

Abstract
The CRMP2 and CRMP4 proteins are strongly expressed in the developing nervous system, mediating neurite outgrowth, neuronal polarity, and axon guidance. In the present study, we demonstrate the interaction of the CRMP2 and CRMP4 proteins with the GluK5 subunit of the kainate (KA) receptor (KAR) and investigated the role of KARs in modulating the development of cultured mouse DRG neurons. We found that KARs modulate neuronal maturation and neurite outgrowth in a bidirectional manner. Accordingly, low concentrations of KA delayed maturation and enhanced neurite outgrowth, whereas maturation was promoted by higher concentrations of KA that attenuated neuritic elongation. The effects of weak KAR activation were prevented by blocking their noncanonical signaling and involved a differential regulation of CRMP2. Whereas the delay in maturation involves PKC-mediated phosphorylation of CRMP2 at T555 leading to a downregulation of membrane Cav2.2, the promotion of neurite outgrowth is achieved by dephosphorylation at T514 at the growth cones, the latter reflecting PKC-driven enhancement of GSK3β phosphorylation at S9. Together, these findings indicate that noncanonical KAR signaling influences neuronal development by modulating CRMP2 activity.
Introduction
Although kainate receptors (KARs) are ubiquitous in the adult nervous system (Bahn et al., 1994), unlike AMPA and NMDA receptors, they contribute to the excitatory drive in only a limited number of central synapses and generate small-amplitude EPSCs (Castillo et al., 1997). Growing evidence suggests that the role of KARs is much broader than the simple information transfer at synapses and that they are probably involved in modulating synaptic function and in regulating the activity of neuronal networks. Indeed, KARs have been implicated in events as diverse as presynaptic modulation of excitatory and inhibitory transmission, postsynaptic depolarization, and the regulation of intrinsic neuronal excitability (Lerma, 2006; Pinheiro and Mulle, 2006; Contractor et al., 2011). The prominent neuromodulatory role of KARs is further supported by their ability to signal via noncanonical G-protein-coupled pathways (Rodriguez-Moreno and Lerma, 1998) in a manner that is independent of ion flux (Rozas et al., 2003), in addition to their conventional ionotropic activity (for review, see Rodrigues and Lerma, 2012), a capacity that was first observed in the hippocampus and later confirmed in other preparations.
The distinct roles and signaling capabilities of KARs are probably mediated by a number of different interacting proteins. To identify those proteins, we performed a proteomic analysis of GluK5, a high-affinity KAR subunit, in which we identified the Collapsin Response Mediator Proteins 2 and 4 (CRMP2 and CRMP4) as potential partners of this subunit. These phosphoproteins are strongly expressed in the developing nervous system and play important roles in neuronal maturation (Yamashita and Goshima, 2012). CRMP2, in particular, is implicated in neurite outgrowth, axonal guidance, neuronal polarity, and neuronal migration (Goshima et al., 1995; Inagaki et al., 2001; Arimura et al., 2005; Uchida et al., 2005; Yoshimura et al., 2005; Ip et al., 2012). KARs are also expressed highly during development, transducing signals through multiple pathways (Lauri and Taira, 2011, 2012). Indeed, there is a growing body of evidence implicating KARs in the development of synaptic connectivity and in the maturation of neuronal networks. However, the role of KARs in these phenomena remains poorly understood and the specific mechanisms underlying KAR activity during development wait to be defined.
In this study, we demonstrate that KARs can bidirectionally modulate neuronal maturation and neurite outgrowth using cultured sensory neurons as a model. This is achieved by a dual KAR-mediated signaling module that adjusts the activity of the microtubule-associated protein CRMP2, directly linking KAR activation and the modulation of cytoskeletal dynamics during neuronal maturation. Our results reveal a number of emergent roles of KARs that are mediated by noncanonical signaling pathways and play a crucial role in neuronal development.
Materials and Methods
All of the procedures relating to the handling of animals were performed in accordance with the guidelines of the European Commission (86/609/CEE) and were supervised by the veterinary officer at the Instituto de Neurociencias de Alicante.
2D electrophoresis and identification of proteins by MALDI-TOF mass spectrometry.
Rabbit anti-GluK5 antibody (C8; Darstein et al., 2003 Herb et al., 1992) and normal rabbit IgG (Sigma) were linked to protein-A Sepharose (GE Healthcare) with 20 mm dimethyl pimelimidate (DMP, Sigma) during 30 min at room temperature (RT) in a 1:1 ratio (1 μg of antibody per each 1 μg of packed beads). The coupling efficiency of both antibodies was monitored using 5 μl of protein A-bound antibody sample taken before and after the covalent coupling, followed by the SDS-PAGE and Coomassie staining of the respective gels. Immunoprecipitations were performed by first incubating membrane preparations with 100 μl of a 50% protein A-Sepharose slurry for 2 h at 4°C to eliminate nonspecific binding. Precleared supernatants containing 10 mg of protein were incubated with 100 μg of DMP-coupled antibodies overnight at 4°C in the presence of 1% BSA to further block nonspecific binding. Immunoprecipitated proteins were eluted with 350 μl of electrofocusing medium containing 7 m urea, 2 m thiourea, 4% CHAPS, ampholines (preblended, pH 3.5–9.5, 8 mg/ml; GE Healthcare), 100 mm DTT, 0.2% tergitol NP7 (Sigma), and traces of bromophenol blue. Proteins were then separated according to their isoelectric point along linear immobilized pH-gradient (IPG) strips (pH, 3–10, 18 cm long; GE Healthcare). Sample loading for the first dimension was performed by passive in-gel swelling. The IPG strips were then equilibrated for 10 min in a buffer containing 6 m urea, 50 mm Tris-HCl, pH 6.8, 30% glycerol, 2% SDS, 10 mg/ml DTT, and bromophenol blue and then equilibrated for 15 min in the same buffer containing 15 mg/ml iodoacetamide instead of the DTT. For the second dimension, the strips were loaded onto vertical 12.5% SDS polyacrylamide gels. Gels were silver stained (Shevchenko et al., 1996). Coimmunoprecipitations were performed in six independent experiments and only the protein spots present in all six anti-GluK5, but not rabbit IgG (control), gels were excised and digested in gel using trypsin (Promega; Shevchenko et al., 1996).
Digested samples were dehydrated in a vacuum centrifuge, solubilized in 10 μl of formic acid (2%), desalted using C18 ZipTips (Millipore: elution with 10 μl of 0.1% trifluoroacetic acid [TFA] in 50% acetonitrile), and concentrated to a 1 μl volume. Aliquots of 0.3 μl, mixed with the same volume of α-cyano-4-hydroxy-trans-cinnamic acid (10 mg/ml 0.1% TFA in 50% acetonitrile), were deposited on a 384-well matrix-assisted laser desorption/ionization (MALDI) target using the dry droplet procedure and air dried at room temperature. Analyses were performed using an UltraFlex MALDI-time-of-flight (TOF)/TOF mass spectrometer (Bruker-Franzen Analytik) operating in the reflectron mode with a 20 kV accelerating voltage and a 70 ns delayed extraction. Mass spectra were acquired in the automatic mode using the AutoXecute module of Flexcontrol (laser power ranged from 30 to 70%, 500 shots; Bruker-Franzen Analytik). Spectra were analyzed using FlexAnalysis software (Bruker-Franzen Analytik) and auto-proteolysis peptides of trypsin (mass-to-charge ratios 842.51, 1045.56, and 2211.10) were used as internal calibrates. Peptides were selected in the mass range of 900–4000 Da. Identification of proteins was performed using the Mascot software package (version 2.1; Matrixscience) against the Swiss-Prot/Trembl databases (2006-08-01). The following parameters were used for database interrogation: mass tolerance of 50 ppm (although the mass accuracy of our analyses was usually better than 20 ppm); fixed chemical modification: carbamidomethylation of cysteine residues; variable chemical modification: oxidation of methionines; matching peptides with one missed cleavage accepted only when they included two consecutive basic residues or when arginine or lysine residues were followed by one or several acidic residues inside the peptide amino acid sequence and significance threshold, p < 0.05.
DRG cell culture.
DRG from postnatal day 0 (P0)-P2 C57 male and female mice were dissected out and digested for 40 min at 37°C with a mixture of collagenase (0.25%), DNase (5 μg/ml), and trypsin (0.125%) in HBSS medium. DRG neurons were then dissociated with a flame-polished Pasteur pipette and plated onto glass coverslips (for morphological analysis) or plastic Petri dishes (for biochemical studies) that had been pretreated with 50 μg/ml poly-d-lysine and 10 μg/ml laminin. The cells (2500 cells/cm2) were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 g/ml streptomycin, 10 ng/ml NGF, and 20 ng/ml BDNF (DRG medium). For cultures without glia, DRG neurons were treated as described above, preplated in DMEM medium for 2 h, and then resuspended in DRG medium and cultured. For morphological and electrophysiological analysis, 1 h after plating, DRG neurons were infected with a Sindbis virus carrying a copy of GFP mRNA to visualize neuronal processes by adding Sindbis supernatant (1:500 dilution) directly to the culture medium. A similar procedure was used for DRG neurons infection with Sindbis virus encoding GFP-CRMP2, GFP-CRMP2T555A, GFP-CRMP2T555D, GFP-CRMP2T514A, and GFP-CRMP2T514D. Less than 5% of the cells infected were glial cells, as easily identified by the distinct cell morphology and because they presented lower fluorescence intensity (i.e., lower levels of GFP expression). In parallel experiments, this was further corroborated by the absence of β-tubulin III immunoreactivity. Drugs were added to the culture 2 h after plating the cells. DRG neurons were fixed 20 h after infection (i.e., 21 h after plating) for morphological analysis by sequentially replacing 50% of the culture medium in four steps with 4% prewarmed paraformaldehyde to preserve the cytoskeletal structure. For biochemical studies, protein extracts were collected by scraping the cells off the plates 21 h after plating in 100 μl of cell lysis buffer (10 mm Tris-HCl, 10 mm EDTA, 1% Triton X-100, 0.1% SDS, and protease inhibitors, pH 7.4; Roche Diagnostics]). Cell viability and KA-induced currents were also evaluated in DRG neurons held in culture for 21 h. Ca2+ current density was performed in DRG neurons cultured for 20–36 h.
Generation of Sindbis virus.
To generate Sindbis virus carrying GFP, GFP was cloned in the StuI site of pSinRep5 (Invitrogen). For the Sindbis virus encoding GFP-CRMP2, GFP lacking a stop codon was placed at the N-terminal of CRMP2 and cloned between the HindIII and EcoRI sites of the pcDNA3 expression vector. This construct was used to generate several point mutations using the QuiKchange II XL Site-Directed Mutagenesis Kit (Stratagene). Four different single mutations in CRMP2 were generated at amino acid residues 514 and 555 by switching threonine to alanine (A) or aspartic acid (D) to produce the phospho-null and phospho-mimetic mutants, respectively. GFP-CRMP2 and all mutated forms thereof were cloned into the pSinRep5 vector under the control of the Sindbis subgenomic promoter and Sindbis virus was produced using the Sindbis Expression System (Invitrogen). Briefly, pSinRep5 and the helper plasmid were linearized, transcribed to mRNA, and cotransfected into BHK cells to produce virions encoding the different proteins of interest. After 24–48 h, the supernatant was collected and stored at −80°C.
Morphological analysis.
DRG neurons expressing GFP or GFP-CRMP2 (wild-type and mutants) were visualized 20 h after infection on a Leica DM 4000B fluorescence microscope equipped with a MBFCX 9000 camera (MBF Bioscience). The neurons were classified into three groups based on their level of maturation according to morphological criteria (see Results section) and reflecting the natural process going on in culture gauged by time-lapse experiments (data not shown). A few percentage of neurons (<5%) presented morphology in between stages difficult to ascribe to a given stage, further showing maturation as a continuous process. For quantification purposes, these neurons were equally ascribed to the closer stages. Evaluating the branching profile of the neurons by Sholl analysis further validated this qualitative morphological analysis. For this purpose, concentric circles with increasing radius (1 μm increments) were drawn around the soma using Neurolucida software (MBF Bioscience) and the number of times each circle crossed a neurite segment was recorded. Neurons in each stage of maturation presented characteristic branching profiles in addition to distinct morphological features (a minimum of 100 cells per condition per culture were analyzed). Neurite length corresponded to the total length of the longest neurite. The length of the longest neuritic process (measured from the soma to the most distant neurite tip) was always analyzed providing similar results, as expected because no significant changes in the number of branches per neurite were observed upon exposure to KA. A minimum of 25 stage 1, 50 stage 2, and 25 stage 3 neurons per condition per culture were analyzed. Quantitative analysis of neuritic properties was performed using Neurolucida software (MBF Bioscience). This time point was chosen because neurons of each stage were clearly identified. At later times (e.g., 48 h), the majority of cells presented a mature (stage 3) phenotype.
Cell viability.
Cell viability was assessed by Alamar Blue assay (Invitrogen). Twenty hours after plating, 10% (v/v) of Alamar blue dye was added to each well containing cultured DRG neurons. After 4 h, aliquots of the media were collected from each well and absorbance of either reduced (570 nm) or oxidized form (600 nm) was measured. The relative change in reduction was considered equivalent to the relative change in cell viability. This was complemented by counterstaining the cell nucleus with DAPI (Vectashield; Vector Laboratories), allowing evaluation of the percentage of nonviable cells.
Immunocytochemistry.
COS-7 cells cultured in DMEM containing 10% fetal bovine serum were transfected with N-terminal Myc-tagged GluK5, untagged GluK1–2a, and CRMP2 constructs using Lipofectamine 2000 (Invitrogen). COS-7 cells were fixed 48 h after transfection and DRG neurons were fixed 20 h after infection. Both COS-7 and DRG neurons were permeabilized by incubating with PBS + 0.2% Triton X-100 for 10 min. The cells were then blocked with PBS + 3% BSA for 30 min to prevent nonspecific binding and then incubated with the primary antibodies in blocking solution for 1 h at RT. COS-7 cells were incubated with a mouse monoclonal anti-Myc antibody (1:500; Santa Cruz Biotechnology) and a rabbit polyclonal anti-CRMP2 antibody (1:1000; Millipore Bioscience Research Reagents) and DRG neurons were immunolabeled with a rabbit polyclonal antibody against neuron-specific β-tubulin III (1:500; Abcam). Next, the cells were incubated with Alexa Fluor-488-conjugated donkey anti-mouse (COS-7 cells) and Alexa Fluor-555-conjugated donkey anti-rabbit (COS-7 cells and DRG neurons; 1:400; Invitrogen) in blocking solution for 1 h at RT. Coverslips were mounted on slides using DAPI-supplemented mounting medium (Vectashield; Vector Laboratories). Although COS-7 cells were visualized on a Leica confocal microscope, DRG neurons were visualized using a Leica DM 4000B fluorescence microscope equipped with a MBFCX 9000 camera (MBF Bioscience) and analyzed using Neurolucida software (MBF Bioscience). For growth cone immunocytochemistry, DRG cells were incubated for 20 min with Alexa Fluor-488-conjugated phalloidin (1:500; Invitrogen) at room temperature to label the actin cytoskeleton, and then incubated overnight at 4°C with rabbit anti-CRMP2 pT514 antibody (1:250; Cell Signaling Technology) in blocking solution. Cells were then immunolabeled with Alexa Fluor-555-conjugated goat anti-rabbit antibody (1:1000; Invitrogen) in blocking solution for 2 h at RT.
Immunoprecipitation assays.
Mouse brain homogenate (MBH) was obtained from P21-P26 C57 mice as described previously (Selak et al., 2006) and incubated with 50% protein A-Sepharose bead slurry (GE Healthcare) for 3 h at 4°C to eliminate nonspecific binding. After incubation, precleared supernatants containing 1 mg of protein were incubated overnight with rotation at 4°C with a rabbit anti-GluK5 antibody (kindly provided by Dr. Melanie Darstein; Darstein et al., 2003) precoupled to protein A-Sepharose (GE Healthcare) in the presence of 1% BSA (to further block nonspecific binding) and protease inhibitors (Roche Diagnostics). The beads were washed 3 times with isolation buffer (150 mm KCl, 20 mm MOPS, 1% Triton X-100, pH 7.4) and resuspended in 6× diluted SDS-PAGE sample buffer (0.35 m Tris, 30% glycerol, 10% SDS, 0.6 m DTT, 0.012% bromophenol blue, pH 6.8). Immune complexes were dissociated from the beads, denatured by heating to 95°C for 5 min, and then analyzed by Western blot. A negative control containing the same amount of rabbit polyclonal IgG instead of the rabbit anti-GluK5 antibody was run in parallel for each experiment.
GST protein-binding assays.
GST-GluK5825–979 recombinant protein was expressed in the protease-deficient BL21 E. coli strain and the bacterial cells were then lysed by sonication in the presence of 0.3 mg/ml lysozyme to purify the GST fusion proteins by incubation for 2 h at 4°C with glutathione Sepharose 4B beads (GE Healthcare). Purified GST proteins were incubated with 400 μg of MBH in buffer N (25 mm HEPES-KOH, pH 7.3, 150 mm KCl, 1% Triton X-100, 10% glycerol, 0.2% BSA, 1 mm DTT) and the fusion protein was washed extensively in ice-cold buffer containing 20 mm Tris, pH 7.5, 0.25 m NaCl, 2 mm EDTA, 2 mm EGTA, and 0.02% Tween 20. After the last wash, the proteins were eluted in the same buffer containing 20 mm glutathione, pH 8.0, and the purified protein was dialyzed against PBS buffer in mini dialysis units (Slide-A-Lyzer; Pierce). Bound proteins were separated by SDS-PAGE and analyzed in immunoblots.
Western blotting.
Protein extracts from DRG neurons, immunoprecipitates, or GST-pull-down assays were diluted in 6× SDS-PAGE sample buffer, boiled at 95°C for 10 min, and then separated by 10% SDS-PAGE electrophoresis. Phosphorylated forms of the CRMP2 protein were detected using rabbit antibodies directed against p-CRMP2T555 (1:200; ECM Biosciences) and p-CRMP2T514 (1:500; Cell Signaling Technology) diluted in TBS-T containing 5% BSA. Phosphorylated forms of GSK3β were detected using rabbit antibodies directed against p-GSK3βY216 (1:1000; Abcam) and p-GSK3βS9 (1:1000; Abcam) diluted in TBS-T containing 5% PhosphoBLOCKER blocking reagent (Cell Biolabs). Total CRMP2 protein was detected using a rabbit antibody directed against CRMP2 (1:4000; Abcam) diluted in TBS-T containing 5% dried milk. Total GSK3β was detected using a rabbit antibody directed against GSK3β (1:500; Abcam) in TBS-T containing 5% dried milk. Cav2.2 was detected using a rabbit antibody directed against CACNA1B (N-type, 1:200; Abcam) diluted in TBS-T containing 5% dried milk. A rabbit antibody directed against GAPDH (1:1000; Abcam) was used as a loading control. The membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibodies (1:5000; Pierce) and then with SuperSignal West Pico Chemiluminescent Substrate (Pierce) to detect total CRMP2, GAPDH, and p-GSK3βY216, or with Luminata Forte Western HRP Substrate (Millipore) to detect p-CRMP2T555, p-CRMP2T514, GSK3β, and p-GSK3βS9 and CACNA1B (N-type). The results were analyzed using a BioImage apparatus and software (Fuji). Striping primary antibodies was not efficient, making impossible a clean signal from the reblotting with different antibodies. Therefore, to determine the ratios pCRMP2/CRMP2 and pGSK3β/GSK3β, gels were run in parallel for total CRMP2 and GSK3β and for each phosphorylated form. The immunoreactivity in each gel was normalized by GAPDH signal in each case and the ratio evaluated by (p-protein1/GAPDH1)/(protein2/GAPDH2) and then normalized to control (untreated) conditions.
Electrophysiological recordings.
After 20 h in culture, KA-induced currents were recorded in DRG neurons at −60 mV by whole-cell patch-clamping using a List EPC-7 amplifier as described previously (Rivera et al., 2007). The internal solution contained the following (in mm): 127 CsMeSO4, 10 CsCl, 1 EGTA, 10 NaCl, and 10 HEPES, pH 7.4. The external solution contained the following (in mm): 140 NaCl, 2.5 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, and 15 glucose, pH 7.4. Cells were rapidly perfused as described previously (Paternain et al., 2003). Borosilicate glass patch pipettes were used with resistance of 2–5 MΩ and series resistance was compensated by 60–80%. Whole-cell voltage-clamp recordings of Ca2+ currents were performed 20–36 h after infection with Sindbis virus encoding GFP, GFP-CRMP2, GFP-CRMP2T555A, or GFP-CRMP2T555D, as described previously (Chi et al., 2009). Neurons were perfused with the following (in mm): 110 NaCl, 2 CaCl2, 30 TEA-Cl, 10 HEPES, 10 glucose, and 1 μm TTX. To remove the TTX-resistant Na+ current and to isolate Ca2+ currents, NaCl was substituted by N-methyl glucamine. The recording pipettes (2–5 MΩ resistance) contained the following (in mm): 140 CsMeSO4, 10 CsCl, 10 HEPES, 5 Mg-ATP, 5 BAPTA, adjusted to pH 7.2 with KOH. All experiments were performed at RT (22–25°C). The currents were filtered at 1 kHz (2-pole Butterworth filter, −12 dB/octave) and digitized at 20 kHz with a standard P/4 leak subtraction protocol to a personal computer to be analyzed with pClamp software (AXON Instruments).
Statistical analysis.
All data are presented as the mean ± SEM of n experiments. The distribution of neurons at each stage of maturation is represented as a percentage quantified in a minimum of 100 cells per condition per culture. The effects of the various interventions on DRG neuron maturation were analyzed by two-way ANOVA. Where significant interactions were detected, post hoc comparisons for each stage were performed using Bonferroni's post hoc test. Comparison of branching profiles in neurons exposed to increasing concentrations of KA was determined by Sholl analysis and analyzed by two-way ANOVA, followed by Bonferroni's post hoc test. Comparison of nonlinear fits (null hypothesis defined as one curve for all datasets) and one-way ANOVA analysis of the area under curve revealed similar qualitative results. Neurite length and the changes in phosphorylation determined in Western blots were expressed as the fold change relative to the controls. Relative differences were analyzed by one-sample t test by comparison with a hypothetical value of 1 (control condition). Modifications in Ca2+ current density, were quantified as the current density (pA/pF) from a minimum of six cells per condition per culture (also expressed as relative Δ values of current density) and analyzed using a one-sample t test by comparison with a hypothetical value of 0 (no effect). Group differences were analyzed using an unpaired t test (comparison of two independent groups) or one-way ANOVA (comparison of several independent groups). Where ANOVA revealed significant general group effects, Dunnet's post hoc test was used to compare different groups with the control group and Tukey's post hoc test for multiple comparisons. In all analyses performed, a familywise 95% confidence level (p < 0.05) was applied. All data processing and analyses were performed using Prism 5.0 software (GraphPad).
Results
CRMP2 and CRMP4 are KAR-interacting proteins
To identify proteins interacting with the GluK5 KAR subunit, we immunoprecipitated detergent solubilized membrane fractions from mouse brain homogenates using a well characterized rabbit anti-GluK5 antibody that recognizes the last eight amino acids of the GluK5 C terminus (C8; kindly provided by Malanie Darstein; Herb et al., 1992; Darstein et al., 2003). Immunoprecipitated protein complexes were separated by 2D electrophoresis and the protein spots recruited by the anti-GluK5 antibody, but not by the control rabbit Ig, were excised and identified by MALDI-TOF mass spectrometry. Three proteins were unambiguously and reproducibly identified in GluK5 immunoprecipitates: DPYL2 (CRMP2), DPYL3 (CRMP4), and a microtubule-associated protein RP/EB family member (MARE-3; Fig. 1A). To confirm these results, the material immunoprecipitated was probed in immunoblots with the antibodies specifically recognizing CRMP2 and CRMP4 (Fig. 1B). Both CRMP2 and CRMP4 were readily detected in complexes immunoprecipitated with the anti-GluK5 antibody, but not with the control rabbit IgG (Fig. 1B). In addition, exogenously expressed GluK5 and CRMP2 colocalized at the plasma membrane of the transfected COS-7 cells (Fig. 1C) and CRMP2 was readily retained in GST pull-down assays using the C-terminal domain of GluK5 (Fig. 1D). These data demonstrate that CRMP2 and probably CRMP4 interact with the C terminus of the GluK5 KAR subunit.
Figure 1.
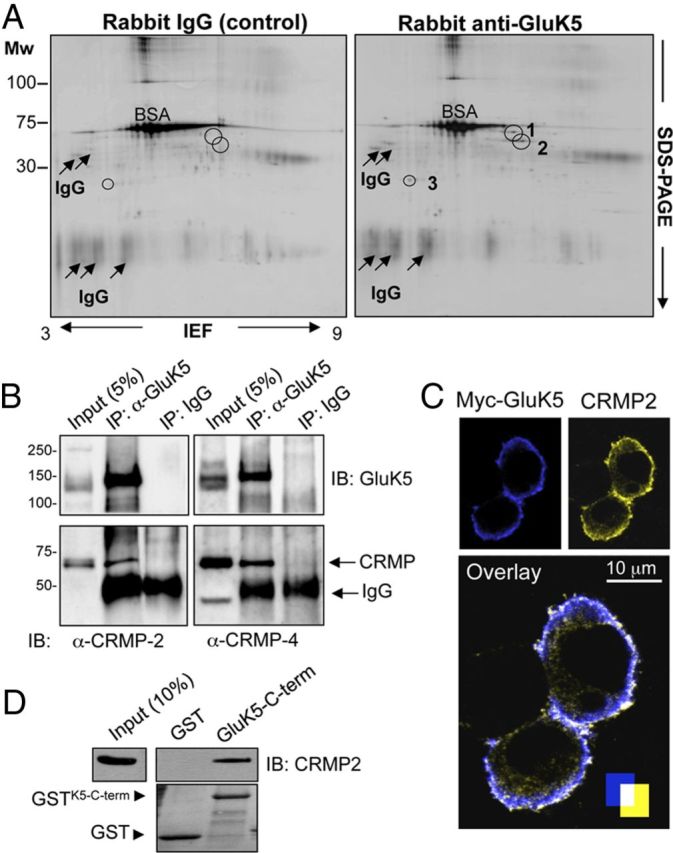
Identification of KAR interacting proteins. A, Proteins interacting with the GluK5 KAR subunit were purified by immunoprecipitation of detergent-solubilized membrane fractions from mouse brain homogenate using rabbit anti-GluK5 antibody and separated by 2D electrophoresis. MW, molecular weight; IEF, isoelectric focusing. The specific protein spots recruited by rabbit anti-GluK5, identified by MALDI-TOF mass spectrometry, corresponded to DPYL2 or CRMP2 (spot 1), DPYL3 or CRMP4 (spot 2), and MARE-3 (spot 3). B, GluK5 antibody immunoprecipitates CRMP2 and CRMP4 in mouse brain extracts. C, Exogenously expressed CRMP2 and GluK5 colocalize in the plasma membrane of COS-7 cells transfected with N-terminal Myc-tagged GluK5 (blue), untagged GluK1-a, and a CRMP2 construct (yellow). Colocalization is shown in white. D, GST-pull-down analysis of CRMP2 from mouse brain extracts targeting the C-terminal domain of GluK5.
KARs bidirectionally modulate neuronal maturation
Given the interaction observed between KARs and the microtubule-interacting proteins involved in developmental and maturation processes, we investigated the role of KARs in neuronal development. For this purpose, we used DRG neurons because these neurons exclusively express GluK1 and GluK5 subunits of KARs, lack AMPA and NMDA receptors (Bettler et al., 1990; Bahn et al., 1994), and exhibit both canonical and noncanonical KAR signaling (Rozas et al., 2003). To visualize the entire neuronal processes of DRG neurons, they were infected with Sindbis virus carrying a copy of GFP mRNA. Although maturation is a continuous process, for simplification purposes, DRG neurons were classified 20 h after infection into one of three different maturation stages based on morphological criteria and by Sholl analysis, which indicated distinct branching profiles for each group (see Materials and Methods), providing strong, unbiased support for this classification (Fig. 2A). Stage 1 neurons are immature cells characterized by the presence of very short neurites with profuse filopodia, with lamellipodia present close to the soma and in the growth cones. Stage 3 neurons are mature neurons lacking filopodia and lamellipodia, yet with long, clean neurites with few ramifications. Intermediate-stage neurons, characterized by ramified, intermediate-length neurites with numerous branching points near the soma and fewer filopodia and lamellipodia not fitting in general within either stage, were classified as stage 2 neurons (Fig. 2A).
Figure 2.

KARs bidirectionally modulate neuronal maturation and neuritic morphology in DRG neurons. A, Representative images of DRG neurons 20 h after infection with a Sindbis virus encoding GFP, showing neurons classified according to morphological criteria: immature stage 1 neurons, intermediate stage 2 neurons, and mature stage 3 neurons. Scale bars, 50 μm. Neurons at each stage of maturation exhibit distinguishable characteristics and branching profiles, as determined by Sholl analysis (graph, right). B, In the absence of glial cells, the selective antagonist of AMPAR/KAR CNQX (30 μm) delayed neuronal maturation. Left: In neurons cultured with glia, the tonic activation of KARs was prevented, as indicated by the absence of effect of CNQX. Right: Bidirectional effect of KA on the maturation of GFP+-DRG neurons. KA delays maturation at low concentrations, promoting maturation at higher concentrations. This biphasic effect is prevented by the selective AMPAR/KAR antagonist CNQX (30 μm) and by LY382884 (10 μm), an antagonist of GluK1-containing KARs, yet not by the selective AMPAR antagonist LY303070 (25 μm). Data are expressed as the percentage of neurons (mean ± SEM) at each stage of maturation quantified from 4–5 different cultures with a minimum of 100 neurons analyzed per condition and culture. **p < 0.01 and ***p < 0.001, two-way ANOVA with Bonferroni's post hoc test for comparisons versus control conditions. C, Bidirectional modulation of the neuritic morphology of DRG neurons (Sholl analysis) after exposure to KA. The data are expressed as the number of intersections (mean ± SEM) in 1 μm incremental radii from the soma quantified from four different cultures analyzing 25–50 neurons per stage, condition, and culture: *p < 0.05 and ***p < 0.001, two-way ANOVA.
The effect of tonic KAR activity on neuronal maturation was investigated by incubating DRG neurons with CNQX, a selective AMPAR/KAR antagonist. CNQX (30 μm) appeared to delay neuronal maturation when added 2 h after plating, reducing the proportion of mature neurons (stage 3, ∼25%) while augmenting that of immature neurons (stage 1) by a comparable extent (Fig. 2B). In all cases in which a significant effect on maturation was observed, post hoc analyses revealed significant alterations in the number of neurons in stages 1 and/or 3. In contrast, stage 2 neurons were present in similar proportions. Consistent with the fact that DRG neurons lack AMPARs, the selective AMPAR antagonist LY303070 failed to produce any effects on neuronal maturation (data not shown). Together, these results suggested that tonic activation of KARs by endogenous glutamate may affect neuronal maturation in culture. Coculturing neurons with glia prevented tonic activation of KARs by endogenous glutamate, evident through the failure of CNQX to influence the maturation of these neurons (Fig. 2B). Therefore, we used this condition (coculturing DRG neurons with glial cells) to prevent any interference by endogenous glutamate when analyzing the effects of activating KARs with different concentrations of KA added 2 h after plating.
To our surprise, KA exerted a bidirectional effect on neuronal maturation as a function of the concentration used. Accordingly, neuronal maturation was delayed by low KA concentrations (300 nm), whereas it was enhanced at higher concentrations (3 and 10 μm; Fig. 2B). Both the inhibitory and stimulatory effects of KA on neuronal maturation were blocked by CNQX and by LY382884 (10 μm), a selective antagonist of GluK1-containing receptors, but not by the AMPAR antagonist LY303070 (25 μm; Fig. 2B). To discard a cytotoxic effect of KA exposure, cell viability was assessed 20 h after plating by Alamar blue assay. No differences were found in KA-treated cultures compared with untreated cultures (data not shown), indicating that KA was not toxic under our experimental conditions. This is consistent with the suggestion that KA-induced activation of AMPARs, rather than KARs, is mostly responsible for KA excitotoxicity (Tomita et al., 2007). We also measured KA-induced currents in the three stages of maturation and found that KA (30 μm; −60 mV of holding potential) induced ionotropic responses in ∼70–75% of cells studied with amplitudes that were not significantly different among the three different maturation stages. Pooling out all cells from the 3 stages, these were 3.2 ± 0.8 pA/pF (n = 25; 3 different cultures) for untreated neurons and 3.4 ± 0.5 pA/pF (n = 39; 3 different cultures) for cultures treated with 300 nm KA. These results demonstrate that GluK1-containing KARs modulate the maturation of cultured DRG neurons in a bidirectional manner.
KARs bidirectionally modulate neurite outgrowth
The results described above demonstrate a clear action of KAR activity on the degree of neuron maturation and suggest that neuritic outgrowth could change accordingly. To test this, we measured the branching profile of DRG neurons treated with low and high doses of KA. Sholl analysis of cultured neurons revealed that low concentrations of KA (300 nm) modified the neurite branching profile at the different stages of neuronal developmental (Fig. 2C), whereas higher concentrations (3–10 μm) significantly modified the branching profile of stage 2 and 3 neurons, but not of stage 1 neurons (Fig. 2C). These modifications in neuritic morphology induced by KA were not due to changes in the number of branches because no significant alterations were observed in the number of neurite tips per neuron (data not shown). Rather, KA affected neurite elongation at different stages of maturation in a concentration-dependent manner in which the length of the longest neurite increased at all stages in the presence of 300 nm KA but decreased significantly at higher concentrations (3–10 μm) in stage 2 and 3 neurons (Fig. 3A,B). The effects of KA on neurite length were blocked by CNQX in neurons at all stages, whereas the selective AMPAR antagonist LY303070 (25 μm) had no effect (Fig. 3C). The bidirectional action of KA on neuritic growth was also blocked by the GluK1-specific antagonist LY382884 (Fig. 3C). These findings indicate that GluK1-containing KARs modulate neurite outgrowth in DRG neurons in a bidirectional manner.
Figure 3.

KARs bidirectionally modulate neurite outgrowth in DRG neurons. Low KA concentrations (300 nm) enhanced neurite elongation in DRG neurons 20 h after infection with Sindbis virus encoding GFP, whereas higher concentrations (3 μm) reduced neurite length in DRG neurons at stages 2 and 3. A, Representative examples at stages 1 and 3. In addition to GFP fluorescence (blue), neurons were immunolabeled with an anti-β-tubulin III antibody (yellow). Colocalization appears as white. Scale bars, 50 μm. B, Data quantification of the action of KA on these cultures. C, Bidirectional modulation of neurite length was blocked by the AMPAR/KAR antagonist CNQX (30 μm) and by the selective antagonist of GluK1-containing KARs, LY382884 (10 μm), yet not by the selective AMPAR antagonist LY303070 (25 μm). Data are expressed as the fold change (mean ± SEM) relative to the untreated cultures (B) or the cultures exposed to each antagonist alone (C), quantified from four different cultures and analyzing 25–50 neurons per stage, condition, and culture: *p < 0.05, one-sample t test.
Bidirectional modulation of neuronal maturation and neurite outgrowth is mediated by dual KAR signaling pathways
We hypothesized that the bidirectional regulation of neuronal maturation and neurite elongation by KARs may be mediated by the dual signaling capacity of these receptors (Rozas et al., 2003). Indeed, the ionotropic activity of KARs in DRG neurons was only detected at micromolar concentrations of KA, as evident in patch-clamp recordings (Fig. 4A), and these inward currents were insensitive to the AMPAR antagonist LY303070 (25 μm), indicating that are exclusively mediated by KAR activation.
Figure 4.

Bidirectional modulation of neuronal maturation and neurite outgrowth is mediated by dual KAR-mediated signaling pathways. A, Representative traces of KA-induced inward currents in DRG neurons. Net currents were only observed at KA concentrations in the micromolar range (>300 nm; top) and were resistant to AMPA receptor antagonism (bottom). B, The delay in neuronal maturation induced by 300 nm KA was blocked by bisindolylmaleimide (Bisind, 0.5 μm) and by pertussis toxin (PTx, 0.2 μg/ml), but persisted in the presence of TTX (50 nm). C, The promotion of neuronal maturation induced by 3 μm KA was unaffected by the presence of Bisind or PTx, but was abrogated in TTX-treated cells. Data are expressed as the percentage of neurons (mean ± SEM) in each stage of maturation quantified from six different experiments with a minimum of 100 neurons analyzed per condition and culture. **p < 0.01 and ***p < 0.001, two-way ANOVA followed by Bonferroni's post hoc test. D, The increase in neurite length induced by 300 nm KA in Stage 1, 2 and 3 neurons was prevented by Bisind and PTx, yet was unaffected by TTX. In contrast, the decrease in neurite length induced by 3 μm KA was unaffected by bisindolylmaleimide or PTx, yet was abolished by TTX. The data are expressed as the fold change (mean ± SEM) relative to untreated neurons or cells exposed to Bisind, PTx, or TTX alone quantified from four different cultures analyzing 25–50 neurons per stage, condition, and culture. *p < 0.05 and **p < 0.01, one-sample t test.
We next characterized the signaling underlying the bidirectional effects of KAR activation on neuronal maturation and neurite outgrowth. The inhibitory effect of 300 nm KA on neuronal maturation in culture was prevented by the blockade of either Gi/o-proteins (Pertussis toxin, 0.2 μg/ml) or PKC (bisindolylmaleimide, 0.5 μm; Fig. 4B), whereas the stimulatory effect of 3 μm KA on such neuronal maturation was unaffected by either treatment, although it was blocked by TTX (Fig. 4C). An identical pharmacological profile was observed for the effects of KA on neurite elongation (Fig. 4D). These results demonstrate that both the delay in neuronal maturation and the stimulation of neurite outgrowth induced by low concentrations of KA are mediated by a Gi/o-protein-coupled pathway that involves the activation of PKC. In contrast, the stimulation of neuronal maturation and the attenuation of neuritic elongation induced by higher concentrations of KA are independent of G-protein activation and these effects are most likely mediated by canonical ionotropic signaling (i.e., membrane depolarization).
KARs regulate CRMP2 phosphorylation
CRMP2 is a cytosolic phosphoprotein that is expressed strongly in the developing nervous system (Goshima et al., 1995), where it influences multiple processes associated with neuronal development, including axonal guidance and outgrowth (Arimura et al., 2005; Uchida et al., 2005; Yoshimura et al., 2005), neuronal polarity (Inagaki et al., 2001; Yoshimura et al., 2005), and neuronal migration (Ip et al., 2012). Based on the association observed between KARs and CRMP2, we hypothesized that the effects of KARs on neuronal maturation and neurite outgrowth involve the modulation of CRMP2 function.
CRMP2 is known to be negatively regulated by two independent phosphorylation pathways that have been described thus far: one mediated by Cdk5 (priming kinase; Cole et al., 2006) and GSK3β (Uchida et al., 2005; Yoshimura et al., 2005) and another by Rho kinase (Arimura et al., 2000). Both pathways attenuate CRMP2-mediated stimulation of axonal outgrowth by disrupting CRMP2-tubulin binding, and thus microtubule assembly, resulting in growth cone collapse (Fukata et al., 2002; Arimura et al., 2005; Yoshimura et al., 2005). Using Western blot analysis, we studied the effect of KAR activation on the phosphorylation state of CRMP2 at its two inhibitory phosphorylation sites: T514 (GSK3β target) and T555 (Rho kinase target). A mild but consistent decrease in CRMP2 phosphorylation at T514 was evident when cells were exposed to 300 nm but not to 3 μm KA, whereas CRMP2 phosphorylation at T555 was enhanced robustly by 300 nm KA (Fig. 5A).
Figure 5.

KARs regulate the phosphorylation state of CRMP2. A, In Western blots, KAR activation by 300 nm but not 3 μm KA decreased T514 phosphorylation of CRMP2 (pCRMP2T514) and increased the T555 phosphorylation of CRMP2 (pCRMP2T555). CNQX (30 μm) alone had no effect on the phosphorylation of CRMP2. **p < 0.01 and ***p < 0.001, one-sample t test (n = 6). B, The increase in pCRMP2T555 caused by 300 nm KA was blocked by the PKC inhibitor bisindolylmaleimide (Bisind; p = 0.2683 vs Bisind alone, unpaired t test, n = 6), whereas it was unaffected by the Rho-kinase inhibitor GSK429286 (5 μm): ###p < 0.001, unpaired t test (n = 6); *p < 0.05, **p < 0.01, and ***p < 0.001, one-sample t test (n = 6). C, KA at 300 nm but not 3 μm increased the S9 phosphorylation of GSK3β (pGSK3βS9). None of the KA concentrations tested modified the Y216 phosphorylation of GSK3β (pGSK3βY216). CNQX alone had no effect on any of the phosphorylation targets at GSK3β. ***p < 0.001, one-sample t test (n = 6). D, Bisindolylmaleimide prevented the alterations to pGSK3βS9 and pCRMP2T514 caused by 300 nm KA (p > 0.2 vs Bisind alone in both cases; unpaired t test, n = 6). *p < 0.05, **p < 0.01, and ***p < 0.001, one-sample t test (n = 6). The data are expressed as the fold change (mean ± SEM) in the ratio of phosphorylated to total protein compared with untreated or DMSO (0.1%)-treated cells. Representative Western blots are shown above the graphs.
According to the sequence of CRMP2, T555 may be also targeted by PKC because this residue is located in a PKC consensus site (RRTTQR) and has been identified as a putative PKC substrate (Kang et al., 2007). We investigated the roles of PKC and Rho kinase in the phosphorylation of T555 induced by KA in DRG neurons. The phosphorylation of T555 induced by KA (300 nm) was prevented by the PKC inhibitor bisindolylmaleimide, but not by GSK429286, a blocker of Rho kinase, indicating that upon KAR activation T555 is directly phosphorylated by PKC not involving Rho kinase (Fig. 5B).
T514 is a substrate for GSK3β, a kinase that is inactivated by phosphorylation at S9 (Grimes and Jope, 2001). Because a downregulation of GSK3β activity could in turn diminish CRMP2T514 phosphorylation, we investigated whether KAR-mediated PKC activation could also result in GSK3βS9 phosphorylation, as was indicated previously (Frame et al., 2001; Cole et al., 2006). GSK3βS9 phosphorylation increased significantly upon KAR activation by low KA concentrations (Fig. 5C), an effect prevented by the PKC inhibitor bisindolylmaleimide (Fig. 5D). As expected, the concomitant reduction of CRMP2T514 phosphorylation was not observed when upstream phosphorylation at GSK3βS9 was blocked pharmacologically (Fig. 5D). Furthermore, the phosphorylation of GSK3βY216 was unaffected by KA administration (Fig. 5C).
We further determined whether the modest reduction of T514 phosphorylation observed after treatment with low concentrations of KA would be more notorious in compartments relevant for neuritic extension (e.g., growth cones), Therefore, immunocytochemical detection of pCRMPT514 was done in cultures treated with KA and normalized to the label provided by fluorescent phalloidin to visualize actin filaments (Fig. 6A). Interestingly, KA regulated in a bidirectional manner the degree of T514 phosphorylation at growth cones but not at neuritic shafts (Fig. 6B), leaving other properties of growth cones (i.e., the number of filopodia) unaffected (Fig. 6B).
Figure 6.

KA regulates the phosphorylation of CRMP2T514 in a bidirectional manner at the growth cones. A, Examples of growth cones labeled with phalloidin (cyan) and an antibody recognizing pCRMP2T514 (yellow) and superimposed (calibration bars are 25 μm). B, The amount of pCRMP2T514 immunoreactivity was referred to the phalloidin signal and quantified in growth cones and neuritic shafts. The number of filopodia per growth cone was also measured, showing no alteration after KA treatment. **p < 0.01, one-way ANOVA with Dunnet post hoc test (n = 27–29 growth cones per condition).
Together, these findings indicate that the activation of noncanonical KAR signaling modulates the phosphorylation state of CRMP2 in a PKC-dependent manner and that this modulation takes place at subcellular structures relevant for neuritic extension, such as growth cones.
KAR-induced inhibition of neuronal maturation is mediated by enhanced CRMP2T555 phosphorylation
Because phosphorylation at T555 depresses CRMP2 activity (Arimura et al., 2005), we proposed that this signaling module may be involved in the delay of neuronal maturation induced by low KA concentrations. Conversely, because reduced T514 phosphorylation enhances CRMP2 activity, we proposed a role for this module in the KA-induced stimulation of neurite growth. We analyzed the effect of 300 nm KA on DRG neurons infected with Sindbis virus carrying phospho-null and phospho-mimetic mutants of CRMP2T514 and CRMP2T555 (T514A, T514D, T555A, and T555D). Overexpression of CRMP2 and phospho-null CRMP2 mutants promoted by themselves neuronal maturation. However, expression of the phospho-mimetic mutants had no effect (Fig. 7A). In DRG cultures that expressed either the CRMP2T514A or CRMP2T514D mutants, 300 nm KA was still able to delay neuronal maturation, whereas expression of CRMP2T555A or CRMP2T555D blocked the action of KA (Fig. 7B). Together, these results demonstrate that the inhibition of neuronal maturation induced by 300 nm KA involves the phosphorylation of CRMP2 at T555 but not at T514. Indeed, the inhibitory effect of KA on neuronal maturation was unaffected by the presence of the GSK3β inhibitor SB216763 (5 μm) or by the inhibition of Rho kinase activity (GSK429286, 5 μm; Fig. 7C), further evidence that PKC triggers directly the inhibition of neuronal maturation induced by the increased CRMP2T555 phosphorylation provoked by KA.
Figure 7.
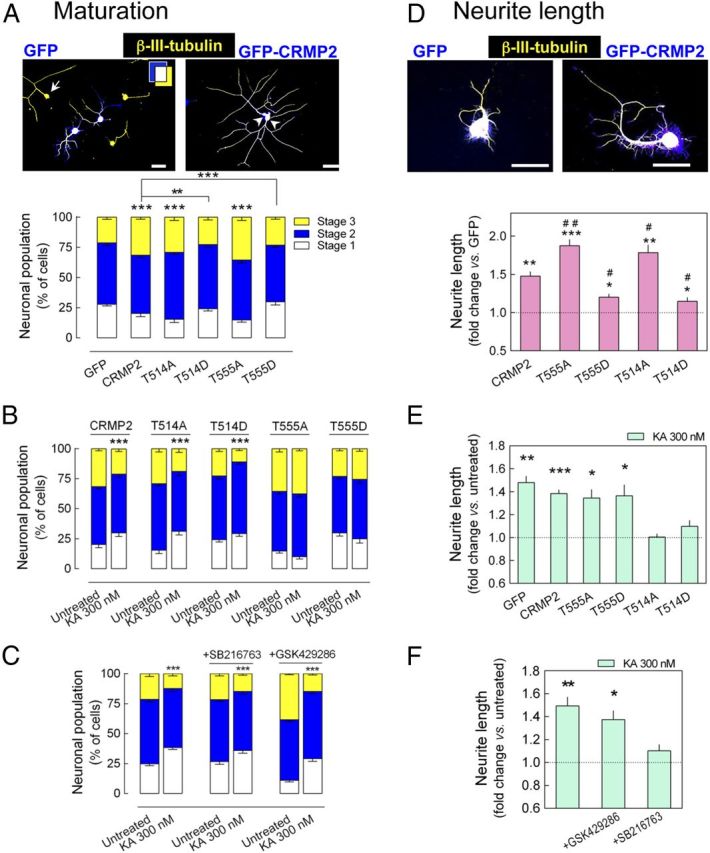
KAR-induced delayed neuronal maturation is mediated by increased T555 phosphorylation of CRMP2, whereas the KAR-induced increase in neurite outgrowth is mediated by decreased phosphorylation of T514 of CRMP2. DRG neurons were infected with Sindbis virus encoding GFP-CRMP2 and phospho-null or phospho-mimetic mutants of CRMP2T514 and CRMP2T555 (T514A, T514D, T555A, and T555D). Subsequently, neuronal maturation level and neurite length of GFP+-DRG neurons cultured in the presence or in the absence of 300 nm KA were analyzed 20 h after infection. A, Effect on neuronal maturation of expression of CRMP2 and phospho-null and phospho-mimetic mutants. Top: Arrow marks a noninfected stage 3 neuron and arrowheads point to two stage 2 neurons. B, The inhibition of neuronal maturation caused by 300 nm KA observed in CRMP2-expressing cells was still observed in DRG neurons expressing either T514A or T514D mutants, but not in those expressing T555A or T555D mutants. C, Neither the GSK3β inhibitor SB216763 (5 μm) nor the Rho kinase inhibitor GSK 429286 (5 μm) prevented the inhibitory effect of 300 nm KA on neuronal maturation. The data are expressed as the percentage of neurons (mean ± SEM) at each stage of maturation quantified from five to six different cultures with a minimum of 100 neurons analyzed per condition and culture. ***p < 0.001, two-way ANOVA followed by Bonferroni's post hoc test. D, Effect on neurite growth of expression of CRMP2, phospho-null, and phospho-mimetic mutants. E, The enhancement in neurite length induced by 300 nm KA either in GFP- or in CRMP2-expressing DRG neurons was unchanged in cells expressing CRMP2T555A and CRMP2T555D, whereas it was impaired in DRG neurons expressing CRMP2T514A or CRMP2T514D. F, The increase in neurite elongation caused by 300 nm KA was prevented by the GSK3β inhibitor SB216763 (5 μm) but not by the Rho kinase inhibitor GSK429286 (5 μm). The data are expressed as the fold change (mean ± SEM) relative to GFP-expressing cells in D, untreated cells in E, or those treated with inhibitor alone in F, each quantified from four to five different cultures analyzing 25–50 neurons per stage, condition, and culture. Neurons from all stages of maturation were pooled in D, E, and F because similar effects were observed for each group. *p < 0.05, **p < 0.01, and ***p < 0.001, one-sample t test. #p < 0.05 and ##p < 0.01, one-way ANOVA with Dunnet's post hoc test versus the effect of CRMP2.
Enhanced neurite outgrowth induced by KAR is mediated by reduced CRMP2T514 phosphorylation
In the light of the above, we investigated whether the enhancement of neuritic elongation induced by 300 nm KA also involves the modulation of CRMP2 phosphorylation. Overexpression of CRMP2 enhanced neuritic elongation at all stages of maturation at a similar extent. Therefore, data from the three stages were pooled. In this pooled group, neurite outgrowth was enhanced by the expression of CRMP2T514A or CRMP2T555A mutants, but it diminished with respect to that induced by CRMP2 expression when the phospho-mimetic mutants T514D or T555D were expressed in the cells (Fig. 7D). Although the stimulatory effect of 300 nm KA on neurite elongation remained unchanged in neurons expressing CRMP2T555A or CRMP2T555D mutants, it was completely blocked in DRG neurons expressing CRMP2T514A or CRMP2T514D (Fig. 7E). Therefore, unlike its inhibitory effect on neuronal maturation, the stimulation of 300 nm KA on neuritic elongation appears to involve the regulation of CRMP2 phosphorylation at T514. Accordingly, the GSK3β inhibitor SB216763 (5 μm) completely blocked the stimulation of neuritic elongation by 300 nm KA, whereas inhibition of Rho kinase (5 μm GSK429286) had no such effect (Fig. 7F).
Overall, these results demonstrate that KAR activation with 300 nm KA stimulates neurite outgrowth in DRG neurons by enhancing the PKC-dependent phosphorylation of GSK3βS9, which results in a reduction of the phosphorylation of CRMP2T514 and the subsequent derepression of CRMP2 activity.
KAR-induced inhibition of neuronal maturation involves the downregulation of voltage-gated calcium channels
Ca2+ plays a key role in neuronal development and, in both developing hippocampal and DRG neurons, cell surface Cav2.2 expression increases in response to CRMP2 overexpression and decreases after CRMP2 knock-down (Brittain et al., 2009; Chi et al., 2009). This effect has been shown recently to be regulated by phosphorylation of CRMP2 (Brittain et al., 2012). Because 300 nm KA inhibits neuronal maturation by increasing CRMP2 phosphorylation at T555, we investigated whether a reduction in cell surface expression of Ca2+ channels might be involved in this action. Indeed, inhibition of N-type Ca2+ channel activity with ϖ-conotoxin (50 nm) did occlude the KA-induced delay in neuronal maturation (Fig. 8A). When Ca2+ current density was measured in whole-cell patch-clamp recordings, a reduction was observed in the density of Ca2+ currents elicited by the application of 50 ms voltage steps to 0 mV from a holding potential of −80 mV in GFP-expressing cells cultured in the presence of 300 nm KA (−33.0 ± 6.57% vs untreated cultures, n = 5, p = 0.0074; Fig. 8B). Cells expressing CRMP2 or CRMP2T555A presented a greater basal Ca2+ current density compared with GFP-expressing cells (Fig. 8C; see also Chi et al., 2009). A similar reduction in Ca2+ current density by 300 nm KA was observed in cells expressing GFP-CRMP2 (Fig. 8D). However, the KA-induced decrease in Ca2+ current density was prevented in cells expressing GFP-CRMP2T555A (Fig. 8D). Similarly, the action of KA was occluded in cells expressing GFP-CRMP2T555D (Fig. 8D), which already had a reduced basal Ca2+ current density in relation to CRMP2-expressing cells (Fig. 8C). Together, these observations show that KARs delay neuronal maturation by downregulating the density of Ca2+ channels at the neuronal membrane through the phosphorylation of CRMP2 at T555, thereby reducing overall Ca2+ activity.
Figure 8.

KAR-induced delay of DRG neuronal maturation involves the downregulation of Cav2.2 channel density. A, The Cav2.2 inhibitor ω-conotoxin (50 nm) occluded the inhibitory effect of 300 nm but not the facilitatory effect of 3 μm KA on neuronal maturation. Data are expressed as the percentage of neurons (mean ± SEM) at each stage of maturation quantified from five different cultures with a minimum of 100 neurons analyzed per condition and culture. ***p < 0.001, two-way ANOVA with Bonferroni's post hoc test. B, KA exposure (300 nm, 20–36 h) reduced N-type Ca2+ currents elicited by the application of 50 ms voltage steps (10 mV increments) from a holding potential of −80 mV in patch-clamped GFP+-DRG neurons. C, DRG neurons overexpressing GFP-CRMP2 presented a significant higher peak Ca2+ current density (pulses from −80 to 0 mV) compared with GFP-expressing cells. Ca2+ current density was similar in cells overexpressing GFP-CRMP2 and the phospho-null mutant GFP-CRMP2T555A, but significantly lower in cells expressing the phospho-mimic mutant GFP-CRMP2T514D. The data are mean ± SEM of peak Ca2+ current density (pA/pF) of 30–37 cells from five to six different cultures. **p < 0.01, one-way ANOVA with Tukey's multiple-comparisons test. Dots represent single current density values from each individual cell. D, Summary of the alterations in Ca2+ current density induced by exposure to 300 nm KA in cells expressing GFP, GFP-CRMP2, GFP-CRMP2T555A, and GFP-CRMP2T555D. The data are expressed as the percentage change relative to the untreated neurons (five to six different cultures, six neurons minimum per condition per culture). **p < 0.01, one-sample t test.
Discussion
In the present study, we demonstrate that KARs modulate neuronal development in DRG neurons in a bidirectional manner that is mediated by the activation of two distinct signaling pathways. Weak activation of KARs at low agonist concentrations delays maturation and promotes neurite outgrowth, whereas moderate to strong KAR activation promotes the maturation of neurons while limiting neuritic growth. Accordingly, low-level KAR activity would promote neurite growth in immature neurons, allowing such developing cells to find their targets. This effect is mediated by noncanonical signaling and it involves the activation of both Gi/o-proteins and PKC. In contrast, strong KAR stimulation promotes neuronal maturation and the arrest of neurite growth, likely by facilitating growth cone collapse, a process mediated by the canonical ionotropic activity of these receptors.
We also demonstrate that KAR-stimulated PKC activity delays maturation and stimulates neurite outgrowth through the differential phosphorylation of CRMP2, a protein that interacts with the KAR subunit GluK5. PKC phosphorylates CRMP2T555, a known target of the Rho kinase (Arimura et al., 2000), directly and it decreases the phosphorylation of CRMP2T514. The latter effect is most likely triggered by PKC-mediated phosphorylation of GSK3βS9, which inactivates GSK3β kinase activity (Grimes and Jope, 2001), subsequently diminishing p-CRMP2T514, as described previously (Yoshimura et al., 2005). It is well known that phosphorylation of CRMP2 at either T555 or T514 blocks the binding of CRMP2 to tubulin dimers, which in turn prevents microtubule polymerization, leading to growth cone collapse and the arrest of neurite outgrowth (Fukata et al., 2002; Arimura et al., 2005; Uchida et al., 2005; Yoshimura et al., 2005). This suggested that the KA-induced promotion of neurite elongation should involve the decrease in the phosphorylation of CRMP2T514, enhancing CRMP2 activity. Accordingly, the increase in neurite outgrowth induced by low-level KAR activity was prevented by the expression of CRMP2T514 phospho-mimetic mutants (CRMP2T514D) and occluded by phospho-null mutants (CRMP2T514A), whereas it was unaffected by the expression of T555 mutants. In contrast, the delay in maturation induced by low-level KAR activity was prevented by a CRMP2T555 phospho-mimetic (CRMP2T555D) and occluded by phospho-null mutants (CRMP2T555A), whereas it was unaffected by the T514 mutants. Moreover, a GSK3β inhibitor abrogated the increase in neurite outgrowth induced by KAR stimulation, although it had no effect on the KAR-induced delay in maturation. The contribution of other modulators, such as Rho-kinase, to these effects was discarded because the presence of Rho-kinase inhibitors did not affect the action of KA. The most plausible explanation for these findings is that the enhanced neurite outgrowth induced by weak KAR activation involves an increase in the activity of CRMP2 due to a reduction in the phosphorylation at CRMP2T514. This effect is most likely driven by PKC-mediated phosphorylation of GSK3βS9, whereas the delay in maturation is driven by a PKC-mediated increase in p-CRMP2T555 levels. Moreover, our data indicate that the delay in maturation also involves the downregulation of CRMP2-mediated Cav2.2 trafficking to the membrane through the increase in the phosphorylation of CRMP2T555.
This interpretation implies the segregation of CRMP2 activity at the cellular level that is differentially modulated by KARs at these two phosphorylation sites. Because CRMP2 affects neurite outgrowth at the growth cone (Arimura et al., 2005; Yoshimura et al., 2005), the changes in phosphorylation induced by KA at T514 are expected to take place in this structure. For this to occur, KARs must be expressed in the growth cones of DRG neurons and there is indeed evidence that both metabotropic and conventional KARs do not accumulate throughout the entire cell, but rather are expressed at specific sites of cell soma and along neurites, including growth cones and neurite bifurcation points (Rozas et al., 2003). Ca2+-imaging experiments revealed that KA administration induces an increase of Ca2+ in the growth cones of DRG neurons directly, probably via Ca2+-permeable KARs (Joseph et al., 2011), and also inhibits Ca2+ entry via voltage-gated calcium channels, an effect mediated by noncanonical KAR signaling (Rozas et al., 2003). Therefore, not only are KARs present in the growth cone, but they can also trigger metabotropic signaling to modulate CRMP2 and GSK3β activity within this structure. Indeed, we observed that 300 nm KA induces the dephosphorylation of CRMP2 at T514 precisely at the growth cones. Unfortunately, we could not detect CRMP2 phosphorylated at T555 by immunocytochemistry due to the poor signal obtained with the commercially available antibodies. However, the increase in CRMP2T555 phosphorylation observed after KA treatment that delays maturation likely occurs throughout the cell and not specifically in the growth cone, likely involving the downregulation of Cav2.2 in the soma and neurites, as indicated by data presented in Figure 8.
Regarding the mechanisms underlying the effects of moderate to strong KAR activation, our data indicate an involvement of ionotropic signaling, predominantly relying on induced electrical activity. The increase in neuronal maturation provoked by this level of activation was TTX sensitive, indicating the involvement of depolarization-induced action potentials that are critical in the development of DRG neurons (Fields, 1998). The reduction in neurite elongation induced by higher KA concentrations was also sensitive to TTX, indicating that it involves substantial depolarization provided by Na+ channel activation, as was shown previously in hippocampal neurons (Cohan and Kater, 1986; Ibarretxe et al., 2007). Such a depolarization would convey Ca2+ channel activation and Ca2+ entry in growth cones, likely arresting growth cone motility and axon extension (Cohan et al., 1987; Gomez and Spitzer, 1999). Moreover, canonical KAR signaling could arrest neurite outgrowth by mediating Ca2+ entry directly (Joseph et al., 2011).
Our findings help improve our understanding of the role of KARs in neuronal development and maturation, for the first time providing a direct link between KAR activation and the modulation of neuronal cytoskeleton dynamics. KARs are strongly expressed during development in a complex, cell-type-specific manner, and endowed with several properties that allow them to modulate GABAergic and glutamatergic drive during development (for review, see Lauri and Taira, 2011 and 2012). Therefore, KARs can determine the spontaneous rhythmic activity in the immature network (Lauri et al., 2005), which appears to be instrumental in the development of synaptic connectivity (Zhang and Poo, 2001). Indeed, tonic KAR activity in immature neurons may be essential for the formation and stabilization of synaptic contacts (Vesikansa et al., 2007), and activity-dependent downregulation of KAR activity (Kidd and Isaac, 1999; Lauri et al., 2006) is crucial for the functional maturation of glutamatergic synapses (Marchal and Mulle, 2004; Lauri et al., 2006). KARs can also regulate growth cone motility (Tashiro et al., 2003; Ibarretxe et al., 2007), a crucial event that allows the axon to explore the environment and to detect appropriate synaptic targets (Goda and Davis, 2003). These observations support a role for KARs in the early stages of contact formation and rearrangement during the development of neuronal connectivity. In the developing hippocampus, KARs bidirectionally regulate the motility of hippocampal mossy fiber filopodia in a developmentally regulated and concentration-dependent manner (Tashiro et al., 2003). Together, these data describe a scenario in which low concentrations of glutamate in early developmental stages enhance neuron motility and neurite outgrowth through KAR-mediated modulation of CRMP2 to promote synaptic target finding while keeping the neuron immature until proper targets are found. Upon the formation of the nascent synapse, the increase in glutamate concentration resulting from enhanced release and/or the reduction in extracellular volume may lead to moderate to strong KAR activation, which reduces filopodia motility (Chang and De Camilli, 2001; Tashiro et al., 2003) and inhibits neurite outgrowth (present data and Ibarretxe et al., 2007; Joseph et al., 2011), thereby promoting the transition of filopodia into presynaptic structures (Goda and Davis, 2003).
Understanding KAR signaling during development has significant physiological and pathological implications. The ability of KARs to modulate CRMP2 signaling is particularly interesting because abnormal CRMP2 activity may give rise to important neurological disorders. Because CRMP2 is involved in the formation of axonal pathways and synaptic connections during nervous system development, deregulation of CRMP2 signaling due to aberrant KAR activity may compromise normal functions in adults (Yoshimura et al., 2006). KARs have been implicated in temporal lobe epilepsy (Mulle et al., 1998), in which axonal sprouting is involved, and it is tempting to speculate that in these circumstances, abnormal KAR activation may provoke CRMP2 deregulation. CRMP2 activity may also contribute to the pathogenesis of Alzheimer′s disease because transgenic mice overexpressing the intracellular domain of amyloid precursor protein exhibit elevated levels of phosphorylated CRMP2, probably due to increased GSK3β activity (Cole et al., 2007). The KAR-CRMP2 signaling module described here may also be implicated in the pathogenesis of other disorders, such as neuropathic pain (Brittain et al., 2011) and schizophrenia (Hensley et al., 2011), although the degree to which KAR contributes to these pathological processes remains to be defined. Nonetheless, the present data suggest that CRMP2 tethers KAR activity (i.e., glutamatergic signaling) to such events. Therefore, a greater understanding of the interplay between KARs and CRMP2 is required to elucidate their role in physiological and pathological processes, and ultimately in pharmacotherapeutic strategies.
Footnotes
This work was supported by grants to J.L. from the Spanish Ministerio de Ciencia e Innovación (BFU2006-07138 and BFU2011-24084), CONSOLIDER (CSD2007-00023), and Prometeo/2011/086. J.M.M. was supported by a doctoral fellowship from the Portuguese Fundação para a Ciência e a Tecnologia and R.J.R. was supported by a Juan de la Cierva fellowship. S.V. is an FPI fellow of the Ministry of Economy and Competitivity. We thank Prof. Masaharu Noda for CRMP plasmids, M. Llinares for excellent technical assistance, and Dr. G. Lopez-Bendito for critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Arimura N, Inagaki N, Chihara K, Ménager C, Nakamura N, Amano M, Iwamatsu A, Goshima Y, Kaibuchi K. Phosphorylation of collapsing mediator protein-2 by rho-kinase: evidence for two separate signalling pathways for growth cone collapse. J Biol Chem. 2000;275:23973–23980. doi: 10.1074/jbc.M001032200. [DOI] [PubMed] [Google Scholar]
- Arimura N, Ménager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M, Morone N, Usukura J, Kaibuchi K. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol. 2005;25:9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahn S, Volk B, Wisden W. Kainate receptor gene expression in the developing rat brain. J Neurosci. 1994;14:5525–5547. doi: 10.1523/JNEUROSCI.14-09-05525.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettler B, Boulter J, Hermans-Borgmeyer I, O'Shea-Greenfield A, Deneris ES, Moll C, Borgmeyer U, Hollmann M, Heinemann S. Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron. 1990;5:583–595. doi: 10.1016/0896-6273(90)90213-Y. [DOI] [PubMed] [Google Scholar]
- Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, Fehrenbacher JC, Fitz SD, Khanna M, Park CK, Schmutzler BS, Cheon BM, Due MR, Brustovetsky T, Ashpole NM, Hudmon A, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+-channel complex. Nat Med. 2011;17:822–829. doi: 10.1038/nm.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2(CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. 2009;284:31375–31390. doi: 10.1074/jbc.M109.009951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Wang Y, Eruvwetere O, Khanna R. Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett. 2012;586:3813–3818. doi: 10.1016/j.febslet.2012.09.022. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Chang S, De Camilli P. Glutamate regulates actin-based motility in axonal filopodia. Nat Neurosci. 2001;4:787–793. doi: 10.1038/90489. [DOI] [PubMed] [Google Scholar]
- Chi XX, Schmutzler BS, Brittain JM, Wang Y, Hingtgen CM, Nicol GD, Khanna R. Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsing response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci. 2009;122:4351–4362. doi: 10.1242/jcs.053280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohan CS, Kater SB. Suppression of neurite elongation and growth cone motility by electrical activity. Science. 1986;232:1638–1640. doi: 10.1126/science.3715470. [DOI] [PubMed] [Google Scholar]
- Cohan CS, Connor JA, Kater SB. Electrical and chemically mediated increases in intracellular calcium in neuronal growth cones. J Neurosci. 1987;7:3588–3599. doi: 10.1523/JNEUROSCI.07-11-03588.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Causeret F, Yadirgi G, Hastie CJ, McLauchlan H, McManus EJ, Hernández F, Eickholt BJ, Nikolic M, Sutherland C. Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J Biol Chem. 2006;281:16591–16598. doi: 10.1074/jbc.M513344200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Noble W, van Aalten L, Plattner F, Meimaridou R, Hogan D, Taylor M, LaFrancois J, Gunn-Moore F, Verkhratsky A, Oddo S, LaFerla F, Giese KP, Dineley KT, Duff K, Richardson JC, Yan SD, Hanger DP, Allan SM, Sutherland C. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer's disease progression. J Neurochem. 2007;103:1132–1144. doi: 10.1111/j.1471-4159.2007.04829.x. [DOI] [PubMed] [Google Scholar]
- Contractor A, Mulle C, Swanson GT. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 2011;34:154–163. doi: 10.1016/j.tins.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darstein M, Petralia RS, Swanson GT, Wenthold RJ, Heinemann SF. Distribution of kainate receptor subunits at hippocampal mossy fiber synapses. J Neurosci. 2003;23:8013–8019. doi: 10.1523/JNEUROSCI.23-22-08013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD. Effects of ion channel activity on development of dorsal root ganglion neurons. J Neurobiol. 1998;37:158–170. doi: 10.1002/(SICI)1097-4695(199810)37:1<158::AID-NEU12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/S1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Ménager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- Goda Y, Davis GW. Mechanisms of synapse assembly and disassembly. Neuron. 2003;40:243–264. doi: 10.1016/S0896-6273(03)00608-1. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Spitzer NC. In vivo regulation of axon extension and pathfinding by growth-cone calcium transients. Nature. 1999;397:350–355. doi: 10.1038/16927. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 1995;376:509–514. doi: 10.1038/376509a0. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/S0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Hensley K, Venkova K, Christov A, Gunning W, Park J. Collapsin Response Mediator Protein-2: An Emerging Pathologic Feature and Therapeutic Target for Neurodisease Indications. Mol Neurobiol. 2011;43:180–191. doi: 10.1007/s12035-011-8166-4. [DOI] [PubMed] [Google Scholar]
- Herb A, Burnashev N, Werner P, Sakmann B, Wisden W, Seeburg PH. The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron. 1992;8:775–785. doi: 10.1016/0896-6273(92)90098-X. [DOI] [PubMed] [Google Scholar]
- Ibarretxe G, Perrais D, Jaskolski F, Vimeney A, Mulle C. Fast regulation of axonal growth cone motility by electrical activity. J Neurosci. 2007;27:7684–7695. doi: 10.1523/JNEUROSCI.1070-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Chihara K, Arimura N, Ménager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K. CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci. 2001;4:781–782. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- Ip JP, Shi L, Chen Y, Itoh Y, Fu WY, Betz A, Yung WH, Gotoh Y, Fu AK, Ip NY. α2-chimaerin controls neuronal migration and functioning of the cerebral cortex through CRMP-2. Nat Neurosci. 2012;15:39–47. doi: 10.1038/nn.2972. [DOI] [PubMed] [Google Scholar]
- Joseph DJ, Williams DJ, MacDermott AB. Modulation of neurite outgrowth by activation of calcium-permeable kainite receptors expressed by rat nociceptive-like dorsal root ganglion neurons. Dev Neurobiol. 2011;71:818–835. doi: 10.1002/dneu.20906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JH, Jiang Y, Toita R, Oishi J, Kawamura K, Han A, Mori T, Niidome T, Ishida M, Tatematsu K, Tanizawa K, Katayama Y. Phosphorylation of Rho-associated kinase (Rho-kinase/ROCK/ROK) substrates by protein kinases A and C. Biochimie. 2007;89:39–47. doi: 10.1016/j.biochi.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- Lauri S, Taira T. Kainate receptors in developing presynaptic terminals. WIREs Membrane Transport and Signaling. 2012;1:45–55. doi: 10.1002/wmts.3. [DOI] [Google Scholar]
- Lauri SE, Taira T. Role of kainate receptors in network activity during development. Adv Exp Med Biol. 2011;717:81–91. doi: 10.1007/978-1-4419-9557-5_8. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Segerstråle M, Vesikansa A, Maingret F, Mulle C, Collingridge GL, Isaac JT, Taira T. Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus. J Neurosci. 2005;25:4473–4484. doi: 10.1523/JNEUROSCI.4050-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Vesikansa A, Segerstråle M, Collingridge GL, Isaac JT, Taira T. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron. 2006;50:415–429. doi: 10.1016/j.neuron.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Lerma J. Kainate receptor physiology. Curr Opin Pharmacol. 2006;6:89–97. doi: 10.1016/j.coph.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Marchal C, Mulle C. Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: a potential role for kainate receptors. J Physiol. 2004;561:27–37. doi: 10.1113/jphysiol.2004.069922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Pérez-Otaño I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Cohen A, Stern-Bach Y, Lerma J. A role for extracellular Na+ in the channel gating of native and recombinant kainate receptors. J Neurosci. 2003;23:8641–8648. doi: 10.1523/JNEUROSCI.23-25-08641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res. 2006;326:457–482. doi: 10.1007/s00441-006-0265-6. [DOI] [PubMed] [Google Scholar]
- Rivera R, Rozas JL, Lerma J. PKC-dependent autoregulation of membrane kainate receptors. EMBO J. 2007;26:4359–4367. doi: 10.1038/sj.emboj.7601865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues RJ, Lerma J. Metabotropic signaling by kainate receptors. WIREs Membrane Transport and Signaling. 2012;1:399–410. doi: 10.1002/wmts.35. [DOI] [Google Scholar]
- Rodríguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/S0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Rozas JL, Paternain AV, Lerma J. Noncanonical signaling by ionotropic kainate receptors. Neuron. 2003;39:543–553. doi: 10.1016/S0896-6273(03)00436-7. [DOI] [PubMed] [Google Scholar]
- Selak S, Paternain AV, Fritzler MJ, Lerma J. Human autoantibodies against early endosome antigen-1 enhance excitatory synaptic transmission. Neuroscience. 2006;143:953–964. doi: 10.1016/j.neuroscience.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Dunaevsky A, Blazeski R, Mason CA, Yuste R. Bidirectional regulation of hippocampal mossy fiber filopodial motility by kainate receptors: a two-step model of synaptogenesis. Neuron. 2003;38:773–784. doi: 10.1016/S0896-6273(03)00299-X. [DOI] [PubMed] [Google Scholar]
- Tomita S, Byrd RK, Rouach N, Bellone C, Venegas A, O'Brien JL, Kim KS, Olsen O, Nicoll RA, Bredt DS. AMPA receptors and stargazin-like transmembrane AMPA receptor-regulatory proteins mediate hippocampal kainate neurotoxicity. Proc Natl Acad Sci U S A. 2007;104:18784–18788. doi: 10.1073/pnas.0708970104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y. Semaphorin 3A signalling is mediated via sequential Cdk5 and GSK3β phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes to Cells. 2005;10:165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- Vesikansa A, Sallert M, Taira T, Lauri SE. Activation of kainate receptors controls the number of functional glutamatergic synapses in the area CA1 of rat hippocampus. J Physiol. 2007;583:145–157. doi: 10.1113/jphysiol.2007.133975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N, Goshima Y. Collapsin response mediator proteins regulate neuronal development and plasticity by switching their phosphorylation status. Mol Neurobiol. 2012;45:234–246. doi: 10.1007/s12035-012-8242-4. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kaibuchi K. Molecular mechanisms of axon specification and neuronal disorders. Ann N Y Acad Sci. 2006;1086:116–125. doi: 10.1196/annals.1377.013. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo MM. Electrical activity and development of neural circuits. Nat Neurosci. 2001;4:1207–1214. doi: 10.1038/nn753. [DOI] [PubMed] [Google Scholar]