorylates the regulatory-associated protein of mTOR (Raptor, an mTOR binding partner), thus inducing its binding to 14-3-3 protein and inhibition of mTOR activity (Kimura et al., 2003; Gwinn et al., 2008; Hardie, 2008). Therefore, AKT/mTOR and AMPK/Raptor pathways associate tightly with mTOR activity, and further the regulation of autophagy and Aβ clearance. In addition, our previous work also indicated that arctigenin increased AMPK phosphorylation in C2C12 and H9C2 cells (Tang et al., 2011). Thus, we performed the related assays for evaluation of arctigenin in the regulation of AKT/mTOR and AMPK/Raptor pathways.
Primary astrocyte-based assay.
As shown in Figure 3F, arctigenin dose-dependently suppressed AKT phosphorylation at serine 473. Next, we investigated the effect of arctigenin on mTOR phosphorylation at serine 2448, a direct AKT phosphorylation site. As shown also in Figure 3F, arctigenin effectively inhibited mTOR phosphorylation, indicative of the inhibition of mTOR activity. Subsequently, we examined the potential regulation of arctigenin on AMPK/Raptor pathway. As indicated in Figure 3G, arctigenin dose-dependently increased the phosphorylation of either AMPK or Raptor without any influences on total AMPK or Raptor.
HEK293-APPswe cell-based assay.
Similarly, the regulation of arctigenin in AKT/mTOR or AMPK/Raptor pathway was also evaluated in HEK293-APPswe cells. As shown in Figure 4, B and C, arctigenin obviously decreased the phosphorylation of AKT or mTOR, and increased the phosphorylation of AMPK or Raptor.
Primary neuronal and BV2 cell-based assays.
Finally, primary neuronal and BV2 cell-based assays were also carried out. The results demonstrated that arctigenin activated AMPK/Raptor and inhibited AKT/mTOR pathways in primary neuronal cells (Fig. 4E,F), similar to the cases for arctigenin in HEK293-APPswe cells and primary astrocytes. However, arctigenin could only activate the AMPK/Raptor pathway (Fig. 4H), but failed to affect the AKT/mTOR pathway in BV2 cells (data not shown), reflecting again the cell-selective feature of arctigenin.
Arctigenin effectively ameliorated memory impairment in APP/PS1 AD mice
Given that arctigenin has been determined to be very effective in both inhibiting Aβ production and enhancing its clearance in cells, we next performed the MWM assay to further examine the potential of this natural product in the amelioration of memory impairment against the double transgenic APP/PS1 mice with age-related Aβ accumulation and deposition in the brain (Wenk, 2004). As shown in Figure 5, A and B, in the training trials, the path lengths and escape latencies to find the platform for the APP/PS1 transgenic mice were significantly longer than those for the nontransgenic mice. However, the prolongation of length and latency was significantly reversed by 3 mg/kg/d arctigenin administration at day 5. In the probe trial assay, the arctigenin-administrated (3 mg/kg/d) transgenic mice crossed over the hidden location of the platform more frequently compared with the vehicle-administrated (5% Tween 80) transgenic mice, and the crossing frequency is even similar to that of the vehicle-administrated nontransgenic mice (Fig. 5C,D). Furthermore, the swimming distance and time spent in all quadrants were analyzed. The results demonstrated that the distance and time within Quandrant 3, the location of the hidden platform, were shorter for the APP/PS1 transgenic mice than those for the nontransgenic mice. Obviously, arctigenin administration reversed the shortcomings of these transgenic mice (Fig. 5E,F). Additionally, arctigenin treatment would not induce any nonspecific changes, such as reduced/increased swimming speeds, floating, and thygmotaxis (data not shown).
Figure 5.

Arctigenin ameliorated memory impairment in APP/PS1 AD mice. MWM test was performed to evaluate spatial memory in mice. A, B, In 5 d training trials, the mice swimming paths and the latencies were measured to assess the mouse memory ability. C, D, In the probe trial, the mice swimming paths and the times for mice crossing the area where the submerged platform was placed in training trails were recorded. E, F, In the probe trial, the swimming distance and time spent in all quadrants were analyzed. T-V, Transgenic mice administrated with vehicle; NT-V, nontransgenic mice administrated with vehicle; T-ATG3, transgenic mice administrated with 3 mg/kg/d arctigenin; ATG3, nontransgenic mice administrated with 3 mg/kg/d arctigenin; V, nontransgenic mice administrated with vehicle. Values are means ± SE, *p < 0.05, **p < 0.01, n = 10 T-ATG3 compared with T-V, #p < 0.05, n = 10 NT-V compared with T-V.
Therefore, our results indicated that arctigenin ameliorated memory impairment in APP-PS1 transgenic mice.
Arctigenin reduced senile plaque formation in APP/PS1 AD mice
As reported, Aβ-formed senile plaque is characteristic feature of AD and also an important criterion of the neuropathological-histological verification of AD (Araujo and Cotman, 1992). We thus evaluated whether arctigenin reduced the amyloid plaque formation in mice. In the assay, after the MWM test, mice were killed and the left hemispheres were fixed in 4% paraformaldehyde for histochemical examination. As shown in Figure 6, A and B, the thioflavine S-stained senile plaque burdens in hippocampus and cortex of transgenic mice were remarkably raised compared with those in nontransgenic mice, while treatment of arctigenin by dose of 3 mg/kg/d obviously reversed high senile plaque formation in transgenic mice. Subsequently, we also used Congo red staining assay to further verify arctigenin's function in senile plaque formation. As expected, arctigenin-administrated (3 mg/kg/d) mice significantly decreased Congo red-stained senile plaque numbers in comparison with the vehicle-administrated transgenic mice (Fig. 6C,D).
Figure 6.

Arctigenin reduced amyloid plaque formation in APP/PS1 AD mice. Mouse brain coronal paraffin sections were stained with thioflavine S and Congo red. A, Images of thioflavine S-stained amyloid plaques in hippocampus. B, Statistics of thioflavine S-stained amyloid plaques in hippocampus and cortex. C, Images of Congo red-stained amyloid plaques in cortex. D, Statistics of Congo red-stained amyloid plaques in hippocampus and cortex. T-V, Transgenic mice administrated with vehicle; NT-V, nontransgenic mice administrated with vehicle; T-ATG3, transgenic mice administrated with 3 mg/kg/d arctigenin. Values are means ± SE, *p < 0.05, ***p < 0.001 n = 10, compared with T-V. Scale bar, 100 μm.
Arctigenin effectively decreased Aβ content in APP/PS1 AD mice
Next, we investigated the effect of arctigenin on Aβ content in vivo, with hippocampus and cortex collected for ELISA test against Aβ40 and Aβ42. As expected, the content of Aβ40/42 in hippocampus or cortex of transgenic mice was remarkably higher compared with those in nontransgenic mice, while treatment of 3 mg/kg/d arctigenin obviously decreased such a high level of Aβ (Fig. 7A,B).
Figure 7.

Arctigenin decreased Aβ content and inhibited BACE1 translation in APP/PS1 AD mice. For Aβ analysis, the hippocampi were lysed in 5 m guanidine hydrochloride, followed by ELISA assay against Aβ40 and Aβ42, n = 10. A, Arctigenin decreased Aβ40 or Aβ42 content in hippocampus. B, Arctigenin decreased Aβ40 or Aβ42 content in cortex. The cortices were lysed in RIPA buffer for Western blot assay, n = 4. C, Arctigenin decreased BACE1 protein level and eIF2α or PERK phosphorylation. D, Densitometry analysis of C. E, RT-PCR assay indicated that arctigenin had no effect on BACE1 transcription in mice. For RT-PCR assay, BACE1 mRNA levels of the cortices were investigated, n = 4. T-V, Transgenic mice administrated with vehicle; NT-V, nontransgenic mice administrated with vehicle; T-ATG3, transgenic mice administrated with 3 mg/kg/d arctigenin. Values are means ± SE, *p < 0.05, **p < 0.01, ***p < 0.001, compared with T-V.
PERK/eIF2α-mediated pathway was involved in the arctigenin-reduced BACE1 effect in APP/PS1 AD mice
As we have determined the involvement of PERK/eIF2α-mediated pathway in the arctigenin-reduced BACE1 translation in cells, we next confirmed such effects in AD model mice. In the assay, cortex extracts were subject to Western blot analyses for BACE1, phosphorylated eIF2α, total eIF2α, phosphorylated PERK, and total PERK. As expected, BACE1 and phosphorylated eIF2α or PERK in the transgenic mice brain were much higher than that in the nontransgenic mice (Fig. 7C,D), which is consistent with the published reports (Ohno et al., 2007; Zhao et al., 2007; Devi and Ohno, 2010; Devi et al., 2010, 2012; Ho et al., 2012). However, the high levels of BACE1, phosphorylated eIF2α, and PERK in transgenic mice were apparently inhibited by arctigenin treatment (Fig. 7C,D). These results thus further supported the idea that the PERK/eIF2α-mediated pathway was involved in the arctigenin-reduced BACE1 effect.
Subsequently, to further explore the question of whether the arctigenin-induced BACE1 decrease was due to the inhibition of translation rather than transcription, we performed the RT-PCR assay to investigate the potential effect of arctigenin on BACE1 mRNA level. As shown in Figure 7E, the mRNA level of BACE1 in transgenic mice was nearly the same as that of nontransgenic mice, and treatment of arctigenin failed to affect BACE1 mRNA level in AD transgenic mice.
Arctigenin effectively enhanced autophagy in APP/PS1 AD mice
Given the achieved result of arctigenin-induced autophagy enhancement in cells, we next investigated such effects of arctigenin in APP/PS1 AD transgenic mice by Western blot assay. As shown in Figure 8, A and B, the level of phosphorylated ULK1 in AD transgenic mice was higher than that in the nontransgenic mice, indicating that the initiation of autophagy in APP/PS1 transgenic mice was impaired. Notably, arctigenin administration (3 mg/kg/d) obviously decreased the phosphorylated ULK1 level, thus confirming that arctigenin efficiently enhanced autophagy in transgenic mice (Fig. 8A,B).
Figure 8.

Arctigenin enhanced autophagy with AMPK/Raptor pathway activation and AKT/mTOR pathway inhibition. For Western blot assay, the cortices were lysed in RIPA buffer. A, Arctigenin decreased ULK1 or P70S6K phosphorylation. B, Densitometry analysis of A. C, Arctigenin decreased AKT or mTOR phosphorylation. D, Densitometry analysis of C. E, Arctigenin increased AMPK or Raptor phosphorylation. F, Densitometry analysis of E. T-V, Transgenic mice administrated with vehicle; NT-V, nontransgenic mice administrated with vehicle; T-ATG3, transgenic mice administrated with 3 mg/kg/d arctigenin. Values are means ± SE, *p < 0.05, **p < 0.01, ***p < 0.001, n = 4, compared with T-V.
mTOR-mediated pathway was involved in arctigenin-regulated autophagy in APP/PS1 AD mice
Similar to the case in cell-based assay for investigation of mTOR-mediated pathway involvement in arctigenin-regulated autophagy, we also evaluated the effect of arctigenin on P70S6K phosphorylation in mice by Western blot. As indicated in Figure 8, A and B, the phosphorylation of P70S6K in transgenic mice was obviously elevated in comparison with that in nontransgenic mice, indicative of high mTOR activity in transgenic mice. As expected, arctigenin administration largely decreased P70S6K phosphorylation in transgenic mice (Fig. 8A,B), further confirming that the mTOR-mediated pathway was involved in arctigenin-regulated autophagy.
AKT/mTOR and AMPK/Raptor pathways were both involved in arctigenin-induced mTOR inhibition in APP/PS1 AD mice
In the above cell-based assays, we have determined the involvement of AKT/mTOR or AMPK/Raptor pathway in arctigenin-induced mTOR inhibition. Here, we confirmed such results in mice by Western blot assays. As indicated in Figure 8, C and D, phosphorylation of AKT or mTOR in transgenic mice was highly elevated compared with that of nontransgenic mice, which is consistent with the published results (Griffin et al., 2005; Caccamo et al., 2011). As expected, arctigenin administration effectively reversed such a high phosphorylation of AKT or mTOR in transgenic mice (Fig. 8C,D).
As shown in Figure 8, E and F, phosphorylation of AMPK or Raptor in transgenic mice was nearly the same as that in nontransgenic mice. However, arctigenin treatment obviously enhanced phosphorylation of either AMPK or Raptor in transgenic mice (Fig. 8E,F).
Therefore, all above results confirmed that AKT/mTOR and AMPK/Raptor pathways were both involved in arctigenin-induced mTOR inhibition in AD transgenic mice.
Discussion
Aβ peptide accumulation functions potently in the pathological cascade of AD, and agents able to inhibit Aβ production and/or enhance Aβ clearance have been confirmed to be effective in slowing AD progression (Yamin et al., 2008; Kurz and Perneczky, 2011). In the current work, after targeting Aβ, we established a “one stone, two birds” strategy to find small molecules that could both inhibit Aβ production and promote Aβ clearance. This strategy led us to the natural product arctigenin, which demonstrated high efficiency in decreasing Aβ content and ameliorating memory impairment in APP/PS1 AD model mice. Our results have demonstrated the usefulness of this strategy in the search for in anti-AD treatments.
Arctigenin, extracted from Arctium lappa (L.), was reported to exhibit some pharmacological actions, including antidiabetes, antitumor, antioxidant, and neuron protective activities, and to exercise mimetic properties (Jang et al., 2002; Predes et al., 2011; Tang et al., 2011; Gu et al., 2012; Huang et al., 2012). We discovered that arctigenin inhibited Aβ production by suppression of BACE1 translation involving eIF2α-mediated pathway. In the past decade, researchers trying to find anti-AD drugs investigated the potential of BACE1 as a rate-limiting enzyme for Aβ production, leading to the development of various BACE1 enzyme inhibitors (May et al., 2011; Cumming et al., 2012). None of these inhibitors, however, has passed clinical trials (Klaver et al., 2010). Current research has supported the idea that downregulation of BACE1 should be a new available strategy for anti-AD treatment as confirmed by elevation of BACE1 in both AD mice and patients (Klaver et al., 2010). Moreover, hyperphosphorylated eIF2α was also currently determined to be strongly associated with the increase of BACE1 translation (Chang et al., 2002; O'Connor et al., 2008; Devi and Ohno, 2010; Mouton-Liger et al., 2012). Here, we found that arctigenin effectively inhibited PERK/eIF2α pathway and decreased BACE1 translation. This result thus highly strengthened the potential of this natural product in anti-AD drug discovery.
Recently, it has been proved that impairment of autophagy contributes much to the abnormal protein accumulation (e.g., Aβ) in several age-dependent neurodegenerative diseases, including AD, Huntington's disease, and Parkinson's disease (Hara et al., 2006; Mizushima et al., 2008; Madeo et al., 2009). Compounds with activity of autophagy activation have been identified in delaying the AD pathology process (Tian et al., 2011; Vingtdeux et al., 2011), and inhibition of mTOR could increase Aβ clearance and rescue memory impairment in AD model mice via enhancing autophagy (Spilman et al., 2010). Arctigenin was determined to inhibit mTOR and trigger autophagy, and the involved pathways of AMPK/Raptor and AKT/mTOR were also completely investigated. Therefore, these results further intensified the potency of arctigenin in anti-AD research. To our knowledge, arctigenin might be the first reported natural product able to both inhibit Aβ production and promote its clearance.
Aging is a principal risk factor for neurodegenerative disease, especially AD, and delaying aging is considered as an applicable strategy in decreasing AD case rate and postponing AD progress (Jirillo et al., 2008; Bhat et al., 2012). There have been reports on proteins and signaling pathways involved in regulation of aging process and longevity. For example, autophagy enhancement induced prolonged lifespan in Drosophila melanogaster (Simonsen et al., 2008), and mTOR inhibitor rapamycin extended lifespan in mice (Harrison et al., 2009). In addition, overexpression of AMPK and its activation extended the lifespan in some species (Apfeld et al., 2004; Onken and Driscoll, 2010). Therefore, our findings that arctigenin could effectively enhance autophagy, inhibit mTOR activity, and increase AMPK activity in mice suggests that arctigenin might have a role in delaying aging.
In summary, we reported that the natural product arctigenin could ameliorate AD mouse memory impairment. The mechanism underlying the anti-AD action of arctigenin has been intensively investigated. As summarized in Figure 9, arctigenin inhibited Aβ production by suppressing BACE1 expression and promoted Aβ clearance by enhancing autophagy through AKT/mTOR signaling inhibition and AMPK/Raptor pathway activation. Our results clearly suggest the potential of arctigenin in anti-AD drug discovery.
Figure 9.

A proposed model illustrating the mechanism of arctigenin-mediated Aβ production inhibition and Aβ clearance promotion. Aβ is generated from a sequent cleavage of APP by BACE1 and γ-secretase. It could be degraded by autophagy. Arctigenin dephosphorylated PERK and eIF2α, causing the suppression of BACE1 translation and further the inhibition of Aβ production. Arctigenin also initiated autophagy with AMPK/Raptor signaling activation and AKT/mTOR pathway inhibition, resulting in enhanced Aβ clearance.
Footnotes
This work was supported by the State Key Program of Basic Research of China (Grant 2010CB912501), the National Natural Science Foundation of China (Grants 81173105, 21021063, 81220108025, 91213306, and 30925040), the Shanghai Basic Research Project from the Shanghai Science and Technology Commission (Grants 11ZR1444500 and 11XD1406100), and the China Postdoctoral Science Foundation Funded Project (Grant 2012M520051).
The authors declare no competing financial interests.
References
- Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo DM, Cotman CW. Beta-amyloid stimulates glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer's disease. Brain Res. 1992;569:141–145. doi: 10.1016/0006-8993(92)90380-R. [DOI] [PubMed] [Google Scholar]
- Bachmeier C, Beaulieu-Abdelahad D, Mullan M, Paris D. Selective dihydropyiridine compounds facilitate the clearance of beta-amyloid across the blood-brain barrier. Eur J Pharmacol. 2011;659:124–129. doi: 10.1016/j.ejphar.2011.03.048. [DOI] [PubMed] [Google Scholar]
- Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C. Astrocyte senescence as a component of Alzheimer's disease. PLoS One. 2012;7:e45069. doi: 10.1371/journal.pone.0045069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on Huntington-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magrí A, Oddo S. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem. 2011;286:8924–8932. doi: 10.1074/jbc.M110.180638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- Chang RC, Wong AK, Ng HK, Hugon J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer's disease. Neuroreport. 2002;13:2429–2432. doi: 10.1097/00001756-200212200-00011. [DOI] [PubMed] [Google Scholar]
- Chen CH, Zhou W, Liu S, Deng Y, Cai F, Tone M, Tone Y, Tong Y, Song W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer's disease. Int J Neuropsychopharmacol. 2011;18:1–14. doi: 10.1038/gt.2010.130. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming JN, Smith EM, Wang L, Misiaszek J, Durkin J, Pan J, Iserloh U, Wu Y, Zhu Z, Strickland C, Voigt J, Chen X, Kennedy ME, Kuvelkar R, Hyde LA, Cox K, Favreau L, Czarniecki MF, Greenlee WJ, McKittrick BA, et al. Structure based design of iminohydantoin BACE1 inhibitors: identification of an orally available, centrally active BACE1 inhibitor. Bioorg Med Chem Lett. 2012;22:2444–2449. doi: 10.1016/j.bmcl.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Devi L, Ohno M. Phospho-eIF2alpha level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS One. 2010;5:e12974. doi: 10.1371/journal.pone.0012974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Alldred MJ, Ginsberg SD, Ohno M. Sex- and brain region-specific acceleration of beta-amyloidogenesis following behavioral stress in a mouse model of Alzheimer's disease. Mol Brain. 2010;3:34. doi: 10.1186/1756-6606-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Alldred MJ, Ginsberg SD, Ohno M. Mechanisms underlying insulin deficiency-induced acceleration of beta-amyloidosis in a mouse model of Alzheimer's disease. PLoS One. 2012;7:e32792. doi: 10.1371/journal.pone.0032792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem. 2005;93:105–117. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- Gu Y, Qi C, Sun X, Ma X, Zhang H, Hu L, Yuan J, Yu Q. Arctigenin preferentially induces tumor cell death under glucose deprivation by inhibiting cellular energy metabolism. Biochem Pharmacol. 2012;84:468–476. doi: 10.1016/j.bcp.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Guglielmotto M, Monteleone D, Giliberto L, Fornaro M, Borghi R, Tamagno E, Tabaton M. Amyloid-beta(4)(2) activates the expression of BACE1 through the JNK pathway. J Alzheimers Dis. 2011;27:871–883. doi: 10.3233/JAD-2011-110884. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of Raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMPK and Raptor: matching cell growth to energy supply. Mol Cell. 2008;30:263–265. doi: 10.1016/j.molcel.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn A, Lund S, Hedtjärn M, Schrattenholz A, Pörzgen P, Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX. 2009;26:83–94. doi: 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- Ho YS, Yang X, Lau JC, Hung CH, Wuwongse S, Zhang Q, Wang J, Baum L, So KF, Chang RC. Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: implication in Alzheimer's disease pathogenesis. J Alzheimers Dis. 2012;28:839–854. doi: 10.3233/JAD-2011-111037. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, Tang J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science. 2000;290:150–153. doi: 10.1126/science.290.5489.150. [DOI] [PubMed] [Google Scholar]
- Huang SL, Yu RT, Gong J, Feng Y, Dai YL, Hu F, Hu YH, Tao YD, Leng Y. Arctigenin, a natural compound, activates AMP-activated protein kinase via inhibition of mitochondria complex I and ameliorates metabolic disorders in ob/ob mice. Diabetologia. 2012;55:1469–1481. doi: 10.1007/s00125-011-2366-3. [DOI] [PubMed] [Google Scholar]
- Jang YP, Kim SR, Choi YH, Kim J, Kim SG, Markelonis GJ, Oh TH, Kim YC. Arctigenin protects cultured cortical neurons from glutamate-induced neurodegeneration by binding to kainate receptor. J Neurosci Res. 2002;68:233–240. doi: 10.1002/jnr.10204. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirillo E, Candore G, Magrone T, Caruso C. A scientific approach to anti-ageing therapies: state of the art. Curr Pharm Des. 2008;14:2637–2642. doi: 10.2174/138161208786264070. [DOI] [PubMed] [Google Scholar]
- Katsouri L, Parr C, Bogdanovic N, Willem M, Sastre M. PPARγ co-activator-1α (PGC-1α) reduces amyloid-β generation through a PPARγ-dependent mechanism. J Alzheimers Dis. 2011;25:151–162. doi: 10.3233/JAD-2011-101356. [DOI] [PubMed] [Google Scholar]
- Kennelly SP, Abdullah L, Paris D, Parish J, Mathura V, Mullan M, Crawford F, Lawlor BA, Kenny RA. Demonstration of safety in Alzheimer's patients for intervention with an anti-hypertensive drug Nilvadipine: results from a 6-week open label study. Int J Geriatr Psychiatry. 2011;26:1038–1045. doi: 10.1002/gps.2638. [DOI] [PubMed] [Google Scholar]
- Kim JY, Hwang JH, Cha MR, Yoon MY, Son ES, Tomida A, Ko B, Song SW, Shin-ya K, Hwang YI, Park HR. Arctigenin blocks the unfolded protein response and shows therapeutic antitumor activity. J Cell Physiol. 2010;224:33–40. doi: 10.1002/JCP.22085. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura N, Tokunaga C, Dalal S, Richardson C, Yoshino K, Hara K, Kemp BE, Witters LA, Mimura O, Yonezawa K. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells. 2003;8:65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- Klaver DW, Wilce MC, Cui H, Hung AC, Gasperini R, Foa L, Small DH. Is BACE1 a suitable therapeutic target for the treatment of Alzheimer's disease? Current strategies and future directions. Biol Chem. 2010;391:849–859. doi: 10.1515/BC.2010.089. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz A, Perneczky R. Amyloid clearance as a treatment target against Alzheimer's disease. J Alzheimers Dis. 2011;24(Suppl 2):61–73. doi: 10.3233/JAD-2011-102139. [DOI] [PubMed] [Google Scholar]
- Kwak YD, Wang R, Li JJ, Zhang YW, Xu H, Liao FF. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener. 2011;6:17. doi: 10.1186/1750-1326-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F, Eisenberg T, Kroemer G. Autophagy for the avoidance of neurodegeneration. Genes Dev. 2009;23:2253–2259. doi: 10.1101/gad.1858009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN, Monk SA, Mathes BM, Mergott DJ, Watson BM, Stout SL, Timm DE, Smith Labell E, Gonzales CR, Nakano M, Jhee SS, Yen M, Ereshefsky L, Lindstrom TD, Calligaro DO, et al. Robust central reduction of amyloid-β in humans with an orally available, nonpeptidic β-secretase inhibitor. J Neurosci. 2011;31:16507–16516. doi: 10.1523/JNEUROSCI.3647-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009;21:656–664. doi: 10.1016/j.cellsig.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton-Liger F, Paquet C, Dumurgier J, Bouras C, Pradier L, Gray F, Hugon J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim Biophys Acta. 2012;1822:885–896. doi: 10.1016/j.bbadis.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–431. [PMC free article] [PubMed] [Google Scholar]
- O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS One. 2010;5:e8758. doi: 10.1371/journal.pone.0008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Predes FS, Ruiz AL, Carvalho JE, Foglio MA, Dolder H. Antioxidative and in vitro antiproliferative activity of Arctium lappa root extracts. BMC Complement Altern Med. 2011;11:25. doi: 10.1186/1472-6882-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer's disease. Genes Brain Behav. 2007;6:54–65. doi: 10.1111/j.1601-183X.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2:760–768. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Zhuang J, Chen J, Yu L, Hu L, Jiang H, Shen X. Arctigenin efficiently enhanced sedentary mice treadmill endurance. PLoS One. 2011;6:e24224. doi: 10.1371/journal.pone.0024224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011;25:1934–1942. doi: 10.1096/fj.10-175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Chandakkar P, Zhao H, d'Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 2011;25:219–231. doi: 10.1096/fj.10-167361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL. Assessment of spatial memory using the radial arm maze and Morris water maze. In: Gerfen C, Holmes A, Sibley D, Skolnick P, Wray S, editors. Current protocols in neuroscience, Unit 8.5A. Somerset, NJ: Wiley; 2004. [DOI] [PubMed] [Google Scholar]
- Wostyn P, van Dam D, Audenaert K, de Deyn PP. Genes involved in cerebrospinal fluid production as candidate genes for late-onset Alzheimer's disease: a hypothesis. J Neurogenet. 2011;25:195–200. doi: 10.3109/01677063.2011.620191. [DOI] [PubMed] [Google Scholar]
- Yamin G, Ono K, Inayathullah M, Teplow DB. Amyloid beta-protein assembly as a therapeutic target of Alzheimer's disease. Curr Pharm Des. 2008;14:3231–3246. doi: 10.2174/138161208786404137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson DW, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy. 2011;7:788–789. doi: 10.4161/auto.7.7.15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Lee SJ, Lee J, Myung CS, Park WK, Lim HJ, Lee GH, Kong JY, Cho H. Sphingosylphosphorylcholine attenuated beta-amyloid production by reducing BACE1 expression and catalysis in PC12 cells. Neurochem Res. 2011;36:2083–2090. doi: 10.1007/s11064-011-0532-0. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Li C, Wang X, Yang Z, Chen J, Hu L, Jiang H, Shen X. 2,2′,4′-trihydroxychalcone from Glycyrrhiza glabra as a new specific BACE1 inhibitor efficiently ameliorates memory impairment in mice. J Neurochem. 2010;114:374–385. doi: 10.1111/j.1471-4159.2010.06751.x. [DOI] [PubMed] [Google Scholar]