

An antibody targeting the CCHFV GP38 molecule protects adult mice against lethal infection in a severe disease model.
Abstract
Crimean-Congo hemorrhagic fever virus (CCHFV) is an important human pathogen. Limited evidence suggests that antibodies can protect humans against lethal CCHFV disease but the protective efficacy of antibodies has never been evaluated in adult animal models. Here, we used adult mice to investigate the protection provided against CCHFV infection by glycoprotein-targeting neutralizing and non-neutralizing monoclonal antibodies (mAbs). We identified a single non-neutralizing antibody (mAb-13G8) that protected adult type I interferon–deficient mice >90% when treatment was initiated before virus exposure and >60% when administered after virus exposure. Neutralizing antibodies known to protect neonatal mice from lethal CCHFV infection failed to confer protection regardless of immunoglobulin G subclass. The target of mAb-13G8 was identified as GP38, one of multiple proteolytically cleaved glycoproteins derived from the CCHFV glycoprotein precursor polyprotein. This study reveals GP38 as an important antibody target for limiting CCHFV pathogenesis and lays the foundation to develop immunotherapeutics against CCHFV in humans.
INTRODUCTION
Crimean-Congo hemorrhagic fever virus (CCHFV) is an enveloped virus in the Nairoviridae family [for a review, see (1, 2)] that is spread in nature by ticks, primarily those of the genus Hyalomma. CCHFV infects a large number of wild and domesticated mammalian species, including bovines and ovines, in addition to some avian species such as ostriches. Infections in these animals are predominantly asymptomatic but can produce a prolonged (>5 days) viremia (3, 4). CCHFV infection in humans, caused by tick bites, exposure to infected animals, or nosocomial infections, can lead to an acute and potentially life-threatening disease termed Crimean-Congo hemorrhagic fever (CCHF) (1, 5). Infection is characterized as a febrile illness with varying degrees of coagulopathy, liver injury, neurological manifestations, respiratory distress, lymphocytopenia, and thrombocytopenia (2). The mortality rate ranges from 3 to 80%, and this large range is theorized to depend on multiple factors including viral strain, route of exposure, speed of diagnosis, and access to emergency health care. There are currently no U.S. Food and Drug Administration–approved drugs to treat CCHF, although there is conflicting evidence that ribavirin protects against lethal human disease (6, 7).
Passive antibody protection has been used in humans to protect against several hemorrhagic fever viruses, including New World arenaviruses and filoviruses (8, 9). Antibody-based therapies composed of human survivor plasma have been used to treat CCHFV-infected humans since the mid-20th century (10–12). CCHF-bulin and CCHF-venin, both produced from plasma of convalescent patients, have been used in Bulgaria (12). These products are delivered intramuscularly or intravenously, respectively. While some evidence suggests that these products can protect against CCHFV, thus far, only a limited number of people have been treated, and controls have not been used to verify the results. Human convalescent serum was used in Dubai during a small nosocomial outbreak, and the five patients receiving the product survived, but two other patients were left untreated and succumbed to disease. These data suggest that antibody therapies can protect against lethality (11). However, other studies have indicated that antibody therapy offers little protective efficacy (10). In general, passive immunotherapy against CCHFV in humans has produced mixed results, with some studies demonstrating protective efficacy and others suggesting that it is not protective. A major issue is the lack of statistical evidence that these therapeutic options are efficacious due to the limited number of cases where patients were treated. Overall, use of convalescent plasma, serum, or purified antibodies has been essentially abandoned because of safety issues regarding human convalescent products and the poorly defined nature of the products.
CCHFV has a tripartite, negative-sense RNA genome consisting of small (S), medium (M), and large (L) segments. The S segment encodes the nucleocapsid (N) protein, and the L segment encodes the RNA-dependent RNA polymerase. The CCHFV glycoproteins encoded by the M segment are expressed as a precursor polyprotein that is proteolytically cleaved along the secretory pathway and eventually produces the two major structural glycoproteins GN and GC, the latter being the only known target of neutralization (13–15). Before the production of the mature proteins, proteolytic processing generates two intermediate molecules termed pre-GN and pre-GC. Pre-GN is further processed by proteases to generate GN and several secreted proteins of unknown function (GP160, GP85, and GP38) and an uncharacterized mucin-like domain (13–17). A panel of murine monoclonal antibodies (mAbs) was produced against the CCHFV strain IbAr 10200, and several of these antibodies target the pre-GN complex or the GC protein. Many of the antibodies targeting GC have neutralizing activity (18). Bertolotti-Ciarlet et al. (18) demonstrated that both the non-neutralizing and neutralizing mAbs protect neonatal mice from lethality. Unfortunately, neonatal mice do not recapitulate CCHF disease, making interpretation of these results difficult. The protective efficacy of glycoprotein-targeting mAbs has never been evaluated in adult animals. Here, we investigated the protective efficacy of these murine mAbs in two relevant adult mouse models of CCHFV infection that more closely recapitulate human viral disease, interferon receptor knockout (KO) mice (IFNAR−/−), and mice in which IFN-I activity was blocked by antibody treatment (19–21). We show that only non-neutralizing antibodies (nNAbs) protected adult mice from CCHFV and that these non-neutralizing mAbs universally targeted the GP38 protein. Our findings demonstrate that antibodies can protect against lethal CCHFV infection in mature animals and provide a basis for further antibody-based CCHFV countermeasure development.
RESULTS
Immunoprotection against lethal CCHFV infection in IFNAR-deficient mice
We investigated whether non-neutralizing and neutralizing murine mAbs reported to target CCHFV pre-GN or GC, respectively (18), protected adult IFNAR−/− mice from lethal infection. We injected groups of mice (n = 10 per group) by the intraperitoneal route with 1 mg of total concentration of the indicated anti-CCHFV antibodies or phosphate-buffered saline (PBS) as a control, 1 day before virus exposure (Fig. 1A). Subsequently, mice were infected with 100 plaque forming units (PFU) of CCHFV strain IbAr 10200, and weight and survival was monitored for 24 days. On day 3, negative control mice began to lose weight, and all mice succumbed to infection by day 5. In contrast, mice treated with mAb-13G8 had less weight loss that was delayed compared to PBS-treated mice. In this group, 6 of 10 mice survived, and the mean time to death (MTD) was significantly delayed compared to the control group (log rank, P < 0.0001). The neutralizing antibody mAb-8A1, which targets GC and robustly neutralizes virus in vitro (18), did not provide significant protection (log rank, P = 0.6004). A combination of mAb-13G8 and mAb-8A1, 0.5 mg of each antibody, provided modest protection as measured by delayed weight loss and a significant delay in the MTD (log rank, P < 0.0001). However, in this group, only a single mouse survived to day 24.
Fig. 1. mAb-13G8 protects against lethal disease in IFNAR−/−mice.
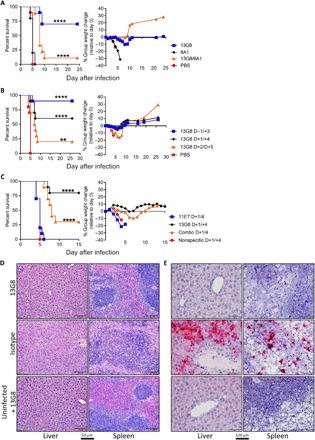
(A) IFNAR−/− mice (n = 10 per group) were infected with CCHFV strain IbAr 10200 by the subcutaneous route. Mice were injected with mAb-13G8, mAb-8A1, or a combination of the two on day −1 (1 mg of total mAb concentration) by the intraperitoneal route. As a control, mice were treated with PBS. Survival and percent group weight change were recorded. Log-rank test, ****P < 0.0001. (B) IFNAR−/− mice (n = 10 per group) were infected with virus as in (A). Mice were treated with two doses of the mAb-13G8 (1 mg per dose) or PBS on days −1/+3, +1/+4, or + 2/+5, and survival and weight were monitored. Log-rank test, ****P < 0.0001, **P = 0.0030. (C) IFNAR−/− mice (n = 10 per group) were infected as in (A). Mice were treated with two doses of mAb-13G8, mAb-11E7, or a combination of both (1 mg of total mAb concentration per dose), and survival and weight were monitored. A nonspecific murine antibody was used as a control. Log-rank test, ****P < 0.0001. (D) Hematoxylin and eosin staining of the livers and spleens from mice infected with CCHFV strain IbAr 10200 and treated with mAb-13G8 or an isotype control antibody. Uninfected mice treated with mAb-13G8 serve as a negative control. (E) In situ hybridization (ISH) staining showing the presence of CCHFV RNA (red) in the liver and spleen of mice taken on day 4 after virus exposure. Cells were counterstained with hematoxylin.
We next determined whether treating mice with multiple doses of mAb-13G8 would increase the protective efficacy. In addition, we examined whether mAb-13G8 could protect mice when treatment was initiated after virus exposure. In this experiment, we treated mice (n = 10 per group) twice with mAb-13G8 either on days −1 and +3, days +1 and +4, or days +2 and +5 relative to infection (Fig. 1B). All PBS control–treated mice succumbed to infection by day 5, after a period of weight loss. Mice treated with mAb-13G8 on day −1/+3 exhibited very little weight loss compared to control mice, and 90% of the mice survived, which was significant (log rank, P < 0.0001). Postexposure treatment of mice on day +1/+4 regimen resulted in 60% survival (log rank, P < 0.0001), and overall, this group had reduced weight loss compared to PBS control–treated mice. Treatment with mAb-13G8 on day +2/+5 conferred modest protection with 20% survival, which was significant (log rank, P = 0.0030) over control animals. However, weight loss in this group was not distinct from PBS-treated mice.
We attempted to enhance the postvirus exposure protective efficacy of mAb-13G8 by also treating mice with the neutralizing antibody mAb-11E7 on day +1 and +4. This mAb was included on the basis of previous data, suggesting both pre- and postexposure efficacy (18). Alone, mAb-11E7 failed to protect mice, and the animals succumbed to infection by day 7 (Fig. 1C). Survival was not statistically distinct from isotype control–treated animals (log rank, P = 0.4243). Similar to the results above, 8 of 10 mice (80%) treated with mAb-13G8 alone survived, which was significant (log rank, P < 0.0001), and mice exhibited little weight loss. A combination of mAb-13G8 and mAb-11E7 (0.5 mg of each antibody per dose; 1 mg total per dose) failed to enhance protection over mAb-13G8 alone, and only 3 of 10 mice survived challenge; however, there was a significant delay in MTD (log rank, P > 0.0001) compared to the control group. Mice treated with mAb-13G8 alone had a statistically significant survival advantage compared to the group receiving the combination of antibodies (log rank, P = 0.0353). Together, these findings indicated that mAb-13G8 protects adult mice against lethal infection, even when administered after virus exposure. However, our findings indicated that anti-GC neutralizing antibodies (mAb-11E7 and the more potently neutralizing mAb-8A1) do not provide any protective benefit when given before or after virus challenge.
mAb-13G8 blocks virus spread to the liver and spleen and prevents liver pathology
The primary targets of CCHFV pathogenesis and viral replication in mice are the liver and spleen (19, 20). Therefore, we evaluated the histopathological effects of CCHFV in these tissues when mice (n = 3 per group) were treated with mAb-13G8 or an isotype control antibody on day −1/+3 (Fig. 1D and table S1). Tissues were examined on day 4, at the peak of disease in this model and 24 hours before mice reaching euthanasia criteria (19, 20). Isotype control–treated mice developed hepatic lesions with inflammation and hepatocellular necrosis with extensive hepatocellular degeneration, and necrosis was present in all animals (Fig. 1D). In addition, periportal oval cell hyperplasia and Kupffer cell hypertrophy in the sinusoids were evident in isotype control–treated, infected animals. Some hepatic vessels also contained fibrin deposits. In marked contrast, livers from mAb-13G8–treated, infected mice had no lesions and were indistinguishable from mAb-13G8–treated, uninfected mice. The spleens of isotype control–treated, infected mice had moderate increases in neutrophils and histiocytes in the splenic red pulp (inflammation), and moderate lymphocytolysis in splenic white pulp were also observed (Fig. 1D). In contrast, splenic lesions were absent in mAb-13G8–treated, infected mice. Hepatic and splenic lesions in CCHFV-infected mice correlated with the presence of CCHFV RNA (Fig. 1E). In the spleen, the in situ hybridization (ISH) signal was most prominent in the red pulp and in the marginal zone of the follicles, suggesting that CCHFV was present within marginal zone macrophages and/or dendritic cells (Fig. 1E). In contrast, we did not detect CCHFV in the liver or spleen of mAb-13G8–treated, infected mice. In addition to viral RNA, we also detected N protein in the liver and spleen by immunohistochemistry (fig. S1). These findings demonstrated that mAb-13G8 prevents damage to and limits viral replication in the liver and spleen when treatment is given on day −1/+3.
GP38 is the target of mAb-13G8
The expression of the CCHFV M segment produces a polyprotein precursor that is proteolytically cleaved into multiple glycoproteins, including GN and GC (Fig. 2A). During this cleavage event, a precursor molecule called pre-GN, consisting of the mucin domain, GP38, and GN, is also formed (14, 15). The target of mAb-13G8 is not well characterized, and initial studies indicated that it and other antibodies, such as mAb-10E11, bound the pre-GN complex. Neither mAb-13G8 nor mAb-10E11 bound purified GN ectodomain when assayed by enzyme-linked immunosorbent assay (ELISA), suggesting that GN is not likely a target of these antibodies (Fig. 2B). Next, we evaluated whether GP38 was the target of mAb-13G8. To this end, we developed a capture ELISA using mAb-13G8 as a capture antibody and rabbit polyclonal CCHFV anti–M-segment antibody as a detection antibody (Fig. 2C). As bait, we designed a secreted molecule by creating a construct consisting of the GP38-coding region with a tissue plasminogen activator (tPA) secretion signal, termed tPA-GP3810200. Secreted tPA-GP3810200 in the supernatant of transfected 293T cells was readily detected when mAb-13G8 was used as the capture antibody, but negative control protein was not detected. These interactions were statistically significant [two-way analysis of variance (ANOVA), P < 0.05]. GP38 was also not detectable by a negative control capture antibody (mAb-QC03). In another experiment, we developed M-segment variants in which the mucin domain (ΔMuc) or the mucin and GP38 (ΔMucΔGP38) domains were removed. By Western blot, the expression of the GN and GC proteins from the ΔMucΔGP38 construct was similar to that from the wild-type M segment at 48 hours; however, the expression of these glycoproteins from the ΔMuc construct was greatly reduced (Fig. 2D). Because the expression of GN and GC was not disrupted from the ΔMucΔGP38 construct, we used it to verify that mAb-13G8 binds to GP38 expressed from the M-segment precursor glycoprotein. We transfected 293T cells with full-length M segment, the M-segment ΔMucΔGP38, tPA-GP3810200, and an irrelevant protein (Junin virus GP1). Supernatants from M segment and tPA-GP3810200 transfected cells both interacted with mAb-13G8 (Fig. 2E). However, no interaction occurred with supernatant produced from ΔMucΔGP38 transfected cells or cells expressing the secreted negative control Junin protein. We also produced a tPA-GP38 construct from CCHFV strain Afg09-2990 and termed it tPA-GP38AF09. In this assay, mAb-13G8 interacted with tPA-GP38AF09, but to a lesser extent, compared to tPA-GP38 from strain IbAr 10200. This was statistically significant at several dilutions (two-way ANOVA, P < 0.05). These data indicated that the target of mAb-13G8 was the GP38 product of the M segment.
Fig. 2. mAb-13G8 interacts with the GP38 molecule.
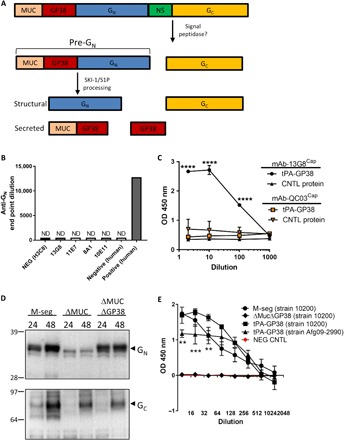
(A) Simplified schematic depicting CCHFV M-segment glycoprotein processing, cleaved by signal peptidases such as subtilisin kexin isozyme-1/site-1 protease (SKI-1/S1P). (B) The indicated mAbs were serially diluted 10-fold (starting at 1:100) and incubated with purified GN ectodomain. H3C8 is an irrelevant murine mAb. Sera from a CCFHV-infected human were used as a positive control along with a negative control human sample. ND, not detected. (C) Capture ELISA was developed using mAb-13G8 or a negative control (CNTL) (mAb-QC03) as capture antibody and anti–M-segment polyclonal rabbit antisera as detection antibody. Two-way ANOVA, ****P < 0.0001. (D) 293T cells were transfected with constructs expressing the indicated proteins and protein expression analyzed by Western blot at 24 and 48 hours. GN was detected using anti-GN 4093 (1:1000), and GC was detected using mAb-11E7 (1:1000). Numbers on the left refer to molecular weight standard (kilodaltons). (E) Capture antibody for GP38 presence in the supernatant. For bait, 293T cells were transfected with plasmids expressing full-length strain IbAr 10200 M segment (M-seg), ΔMucΔGP38, tPA-GP3810200, or a negative control (NEG CNTL) plasmid. Two-way ANOVA, **P < 0.001, ***P < 0.0001.
Passive protection by mAb-13G8 against heterologous CCHFV strains is limited
GP38 exhibits some amino acid heterogeneity between different strains (Fig. 3A). We examined the cross-protective efficacy of mAb-13G8 against a heterologous challenge using the CCHFV strain Afg09-2990 whose GP38 molecule has 25 amino acid differences compared to strain IbAr 10200 (91.6% homology; fig. S2). Similar to strain IbAr 10200, strain Afg09-2990 is lethal in IFN-I–deficient mice, and the kinetics of infection are identical (19). Four groups of IFNAR−/− mice (n = 13 per group) were treated with 1 mg of mAb-13G8 or an isotype control on day −1/+3. Mice were infected with either strain IbAr 10200 or strain Afg09-2990, and the protection was evaluated in 10 mice per group (Fig. 3B), and 3 mice per group were euthanized for histopathology on day 4. As expected, mAb-13G8 treatment of strain IbAr 10200–infected mice resulted in minimal weight loss and 90% survival, which was significant over isotype control mice, which all succumbed to infection by day 5 (log rank, P < 0.0001). In contrast, protection of mAb-13G8 against strain Afg09-2990–infected mice was diminutive and only resulted in 20% survival. However, this was a significant increase compared to isotype control–treated, Afg09-2990–infected mice (log rank, P = 0.0002). Overall mAb-13G8 protected the homologous strain IbAr 10200–infected mice significantly better compared to the heterologous strain Afg09-2990–infected mice (log rank, P = 0.0028).
Fig. 3. Heterologous protection of mice by mAb-13G8.
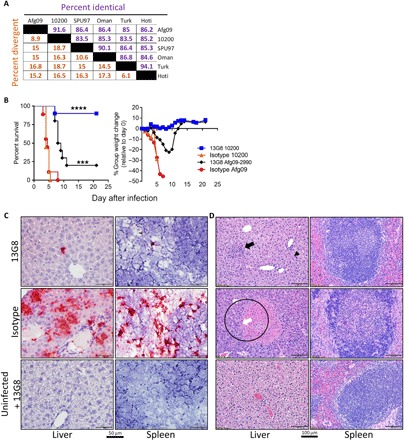
(A) Percent identical and percent divergence of CCHFV GP38 amino acid sequences from the indicated strains were determined using MegAlign Ver. 13.0.0 (DNASTAR software). (B) IFNAR−/− (n = 10 per group) mice were treated with two doses of the mAb-13G8 or an isotype control antibody on day −1/+3 and challenged subcutaneously with either IbAr 10200 or Afg09-2990. Survival and percent weight change were monitored. Log-rank test, ****P < 0.0001, ***P = 0.0002. (C) ISH staining from the livers and spleens from strain Afg09-2990–infected mice harvested on day 4 (n = 3). ISH was conducted as in Fig. 1E. (D) Hematoxylin and eosin of the liver and spleen from Afg09-2990–infected mice taken on day 4 (n = 3). Top left: Focal area of mild inflammation (black arrow) and occasional Kupffer cell hypertrophy (arrowhead). Middle left: A vessel is occluded by a fibrin thrombus (white arrow) and surrounded by an area of coagulative necrosis (circled). These mice were infected at the same time as those in Fig. 1.
We analyzed histopathology and viral load in the livers and spleens of strain Afg09-2990–infected mice (n = 3 per group) on day 4 (Fig. 3, figs. S3 to S5, and table S1). We detected some viral mRNA in both livers and spleens in all three Afg09-2990–infected mice; however, staining was much more intense in isotype control–treated mice (Fig. 3C and fig. S3). This suggests that mAb-13G8 was inhibiting viral replication to some extent at this time point. Despite this, the level of viral RNA in strain Afg09-2990–infected mouse livers and spleens was higher overall compared to those infected with strain IbAr 10200, where staining was absent (Fig. 1E and fig. S3). Liver lesions and inflammatory infiltrates in Afg09-2990–infected mice treated with mAb-13G8 were minimal to mild (Fig. 3D). However, in contrast to IbAr 10200–infected mice, in some areas, neutrophils were the predominant inflammatory cell. A single mouse in this group also exhibited rare Kupffer cell hypertrophy. Similar to the livers, splenic lesions were minimal. However, two animals exhibited minimal reactive lymphoid hyperplasia, and one animal had a single focal area of minimal neutrophilic inflammation affecting splenic white pulp. In contrast, all animals in the isotype control group exhibited microscopic hepatic lesions consistent with CCHF disease including moderate-to-severe inflammation, moderate hepatocellular degeneration and necrosis, and mild Kupffer cell hypertrophy. We also observed lymphoid necrosis in two of the three Afg09-2990–infected isotype control mice (Fig. 3D). These data indicated that mAb-13G8 can partially protect mice against heterologous CCHFV strains, but protection is markedly reduced compared to that against homologous virus.
mAb-13G8 requires complement activity for maximal protection
nNAb can protect against viral infections through Fc-mediated processes such as antibody-mediated cytotoxicity (ADCC) or complement-mediated functions (22–24). We evaluated whether these processes were involved in mAb-13G8–mediated protection using Fc receptor–deficient (FcR−/−) and C3-deficient (C3−/−) mice as mAb-13G8 is an immunoglobulin G2b (IgG2b) isotype, which can mediate these effector functions. These mice are unable to facilitate FcR function or complement-mediated activity, respectively. CCHFV only causes disease in mice when IFN-I signaling is blocked; however, we recently developed a model system using an antibody (mAb-5A3) to block IFN-I signaling, which allows the exploration of CCHFV in essentially any transgenic model system (19, 25). In this system, kinetics of disease are identical to IFN-I receptor KO mice. For this experiment, we subcutaneously injected mAb-13G8 and isotype control antibodies (1 mg per dose) on days −1 and +3 (Fig. 4A). Mice (n = 8 per group) were infected intraperitoneally with CCHFV strain IbAr 10200. On day +1 after infection, we injected mice with mAb-5A3, which disrupted the IFN-I activity. Wild-type and FcR−/− mice were fully protected by mAb-13G8 but not by the isotype control antibody. Protection compared to isotype control–treated FcR−/− mice was significant (log rank, P < 0.0001), as was protection in C57BL/6:129 mice (log rank, P = 0.0004). In contrast, mice lacking C3 (which were on a C57BL/6:129 background) were poorly protected from CCHFV infection, and mAb-13G8 only slightly delayed the MTD, with one of eight mice surviving compared to zero of eight in the isotype control antibody group. However, this protection was significant (log rank; P = 0.0201). Because FcR−/− mice were on a BALB/c background, we verified the lethality of CCHFV in this mouse strain. BALB/c mice were highly susceptible to CCHFV, with lethality occurring 24 hours before that of C57BL/6 mice, and similar to C57BL/6 mice, lethality required blockade of the IFN-I response (fig. S6). These data support a role for complement activity, but no FcR activity in mAb-13G8 facilitated protection.
Fig. 4. Fc domains play a role in mAb-13G8–mediated protection.
(A) FcR−/−, C3−/−, or B6;129 (wild-type) mice (n = 8 per group) were injected subcutaneously with two doses of the mAb-13G8 day −1/+3 or an isotype control (1 mg per dose). IFN-I was blocked on day +1 using mAb-5A3 (1 mg/ml) injected intraperitoneally. Mice were challenged with 100 PFU of CCHFV IbAr 10200 by the intraperitoneal route. Survival and weight were plotted. Log-rank test, ****P < 0.0001, ***P = 0.0004, *P = 0.0201. (B) B6 mice (n = 8) were treated with mAb-10E11, mAb-11E7, or isotype control on day −1/+3 (1 mg per dose) and infected as in (A). Log-rank test, *P = 0.0433.
Murine IgG1 antibodies are unable to efficiently capitalize on Fc-mediated effector functions. To confirm a requirement for Fc domains in complement-mediated protection, we identified an IgG1 subclass antibody that bound GP38 (fig. S7A) and also protected neonatal mice from CCHFV (18). This antibody, mAb-10E11, also competed with mAb-13G8 for binding (fig. S7B) but with an apparent lower affinity based on KD (affinity constant) values (fig. S7C). The ability for this antibody to protect adult mice from lethal infection was evaluated. C57BL/6 mice (n = 8 per group) were injected subcutaneously (1 mg per dose) on day −1/+3 with mAb-10E11 (anti-GP38), the neutralizing antibody mAb-11E7 (anti-GC) or an isotype control antibody (Fig. 4B). Mice were infected intraperitoneally with CCHFV strain IbAr 10200, and IFN-I activity was blocked on day +1 using mAb-5A3. Neither mAb-11E7, mAb-10E11, nor the control antibody protected animals from weight loss or lethality. All mAb-10E11– or mAb-11E7–treated mice succumbed to disease by day 6. A single control animal survived challenge, and this was statistically significant compared to mice treated with mAb-10E11 (log rank, P = 0.0433). Similar results were produced in IFNAR−/− mice treated with mAb-10E11 and infected with IbAr 10200 (fig. S7D). In that experiment, two mAb-10E11 animals survived versus a single control survivor, but this was not significant (log rank, P = 0.113). These results demonstrated that an IgG1 antibody targeting GP38, which competes against mAb-13G8, does not confer protection to infected mice.
GP38 localizes to the viral envelope and plasma membrane
We investigated whether GP38 localized to viral envelope and/or the target cell plasma membrane where it could facilitate complement-mediated lysis of viral particles or infected cells. To determine whether GP38 is present in the viral envelope, we stained CCHF virus–like particles (CCHFVLP) with mAb-13G8 (anti-GP38) or mAb-11E7 (anti-GC) or an irrelevant antibody. We also stained irrelevant VLPs, derived from Venezuelan equine encephalitis virus (VEEV) surface proteins, with the mAb-13G8. Particles were then stained with immunogold-labeled secondary antibodies and were examined using electron microscopy. CCHFVLP, but not VEEVLP, were positive for both GP38 and GC, as indicated by mAb-13G8 and 11E7 staining, respectively (Fig. 5A). This suggested that GP38 can localize to the virus envelope. To evaluate localization of GP38 to cellular plasma membranes, we transfected 293T cells with plasmids encoding the full-length M segment, ΔMucΔGP38, or a negative control and quantitated the surface expression of GP38 and GC by flow cytometry using mAb-13G8 or mAb-11E7 under nonpermeabilized conditions. GP38 was detected in the full-length M segment–expressing cells, but no signal was detected in the ΔMucΔGP38 or negative control cells. GC could be detected in both the full-length M segment and the ΔMucΔGP38-expressing cells, but to a slightly lesser amount in the latter (Fig. 5B). Using confocal microscopy, GP38 could also be detected on the surfaces of VeroE6 cells expressing M segment but not mock-transfected cells or cells expressing ΔMucΔGP38 (Fig. 5C). These data support a conclusion that in addition to being secreted, GP38 can localize to viral and cellular membranes. Despite the presence of GP38 on the VLP surface, mAb-13G8 did not neutralize CCHFVLP in the presence of complement (fig. S8).
Fig. 5. GP38 localization to the viral envelope and plasma membrane.
(A) CCHFVLP or VEEVLP were incubated with the indicated antibodies and then stained with an anti-mouse secondary antibody conjugated to 10-nm gold particles. Samples were then examined by electron microscopy for the presence of GP38 or GC. (B) 293T cells were transfected with full-length M segment (M-seg), ΔMucΔGP38, or an irrelevant protein (arenavirus glycoprotein precursor, NEG). To detect surface GP38 and GC, nonpermeabilized cells were incubated with mAb-13G8 or mAb-11E7, respectively. Cells were then stained with an anti-mouse secondary antibody conjugated with Alexa Fluor 488 and analyzed. Ten thousand cells were counted per sample, and data were plotted with FlowJo software. (C) Vero E6 cells were transfected with the indicated plasmids and incubated with MAb-10E11, then stained with an anti-mouse secondary antibody conjugated with Alexa Fluor 488, and analyzed by confocal microscopy.
GP38 is a target of anti-CCHFV antibody responses in humans
We evaluated the panel of murine mAbs previously described to bind pre-GN (18) for their ability to bind GP38 using a 6-His–tagged GP38 molecule based on the IbAr 10200 strain (GP38his). In addition to mAb-10E11 and mAb-13G8, all of the mAbs previously reported to bind pre-GN interacted with GP38his (Fig. 6A). This includes mAb-6C11, previously shown to interact with GP38 (17, 26). Furthermore, DNA vaccination of rabbits and mice with a plasmid encoding the CCHFV IbAr 10200 M segment elicited anti-GP38 antibodies in both species (fig. S9). Because our data indicated GP38 to be a target of humoral immune responses in CCHFV-infected and vaccinated animals, we investigated whether GP38-targeting antibodies were produced in infected humans. We evaluated convalescent human sera from two CCHF survivors of African origin for the presence of antibodies against GN, GP38, and N protein by ELISA using purified proteins (Fig. 6B). We found that both CCHFV-infected humans produced antibody response against all three targets, whereas human sera from an unexposed human were negative for these same molecules. These findings indicated that GP38 is a natural immune target of antibody responses in CCHFV-infected humans and vaccinated animals. We next conducted a tandem competition using Octet analysis assay to identify putative epitope-binding groups based on the ability of members of this murine mAb library to compete against each other for GP38 binding (Fig. 6C). We used recombinant GP38 as the antigen and interrogated the antibodies in Fig. 6A, along with two nonspecific control antibodies, mAb-11E7 (anti-CCHFV GC) and mAb-H3C8 (Ebola virus glycoprotein). As expected, all antibodies competed efficiently against themselves (permissivity, ≤25), and the nonspecific control mAbs failed to block binding of any of the GP38-targeting mAbs to the GP38 antigen (permissivity, ≥75). With regard to the GP38-targeting mAbs, this binding assay revealed three nonoverlapping antibody competition groups, suggesting that at least three independent epitopes are present on the GP38 molecule (Fig. 6D).
Fig. 6. Characterization of mAb and human convalescent sera interactions with the GP38 molecule.
(A) GP38his was plated in the wells of a 96-well plate (500 ng per well). The indicated mAbs were serially diluted 10-fold (from 1:100) and incubated with purified protein. End point titers were calculated as described in Materials and Methods. Data were plotted as a mean titer for each group. Asterisk indicates that mAb-6B12 was run in a separate assay. The dashed line indicates the limit of detection. (B) Sera from CCHF human survivors or a negative control sample were serially diluted in a purified GP38, GN, or N protein ELISA. The dashed line indicates the limit of detection. (C) Epitope grouping of GP38-reactive mAbs based on competitive ability. Antibodies are grouped on the basis of binding permissivity relative to their competing partner. Colors represent the degree of permissivity, with green denoting low competition (≥76), yellow weak competition (51 to 75), orange intermediate competition (26 to 51), and red high competition (≤25). (D) Schematic depicting putative GP38 antibody competition groups using antibody from (A).
DISCUSSION
Immunotherapeutics are effective treatment options against human viral infections, including orthopoxviruses (27), rabies virus (28), Ebola virus (29), and Junin virus (9). Historically, these products were composed of hyperimmune serum from survivors (or vaccinated individuals), but more recently, there is a greater interest in the use of mAbs or in polyclonal products generated in transgenic animals that express human antibody. Crude antibody–based therapeutics have been developed and used against CCHFV since it first emerged in the 1940s (10–12). An inherent problem, however, was the fact that these products were poorly characterized, had undefined potency, and carry a questionable safety profile due to their derivation from human blood products. Limited human data suggest that antibody-based therapeutics against CCHFV are effective (10) but their use as a treatment option against CCHFV has never been fully explored. This is, in part, due to the historic lack of adult animal models (mice and nonhuman primates) to study CCHF disease and medical countermeasure (MCM) efficacy, which has only recently been rectified (20, 21, 30). In addition, Dowall et al. (31) demonstrated in adult mice that passive transfer of antibodies from mice vaccinated with a CCHFV modified vaccinia Ankara–based vaccine failed to protect naïve adult animals, despite neutralizing antibody activity. These data suggested that not antibody, but rather other factors such as T cells were critical for protection in adult mice. Until our current study, the only animal model showing that antibodies could protect against CCHFV involved neonatal mice (18). That study tested the efficacy of a panel of murine mAbs targeting pre-GN and GC. They found that antibodies against both pre-GN and GC protected 2- to 3-day-old mice, suggesting that both neutralizing (GC-targeting) and GN-targeting antibodies have protective efficacy. Using several of the more protective antibodies identified in this panel, we demonstrate here that most fail to provide protection against lethal CCHFV infection in adult mice. In two adult mouse models, no neutralizing antibody, regardless of IgG subclass, provided even a minor amount of protection (delayed MTD, limited weight loss); thus, neonatal mice do not predict protective efficacy in adult animals. We identified only a single antibody, mAb-13G8, which was highly protective in adult mice. These results suggest that CCHFV immunotherapeutics may successfully treat CCHFV infection and illuminate the importance of nNAbs as potentially the most effective.
Before our study, the target of mAb-13G8 was identified as pre-GN, a region of the unprocessed precursor glycoprotein encoded by the M segment encompassing multiple domains including the mucin-like domain, GP160, GP85, GP38, and GN (Fig. 2A) (15). Our data suggest that the protective target of mAb-13G8 is GP38. On the basis of the understanding that GP38 was a secreted molecule (15, 17), we anticipated that protective efficacy would not require complement and/or FcR function, and mAb-13G8 was blocking an unknown deleterious function(s) of a secreted viral protein. Unexpectedly, complete protection required functional complement activity. Murine IgG1 antibodies poorly facilitate Fc-mediated effector functions. Our finding that complement was involved in anti-GP38 antibody–mediated protection was supported by the inability of an anti-GP38 IgG1 subclass of antibody, mAb-10E11, to protect mice from lethal infection. In neonatal mice, mAb-10E11, similar to mAb-13G8, was extremely protective, and we found that it binds an epitope overlapping with that of mAb-13G8. A caveat to this finding, however, is the fact that retrospectively, neonatal mice failed to predict protection in adult mice and mAb-10E11 does not bind GP38 with as high an affinity as mAb-13G8. Nevertheless, the prospect that complement is required for protection was supported by our previously unknown finding that GP38 not only is secreted but also becomes localized to the viral envelope and cellular plasma membranes. Furthermore, the fact that mAb-13G8 inhibited spread of CCHFV to the liver after subcutaneous infection also supports a model, whereby virus or virally infected cells are actively targeted for clearance. If mAb-13G8 only blocked the function of a secreted viral protein, then one would suspect that virus would traffic to a key tissue targets (i.e., liver), but virulence would be limited because of blockade of GP38 function. Our study has identified a novel mechanism(s) of CCHFV inhibition, and more work will be needed to fully characterize the mechanism of protection of anti-GP38 immunotherapeutics.
Protective nNAbs have been reported for diverse groups of viruses including alphaviruses, HIV, and flaviviruses (23, 24, 32). Passive protection elicited by the nNAbs often involves Fc functionality, and several studies suggest that complement-dependent cytotoxicity and/or ADCC play predominant roles (22). The nNAbs against flaviviruses target the nonstructural protein 1 (NS1) and protect through Fc-dependent and Fc-independent mechanism(s) (32). The NS1 molecule has some similarity to CCHFV GP38, as it is a secreted viral toxin that plays a role in influencing immune responses by activating Toll-like receptor 4, disrupting endothelial barrier function and manipulating complement (33). However, NS1 is also localized to plasma membranes in target cells where its role is poorly characterized (33). The fact that GP38 is a secreted molecule that can localize to both the viral envelope and the plasma membrane and serve as a target of protective antibodies warrants a comprehensive analysis of the multifaceted functions of this protein.
CCHFV is genetically diverse, likely a result of the vast geographical regions where the virus circulates, which includes Africa, Asia, and Europe (2). This genetic diversity affects virulence, and strains from different regions have widely varying degrees of lethality in humans. Because of this genetic diversity, CCHFV is divided into several lineages (or clades) based on M- and S-segment divergence (2). These differences can affect antigenicity of glycoproteins, including the interaction of neutralizing antibodies (34). GP38 similarly exhibits high diversity among lineages (Fig. 3A), and this appears to affect the cross-reactivity of mAb-13G8 (originally produced against strain IbAr 10200) against GP38-derived from Afg09-2990. (Fig. 2E). Therefore, it is not unexpected that the protective efficacy of mAb-13G8 against the heterologous strain Afg09-2990 was weaker compared to that against the homologous strain IbAr 10200. This highlights a general weakness in mAb therapy against viruses that inherently have high degrees of genetic diversity. Certainly, the development of more broadly targeting antibodies against GP38 will be needed to make this a viable therapeutic antiviral approach. As we have identified several other GP38-targeting murine antibodies, it will be of interest to determine what their protective profiles are both against IbAr 10200 and heterologous strains. Moreover, we identified three nonoverlapping regions on GP38 involved in antibody binding (Fig. 6D); however, we investigated the protective efficacy for only one region that was targeted by both the protective antibody mAb-13G8 and the nonprotective antibody mAb-10E11. Accordingly, it will be important to determine whether the other GP38 antibody binding regions are targets of protective antibodies and the extent to which they can provide cross-strain protection. For GP38 to be a viable target against CCHFV, a focus on cross-protection will need to be a priority. Once more broadly cross-protective antibodies are identified, protective efficacy could be evaluated in the recently developed nonhuman primate model, which currently uses the Kosova Hoti strain (30). These studies would help bridge protective efficacy in animal models with human disease.
The pre-GN mAbs were reported by Bertolotti-Ciarlet et al. (18); in our study all of these antibodies bound GP38 in multiple assays, and none of the antibodies bound to GN. As GN-specific antibodies are elicited upon infection and vaccination, a future study should include identification of GN-specific mAbs to determine whether they are protective. We show that GP38 is also targeted in mice and rabbits vaccinated with plasmids expressing the full-length M segment. In addition, human sera taken from a small cohort (n = 2) of CCHF survivors were both positive for GP38, in addition to GN and N protein. Thus, GP38 appears to be a common target of humoral immune responses against CCHFV. Poor IgG responses are nearly universal in fatal cases of CCHF human disease. It would be interesting to determine whether early anti-GP38 responses correlate with decreased disease severity. Conversely, GP38 levels could possibly be predictive of increased disease severity and could therefore serve as a diagnostic marker. This would be similar to NS1, which serves as a diagnostic marker for disease caused by various flaviviruses, including Japanese encephalitis virus, dengue virus, and West Nile virus (35). Our work led to the development of several tools that will be helpful in determining these questions, including multiple reactive antibodies and purified GP38.
GC-targeting antibodies effectively neutralize CCHFV in cell culture, with 80% plaque-reducing neutralizing titers of 2560 (mAb-11E7) and >5120 (mAb-8A1) (18, 34). However, our findings indicated that neutralizing activity does not afford protection in two mouse models when antibodies (mAb-11E7 or mAb-8A1) are administered either before or after challenge (Figs. 1, A and C, and 4B and fig. S7D). In a recent work, we found that an M-segment DNA vaccine produced neutralizing antibodies but the neutralizing antibody titers did not predict survival and did not correlate with protection (25). So, why do neutralizing antibodies fail to protect against CCHFV? The lack of protective efficacy of neutralizing antibodies would support a model whereby dissemination of CCHFV in mice is not predominantly facilitated by free virus. This would suggest that virus may spread within targeted cells, such as neutrophils, dendritic cells, and/or macrophages. Alternatively, some viruses can cloak themselves in exosomes and avoid immune detection (35, 36). Perhaps CCHFV infection of certain cells in vivo leads to the release of virus in a protected form that is not recapitulated in contrived in vitro virus neutralizing assays that use immortalized cell lines. The failure of species-specific neutralizing antibody to block viral infection in vivo presents an interesting aspect to CCHFV biology that will require more exploration.
CCHFV not only is endemic in Africa, Asia, and Europe but also is emerging into new areas with the expansion of its vector, the Hyalomma ssp. tick (2). Most recently autochthonous CCHFV infections were reported in Spain 6 years after CCHFV-positive ticks were identified in Southwestern Europe (37). These human infections included the index fatal case that resulted in fulminate hepatic failure and spread of the virus to a medical caregiver (5). This highlights the need for MCMs that can either prevent CCHFV infection or attenuate disease severity after exposure. Because antibody provides instant immunity, it is an attractive therapeutic option for limiting or preventing viral disease severity in a postexposure setting. For example, treatment of humans infected with Junin virus is successful even up to 8 days after symptoms appear, provided that the supply of convalescent human plasma is of sufficient potency as defined by neutralizing antibody titers (9). Our work demonstrates that antibody may be a viable MCM strategy against CCHFV and warrants the further development of antibody-based products targeting GP38.
MATERIALS AND METHODS
Ethics statement
All animal studies were conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles state in the Guide for the Care and Use of Laboratory Animals, National Research Council (38). All animal experimental protocols were approved by a standing internal Institutional Animal Care and Use Committee. The facilities where this research was conducted are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Animals meeting criteria were humanely euthanized.
All data and human participants research were previously de-identified and given a “research not involving human participants” determination by the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) Office of Human Use and Ethics log number FY18-28.
Viruses and cells
Huh7 and SW13 cells were propagated in Dulbecco’s modified Eagle’s medium with Earle’s salts (Corning), both medias were supplemented with 10% fetal bovine serum (FBS; Gibco), 1% penicillin/streptomycin (Gibco), 1% sodium pyruvate (Sigma-Aldrich), 1% l-glutamine (HyClone), and 1% Hepes (Gibco). Minimally passaged CCHFV strain Afg09-2990 (39) or strain IbAr 10200 (USAMRIID collection) were used for all experiments, as indicated. Strain Afg09-2990 was previously passaged three times in Vero cells and then propagated at USAMRIID two times in Huh7 cells. Strain IbAr 10200 was passaged, as previously described (25). The viruses were collected from clarified cell culture supernatants and stored at −80°C. All CCHFV work was handled in biosafety level 4 (BSL-4) containment at USAMRIID.
Anti-CCHFV and isotype antibodies
Anti-CCHFV murine mAbs are part of the USAMRIID hybridoma collection and have been described elsewhere (18). Antibody for murine challenges was purified in-house using the USAMRIID hybridoma facility. Murine isotype control antibodies for IgG2b and IgG1 were purchased from Bio X Cell. IgG2a isotype antibodies were purified in-house from a mAb-QC03 (Junin GP1) murine hybridoma.
Mice
C57BL/6, IFNAR–/– mice (B6.129S2-Ifnar1tm1Agt/Mmjax), C57BL/6;129 mice, and C3–/– mice (B6;129S4-C3tm1Crr/J) were obtained from the Jackson Laboratory. FcR–/– mice [C.129P2(B6)-Fcer1gtm1Rav N12] were obtained from Taconic. Mice were all female and 6 to 15 weeks in age at the time of challenge.
Passive protection experiments
Mice were challenged with 100 PFU of CCHFV strain IbAr 10200 or Afg09-2990 by the subcutaneous (IFNAR−/−) or intraperitoneal (all other mice) route, as indicated. Virus was diluted in a total volume of 0.2 ml of PBS (Gibco). All mice except IFNAR−/− were intraperitoneally injected with 2.5 mg of anti-IFNAR 1 (mAb-5A3; Leinco Technologies Inc.) diluted in PBS 24 hours after infection in a total volume of 0.4 ml. For antibody injections, as indicated, mice were injected subcutaneously or intraperitoneally with 1 mg per antibody per dose in a total volume of 0.2 ml diluted in PBS.
Histology
Necropsy was performed on the liver and spleen. Tissues were immersed in 10% neutral-buffered formalin for 30 days. Tissue were then trimmed and processed according to standard protocols (40). Histology sections were cut at 5 to 6 μm on a rotary microtome, mounted onto glass slides, and stained with hematoxylin and eosin. Examination of the tissue was performed by a board-certified veterinary pathologist.
In situ hybridization
CCHFV was detected in infected liver samples by ISH probes targeting IbAr 10200 or Afg09-2990 M segment of CCHFV, as previously reported (19). Formalin-fixed, paraffin-embedded liver sections were deparaffinized and peroxidase-blocked.
Confocal microscopy
Vero E6 cell monolayers were transfected in a 96-well Corning cyclic olefin copolymer polymer plate with the indicated plasmids using FuGENE 6. Transfected cells were incubated for 72 hours in a 37°C incubator with 5% CO2. Cells were rinsed with PBS and fixed in 3.7% formalin for 10 min at room temperature. Fixed wells were subsequently rinsed with PBS and blocked with 7.5% bovine serum albumin (Sigma-Aldrich) in PBS (blocking buffer) overnight at 4°C. Samples were then incubated with mAb-10E11 (5 μg/μl) diluted 1:200 in blocking buffer overnight at 4°C and then rinsed three times with PBS. Following primary antibody incubation, PBS-washed sections were incubated with Alexa Fluor 488–conjugated goat anti-mouse IgG antibody (Invitrogen) diluted (1:1000) in blocking buffer for 1 hour at room temperature. Samples were then incubated with 4′,6-diamidino-2-phenylindole–PBS for 10 min at room temperature, rinsed in PBS, and imaged.
Microscopy
Images were acquired using a Zeiss LSM 700, Zeiss LSM 880 confocal system, or Olympus BX46. Images were processed using Zeiss Zen confocal software, CellSens software, or ImageJ software.
Cloning
All GP38 constructs were produced through de novo synthesis (GENEWIZ, Germantown, MD). tPA-GP38 strain IbAr 10200 (NC_005300) or strain Afg09-2990 (HM452306.1) was produced by the addition of the tPA secretion signal (MDAMKRGLCCVLLLCGAVFVSPS). Genes were cloned into the NotI and BglII sites of the pWRG7077 vector and verified by sequence analysis. For the histidine-tagged version of tPA-GP38 from strain IbAr 10200, six histidine residues were added to the C-terminal domain of the protein by de novo synthesis and cloned into the HindIII and XhoI site of pBFksr-HCacc-MCS, which contains a cytomegalovirus promotor (Biofactora).
The codon-optimized full-length M segment used was previously reported (25). The modified M segments lacking the mucin and/or GP38 regions were produced by polymerase chain reaction (PCR). ΔMUC was produced using the forward primer 5′-GATCTCCATCTTCAGGTTGTGGCTGCCGTGGGTCTC-3′ and reverse primer 3′-GAGACCCACGGCAGCCACAACCTGAAGATGGAGATC-5′, which removed the mucin-coding region in nucleotide regions 120 to 729. ΔMUCΔGP38 was produced using the forward primer 5′-ATCGCTGGGCTCCTCGCTGTGGCTGCCGTGGGTCTC-3′ and reverse primer 3′-GAGACCCACGGCAGCCACAGCGAGGAGCCCAGCGAT-5′, which removed nucleotide regions 120 to 1545. Both ΔMUC and ΔMUCΔGP38 constructs retained the signal sequences 1 to 117. All PCR reactions were performed using the Phusion polymerase (Invitrogen). Following PCR, fragments were digested with NotI and BglII and ligated into the pWRG7077 vector. Sequence analysis was used to verify that the changes had been successfully incorporated into the gene.
GP38 purification
Production of recombinant IbAr 10200 GP38his was accomplished by transient transfection of HEK293Ts (American Type Culture Collection) with the tPA-GP38his plasmid using FuGENE 6 according to the manufacturer’s instructions. Supernatants were collected 48 and 72 hours after transfection (with media replacement after 48 hours). Supernatants were clarified and mixed with protease inhibitors. Supernatants were then run over a Ni2+ HisTrap FF column (GE Healthcare) using an ÄKTA high-performance liquid chromatography system. The column was then washed with five-column volumes of binding buffer [500 mM NaCl, 10 mM imidazole, and 20 mM sodium phosphate (pH 7.4)]. Subsequently, captured GP38his was eluted off using elution buffer (binding buffer with addition of 500 mM imidazole). Fractions with the highest absorbance (at 280 nm) were then pooled and concentrated through a Centriprep 10-kDa filter device (Millipore). Final product protein concentration was determined by BCA protein assay (Thermo Fisher Scientific).
Flow cytometry
293T cell monolayers were transfected in T25 flasks with the indicated plasmids using FuGENE 6 (Promega). Transfected cells were incubated for 72 hours in a 37°C incubator with 5% CO2. Cells were detached with gentle tapping, were pelleted by centrifugation at 750g, and resuspended in 200 μl of fluorescence-activated cell sorting (FACS) buffer (PBS and 5% FBS). Cells were incubated (1:100 dilution) with mAbs and incubated for 1 hour at room temperature. Cells were then pelleted by centrifugation at 750g and washed three times with FACS buffer. Cells were then incubated with goat anti-mouse Alexa Fluor 488–conjugated secondary antibody (1:500; Invitrogen) for 30 min at room temperature, washed three times, and resuspended in 1 ml of FACS buffer. Flow cytometry was performed on a FACSCalibur flow cytometer (Becton Dickinson). Data were collected and analyzed using FlowJo software (Tree Star Inc., Ashland, OR). A total of 10,000 cells were analyzed for each sample using a live gate.
GP38 capture ELISA
mAb-13G8 or mAb-QC03 (2.5 μg/ml) were diluted in 0.1 M carbonate buffer (pH 9.6), plated on a high-binding 96-well plate (Corning, Corning, NY) and incubated overnight at 4°C. Plates were blocked for 2 hours in blocking buffer [PBS with 0.05% Tween 20 (PBST) containing 3% milk/3% goat sera] at 37°C. Plates were washed four times in PBST and incubated with supernatant from transfected 293T cells at the indicated dilution in blocking buffer for 2 hours at 37°C. Plates were washed four times in PBST and incubated with an anti–M-segment antisera from DNA-vaccinated rabbits (diluted 1:1200) in blocking buffer and incubated at 37°C for 1 hour. Plates were washed four times in PBST and incubated with anti-rabbit IgG conjugated to horseradish peroxidase (1:1000; KPL) for 1 hour at 37°C. Plates were washed again four times in PBST, and 100 μl of SureBlue Reserve TMB microwell peroxidase 1-component (KPL) was added to each well. Reactions were stopped by adding 100 μl of TMB stop solution (KPL). The optical density (OD) at 450 nm was read on a Tecan microplate reader (Tecan Group Ltd.).
GP38his ELISA
GP38his was diluted in 0.1 M carbonate buffer (pH 9.6) and plated in duplicate in the wells of a high-binding 96-well plate (Corning). Plates were blocked for 1 hour with PBST and 5% milk. Murine mAbs (ascites fluid) were serially diluted 10-fold (starting from 1:100) in PBST containing 5% milk. Murine mAb dilutions were incubated with GP38his 1 hour at 37°C. Plates were washed four times in PBST and incubated with an anti-mouse IgG conjugated to horseradish peroxidase (1:1000; Sigma-Aldrich) for 1 hour at 37°C. Plates were washed again four times in PBST, and 100 μl of 2,2′-azinobis-3-ethylbenzthiazoline-6-sulfonic acid (ABTS) substrate (KPL) was added to each well. Reactions were stopped by adding 100 μl of ABTS stop solution of 5% (w/v) SDS. The OD at 405 nm was read on a Tecan microplate reader. End point titers were determined as the highest dilution with an absorbance value greater than the mean absorbance value from negative control antibodies (mouse ELISA, mAb-11E7, and anti-Junin virus GP1 mAb-QC03; or human ELISA and convalescent sera from an Ebola survivor) plus 3 SDs. Mean titers were plotted using GraphPad Prism 7 software.
For analysis of human sera, high-binding 96-well plates were coated with 500 ng per well of recombinant GP38, recombinant GN (Native Antigen), or recombinant N protein (Native Antigen) O/N, as described. The following morning, plates were blocked with Neptune blocking buffer (ImmunoChemistry Technologies) for 2 hours at 37°C. Plates were then washed and probed with half-log dilutions (starting at 1:50) of sera from convalescent human survivors of CCHF in Neptune blocking buffer for 1 hour at 37°C. After washing, anti-human IgG conjugated to horseradish peroxidase (1:1000; Sigma-Aldrich) was added for 1 hour at 37°C. Following a final wash, plates were treated with TMB substrate (SeraCare) at room temperature, and reactions were arrested using TMB stop solution. The OD at 450 nm was read on a Tecan ELISA plate reader, and mean titers were determined as stated above.
DNA vaccination
Anti–M-segment rabbit sera were produced by DNA vaccination of rabbits using a disposable syringe jet injection device (PharmaJet), as previously described (41). Rabbits were vaccinated with the full-length M segment (pWRG7077/CCHFV–M segmentopt IbAr 10200) at a concentration of 1 mg per dose of plasmid diluted in PBS in a total volume of 0.5 ml per injection. Rabbits were vaccinated three times at 3-week intervals. Four weeks after the final vaccination, sera were collected.
Western blot
293T cells were transfected with plasmids encoding the IbAr 10200 M segment or ΔMUCΔGP38-M using FuGENE HD (Promega). Transfected 293T cells incubated at 37°C for 24 or 48 hours, after which cells were collected by low-speed centrifugation and lysed in tris lysis buffer [10 mM tris (pH 7.5), 2.5 mM MgCl2, 100 NaCl, 0.5% Triton X-100, leupeptin (5 μg/μl; Sigma-Aldrich), and 1 mM phenylmethylsulfonyl fluoride]. Twenty microliters of sample were mixed with 4× protein sample buffer [0.125 M tris (pH 8.0), 1% SDS, 0.01% bromophenol blue, and 10% sucrose] with the addition 10× reducing buffer. Protein samples then were analyzed by 10% SDS–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories). PVDF membranes were blocked for 2 hours in PBS [10 mM tris (pH 8.0), 150 mM NaCl, and 0.05% Tween 20] containing 5% nonfat dry milk, rinsed with PBST, and incubated with rabbit polyclonal anti-GN antibody (25) (1:1000), and GC was detected using mAb-11E7 (1:1000). Membranes were subsequently washed with PBST and incubated for 1 hour with horseradish peroxidase–conjugated anti-rabbit (anti-GN; 1:5000 in PBST) or anti-mouse (mAb-11E7) (1:10000 in PBST; Amersham). Bound antibody was detected by treating the PVDF with SuperSignal West Femto chemiluminescent substrate detection reagents (Pierce) and photographed on a G-box (Syngene).
VLP production
CCHFVLP and VEEVLP were produced as previously described (25, 42).
Immunogold staining and electron microscopy of VLPs
Five microliters of CCHFVLP or VEEVLP were applied to formvar-coated 200-mesh nickel grids and incubated for 15 to 20 min. VLP grids were blocked with 4% normal goat serum for 5 min and then wicked dry. Samples were then incubated for 20 to 30 min with either mAb-13G8 (1:500), mAb-11E7 (1:1000), or a negative control antibody H3C8 (1:1000). A control with buffer solution (without primary antibody) was prepared in parallel. Samples were rinsed with buffer for 5 min. Secondary antibody (10-nm gold; goat anti-mouse; 1:25) was added to all samples, incubated for 15 min, washed for 5 min with buffer, then fixed with 1% glutaraldehyde for 1 min, and rinsed in Milli-Q water. Samples were negative stained for 1 min with 1% uranyl acetate and subsequently imaged on a Jeol 1011 transmission electron microscope at various magnifications.
Antibody competition
Tandem competition assays were used for binning mAbs to CCHFV GP38 recombinant protein. Nickel-charged tris-nitrilotriacetic acid (Pall ForteBio part number 18-5101) sensors were loaded with rGP38his recombinant protein, equilibrated for 10 min in water, then 10 mM nickel chloride for 60 s, and washed for 60 s in PBS. Sensors were then loaded with rGP38his recombinant protein (10 μg/ml) by 5-min incubation in 1× kinetics buffer (Pall ForteBio). Baseline readings were determined by equilibrating sensors for 60 s in 1× kinetics buffer. Purified mAbs against GP38 were diluted with 1× kinetics buffer to a concentration of 100 nM. Two null antibodies (EBOV-H3C8 and CCHFV-11E7) were also diluted and tested. One column of the Octet sample plate was used for seven mAbs, and each assay included a no-antibody control well. The eight sensors were incubated in the saturating antibody wells for 5 min before moving the sensors to a baseline well for 60 s in 1× kinetics buffer. The sensors were regenerated by incubating them in solution of 10 mM glycine with a pH of 2.0 for 10 s, followed by 10 s in PBS (pH 7.4). This regeneration cycle was performed three times before moving the sensors to a 1-min PBS wash. After washing, sensors were recharged with a 1-min incubation in a 10 mM nickel chloride solution. The sensors were then stored in water before using in additional assays. The data from the sensors were analyzed using the binning function of the Octet analysis software and competition groups assigned.
Statistical analysis
Weight loss was determined using one-way or two-way ANOVA with the Bonferroni correction. Survival statistics used the log-rank test. Significance levels were set at a P value less than 0.05. All analyses were performed using GraphPad Prism 7 software.
Supplementary Material
Acknowledgments
We thank R. Brocato, D. Bente, and S. Rodriguez for critical review of the manuscript. Funding: This project was funded by the Military Infectious Disease Research Program, Program Area T. Author contributions: J.W.G., C.J.S., and A.R.G. designed the research. J.W.G., C.J.S., A.R.G., M.E.L., X.Z., S.P.D., J.A.W., J.L., K.M.C., S.O., O.F., L.A.A., K.A.K., and C.J.F. performed the research. J.W.G, C.J.S., A.R.G., M.E.L., X.Z., S.P.D., and J.A.W. analyzed the data. C.S.S. supervised the project. J.W.G., C.J.S., and A.R.G. wrote initial drafts of the manuscript. All authors contributed to the final manuscript. Competing interests: J.W.G., C.J.S., and A.R.G. are inventors on a provisional patent application related to this work filed by USAMRIID (no. 62/789,576, filed 08 January 2019). L.A.A. contributed to this work while at USAMRIID, and this work is not necessarily endorsed by his current employer Battelle National Biodefense Institute, the National Biodefense Analysis and Countermeasures Center, or the Department of Homeland Security. Opinions, interpretations, conclusions, and recommendations are ours and are not necessarily endorsed by the U.S. Army or the Department of Defense. All other authors declare that they have no competing interests. Data materials and availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested to the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw9535/DC1
Supplementary Materials and Methods
Fig. S1. Representative immunohistochemistry stain for CCHFV N protein in livers and spleens of mice.
Fig. S2. GP38 amino acid differences between CCHFV strains IbAr 10200 and Afg09-2990.
Fig. S3. Combined ISH analysis of CCHFV strain IbAr 10200– and Afg09-2990–infected mice treated with mAb-13G8.
Fig. S4. Combined hematoxylin and eosin liver analysis of CCHFV strain IbAr 10200– and Afg09-2990–infected mice treated with mAb-13G8.
Fig. S5. Combined hematoxylin and eosin spleen analysis of CCHFV strain IbAr 10200– and Afg09-2990–infected mice treated with mAb-13G8.
Fig. S6. CCHFV lethality in IFN-I–blocked BALB/c and C57BL/6 mice.
Fig. S7. 10E11 binds GP38 but does not protect against lethal disease in IFNAR−/− mice.
Fig. S8. CCHFVLP neutralization by select mAbs with and without complement.
Fig. S9. GP38 binding of mouse and rabbit sera.
Table S1. Histopathology summary.
REFERENCES AND NOTES
- 1.Ergönül Ö., Crimean-Congo haemorrhagic fever. Lancet Infect. Dis. 6, 203–214 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bente D. A., Forrester N. L., Watts D. M., McAuley A. J., Whitehouse C. A., Bray M., Crimean-Congo hemorrhagic fever: History, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antivir. Res. 100, 159–189 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Shepherd A. J., Leman P. A., Swanepoel R., Viremia and antibody response of small African and laboratory animals to Crimean-Congo hemorrhagic fever virus infection. Am. J. Trop. Med. Hyg. 40, 541–547 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Spengler J. R., Estrada-Peña A., Garrison A. R., Schmaljohn C., Spiropoulou C. F., Bergeron É., Bente D. A., A chronological review of experimental infection studies of the role of wild animals and livestock in the maintenance and transmission of Crimean-Congo hemorrhagic fever virus. Antivir. Res. 135, 31–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Negredo A., de la Calle-Prieto F., Palencia-Herrejón E., Mora-Rillo M., Astray-Mochales J., Sánchez-Seco M. P., Bermejo Lopez E., Menárguez J., Fernández-Cruz A., Sánchez-Artola B., Keough-Delgado E., Ramírez de Arellano E., Lasala F., Milla J., Fraile J. L., Ordobás Gavín M., Martinez de la Gándara A., López Perez L., Diaz-Diaz D., López-García M. A., Delgado-Jimenez P., Martín-Quirós A., Trigo E., Figueira J. C., Manzanares J., Rodriguez-Baena E., Garcia-Comas L., Rodríguez-Fraga O., García-Arenzana N., Fernández-Díaz M. V., Cornejo V. M., Emmerich P., Schmidt-Chanasit J., Arribas J. R., Autochthonous Crimean-Congo hemorrhagic fever in Spain. N. Engl. J. Med. 377, 154–161 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Ergönül Ö., Çelikbaş A., Dokuzoğuz B., Eren Ş., Baykam N., Esener H., Characteristics of patients with Crimean-Congo hemorrhagic fever in a recent outbreak in Turkey and impact of oral ribavirin therapy. Clin. Infect. Dis. 39, 284–287 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Koksal I., Koksal I., Yilmaz G., Aksoy F., Aydin H., Yavuz I., Iskender S., Akcay K., Erensoy S., Caylan R., Aydin K., The efficacy of ribavirin in the treatment of Crimean-Congo hemorrhagic fever in Eastern Black Sea region in Turkey. J. Clin. Virol. 47, 65–68 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Hiatt A., Pauly M., Whaley K., Qiu X., Kobinger G., Zeitlin L., The emergence of antibody therapies for Ebola. Hum. Antibodies 23, 49–56 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Maiztegui J. I., Fernandez N. J., de Damilano A. J., Efficacy of immune plasma in treatment of Argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet 2, 1216–1217 (1979). [DOI] [PubMed] [Google Scholar]
- 10.Keshtkar-Jahromi M., Kuhn J. H., Christova I., Bradfute S. B., Jahrling P. B., Bavari S., Crimean-Congo hemorrhagic fever: Current and future prospects of vaccines and therapies. Antivir. Res. 90, 85–92 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Suleiman M. N., Muscat-Baron J. M., Harries J. R., Satti A. G., Platt G. S., Bowen E. T., Simpson D. I., Congo/Crimean haemorrhagic fever in Dubai. An outbreak at the Rashid Hospital. Lancet 2, 939–941 (1980). [PubMed] [Google Scholar]
- 12.Vassilenko S. M., Vassilev T. L., Bozadjiev L. G., Bineva I. L., Kazarov G. Z., Specific intravenous immunoglobulin for Crimean-Congo haemorrhagic fever. Lancet 335, 791–792 (1990). [DOI] [PubMed] [Google Scholar]
- 13.Bergeron É., Vincent M. J., Nichol S. T., Crimean-Congo hemorrhagic fever virus glycoprotein processing by the endoprotease SKI-1/S1P is critical for virus infectivity. J. Virol. 81, 13271–13276 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez A. J., Vincent M. J., Nichol S. T., Characterization of the glycoproteins of Crimean-Congo hemorrhagic fever virus. J. Virol. 76, 7263–7275 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zivcec M., Scholte F. E. M., Spiropoulou C. F., Spengler J. R., Bergeron É., Molecular insights into Crimean-Congo hemorrhagic fever virus. Viruses 8, 106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altamura L. A., Bertolotti-Ciarlet A., Teigler J., Paragas J., Schmaljohn C. S., Doms R. W., Identification of a novel C-terminal cleavage of Crimean-Congo hemorrhagic fever virus PreG(N) that leads to generation of an NSM protein. J. Virol. 81, 6632–6642 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanchez A. J., Vincent M. J., Erickson B. R., Nichol S. T., Crimean-Congo hemorrhagic fever virus glycoprotein precursor is cleaved by Furin-like and SKI-1 proteases to generate a novel 38-kilodalton glycoprotein. J. Virol. 80, 514–525 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertolotti-Ciarlet A., Smith J., Strecker K., Paragas J., Altamura L. A., McFalls J. M., Frias-Stäheli N., García-Sastre A., Schmaljohn C. S., Doms R. W., Cellular localization and antigenic characterization of Crimean-Congo hemorrhagic fever virus glycoproteins. J. Virol. 79, 6152–6161 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindquist M. E., Zeng X., Altamura L. A., Daye S. P., Delp K. L., Blancett C., Coffin K. M., Koehler J. W., Coyne S., Shoemaker C. J., Garrison A. R., Golden J. W., Exploring Crimean-Congo hemorrhagic fever virus-induced hepatic injury using antibody-mediated type I interferon blockade in mice. J. Virol. 92, e01083-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zivcec M., Safronetz D., Scott D., Robertson S., Ebihara H., Feldmann H., Lethal Crimean-Congo hemorrhagic fever virus infection in interferon α/β receptor knockout mice is associated with high viral loads, proinflammatory responses, and coagulopathy. J. Infect. Dis. 207, 1909–1921 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bereczky S., Lindegren G., Karlberg H., Åkerström S., Klingström J., Mirazimi A., Crimean-Congo hemorrhagic fever virus infection is lethal for adult type I interferon receptor-knockout mice. J. Gen. Virol. 91, 1473–1477 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Schmaljohn A. L., Protective antiviral antibodies that lack neutralizing activity: Precedents and evolution of concepts. Curr. HIV Res. 11, 345–353 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Horwitz J. A., Bar-On Y., Lu C. L., Fera D., Lockhart A. A. K., Lorenzi J. C. C., Nogueira L., Golijanin J., Scheid J. F., Seaman M. S., Gazumyan A., Zolla-Pazner S., Nussenzweig M. C., Non-neutralizing antibodies alter the course of HIV-1 infection in vivo. Cell 170, 637–648.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmaljohn A. L., Johnson E. D., Dalrymple J. M., Cole G. A., Non-neutralizing monoclonal antibodies can prevent lethal alphavirus encephalitis. Nature 297, 70–72 (1982). [DOI] [PubMed] [Google Scholar]
- 25.Garrison A. R., Shoemaker C. J., Golden J. W., Fitzpatrick C. J., Suschak J. J., Richards M. J., Badger C. V., Six C. M., Martin J. D., Hannaman D., Zivcec M., Bergeron E., Koehler J. W., Schmaljohn C. S., A DNA vaccine for Crimean-Congo hemorrhagic fever protects against disease and death in two lethal mouse models. PLOS Negl. Trop. Dis. 11, e0005908 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergeron E., Zivcec M., Chakrabarti A. K., Nichol S. T., Albariño C. G., Spiropoulou C. F., Recovery of recombinant Crimean Congo hemorrhagic fever virus reveals a function for non-structural glycoproteins cleavage by furin. PLOS Pathog. 11, e1004879 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao X., Feng Y., Zhu Z., Dimitrov D. S., Identification of a putative Crimean-Congo hemorrhagic fever virus entry factor. Biochem. Biophys. Res. Commun. 411, 253–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilde H., Chomchey P., Punyaratabandhu P., Phanupak P., Chutivongse S., Purified equine rabies immune globulin: A safe and affordable alternative to human rabies immune globulin. Bull. World Health Organ. 67, 731–736 (1989). [PMC free article] [PubMed] [Google Scholar]
- 29.Mupapa K., Massamba M., Kibadi K., Kuvula K., Bwaka A., Kipasa M., Colebunders R., Muyembe-Tamfum J. J., Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. International Scientific and Technical Committee. J. Infect. Dis. 179(suppl. 1), S18–S23 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Haddock E., Feldmann F., Hawman D. W., Zivcec M., Hanley P. W., Saturday G., Scott D. P., Thomas T., Korva M., Avšic-Županc T., Safronetz D., Feldmann H., A cynomolgus macaque model for Crimean-Congo haemorrhagic fever. Nat. Microbiol. 3, 556–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dowall S. D., Buttigieg K. R., Findlay-Wilson S. J., Rayner E., Pearson G., Miloszewska A., Graham V. A., Carroll M. W., Hewson R., A Crimean-Congo Haemorrhagic Fever (CCHF) viral vaccine expressing nucleoprotein is immunogenic but fails to confer protection against lethal disease. Hum. Vaccin. Immunother. 12, 519–527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung K. M., Nybakken G. E., Thompson B. S., Engle M. J., Marri A., Fremont D. H., Diamond M. S., Antibodies against West Nile virus nonstructural protein NS1 prevent lethal infection through Fc gamma receptor-dependent and -independent mechanisms. J. Virol. 80, 1340–1351 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rastogi M., Sharma N., Singh S. K., Flavivirus NS1: A multifaceted enigmatic viral protein. Virol. J. 13, 131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zivcec M., Guerrero L. I. W., Albariño C. G., Bergeron É., Nichol S. T., Spiropoulou C. F., Identification of broadly neutralizing monoclonal antibodies against Crimean-Congo hemorrhagic fever virus. Antivir. Res. 146, 112–120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng Z., Hensley L., McKnight K. L., Hu F., Madden V., Ping L. F., Jeong S.-H., Walker C., Lanford R. E., Lemon S. M., A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496, 367–371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng Z., Lemon S. M., Peek-a-boo: Membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 22, 59–64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Estrada-Pena A., Palomar A. M., Santibáñez P., Sánchez N., Habela M. A., Portillo A., Romero L., Oteo J. A., Crimean-Congo hemorrhagic fever virus in ticks, Southwestern Europe, 2010. Emerg. Infect. Dis. 18, 179–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.N. R. Council, Guide for the Care and Use of Laboratory Animals (National Academies Press, ed. 8, 2011). [PubMed] [Google Scholar]
- 39.Ölschläger S., Gabriel M., Schmidt-Chanasit J., Meyer M., Osborn E., Conger N. G., Allan P. F., Günther S., Complete sequence and phylogenetic characterisation of Crimean-Congo hemorrhagic fever virus from Afghanistan. J. Clin. Virol. 50, 90–92 (2011). [DOI] [PubMed] [Google Scholar]
- 40.E. B. Prophet, B. Mills, J. B. Arrington, L. H. Sobin, “Laboratory methods for histotechnology” (Armed Forces Institute of Pathology, Washington, D.C., 1992). [Google Scholar]
- 41.Golden J. W., Maes P., Kwilas S. A., Ballantyne J., Hooper J. W., Glycoprotein-specific antibodies produced by DNA vaccination protect guinea pigs from lethal Argentine and Venezuelan hemorrhagic fever. J. Virol. 90, 3515–3529 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suschak J. J., Bagley K., Six C., Shoemaker C. J., Kwilas S., Spik K. W., Dupuy L. C., Schmaljohn C. S., The genetic adjuvant IL-12 enhances the protective efficacy of a DNA vaccine for Venezuelan equine encephalitis virus delivered by intramuscular injection in mice. Antivir. Res. 159, 113–121 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw9535/DC1
Supplementary Materials and Methods
Fig. S1. Representative immunohistochemistry stain for CCHFV N protein in livers and spleens of mice.
Fig. S2. GP38 amino acid differences between CCHFV strains IbAr 10200 and Afg09-2990.
Fig. S3. Combined ISH analysis of CCHFV strain IbAr 10200– and Afg09-2990–infected mice treated with mAb-13G8.
Fig. S4. Combined hematoxylin and eosin liver analysis of CCHFV strain IbAr 10200– and Afg09-2990–infected mice treated with mAb-13G8.
Fig. S5. Combined hematoxylin and eosin spleen analysis of CCHFV strain IbAr 10200– and Afg09-2990–infected mice treated with mAb-13G8.
Fig. S6. CCHFV lethality in IFN-I–blocked BALB/c and C57BL/6 mice.
Fig. S7. 10E11 binds GP38 but does not protect against lethal disease in IFNAR−/− mice.
Fig. S8. CCHFVLP neutralization by select mAbs with and without complement.
Fig. S9. GP38 binding of mouse and rabbit sera.
Table S1. Histopathology summary.