

Abstract
Background
Trypanosoma cruzi, the protozoan causative of Chagas disease, is classified into six main Discrete Typing Units (DTUs): TcI-TcVI. This parasite has around 105 copies of the minicircle hypervariable region (mHVR) in their kinetoplastic DNA (kDNA). The genetic diversity of the mHVR is virtually unknown. However, cross-hybridization assays using mHVRs showed hybridization only between isolates belonging to the same genetic group. Nowadays there is no methodologic approach with a good sensibility, specificity and reproducibility for direct typing on biological samples. Due to its high copy number and apparently high diversity, mHVR becomes a good target for typing.
Methodology/Principal findings
Around 22 million reads, obtained by amplicon sequencing of the mHVR, were analyzed for nine strains belonging to six T. cruzi DTUs. The number and diversity of mHVR clusters was variable among DTUs and even within a DTU. However, strains of the same DTU shared more mHVR clusters than strains of different DTUs and clustered together. In addition, hybrid DTUs (TcV and TcVI) shared similar percentages (1.9–3.4%) of mHVR clusters with their parentals (TcII and TcIII). Conversely, just 0.2% of clusters were shared between TcII and TcIII suggesting biparental inheritance of the kDNA in hybrids. Sequencing at low depth (20,000–40,000 reads) also revealed 95% of the mHVR clusters for each of the analyzed strains. Finally, the method revealed good correlation in cluster identity and abundance between different replications of the experiment (r = 0.999).
Conclusions/Significance
Our work sheds light on the sequence diversity of mHVRs at intra and inter-DTU level. The mHVR amplicon sequencing workflow described here is a reproducible technique, that allows multiplexed analysis of hundreds of strains and results promissory for direct typing on biological samples in a future. In addition, such approach may help to gain knowledge on the mechanisms of the minicircle evolution and phylogenetic relationships among strains.
Author summary
Chagas disease is an important public health problem in Latin America showing a wide diversity of clinical manifestations and epidemiological patterns. It is caused by the parasite Trypanosoma cruzi. This parasite is genetically diverse and classified into six main lineages. However, the relationship between intra-specific genetic diversity and clinical or epidemiological features is not clear, mainly because low sensitivity for direct typing on biological samples. For this reason, genetic markers with high copy number are required to achieve sensitivity. Here, we deep sequenced and analyzed a DNA region present in the large mitochondria of the parasite (named as mHVR, 105 copies per parasite) from strains belonging to the six main lineages in order to analyze mHVR diversity and to evaluate its usefulness for typing. Despite the high sequence diversity, strains of the same lineage shared more sequences than strains of different lineages. Curiously, hybrid lineages shared mHVR sequences with both parents suggesting that mHVR (and DNA minicircles from the mitochondria) are inherited from both parentals. The mHVR amplicon sequencing workflow proposed here is reproducible and, potentially, it would be useful for typing hundreds of biological samples at time. It also provides a valuable approach to perform evolutionary and functional studies.
Introduction
The protozoan parasite Trypanosoma cruzi (Kinetoplastea: Trypanosomatidae) is the causative agent of Chagas disease. This parasite infects millions of people throughout its distribution in Latin America. Chagas disease can display a broad pathological spectrum, including potentially fatal cardiological and gastrointestinal dysfunctions [1].
T. cruzi is a monophyletic taxon showing a remarkable genetic heterogeneity, with at least six phylogenetic lineages formally recognised as Discrete Typing Units (DTUs), TcI–TcVI [2, 3]; and a seventh lineage, named TcBat [4–6]. The genetic diversity of T. cruzi was firstly revealed by Multilocus Enzyme Electrophoresis [7, 8] and posteriorly by very diverse techniques including Multilocus Sequence Typing (MLST) [9–12], microsatellite typing (MLMT) [13–18], target-specific PCR [19–21], PCR-RFLP [22, 23], PCR-DNA blotting with hybridization assays [24–26], and recently by amplicon deep sequencing [27, 28]. The different approaches have their own advantages and disadvantages and bring out the genetic diversity of T. cruzi at different levels. Approaches that allow direct typing from biological samples (blood, tissues, etc.), avoiding parasite culture, are more suitable for clinical and epidemiological studies. However, nowadays there is no methodologic approach with a good sensibility, specificity and reproducibility for direct typing on biological samples.
Because there is usually a low number of parasites in infected tissues or blood samples, genetic markers with high number of copies are required to achieve good sensitivity of detection [29]. In this regard, T. cruzi, as all the kinetoplastids, has a unique and large mitochondrion which contains a complex network of DNA, the kinetoplastic DNA (kDNA). The kDNA represents approximately 20–25% of the total cellular DNA in T. cruzi and consists of two kind of circular DNA molecules: maxicircles and minicircles. Maxicircles contain mitochondrial genes characteristic of other eukaryotes [30]. Minicircles are present in tens of thousands of copies [31]. Each of them is organized into four highly conserved regions located 90° apart each other, and an equal number of hypervariable regions (mHVRs) interspersed between the conserved regions [32]. The highly conserved regions of minicircles have been widely used as targets for molecular detection of T. cruzi DNA. The used primers show a good sensitivity and specificity [29] and amplify a region of about 330 bp that totally include the mHVRs present between conserved regions. This amplified region has been used in hybridization assays (mHVR probes) and DTU-specific hybridization was observed only between isolates belonging to the same genetic group [25, 26, 33–35]. This specificity observed in hybridization assays suggests the presence of DTU specific sequences and even genotype-specific sequences (i.e. sequences showing specificity at intra-DTU level). However, technical limitations that existed until a few years ago for sequencing these highly variable kDNA regions, prevented the identification of the sequences in which the specificity relies. Some attempts were made by cloning and sequencing some mHVRs [36, 37] but the limited number of studied sequences were not enough to obtain a complete picture of the genetic diversity of these sequences. Thus, the observed hybridization patterns between mHVRs continue being a black box system and the sequence diversity of T. cruzi mHVRs virtually unknown.
Beyond the potential utility for strain typing, studying mHVR diversity is also interesting because these sequences are involved in functions that are only known in kinetoplastids and in no other eukaryotic organism. mHVRs code for short RNAs called guide RNAs (gRNAs). gRNAs are involved on edition of several mitochondrially-coded mRNAs. This edition varies from addition of some Us to building almost the full open reading frame of the mRNA [38, 39]. In this sense, gRNAs can be inferred from sequences of the mitochondrial mRNAs and diversity on edition among strains can be addressed [40]. In addition, studying mHVR diversity can shed light on how such sequences evolve and how they are inherited.
Here, we propose an amplicon deep sequencing approach that allows an accurate knowledge of the sequence diversity of the hypervariable region of kDNA minicircles of T. cruzi and opens the possibility of functional and evolutionary studies. This approach can be also used as a typing method for hundreds of samples at time.
Materials and methods
Strains
DNA from nine cloned T. cruzi strains belonging to the six main DTUs was examined in this study (Table 1). All the strains were typified by using an optimized Multilocus Sequence Typing scheme based on four gene fragments (HMCOAR, GPI, TcMPX and RHO1) according to Diosque et al. [7], in order to confirm DTU for each strain.
Table 1. Strains used in this study.
| Strain | DTU | Origin | Host |
|---|---|---|---|
| 1. PalDa20cl3 | TcI | El Palmar, Argentina | Didelphis albiventris |
| 2. TEV55cl1 | TcI | Tres Estacas, Argentina | Triatoma infestans |
| 3. Esmeraldo | TcII | Sao Felipe, Brazil | Homo sapiens |
| 4. TU18cl93 | TcII | Potosí, Bolivia | Triatoma infestans |
| 5. X109/2 | TcIII | Makthlawaiya, Paraguay | Canis familiaris |
| 6. CANIIIcl1 | TcIV | Belém, Brazil | Homo sapiens |
| 7. MNcl2 | TcV | Región IV, Chile | Homo Sapiens |
| 8. LL014R1 | TcV | Las Leonas, Argentina | Triatoma infestans |
| 9. LL015P68R0cl4 | TcVI | Las Leonas, Argentina | Canis familiaris |
Primer design and library construction
In order to amplify the minicircles hypervariable region, kDNA specific primers 121 (5’-ACACTCTTTCCCTACACGACGCTCTTCCGATCTAAATAATGTACGGG(T/G)GAGATGCATGA-3’) and 122 (5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGGTTCGATTGGGGTTGGTGTAATATA-3’) were modified by adding an oligo adapter to be used in an Illumina platform. The mHVR libraries were generated by a one-step PCR performed in 5 μl reaction volumes containing 5 ng of DNA, 250 nM of each primer, 2 μM of barcode primers, 5 U of Fast Start High Fidelity Enzyme Blend (Roche), 0.50 μl of 10X buffer (supplied with the Fast Start High Fidelity Enzyme Blend), 25 nM of MgCl2 (Roche), 0.25 μl of DMSO (Roche), 10 mM of PCR grade nucleotide mix (Roche). The PCR reaction was carried out on a Veriti Thermal Cycler (Life Technologies) and ran as follow: an initial denaturation step (10 min at 95°C), 10 cycles (95°C for 15 seconds, 60°C 30 seconds, 72°C 1 min), 2 cycles (95°C for 15 seconds, 80°C 30 seconds, 60°C 30 seconds, 72°C 1 min), 8 cycles (95°C for 15 seconds, 60°C 30 seconds, 72°C 1 min), 2 cycles (95°C for 15 seconds, 80°C 30 seconds, 60°C 30 seconds, 72°C 1 min), 8 cycles (95°C for 15 seconds, 60°C 30 seconds, 72°C 1 min) and 5 cycles (95°C for 15 seconds, 80°C 30 seconds, 60°C 30 seconds, 72°C 1 min). Amplicons were then purified using the magnetic beads Agencourt AMPure XP-PCR Purification (Beckman Genomics, USA). The concentration of the purified amplicons was controlled using Qubit Fluorometer 2.0 (Invitrogen, USA). All libraries were validated using the Fragment Analyzer system (Advanced Analytical Technologies, USA). The average size of the mHVR amplicons was ~480bp. All samples were then pooled and prepared according to the manufacturer's recommendations (Illumina Protocols: Sequencing Library Preparation) and sequenced on an Illumina MiSeq using a 500 cycle v2 kit (Illumina, San Diego, USA) to produce amplicons of approximately ~480 bp in length (250 bp paired-end reads).
Bioinformatics
Read pre-processing
Reads were demultiplexed and adaptors were removed using Illumina Miseq Reporter, according to the manufacture recommendations. Raw sequence reads for all samples were quality filtered using the pair-end mode of Trimmomatic v0.36 [41]. This software was used to remove low quality bases from the beginning and end of sequence reads pairs (trimming). Also, a sliding window of 8 bases from left to right was performed. Sequence reads were cut whenever the average quality into the window fell below the threshold (<15, Phred score) and the right side of the read sequence was deleted. Sequences with a minimum read length of 150 nt, were retained. Then, the retained paired-reads were merged into a consensus sequence with its associated corrected base quality scores and chimeras were removed using LeeHom software [42] with default parameters.
Clustering
The following steps in the workflow were all performed using QIIME v1.9.1 [43]. A further quality check of sequence reads was carried with the “split_fastq_libraries.py” script. Default parameters were used, except for the quality threshold for trimming, which was raised to 25. Then, preprocessed sequences were clustered with the “pick_de_novo_otus.py” script. The de novo approach groups sequences based on sequence identity using the uclust algorithm [44]. Default parameters were used, and sequences were clustered according to three different identity thresholds—85%, 90% and 95%—in order to determine different mHVR clusters. The terms “minicircle class” or “mHVR class” are used in the bibliography without any clear definition and sometimes referring to mHVRs that codes the same gRNA. Here, we used the term “mHVR cluster” defined as a group of mHVRs that share a minimum sequence identity percentage with a cluster centroid without considering if a gRNA is coded by it. The mHVR cluster size is defined as the number of reads that belongs to such cluster. The following analyses were performed for these three identity thresholds.
Output tables were filtered at 0.005% using the “filter_otus_from_otu_table.py” script, in order to discard mHVR clusters with low abundance which are more probably sequencing artifacts [45], remaining parameters were used by default. The presence of a cluster into a strain was discarded when its abundance was lower than 20 read sequences. Diversity was measured by subsampling mHVR clusters tables using the “multiple_rarefactions.py” script. Clusters tables were rarefied including a maximum of 10,000 reads/sample in order to determine the minimum number of reads needed to detect all the clusters of mHVR. Alpha diversity measures—Simpson index and observed clusters—were estimated to determine the composition of each strain in sampling units using the “alpha_diversity.py" script. The output files of “alpha_diversity.py” were concatenated into a single file for generating rarefaction curves with the “collate_alpha.py” script followed by the “make_rarefaction_plots.py” script. In order to estimate compositional dissimilarity among strains, the “jackknifed_beta_diversity.py” script was used. Default parameters and the Bray-Curtis measure were chosen. The jackknifed beta diversity workflow calculates the beta diversity between each pair of previously resampled input strains, forming a distance matrix. The distance matrix then was visualized using UPGMA and Principal Coordinate Analysis (PCoA).
Rarefactions
In order to determine the minimum number of reads required to obtain the correct assignment of DTU for each strain, beta diversity was estimated at different subsampling levels. For each of the identity thresholds, the resampling of mHVR clusters was performed using decreasing amount of sequence reads, from 820,000 to 10,000 reads, with intervals of 10,000 reads and for 100 replications at each subsample.
Reproducibility assessment
To evaluate the reproducibility of the mHVR amplicon sequencing, two independent amplifications (PCR1 and PCR2) for LL015P68R0cl4 strain were performed. The data obtained were processed following the pipeline previously described. Clusters shared between PCR1 and PCR2 were evaluated, the Pearson’s correlation coefficient was calculated and the linear regression curve that best fitted the data was estimated.
Accession numbers
The raw data set has been deposited in the NCBI SRA database (BioProject ID: PRJNA514922).
Results
mHVR abundance and diversity
A total of 22,092,382 paired reads were obtained by amplicon sequencing of the mHVR from nine strains belonging to six DTUs. A total of 14,766,753 sequences were retained (an average of ≈1.4 million of sequences per strain) after trimming low quality ends, merging paired reads (forward and reverse), elimination of chimeric reads and filtering by base quality (S1 Table). Surviving sequences were clustered according to different identity thresholds (85%, 90% and 95%) (Table 2, S2 and S3 Tables). The number of mHVR clusters for each strain was very similar using different thresholds (with differences less than 10% in all comparisons between 85% and 95% thresholds). However, clustering at 85% threshold returned few more mHVR clusters than clustering at 90% and 95% identity (See Table 2, S2 and S3 Tables). In addition, most clusters were highly divergent among them (S1 Fig). At any threshold, the number of mHVR clusters was variable among strains and DTUs (Table 2), ranging from 71 (Mncl2 –TcV) to 373 (X109/2 –TcIII) clusters. Additionally, strong intra-DTU variations in the number of clusters were observed in strains of TcI and TcII (Table 2). Finally, rarefactions of each dataset discarded that these differences among strains are the effect of different sequencing depths (Table 2, S2 and S3 Tables).
Table 2. Number of mHVR clusters defined at a threshold of 85% sequence identity for different strains.
| TcI | TcII | TcIII | TcIV | TcV | TcVI | ||||
|---|---|---|---|---|---|---|---|---|---|
| PalDa20 cl3 |
TEV55cl1 | Esmeraldo | Tu18 cl93 |
X109/2 | CANIIIcl1 | LL014 R1 |
MNcl2 | LL015 P68R0 cl4 |
|
| mHVR clusters | 324 | 234 | 347 | 151 | 373 | 149 | 72 | 71 | 108 |
| Rarefaction at 820,000 sequences | |||||||||
| mHVR clusters | 324 | 233.7 | 346.8 | 151 | 369 | 144.4 | 72 | 69.4 | 108 |
| Simpson Index(diversity)* | 0.991 | 0.989 | 0.994 | 0.978 | 0.994 | 0.885 | 0.827 | 0.902 | 0.942 |
* Average over 10 replications
Strains belonging to TcIV, TcV and TcVI showed some dominant clusters containing a high proportion of reads (i.e. the cluster size) (Fig 1). The sum of the six most abundant clusters in TcIV, TcV and TcVI represent in all cases more than 50% of the clustered sequences (80.9% and 69.1% in the TcV strains LL014R1 and MNcl2, respectively; 58.7% in TcIV strain CANIIIcl1; and 52.5% in the TcVI strain LL015P68R0cl4). Even more, in LL014R1 and MNcl2 (TcV strains) the most abundant cluster represented the 29.7% and 17.8% of the total mHVR, respectively. Instead, none of the clusters present in TcI, TcII and TcIII strains represented more than 5.2%. This higher diversity in TcI, TcII and TcIII is also revealed by a higher Simpson diversity index than other DTUs (Table 2). Moreover, intra-DTU differences in mHVR cluster diversity were observed in TcII. Particularly, Tu18cl93 had relatively less cluster diversity than Esmeraldo (Table 2 and Fig 1).
Fig 1. mHVR clusters distributed by size in nine strains.

X-axis represent mHVR clusters ordered by decreasing size. The y-axis indicates the mHVR cluster size (i.e. number of reads in the cluster). The cluster size was standardized assuming a total of 120,000 mHVR sequences per parasite (i.e. the value represents the expected cluster size in a kDNA network with 120,000 mHVRs) in order to compare strains with different sequencing depths. Clusters with more than 6,000 sequences were observed for MNcl2, LL014R1, LL015P68R0cl4 and CANIIIcl1 but the bars were cut at this value in order to a clearer comparison among strains.
Shared and non-shared mHVR clusters at intra- and inter- DTU level
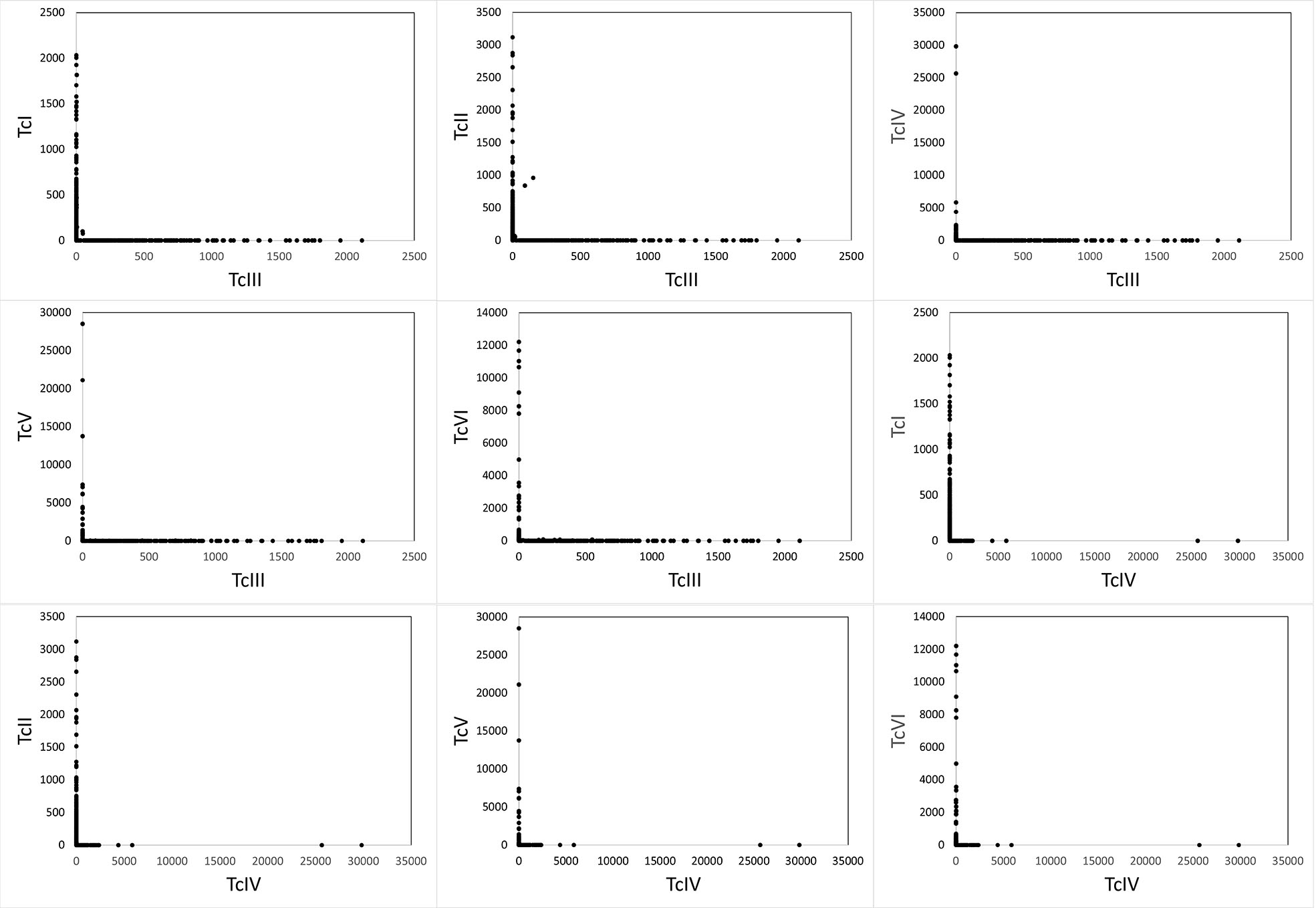
As expected, shared mHVR clusters were mostly observed in strains belonging to the same DTU. However, the percentage of shared clusters was highly variable depending on DTU. TcV strains (LL014R1 and MNcl2) showed the higher proportion of shared clusters (97.3%; 72/74). However, we observed strong differences in the cluster sizes (Fig 2C) although a positive correlation was detected (correlation coefficient, r = 0.75) and some shared clusters were highly abundant in both strains (Fig 2C). TcI strains (PalDa20cl3 and TEV55cl1) shared 17.5% (83/475), and TcII strains (Tu18cl93 and Esmeraldo) shared 7.1% (33/466). Conversely, when we look for shared mHVR clusters between strains belonging to different DTUs, we detected none or few shared clusters (Fig 2D–2I and S2 Fig).
Fig 2. Strains belonging to the same DTU share more abundant mHVR clusters than strains of different DTUs (85% identity threshold).

Each dot in the graph represents a mHVR cluster and the coordinates represent its standardized size in different strains (A-C) and in different DTUs of epidemiologic relevance (D-I). Dots that do not localize in the axes represent shared clusters. mHVR clusters for: TcI (A), TcII (B), TcV (C), TcI vs TcII (D), TcI vs TcV (E), TcI vs TcVI (F), TcII vs TcV (G), TcII vs TcVI (H) and TcV vs TcVI (I).
Strain clustering based on mHVR supports DTU-based classification and supports the hypothesis of biparental inheritance of minicircles in TcV and TcVI
The Bray-Curtis dissimilarity between strains was calculated using mHVR clusters conformed at the different identity thresholds (85%, 90% and 95%). Such dissimilarities were used to analyze principal coordinates (PCoA) and to build UPGMA trees (Fig 3 and S3 Fig). Strains from the same DTU clustered together (Fig 3) despite the high dissimilarities between strains belonging to the same DTU (Fig 3C). These high dissimilarities between strains belonging to the same DTU determine that the three first axis in the PCoA explain just 49.1% of the variance. TcV strains clustered distant from other DTUs. TcIII and TcIV strains clustered near to each other. Interestingly, TcVI strain was placed between TcII and TcIII in the PCoA. Moreover, TcVI was clustered with TcII in the UPGMA tree (Fig 3C). Such results are not in agreement with the hypothesis of uniparental inheritance of the minicircles in the hybrid TcVI, which comes from hybridization between TcII and TcIII. Consequently, we analyzed shared clusters between TcII, TcIII and the hybrids DTUs (TcV and TcVI) in order to analyze the hypotheses of uniparental or biparental inheritance of minicircles. We used a 90% identity threshold in order to be more confident about the identity by descendance of the clusters. We observed that TcV and TcVI share 11/530 and 19/559 mHVR clusters with TcII, respectively. Likewise, TcV and TcVI shared 12/429 and 9/469 mHVR clusters with TcIII, respectively (Fig 4). Instead, TcII and TcIII share only 2 mHVR clusters between them out of a total number of clusters of 842 combining TcII and TcIII. These results suggest that minicircle inheritance is biparental. In addition, TcV and TcVI shared more mHVR clusters with their parental DTUs than between them (Fig 4) which is concordant with the hypothesis of independent origins of TcV and TcVI.
Fig 3. Principal coordinates analysis and UPGMA clustering.

Both analyses were based on the mHVR clusters identified at a threshold of 85% for each strain. (A) 2D graphs combining two out of the three first axes resulting from PCoA. (B) Graph representing the three first axes of the PCoA. (C) Consensus UPGMA based on 10 rarefactions of the mHVR clusters at 820,000 sequences.
Fig 4. Shared mHVR clusters between parental (TcII and TcIII) and hybrids (TcV and TcVI) strains suggest biparental inheritance of minicircles in hybrids.

Each dot represents a shared mHVR cluster (i.e. abundance > 0 in both analyzed DTUs). TcII, combination of Esmeraldo and Tu18cl93. TcIII, X109/2 strain. TcV, combination of clusters of LL014R1 and MNcl2. TcVI, LL015P68R0cl4 strain.
Potential suitability of the amplicon sequencing for NGS-based typing of T. cruzi
In order to test if parallel amplicon sequencing would be useful for simultaneous typing of hundreds of strains, we first evaluated rarefaction curves. In general, the minimum number of reads required to detect at least 95% of the observed clusters was 20,000 filtered reads. The only exception was MNcl2, which required 40,000 filtered reads. Increasing the number of reads per sample beyond 20,000 slightly increased the number of detected mHVR clusters (Fig 5A). In addition, we evaluated the minimum number of reads required to observe the right DTU assignment described in Fig 2. As few as 10,000 reads were enough to accurate clustering of the strains (Fig 5B and 5C) at 100% of the rarefactions.
Fig 5. Suitability of amplicon sequencing of the mHVR for typing.

(A) Proportion of clusters in relation to the maximum observed in Table 2 at different sequencing depths. (B) Principal Coordinates analysis based on Bray-Curtis dissimilarities showing the first three axes at a sequencing depth of 10,000 reads. (C) UPGMA based on Bray-Curtis distance showing relationships among strains at a sequencing depth of 10,000 reads. (D) Correlation between mHVR cluster sizes in two independent PCRs from the strain LL015P68R0cl4 (TcVI).
Amplicon sequencing of the mHVR could be useful to identify intra-DTU clusters, particularly in TcV or TcVI where strains may have the same composition of mHVR clusters but with high differences in abundance of each one. In order to develop future methods to assign strains to intra-DTU clusters is pre-requisite that amplicon sequencing can be reproducible to determine mHVR cluster abundance. Consequently, we assessed reproducibility by sequencing and comparing two independent PCR reactions of the mHVR in LL015P68R0cl4 strain (TcVI). High correlation in cluster abundances in different PCRs of the same sample was observed (r = 0.999 for the three different identity thresholds) (Fig 5D).
Discussion
Here, we made a deep amplicon sequencing of the hypervariable region of kDNA minicircles in the six main lineages (DTUs) of T. cruzi. To the best of our knowledge, this is the first time that these kDNA regions were sequenced at millions of reads of depth. Our results shed light on different and very interesting aspects of these intriguing DNA sequences. We accurately show the level of sequence diversity of mHVR within strains, between strains belonging to the same DTU, and between strains belonging to different DTUs. Although it was already known that mHVR were highly diverse [36], the magnitude of this diversity at the intra- and inter-DTU level has not been demonstrated with the high precision provided by an NGS approach, as we made here.
We propose a method for typing/elucidating intra-specific diversity of T. cruzi based on the deep sequencing of the hypervariable region of kDNA minicircles. The idea is based on the outdated but highly sensitive method of mHVR probes [25, 26, 35, 46–48]. Such probes are useful to detect T. cruzi diversity in biological samples. However, this methodology has the disadvantages of being technically cumbersome, relying on visual interpretation of bands and requiring representative strains of the diversity of T. cruzi in every assay (used as probes). The deep amplicon sequencing approach proposed here is reproducible and based on objective sequence data which can be stored in online databases. Also, the method is multiplexable for hundreds of samples at time and it would be directly applied to biological samples as the mHVR probes. The method may be potentially useful to address epidemiological questions about associations between intra-specific diversity and variability in clinical manifestations of the chronic disease or the different rates of congenital transmission in different endemic areas. Such questions have been unsuccessfully addressed using molecular markers with low resolution and/or low sensitivity on biological samples. We determined that around 20,000 filtered reads are enough to reveal most mHVR diversity in a strain and theoretically allowing for running hundreds of samples in a single run of a MiSeq with costs similar or lower than MLST. However, a wider set of strains belonging to the six main lineages must be studied. In addition, new bioinformatic methods of analysis will be required for a direct application of the method to biological samples.
In order to develop such typing method, we preliminarily analyzed and compared the diversity of mHVR sequences in reference strains of six DTUs and at millions of reads of sequencing depth. We observed that strains of the same DTU share more mHVR clusters than strains of different DTUs. However, unprecedented high differences in mHVR cluster composition was observed for strains of the same DTU with less than 20% of shared mHVR clusters in TcI and TcII. Instead, almost all mHVR clusters were shared between different TcV strains. In addition, the patterns of DTU specificity observed by using mHVR probes may be explained in TcV and TcVI by the presence of some shared and abundant clusters. Instead, considering the higher diversity and low abundance of clusters in TcI, TcII and TcIII, the global pattern of sequences is probably the responsible of specificity in the hybridization assays involving these DTUs.
Interestingly, our data revealed that diversity of mHVR sequences was variable even within a DTU. This was particularly evident in TcII, where the number of mHVR clusters in Esmeraldo strain doubled that of Tu18cl93. Such differences may be caused by long times in culture as it has been observed for other trypanosomatids [40, 49]. However, both strains were isolated in the eighties and although it is possible that they had different times in culture, such times would be not very different (i.e. not in the order of decades). According to this, we suppose that the observed difference in mHVR diversity between the two TcII strains is not due to long time in culture. In support of the hypothesis of no influence of the time in culture, we observed no differences in mHVR diversity between the two TcV strains examined, despite they have very different times of isolation and maintenance mode in the laboratory. One of them was isolated in the 1980s and subjected to long periods of maintenance in culture (Mncl2); and the other TcV strain (LL014R1) was isolated in 2008 and maintained in triatomine-mouse passages.
Our results also shed some light on the evolutionary mechanism determining the large genetic distances in mHVR sequences among strains and DTUs. The focus should be first placed on TcV strains which are identical according to MLST and which shared most mHVR clusters. Despite this, they strongly varied in relative frequencies of mHVR clusters. Such variations cannot be attributed to simple stochasticity of the PCR amplification because we observed good correlation between different PCR reactions from the same sample (Fig 5D). Consequently, it is probable that minicircle diversity is mainly driven by genetic drift. We propose that when two strains diverge, the frequencies of mHVR cluster varies stochastically, some clusters increasing their relative frequency and other decreasing it. The next step can be seen in strains of TcI which are more genetically distant than the TcV ones. Such TcI strains show clusters with high abundance in one strain and with very low (or null) abundance in the other one (look at most clusters located on the axes in Fig 2). Therefore, some clusters will be lost if such lost is not deleterious (i.e. replaced by a different mHVR class that codes a gRNA editing the same mRNA fragment). Thus, strains would diverge by variations in frequency of the mHVR classes faster than by changes in their sequences. These variations in the frequency of mHVR classes probably are not under selective pressure. mHVR frequency variations are apparently allowed because the effective edition of the mRNA is not dependent on the abundance of a minicircle [50, 51]. Variations in the frequency of mHVR classes have been also inferred for T. brucei and Leishmania [52] and by a theoretical study assuming random or partially random segregation of minicircles [53].
With the purpose of developing in the future DTU specific PCRs, we analyzed if different DTUs share common mHVR clusters. Telleria et al. [36] did not detected shared sequences between DTUs probably because the low sequencing depth. With a different approach, Velazquez et al. [37] detected that most abundant mHVR classes in CL-Brener (TcVI) were also present in other DTUs but in a considerably lower frequency. We detected shared mHVRs between different DTUs but we did not detect any sequence shared by the six DTUs. Interestingly, we observed shared clusters between TcVI and TcIII (2.1%). This is expected considering that TcIII is a parental DTU of the hybrid TcVI and maxicircle sequences of TcIII are closely related to the TcVI ones [54–58]. However, the TcVI strain also shared 2.5% of mHVR clusters with Esmeraldo strain (belonging to TcII, the other parental DTU of TcVI). Something similar is observed for the also hybrid DTU TcV (Fig 3). Instead, only 2 mHVR clusters were shared between TcII and TcIII strains (0.2%). This clearly suggests that although maxicircles have apparently uniparental inheritance in TcV and TcVI, minicircles were probably inherited from both parentals and some of them persisted for 60,000 years since hybridization [59]. Biparental inheritance of minicircles and maxicircles has been proposed for Trypanosoma brucei hybrids [60–62]. In this parasite, it has been observed that maxicircle and minicircle inheritance is biparental in hybrids. However, maxicircles (20–50 copies) are homogenized by genetic drift resulting in the loss of whole maxicircles of one parental in few generations. However, minicircles have much more copies and they resist the fixation effect of genetic drift for more time. Consequently, maxicircle inheritance is biparental and just seems to be uniparental due to genetic drift. As consequence of the biparental inheritance of minicircles, it has been proposed that such inheritance may help to preserve mHVR diversity in T. brucei preventing the effect of the drift, and even that T. brucei requires genetic exchange to prevent the deleterious effect of loss of essential minicircle classes [53]. Nevertheless, genetic exchange has remained elusive to be detected in T. cruzi. Experimental hybrids obtained by Gaunt and coworkers showed that maxicircles are from one parental but minicircles were not analyzed [63] and kDNA inheritance was still not addressed in more recent experimental hybrids [64]. In addition, the frequency of genetic exchange may be variable among different DTUs. TcV and TcVI (which display a clearly clonal genetic structure at population level) [9, 10, 12, 57] have very low mHVR diversity. Instead, TcI, TcII and TcIII, for which genetic exchange has been proposed in the nature [11, 13, 15, 65], have higher mHVR diversity.
Moreover, our data may help elucidate the origin of hybrid DTUs. It has been proposed that TcV and TcVI are the result of a single hybridization event between TcII and TcIII and both DTUs diverged posteriorly [66, 67]. However, the alternative hypothesis (two independent hybridization) gain weight in the last years. Particularly, Multilocus Microsatellite Typing (MLMT) and Multilocus Sequence Typing (MLST) analyses favored the two independent hybridizations hypothesis [57, 59]. Considering biparental inheritance, and assuming a single hybridization event, the two hybrid DTUs (TcV and TcVI) should share more mHVR classes between them than with the parentals. However, our analyses show the contrary with very few classes shared between TcV and TcVI (Fig 4). This result supports independent hybridizations for the origin of TcV and TcVI. Alternatively, because both DTUs would have lost many mHVR clusters, the high divergence among them may have been caused by simple stochasticity, although is less likely. Interestingly, if minicircle are biparentally inherited it is expected that they will behave like the nuclear genes. So, it is expected that nuclear phylogenies will be similar to the mHVR phylogeny and both discordant to maxicircle phylogeny in cases of hybridization or introgression. However, some hypotheses about events that occurred very distant in time (e.g. mitochondrial introgression in the origin of TcIII [57–58]) might not be addressed by mHVR-based phylogenies because the almost null number of shared mHVR clusters between some DTUs.
Concluding, massive amplicon sequencing of the mHVR is reproducible and suitable for typing hundreds of T. cruzi strains at time because few thousands of reads are required per sample. However, some drawbacks still need solution. The main problem in biological samples are mixed infections of different genotypes or DTUs which are very frequent [48]. However, such problem can be overpassed by developing new bioinformatic methods comparing mHVR composition of a sample against a reference mHVR database which should collect information about the diversity in the DTUs of T. cruzi. In addition, the develop of an online database where mHVR representative sequences are stored is needed. We are currently working on such items. In addition, some rare events of mitochondrial introgression observed in natural populations of T. cruzi lead to discordant typing between nuclear and maxicircle markers [16, 68, 69]. However, it is unknown the effect of mitochondrial introgression on minicircles. In this sense, a Multilocus deep Sequence Typing (MLdST) may be good alternative and a second step. The deep sequencing of amplicons of the mHVR plus satDNA (a 195 bp sequence with 105 sequences per genome) [70] may help elucidate such rare events and may increase sensitivity for typing on biological samples.
Supporting information
(PDF)
(PDF)
(PDF)
One representative mHVR sequence from each cluster was selected and aligned using MEGA v7 with default parameters. Uncorrected p-distances were used, and gaps were ignored in pairwise comparisons. Value above branches indicates their length and values under branches indicates their support calculated by 100 bootstrap replications.
(TIFF)
Dots that do not localize in the axes represent shared clusters. The axis scales are different among Figs and they were set according the mHVR cluster with higher number of sequences.
(JPG)
{kind=link}
(JPG)
{kind=link}
Acknowledgments
We would like to thanks Pablo Alfredo Vera, Veronica Nishinakamasu and Marianne Muñoz (Instituto de Biotecnología, Centro de Investigaciones en Ciencias Agronómicas y Veterinarias, Instituto Nacional de Tecnología Agropecuaria) for their technical assistance in this project.
Data Availability
The raw data set has been deposited in the NCBI SRA database (BioProject ID: PRJNA514922) with the accession codes SAMN10737665, SAMN10737666, SAMN10737667, SAMN10737668, SAMN10737669, SAMN10737670, SAMN10737671, SAMN10737672 and SAMN10737673.
Funding Statement
The current study is funded by Bunge and Born foundation and the National Scientific and Technical Research Council (CONICET, Argentina) to Patricio Diosque. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bonney KM. Chagas disease in the 21st century: a public health success or an emerging threat? Parasite. 2014;21:11 10.1051/parasite/2014012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zingales B, Andrade SG, Briones MR, Campbell DA, Chiari E, Fernandes O, et al. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz. 2009;104(7):1051–4. Epub 2009/12/23. S0074-02762009000700021 [pii]. 10.1590/s0074-02762009000700021 . [DOI] [PubMed] [Google Scholar]
- 3.Zingales B, Miles MA, Campbell DA, Tibayrenc M, Macedo AM, Teixeira MM, et al. The revised Trypanosoma cruzi subspecific nomenclature: rationale, epidemiological relevance and research applications. Infect Genet Evol. 2012;12(2):240–53. Epub 2012/01/10. S1567-1348(11)00456-4 [pii] 10.1016/j.meegid.2011.12.009 . [DOI] [PubMed] [Google Scholar]
- 4.Lima L, Espinosa-Alvarez O, Ortiz PA, Trejo-Varon JA, Carranza JC, Pinto CM, et al. Genetic diversity of Trypanosoma cruzi in bats, and multilocus phylogenetic and phylogeographical analyses supporting Tcbat as an independent DTU (discrete typing unit). Acta Trop. 2015;151:166–77. Epub 2015/07/23. 10.1016/j.actatropica.2015.07.015 S0001-706X(15)30063-2 [pii]. . [DOI] [PubMed] [Google Scholar]
- 5.Marcili A, Lima L, Cavazzana M, Junqueira AC, Veludo HH, Maia Da Silva F, et al. A new genotype of Trypanosoma cruzi associated with bats evidenced by phylogenetic analyses using SSU rDNA, cytochrome b and Histone H2B genes and genotyping based on ITS1 rDNA. Parasitology. 2009;136(6):641–55. Epub 2009/04/17. 10.1017/S0031182009005861 S0031182009005861 [pii]. . [DOI] [PubMed] [Google Scholar]
- 6.Pinto CM, Kalko EK, Cottontail I, Wellinghausen N, Cottontail VM. TcBat a bat-exclusive lineage of Trypanosoma cruzi in the Panama Canal Zone, with comments on its classification and the use of the 18S rRNA gene for lineage identification. Infect Genet Evol. 2012;12(6):1328–32. Epub 2012/05/01. 10.1016/j.meegid.2012.04.013 S1567-1348(12)00145-1 [pii]. . [DOI] [PubMed] [Google Scholar]
- 7.Barnabe C, Brisse S, Tibayrenc M. Population structure and genetic typing of Trypanosoma cruzi, the agent of Chagas disease: a multilocus enzyme electrophoresis approach. Parasitology. 2000;120 (Pt 5):513–26. Epub 2000/06/07. . [DOI] [PubMed] [Google Scholar]
- 8.Brisse S, Barnabe C, Tibayrenc M. Identification of six Trypanosoma cruzi phylogenetic lineages by random amplified polymorphic DNA and multilocus enzyme electrophoresis. Int J Parasitol. 2000;30(1):35–44. Epub 2000/02/17. S0020-7519(99)00168-X [pii]. . [DOI] [PubMed] [Google Scholar]
- 9.Diosque P, Tomasini N, Lauthier JJ, Messenger LA, Monje Rumi MM, Ragone PG, et al. Optimized Multilocus Sequence Typing (MLST) Scheme for Trypanosoma cruzi. PLoS Negl Trop Dis. 2014;8(8):e3117 10.1371/journal.pntd.0003117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lauthier JJ, Tomasini N, Barnabe C, Rumi MM, D'Amato AM, Ragone PG, et al. Candidate targets for Multilocus Sequence Typing of Trypanosoma cruzi: validation using parasite stocks from the Chaco Region and a set of reference strains. Infect Genet Evol. 2012;12(2):350–8. Epub 2012/01/03. S1567-1348(11)00455-2 [pii] 10.1016/j.meegid.2011.12.008 . [DOI] [PubMed] [Google Scholar]
- 11.Tomasini N, Lauthier JJ, Monje Rumi MM, Ragone PG, Alberti D'Amato AM, Brandan CP, et al. Preponderant clonal evolution of Trypanosoma cruzi I from Argentinean Chaco revealed by Multilocus Sequence Typing (MLST). Infect Genet Evol. 2014;27C:348–54. 10.1016/j.meegid.2014.08.003 . [DOI] [PubMed] [Google Scholar]
- 12.Yeo M, Mauricio IL, Messenger LA, Lewis MD, Llewellyn MS, Acosta N, et al. Multilocus sequence typing (MLST) for lineage assignment and high resolution diversity studies in Trypanosoma cruzi. PLoS Negl Trop Dis. 2011;5(6):e1049 Epub 2011/06/30. 10.1371/journal.pntd.0001049 10-PNTD-RA-1407R2 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Llewellyn MS, Lewis MD, Acosta N, Yeo M, Carrasco HJ, Segovia M, et al. Trypanosoma cruzi IIc: phylogenetic and phylogeographic insights from sequence and microsatellite analysis and potential impact on emergent Chagas disease. PLoS Negl Trop Dis. 2009;3(9):e510 Epub 2009/09/02. 10.1371/journal.pntd.0000510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llewellyn MS, Miles MA, Carrasco HJ, Lewis MD, Yeo M, Vargas J, et al. Genome-scale multilocus microsatellite typing of Trypanosoma cruzi discrete typing unit I reveals phylogeographic structure and specific genotypes linked to human infection. PLoS Pathog. 2009;5(5):e1000410 Epub 2009/05/05. 10.1371/journal.ppat.1000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macedo AM, Pimenta JR, Aguiar RS, Melo AI, Chiari E, Zingales B, et al. Usefulness of microsatellite typing in population genetic studies of Trypanosoma cruzi. Mem Inst Oswaldo Cruz. 2001;96(3):407–13. Epub 2001/04/21. S0074-02762001000300023 [pii]. 10.1590/s0074-02762001000300023 . [DOI] [PubMed] [Google Scholar]
- 16.Messenger LA, Llewellyn MS, Bhattacharyya T, Franzen O, Lewis MD, Ramirez JD, et al. Multiple mitochondrial introgression events and heteroplasmy in Trypanosoma cruzi revealed by maxicircle MLST and next generation sequencing. PLoS Negl Trop Dis. 2012;6(4):e1584 Epub 2012/04/17. 10.1371/journal.pntd.0001584 PNTD-D-11-00969 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ocana-Mayorga S, Llewellyn MS, Costales JA, Miles MA, Grijalva MJ. Sex, subdivision, and domestic dispersal of Trypanosoma cruzi lineage I in southern Ecuador. PLoS Negl Trop Dis. 2010;4(12):e915 Epub 2010/12/24. 10.1371/journal.pntd.0000915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliveira RP, Broude NE, Macedo AM, Cantor CR, Smith CL, Pena SD. Probing the genetic population structure of Trypanosoma cruzi with polymorphic microsatellites. Proc Natl Acad Sci U S A. 1998;95(7):3776–80. Epub 1998/05/09. 10.1073/pnas.95.7.3776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brisse S, Verhoef J, Tibayrenc M. Characterisation of large and small subunit rRNA and mini-exon genes further supports the distinction of six Trypanosoma cruzi lineages. Int J Parasitol. 2001;31(11):1218–26. Epub 2001/08/22. S0020-7519(01)00238-7 [pii]. . [DOI] [PubMed] [Google Scholar]
- 20.Burgos JM, Diez M, Vigliano C, Bisio M, Risso M, Duffy T, et al. Molecular identification of Trypanosoma cruzi discrete typing units in end-stage chronic Chagas heart disease and reactivation after heart transplantation. Clin Infect Dis. 2010;51(5):485–95. Epub 2010/07/22. 10.1086/655680 . [DOI] [PubMed] [Google Scholar]
- 21.Falla A, Herrera C, Fajardo A, Montilla M, Vallejo GA, Guhl F. Haplotype identification within Trypanosoma cruzi I in Colombian isolates from several reservoirs, vectors and humans. Acta Trop. 2009;110(1):15–21. Epub 2009/01/13. 10.1016/j.actatropica.2008.12.003 S0001-706X(08)00350-1 [pii]. . [DOI] [PubMed] [Google Scholar]
- 22.Cosentino RO, Aguero F. A simple strain typing assay for Trypanosoma cruzi: discrimination of major evolutionary lineages from a single amplification product. PLoS Negl Trop Dis. 2012;6(7):e1777 Epub 2012/08/04. 10.1371/journal.pntd.0001777 PNTD-D-11-00534 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgos JM, Altcheh J, Bisio M, Duffy T, Valadares HM, Seidenstein ME, et al. Direct molecular profiling of minicircle signatures and lineages of Trypanosoma cruzi bloodstream populations causing congenital Chagas disease. Int J Parasitol. 2007;37(12):1319–27. Epub 2007/06/16. S0020-7519(07)00138-5 [pii] 10.1016/j.ijpara.2007.04.015 . [DOI] [PubMed] [Google Scholar]
- 24.Venegas J, Conoepan W, Pichuantes S, Miranda S, Apt W, Arribada A, et al. Differential distribution of Trypanosoma cruzi clones in human chronic chagasic cardiopathic and non-cardiopathic individuals. Acta Trop. 2009;109(3):187–93. Epub 2008/12/09. 10.1016/j.actatropica.2008.11.007 S0001-706X(08)00325-2 [pii]. . [DOI] [PubMed] [Google Scholar]
- 25.Diez C, Lorenz V, Ortiz S, Gonzalez V, Racca A, Bontempi I, et al. Genotyping of Trypanosoma cruzi sublineage in human samples from a North-East Argentina area by hybridization with DNA probes and specific polymerase chain reaction (PCR). Am J Trop Med Hyg. 2010;82(1):67–73. 10.4269/ajtmh.2010.09-0391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rumi MM, Perez Brandan C, Gil JF, D'Amato AM, Ragone PG, Lauthier JJ, et al. Benznidazole treatment in chronic children infected with Trypanosoma cruzi: serological and molecular follow-up of patients and identification of Discrete Typing Units. Acta Trop. 2013;128(1):130–6. 10.1016/j.actatropica.2013.07.003 . [DOI] [PubMed] [Google Scholar]
- 27.Llewellyn MS, Messenger LA, Luquetti AO, Garcia L, Torrico F, Tavares SB, et al. Deep sequencing of the Trypanosoma cruzi GP63 surface proteases reveals diversity and diversifying selection among chronic and congenital Chagas disease patients. PLoS Negl Trop Dis. 2015;9(4):e0003458 10.1371/journal.pntd.0003458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deep sequencing reveals multiclonality and new discrete typing units of Trypanosoma cruzi in rodents from the southern United States. Journal of Microbiology, Immunology and Infection. 2018. 10.1016/j.jmii.2018.12.004. PRONOVOST2018. [DOI] [PubMed] [Google Scholar]
- 29.Schijman AG, Bisio M, Orellana L, Sued M, Duffy T, Mejia Jaramillo AM, et al. International study to evaluate PCR methods for detection of Trypanosoma cruzi DNA in blood samples from Chagas disease patients. PLoS Negl Trop Dis. 2011;5(1):e931 Epub 2011/01/26. 10.1371/journal.pntd.0000931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simpson L. The mitochondrial genome of kinetoplastid protozoa: genomic organization, transcription, replication, and evolution. Annu Rev Microbiol. 1987;41:363–82. Epub 1987/01/01. 10.1146/annurev.mi.41.100187.002051 . [DOI] [PubMed] [Google Scholar]
- 31.Lukes J, Guilbride DL, Votypka J, Zikova A, Benne R, Englund PT. Kinetoplast DNA network: evolution of an improbable structure. Eukaryot Cell. 2002;1(4):495–502. 10.1128/EC.1.4.495-502.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degrave W, Fragoso SP, Britto C, van Heuverswyn H, Kidane GZ, Cardoso MA, et al. Peculiar sequence organization of kinetoplast DNA minicircles from Trypanosoma cruzi. Mol Biochem Parasitol. 1988;27(1):63–70. Epub 1988/01/01. 0166-6851(88)90025-4 [pii]. . [DOI] [PubMed] [Google Scholar]
- 33.Veas F, Cuny G, Breniere SF, Tibayrenc M. Subspecific kDNA probes for major clones of Trypanosoma cruzi. Acta Trop. 1990;48(1):79–82. . [DOI] [PubMed] [Google Scholar]
- 34.Breniere SF, Bosseno MF, Revollo S, Rivera MT, Carlier Y, Tibayrenc M. Direct identification of Trypanosoma cruzi natural clones in vectors and mammalian hosts by polymerase chain reaction amplification. Am J Trop Med Hyg. 1992;46(3):335–41. 10.4269/ajtmh.1992.46.335 . [DOI] [PubMed] [Google Scholar]
- 35.Solari A, Campillay R, Ortiz S, Wallace A. Identification of Trypanosoma cruzi genotypes circulating in Chilean chagasic patients. Exp Parasitol. 2001;97(4):226–33. 10.1006/expr.2001.4607 . [DOI] [PubMed] [Google Scholar]
- 36.Telleria J, Lafay B, Virreira M, Barnabe C, Tibayrenc M, Svoboda M. Trypanosoma cruzi: sequence analysis of the variable region of kinetoplast minicircles. Exp Parasitol. 2006;114(4):279–88. 10.1016/j.exppara.2006.04.005 . [DOI] [PubMed] [Google Scholar]
- 37.Velazquez M, Diez CN, Mora C, Diosque P, Marcipar IS. Trypanosoma cruzi: an analysis of the minicircle hypervariable regions diversity and its influence on strain typing. Exp Parasitol. 2008;120(3):235–41. 10.1016/j.exppara.2008.07.016 . [DOI] [PubMed] [Google Scholar]
- 38.Zimmer SL, Simpson RM, Read LK. High throughput sequencing revolution reveals conserved fundamentals of U-indel editing. Wiley Interdisciplinary Reviews: RNA. 2018;9(5):e1487 10.1002/wrna.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hajduk S, Ochsenreiter T. RNA editing in kinetoplastids. RNA biology. 2010;7(2):229–36. 10.4161/rna.7.2.11393 . [DOI] [PubMed] [Google Scholar]
- 40.Simpson L, Douglass SM, Lake JA, Pellegrini M, Li F. Comparison of the Mitochondrial Genomes and Steady State Transcriptomes of Two Strains of the Trypanosomatid Parasite, Leishmania tarentolae. PLoS Negl Trop Dis. 2015;9(7):e0003841 10.1371/journal.pntd.0003841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. Epub 2014/04/04. 10.1093/bioinformatics/btu170 btu170 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renaud G, Stenzel U, Kelso J. leeHom: adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res. 2014;42(18):e141 Epub 2014/08/08. 10.1093/nar/gku699 gku699 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr Protoc Microbiol. 2012;Chapter 1:Unit 1E 5. Epub 2012/11/28. 10.1002/9780471729259.mc01e05s27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1. 10.1093/bioinformatics/btq461 . [DOI] [PubMed] [Google Scholar]
- 45.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4516–22. 10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solari A, Munoz S, Venegas J, Wallace A, Aguilera X, Apt W, et al. Characterization of Chilean, Bolivian, and Argentinian Trypanosoma cruzi populations by restriction endonuclease and isoenzyme analysis. Exp Parasitol. 1992;75(2):187–95. . [DOI] [PubMed] [Google Scholar]
- 47.Breniere SF, Bosseno MF, Telleria J, Carrasco R, Vargas F, Yaksic N, et al. Field application of polymerase chain reaction diagnosis and strain typing of Trypanosoma cruzi in Bolivian triatomines. Am J Trop Med Hyg. 1995;53(2):179–84. 10.4269/ajtmh.1995.53.179 . [DOI] [PubMed] [Google Scholar]
- 48.Monje-Rumi MM, Brandan CP, Ragone PG, Tomasini N, Lauthier JJ, Alberti D'Amato AM, et al. Trypanosoma cruzi diversity in the Gran Chaco: Mixed infections and differential host distribution of TcV and TcVI. Infect Genet Evol. 2015;29(0):53–9. 10.1016/j.meegid.2014.11.001 . [DOI] [PubMed] [Google Scholar]
- 49.Thiemann OH, Maslov DA, Simpson L. Disruption of RNA editing in Leishmania tarentolae by the loss of minicircle-encoded guide RNA genes. EMBO J. 1994;13(23):5689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maslov DA, Simpson L. The polarity of editing within a multiple gRNA-mediated domain is due to formation of anchors for upstream gRNAs by downstream editing. Cell. 1992;70(3):459–67. . [DOI] [PubMed] [Google Scholar]
- 51.Arts GJ, van der Spek H, Speijer D, van den Burg J, van Steeg H, Sloof P, et al. Implications of novel guide RNA features for the mechanism of RNA editing in Crithidia fasciculata. EMBO J. 1993;12(4):1523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simpson L. The genomic organization of guide RNA genes in kinetoplastid protozoa: several conundrums and their solutions. Mol Biochem Parasitol. 1997;86(2):133–41. . [DOI] [PubMed] [Google Scholar]
- 53.Savill NJ, Higgs PG. A theoretical study of random segregation of minicircles in trypanosomatids. Proc Biol Sci. 1999;266(1419):611–20. 10.1098/rspb.1999.0680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Machado CA, Ayala FJ. Nucleotide sequences provide evidence of genetic exchange among distantly related lineages of Trypanosoma cruzi. Proc Natl Acad Sci U S A. 2001;98(13):7396–401. Epub 2001/06/21. 10.1073/pnas.121187198 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flores-Lopez CA, Machado CA. Analyses of 32 loci clarify phylogenetic relationships among Trypanosoma cruzi lineages and support a single hybridization prior to human contact. PLoS Negl Trop Dis. 2011;5(8):e1272 Epub 2011/08/11. 10.1371/journal.pntd.0001272 PNTD-D-10-00294 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Freitas JM, Augusto-Pinto L, Pimenta JR, Bastos-Rodrigues L, Goncalves VF, Teixeira SM, et al. Ancestral genomes, sex, and the population structure of Trypanosoma cruzi. PLoS Pathog. 2006;2(3):e24 Epub 2006/04/13. 10.1371/journal.ppat.0020024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tomasini N, Diosque P. Evolution of Trypanosoma cruzi: clarifying hybridisations, mitochondrial introgressions and phylogenetic relationships between major lineages. Mem Inst Oswaldo Cruz. 2015;110(3):403–13. 10.1590/0074-02760140401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tomasini N. Introgression of the Kinetoplast DNA: An Unusual Evolutionary Journey in Trypanosoma cruzi. Curr Genomics. 2018;19(2):133–9. Epub 2018/03/02. 10.2174/1389202918666170815124832 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewis MD, Llewellyn MS, Yeo M, Acosta N, Gaunt MW, Miles MA. Recent, independent and anthropogenic origins of Trypanosoma cruzi hybrids. PLoS Negl Trop Dis. 2011;5(10):e1363 Epub 2011/10/25. 10.1371/journal.pntd.0001363 PNTD-D-11-00039 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turner CM, Hide G, Buchanan N, Tait A. Trypanosoma brucei: inheritance of kinetoplast DNA maxicircles in a genetic cross and their segregation during vegetative growth. Exp Parasitol. 1995;80(2):234–41. Epub 1995/03/01. S0014-4894(85)71029-6 [pii] 10.1006/expr.1995.1029 . [DOI] [PubMed] [Google Scholar]
- 61.Gibson W, Peacock L, Ferris V, Williams K, Bailey M. The use of yellow fluorescent hybrids to indicate mating in Trypanosoma brucei. Parasit Vectors. 2008;1(1):4 Epub 2008/02/27. 10.1186/1756-3305-1-4 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibson W, Garside L. Kinetoplast DNA minicircles are inherited from both parents in genetic hybrids of Trypanosoma brucei. Mol Biochem Parasitol. 1990;42(1):45–53. Epub 1990/08/01. 0166-6851(90)90111-X [pii]. . [DOI] [PubMed] [Google Scholar]
- 63.Gaunt MW, Yeo M, Frame IA, Stothard JR, Carrasco HJ, Taylor MC, et al. Mechanism of genetic exchange in American trypanosomes. Nature. 2003;421(6926):936–9. Epub 2003/02/28. 10.1038/nature01438 nature01438 [pii]. . [DOI] [PubMed] [Google Scholar]
- 64.Alves CL, Repoles BM, da Silva MS, Mendes IC, Marin PA, Aguiar PHN, et al. The recombinase Rad51 plays a key role in events of genetic exchange in Trypanosoma cruzi. Scientific reports. 2018;8(1):13335 10.1038/s41598-018-31541-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baptista Rde P, D'Avila DA, Segatto M, do Valle IF, Franco GR, Valadares HM, et al. Evidence of substantial recombination among Trypanosoma cruzi II strains from Minas Gerais. Infect Genet Evol. 2014;22:183–91. 10.1016/j.meegid.2013.11.021 . [DOI] [PubMed] [Google Scholar]
- 66.Sturm NR, Vargas NS, Westenberger SJ, Zingales B, Campbell DA. Evidence for multiple hybrid groups in Trypanosoma cruzi. Int J Parasitol. 2003;33(3):269–79. Epub 2003/04/03. S0020751902002643 [pii]. . [DOI] [PubMed] [Google Scholar]
- 67.Westenberger SJ, Barnabe C, Campbell DA, Sturm NR. Two hybridization events define the population structure of Trypanosoma cruzi. Genetics. 2005;171(2):527–43. Epub 2005/07/07. genetics.104.038745 [pii] 10.1534/genetics.104.038745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roellig DM, Savage MY, Fujita AW, Barnabe C, Tibayrenc M, Steurer FJ, et al. Genetic variation and exchange in Trypanosoma cruzi isolates from the United States. PLoS One. 2013;8(2):e56198 Epub 2013/03/05. 10.1371/journal.pone.0056198 PONE-D-11-18280 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barnabe C, Breniere SF. Scarce events of mitochondrial introgression in Trypanosoma cruzi: new case with a Bolivian strain. Infect Genet Evol. 2012;12(8):1879–83. 10.1016/j.meegid.2012.08.018 . [DOI] [PubMed] [Google Scholar]
- 70.Elias MC, Vargas NS, Zingales B, Schenkman S. Organization of satellite DNA in the genome of Trypanosoma cruzi. Mol Biochem Parasitol. 2003;129(1):1–9. Epub 2003/06/12. S0166685103000549 [pii]. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
(PDF)
One representative mHVR sequence from each cluster was selected and aligned using MEGA v7 with default parameters. Uncorrected p-distances were used, and gaps were ignored in pairwise comparisons. Value above branches indicates their length and values under branches indicates their support calculated by 100 bootstrap replications.
(TIFF)
Dots that do not localize in the axes represent shared clusters. The axis scales are different among Figs and they were set according the mHVR cluster with higher number of sequences.
(JPG)
(JPG)
Data Availability Statement
The raw data set has been deposited in the NCBI SRA database (BioProject ID: PRJNA514922) with the accession codes SAMN10737665, SAMN10737666, SAMN10737667, SAMN10737668, SAMN10737669, SAMN10737670, SAMN10737671, SAMN10737672 and SAMN10737673.