

The catalytic Elp123 subcomplex of Elongator specifically recognizes and binds tRNAs in its active site.
Abstract
The highly conserved Elongator complex modifies transfer RNAs (tRNAs) in their wobble base position, thereby regulating protein synthesis and ensuring proteome stability. The precise mechanisms of tRNA recognition and its modification reaction remain elusive. Here, we show cryo–electron microscopy structures of the catalytic subcomplex of Elongator and its tRNA-bound state at resolutions of 3.3 and 4.4 Å. The structures resolve details of the catalytic site, including the substrate tRNA, the iron-sulfur cluster, and a SAM molecule, which are all validated by mutational analyses in vitro and in vivo. tRNA binding induces conformational rearrangements, which precisely position the targeted anticodon base in the active site. Our results provide the molecular basis for substrate recognition of Elongator, essential to understand its cellular function and role in neurodegenerative diseases and cancer.
INTRODUCTION
Catalytic reactions and regulatory mechanisms of transfer RNA (tRNA) modification enzymes have a profound influence on intracellular protein synthesis and in most of the cellular processes in all three domains of life (1, 2). In yeast, 11 of the 13 tRNAs carrying a uridine base in the wobble position (U34) are carboxymethylated (cm5) by the Elongator complex (3–5). This pivotal C5 substitution of U34 subsequently leads to the synthesis of 5-methoxycarbonylmethyl (mcm5), 5-carbamoylmethyl (ncm5), or 5-methoxy-carbonyl-methyl-2-thio (mcm5s2) by other enzymatic cascades (6). These modifications fine-tune the dynamic processes of tRNA binding, recognition, and rejection in the A site of the ribosome (7–9). Reduced levels of modified anticodons result in a codon-dependent decrease in ribosomal translocation speed during the elongation cycle and affect protein synthesis rates globally (10). Accordingly, cotranslational folding dynamics of nascent polypeptide chains (11–13) are disturbed, ultimately inducing intracellular proteome aggregation and proteotoxic stress (14–16). Therefore, genetic depletion or specific mutations affecting the integrity and activity of the Elongator complex are associated with the onset of severe neurodegenerative diseases, intellectual disabilities, and cancer (13, 17–21).
The eukaryotic Elongator complex is composed of six highly conserved subunits (Elp1 to Elp6) that are all required for its full tRNA modification activity in vivo (22, 23). The fully assembled yeast complex comprises two copies of each of its six subunits that are arranged in the catalytic Elp123 (620 kDa) and the associated Elp456 (230 kDa) subcomplexes (24, 25). Within the symmetric Elp123 subcomplex (26, 27), Elp1 forms a homodimer via its C-terminal domain (CTD) and serves as a scaffold for Elp2 and Elp3 subunits forming two lobes (26–28). The catalytic Elp3 subunit harbors a radical S-adenosyl methionine binding (rSAM) and a lysine acetyl–coenzyme A (CoA) transferase (KAT) domain. Its reaction center is located in the large interface between the two domains, bringing together all components necessary to create an acetyl radical that can be transferred onto the C5 position of U34 (5, 29, 30). In detail, an iron-sulfur cluster (FeS) coordinated in the rSAM domain (4) is necessary to recruit SAM and initially creates a 5′-deoxyadenosyl radical (5′-dA•) by reductive cleavage of SAM. The KAT domain is essential for the binding of acetyl-CoA, which is hydrolyzed upon tRNA binding (5). The heterohexameric Elp456 subcomplex (25) that binds asymmetrically to one Elp123 lobe regulates its interaction with the tRNA via its adenosine triphosphatase (ATPase) activity (25). As both Elp3 and Elp456 bind the tRNA anticodon stem loop (ASL), a sequential order of tRNA binding events during the reaction cycle is assumed (25, 27, 31). Moreover, the transient recruitment of accessory proteins, including kinases and phosphatases, dynamically regulates the Elongator modification cycle (13).
Despite increasing knowledge about the contribution of the individual subunits, high-resolution information of the Elp123 subcomplex and its active site are missing. We and others have previously proposed structural models of the Elp123 subcomplex and Elongator via integrative modeling (26, 27). However, these architectural models were obtained in the absence of substrate tRNA and lacked detailed information of the intersubunit interactions and the active site itself. Moreover, as Elp123 holds two equivalent active sites, but Elp456 subcomplex binds exactly on top of only one Elp3 subunit, it is possible that tRNA binding and binding of Elp456 to the same site are mutually exclusive. Here, we show cryo–electron microscopy (cryo-EM) structures of free and tRNA-bound Elp123 at resolutions of 3.3 and 4.4 Å, respectively. Our results provide the structural framework for understanding substrate recognition and the modification reaction intermediates in molecular detail. Ultimately, it is of utmost importance to understand this highly conserved cellular machine to assess the consequences of clinically relevant Elongator mutations.
RESULTS
Cryo-EM reveals the structure of Elp123 and its active site
Pure, stoichiometric, and homogeneous samples of endogenous yeast Elp123 subcomplex (fig. S1A) were stabilized with low concentrations of glutaraldehyde and vitrified to acquire cryo-EM images (fig. S1B). After classification and refinement, the dataset resulted in a map of Elp123 at a resolution of 4.5 Å (fig. S1C). The structure is composed of two lobes connected by an arch, as previously described by negative-stain EM (26). Flexibility between the two lobes was indicated by the partial alignment of the two-dimensional (2D) class-averages and the decreased local resolution in the arch. Thus, local refinement procedures, using differently masked areas of the lobe with and without the dimerization domain of Elp1 (Elp1-DD), yield improved resolutions of 3.7 Å for the Elp123 lobe including Elp1-DD and 3.3 Å without this region (fig. S1, D to G). Local amplitude scaling (LocScale) (32) of the Elp123 lobe maps was used to build and refine an atomic model of the Elp123 subcomplex (Fig. 1A and table S1).
Fig. 1. Cryo-EM structure of yeast Elp123 showing its active site.
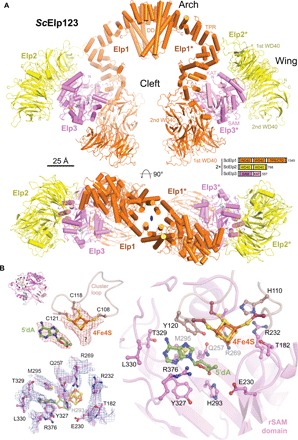
(A) Atomic model of Elp123 shown from front and top. The FeS cluster (orange and yellow) and the 5′dA molecule (green) are highlighted. Elp1, Elp2, and Elp3 domain organization is shown schematically and labeled in the upper panel. Scale bar, 25 Å. (B) Close-up of the active site of Elp3. Model and respective density of the [4Fe4S] cluster and the 5′dA molecule are shown. The coordinating cysteine residues are labeled, and the additional density next to the fourth iron atom is indicated by a question mark (upper left). Residues involved in FeS cluster and SAM coordination are shown with their respective densities (lower left). Organization of the active site including ligands and involved protein residues (right).
The arrangement of the individual Elp1, Elp2, and Elp3 proteins in the Elp123 subcomplex confirms the previously proposed overall architecture (26, 27), although the good quality of the 3.3 Å resolution map allowed us to complete the full-length structure of Elp1 by de novo building (Fig. 1A). We determined the detailed structures of the yet unknown two WD40 domains: parts of the tetratricopeptide repeat (TPR) region and unanticipated loop regions (aa687–707 and aa807–854) absent from previous crystallographic studies (fig. S2A) (28). In addition, the map revealed the tight interaction network between the individual subunits in Elp123 that ensures the integrity of each lobe. In particular, Elp3 is tightly sandwiched between the double WD40 domains of Elp1 and Elp2. The central sheet of the KAT domain of Elp3 forms a continuous β sheet with the seventh blade of the second WD40 domain of Elp1, establishing a core interface that locks the relative orientation of these two modules (fig. S2B). Furthermore, the first WD40 domain of Elp2 connects to the Elp1 TPR region through the interaction of K250, L251, L253, and L254 in a groove formed by residues (L938, Q942, L952, Q956, Q959) in helices α14 and α15 of Elp1 (fig. S2C). As Elp2 shares a large interface with the rSAM domain of Elp3, it appears to be necessary to stabilize Elp3 and its active site throughout the modification cycle.
Next, we compared the structures of the three subunits in the Elp123 complex with the models obtained from the individual proteins in isolation using crystallography. The dimeric CTD of Elp1 shows a very similar conformation as in known crystal structures. The previously identified loop region implicated in tRNA binding (ScElp11176–1251) (33) remained unstructured (fig. S3A). In Elp2 (26, 34), two additional loops (aa242–254 and aa484–532) at the interface of Elp1 and Elp3 subunits become clearly visible in the Elp123 lobe map (fig. S3B). The structure of yeast Elp3 (ScElp3) is highly similar to its bacterial counterpart (DmcElp3) (29) in the relative arrangement of the rSAM and KAT domains (fig. S3C), as well as the positioning of the conserved tRNA binding and KAT active site residues (Y540, Y541) (5, 23). ScElp3, in contrast to DmcElp3, does not harbor any zinc-binding motif (29), as two of the three responsible cysteine residues are absent in eukaryotic Elp3s. Instead, ScElp3 presents a helix (aa391–403) in this conserved region that points toward the central cavity. The FeS cluster organization in ScElp3 within the Elp123 complex is much more similar to other known rSAM enzymes (e.g., RlmN) (35, 36), and ScElp3 does not dimerize unlike DmcElp3 (fig. S3D).
Our reconstruction shows well-defined densities for the entire FeS cluster loop region, the FeS cluster itself, and a bound 5′dA molecule (i.e., product of the reductive SAM cleavage; Fig. 1B). Endogenous Elp123 harbors a cubic [4Fe4S] cluster that is coordinated by three strictly conserved cysteine residues (4, 23), namely, C108, C118, and C121 (Fig. 1B). Three cysteine residues contact three iron atoms of the cluster at an expected distance of ~2.1 Å, whereas an additional density surrounds the fourth iron atom that most likely represents bound methionine originating from SAM cleavage like in RlmN (36). 5′dA is accommodated by conserved residues (Y120, H293, M295, Y327, and L330) that specifically contact the ligand (Fig. 1B). In summary, our data demonstrate that the [4Fe4S] cluster coordination and SAM binding in Elongator are very similar to those in other rSAM proteins (37) and that they can occur in the absence of tRNA binding, as suggested earlier (4, 29).
Elp123 binds tRNA through Elp1 and Elp3
As the previously described tRNA binding sites in Elp1 and Elp3 are fully accessible in our structure, we asked whether tRNA binds directly to the Elp123 subcomplex even in the absence of Elp456. We observed a very strong interaction between endogenous Elp123 and purified in vitro–transcribed Cy5-labeled yeast tRNAAlaUGC using electrophoretic mobility shift assay (EMSA) and microscale thermophoresis (Fig. 2, A and B). Given the high affinity and detrimental effects of glutaraldehyde for the complex formation (Fig. 2A), we incubated endogenous Elp123 with yeast tRNAAlaUGC to image the complex by cryo-EM without additional stabilization. After classification, an extra density matching the size and shape of tRNA was detectable in a fraction of particles (Fig. 2C and fig. S4, A to E). The structure of the Elp123-tRNA lobe solved at an overall resolution of 4.4 Å allowed the unambiguous fit of a tRNAAlaUGC model in the extra density, applying a systematic fitting procedure described previously (fig. S4, F and G) (26). Flexible fitting of the subunits and the tRNAAlaUGC ASL allowed us to build and refine an atomic model of the tRNA-bound Elp123 complex (Fig. 2, C and D). Our structure shows how the tRNA binds to the core of the Elp123 lobe by anchoring its ASL in the Elp3 active site and its T loop to the Elp1 CTD, exposing the partially flexible acceptor stem to the surrounding solvent (Fig. 2D). The comparison between the atomic models of the Elp123 lobe with and without the tRNA showed that Elp2 is slightly displaced toward Elp3 upon tRNA binding and that the first WD40 domain and the DD region of Elp1 move toward the tRNA, leading to a slight compaction of the Elp123 subcomplex (Fig. 3A and movie S1). These conformational changes perfectly coincide with flexible regions showing lower local resolution (fig. S4D). Considering the twofold symmetry of the Elp123 subcomplex, the accessibility of its active sites, and the asymmetric interaction of the Elp456 subcomplex, it remains unknown whether two tRNAs could be bound by Elp123. By performing particle subtraction and classification, we identified a set of particles that show two symmetric densities in both lobes of the Elp123 subcomplex (Fig. 3B and fig. S5, A to C). This pool of particles yielded a C2-symmetrized map at a resolution of 6.7 Å, in which the tRNA position is identical to that observed in the Elp123-tRNA lobe structure. Hence, we show that two tRNAs can bind simultaneously to the Elp123 subcomplex.
Fig. 2. Cryo-EM structure of Elp123 bound to tRNAAlaUGC.
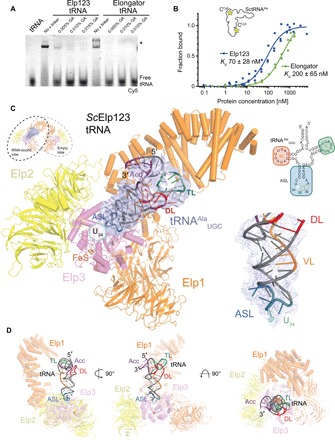
(A) EMSA assay using endogenous Elp123 (left), recombinant Elongator (right), and fluorescently labeled tRNAAlaUGC in the presence of different concentrations of glutaraldehyde. The positions of free tRNA and protein-tRNA complexes (*) are indicated next to the native polyacrylamide gel electrophoresis (PAGE). (B) Representative microscale thermophoresis measurements, respective fits, and calculated dissociation constant (Kd) values for Elp123 (blue) and Elongator (green). In both cases, the Hill coefficient is close to 1, indicating the presence of independent binding sites. n = 3. (C) Atomic model of Elp123 lobe bound to tRNA (Elp1, orange; Elp2, yellow; Elp3, violet) showing additional electron density for tRNA at a resolution of 4.4 Å. The fitted tRNAAlaUGC model is colored in accordance to the scheme on the right [purple, acceptor stem (Acc); red, D-loop (DL); blue, ASL; orange, variable loop (VL); green, T-loop (TL)]. Close-up view highlighting the phosphate backbone of the VL and ASL following the density (bottom right). (D) Atomic model of Elp123-tRNA from different perspectives.
Fig. 3. Elp123 compaction upon tRNA binding.
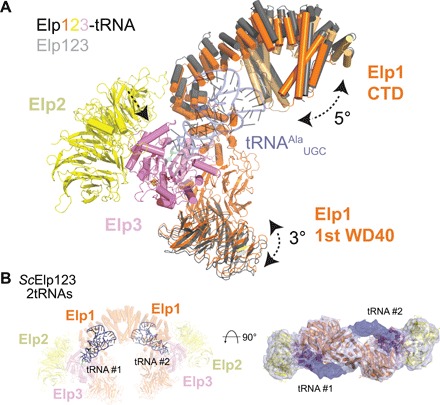
(A) Superimposition of Elp123 (gray) and Elp123-tRNA (orange/light orange, yellow, violet) structures. Observed differences are indicated by arrows. (B) Model (left) and density (right) of Elp123 bound to two tRNA molecules at the same time. Subunits and tRNA molecules (blue) are labeled.
Anticodon interactions on Elp3 active site
Despite the compaction of the Elp123 subcomplex upon tRNA binding, the structure of Elp3 did not change markedly (Fig. 4A). The density for the [4Fe4S] cluster in the Elp123-tRNA complex is very distinct, but the density for SAM (or 5′dA) is harder to assess because the ligand is only partially occupied or even absent. The orientation of most of the residues involved in the FeS cluster and SAM coordination appears unchanged after tRNA binding (Fig. 4B). As much as can be judged at the nominal resolution, only R376, which is located directly between U34 and 5′dA, appears to protrude deeper into the reaction pocket, where it also affects the orientation of Y120 (fig. S5D). However, the bound tRNA molecule undergoes strong structural deformations in the ASL region by anchoring to Elp3 (Fig. 4C). Binding to Elp123 disturbs the typical stair-stepped conformation of the anticodon, positioning the U34 RNA base directly above the channel, leading to the active site (Fig. 4D). These ASL changes are typically found in tRNA structures that bound to modification enzymes (36, 38), guaranteeing specific access to the modified base. The ASL bases are held in position by previously identified basic residues (e.g., R145, R160, K325, R373, R393, and R411) (29), which are highly conserved among Elp3 proteins from different domains of life. Additional densities also contacting the tRNA were identified, but not of good enough quality to unambiguously trace the protein backbone. We assume that they represent a specific loop region in the Elp1-CTD and the N terminus of Elp3, which both might get partially structured upon tRNA binding. In accordance, we show that a purified Elongator complex lacking the Elp1-CTD loop region shows strongly reduced tRNA binding affinity (fig. S5E) and that the purified N terminus of Elp3 (aa1–95) binds to different tRNAs by itself (fig. S5F).
Fig. 4. Structural details of tRNA binding to Elp123.
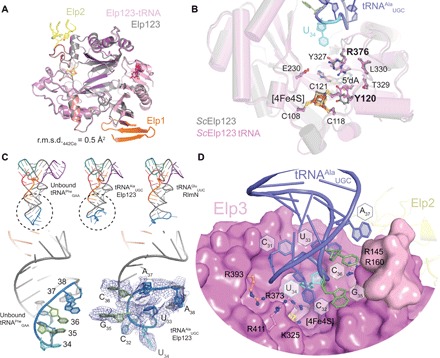
(A) Structural superimposition of tRNA-bound (violet) and unbound Elp3 (gray). The contact points of neighboring Elp1 and Elp2 subunits are shown. r.m.s.d., Root-mean-square deviation. (B) Superimposition and close-up of the active site in the unbound (gray) and tRNA-bound (colored) Elp123 structure, highlighting potentially shifted residues (in bold). (C) Structural comparison between the Elp123-bound tRNAAlaUGC (middle) and different structural models from different templates (tRNAPheGAA PDB ID: 1EHZ, left; tRNAGluUUC PDB ID: 5HR6, right). Below, the differences between the ASL region of tRNAPheGAA (left) and tRNAAlaUGC bound to Elp123 are highlighted. For the Elp123-tRNA structure, the density in the ASL region is shown and the individual bases are labeled. (D) Close-up view of the ASL region bound to the central cavity of Elp3. Conserved basic residues and RNA bases in the anticodon loop are labeled.
Functional characterization of Elp123
Next, we tested whether purified endogenous Elp123 and recombinant Elongator show tRNA-dependent acetyl-CoA hydrolysis activity, similar to archaeal Elp3 (5). Isolated bulk tRNA from wild-type or an elp3 deletion strain (i.e., endogenously modified tRNAs missing only Elongator-associated modifications) strongly increased acetyl-CoA hydrolysis in Elp123, showing a correlation between substrate recognition and induction of acetyl-CoA hydrolysis. However, using in vitro–transcribed tRNAAlaUGC, the hydrolysis activity was almost undetectable, indicating that unmodified tRNAs do not efficiently stimulate acetyl-CoA hydrolysis (Fig. 5A). These results might also point to the interplay of different tRNA modifications in the stimulation of Elongator activity, as suggested for other modification enzymes (39). We thus used the acetyl-CoA hydrolysis assay to monitor one of the first steps of the reaction without the need of other proteins that are crucial for the tRNA modification reaction (e.g., Kti proteins).
Fig. 5. Functional validation of Elp123.
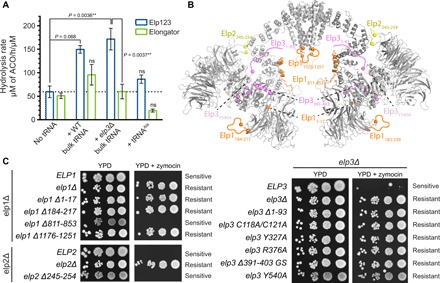
(A) Mean acetyl-CoA (ACO) hydrolysis rate of endogenous ScElp123 (blue) and recombinant Elongator (green) in the presence of different tRNAs. Statistical significance was determined by ordinary two-way analysis of variance (ANOVA; α = 0.05) followed by Tukey’s honestly significant difference test applied to the entire dataset. ns, not significantly different from the protein samples without tRNA (horizontal dotted line); **P < 0.01; mean ± SEM; n = 3; in the case of elp3Δ bulk tRNA, n = 6. (B) Structural overview of all mutations, truncations, and deletions tested in yeast. (C) Phenotypic analyses of various yeast strains carrying variations in Elp1, Elp2, and Elp3 using zymocin resistance assays.
Furthermore, on the basis of our structures, we tested a variety of deleted, truncated, and mutated versions of yeast Elongator using established phenotypical assays sampling tRNA modification defects in vivo (Fig. 5, B and C; fig. S6A; and table S2). In detail, we mutated active site residues on Elp3 (C118S/C121S, Y327, R376A, Y540A; Figs. 1B and 5B) and tRNA binding loops on Elp1 and Elp3 (aa1176–1251 and aa1–93, respectively; Fig. 5B and fig. S5, E and F), two newly described helices in the proximity of tRNA (Elp3, aa391–403; Elp1, aa811–853; Fig. 5B and fig. S3C), the Elp2 loop–contacting Elp1 (aa245–254; fig. S2C), and previously unknown Elp1 regions (aa1–17 and aa184–217; Fig. 5B). Although none of the mutations affected complex integrity (fig. S6B), all the mutants except the Elp1 helix and the loop-connecting Elp2 and Elp1 caused loss of Elongator function in vivo. These results confirm the functional relevance and importance of the residues, which are visible in our structures and evidently involved in tRNA recruitment, SAM binding, [4Fe4S] coordination, and acetyl-CoA hydrolysis. Last, this analysis also rationalizes the importance of previously suggested phosphorylation sites located in the Elp1-CTD and the N terminus of Elp3 for tRNA modification activity (29, 40). However, we cannot exclude that some of the essential regions tested might as well bind to accessory cofactors such as Kti11, Kti12, and Kti14, as they are also involved in the final modification steps in vivo (13).
DISCUSSION
The atomic models of eukaryotic Elp123 and tRNA-bound Elp123 revealed the molecular details of the Elongator active site, showing that the [4Fe4S] cluster bound to the 5′dA molecule. The tRNA-bound Elp123 structure reveals the tRNA anticodon deformation and the positioning of the modifiable U34 base upon interaction with Elp3. Moreover, the tRNA binding stimulates the acetyl-CoA hydrolysis in Elp123 but shows weaker stimulation with full Elongator. Therefore, our structural and functional analyses allow us to suggest a scheme of the modification reaction carried out by the eukaryotic Elongator complex. In detail, the two individual reactions, leading to (i) 5′dA in the rSAM domain and (ii) an activated acetyl group in the KAT domain, can proceed independently and get interconnected by the incoming tRNA substrate (Fig. 6A). The [4Fe4S] cluster supplies the requisite electron for reductive cleavage of SAM, which generates a 5′-dA• radical required for subsequent reaction steps (30). In addition, tRNA binding triggers acetyl-CoA hydrolysis, suggesting that the acetyl group moves from the KAT domain into the proximity of 5′-dA• and U34, where it can get converted into an acetyl radical. In this scenario, highly reactive radical intermediates would be transferred over very short distances, minimizing the risk of unspecific acetylation events and by-products (Fig. 6A). Our proposed reaction scheme provides a structural rationale for the transfer of an acetyl radical to the U34 base located in close proximity.
Fig. 6. Reaction and regulatory mechanisms of Elongator.
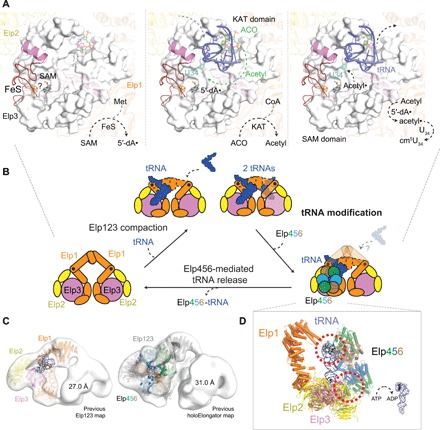
(A) Overview of intermediate steps carried out by Elp3 during the cm5 tRNA modification reaction cycle. The [4Fe4S] cluster (orange/yellow; FeS) bound in the SAM domain of Elp3 and the acetyl-CoA (ACO, green) molecule bound in the KAT domain of Elp3 are labeled. (B) Schematic illustration of Elp123 structural rearrangements upon tRNA binding and Elp456 interaction. (C) Model of the Elp123-tRNA and Elongator lobe bound to tRNA fitted into the negative-stain densities of Elp123 and Elongator. (D) Tilted view of tRNA-bound Elongator lobe model highlighting the two main contact points. The possibility of adenosine triphosphate (ATP)–mediated tRNA release is indicated. ADP, adenosine diphosphate.
We showed that Elp123 is able to bind one or two tRNAs with substantially higher affinity than the Elp456 subcomplex or the full complex, thereby favoring the idea that tRNAs are first attracted to Elp123 alone. In addition, the lower affinity to the full Elongator complex is accompanied by a lower tRNA-induced acetyl-CoA hydrolysis activity of the full complex. The binding of tRNA generates a compaction in the Elp123 subcomplex due to the movement of Elp1 (both DD and first WD40 domain) and Elp2 toward the tRNA, while Elp3 does not seem to change its conformation (see movie S1). The observed rearrangement of Elp1-CTD in both lobes, even if only one lobe is occupied by a tRNA molecule at a time, suggests a parallel regulatory mechanism in both Elp123 lobes. It seems unlikely that the Elp1 CTDs can move independently from each other, but it remains to be shown whether the asymmetrically bound Elp456 ring could mediate such behavior (Fig. 6B).
Using a phenotypical assay, we mapped the specific Elp123 regions that are essential for the tRNA modification in vivo. Moreover, the good agreement between our previous negative-stain reconstructions and new high-resolution cryo-EM maps allowed us to assess the position of an Elp123-bound tRNA in relation to the Elp456 subcomplex within Elongator (Fig. 6C). The proposed interfaces with Elp456 perfectly match previous cross-linking–mass spectrometry results among these regions in Elp3 and Elp1 and several residues of Elp4 and Elp6 (26). In our tRNA-bound Elongator model, the spatial overlap between tRNA and Elp456 is relatively small, but the ring contacts the two anchoring points of the tRNA in Elp123, namely, the Elp3 active site and the Elp1-CTD loop (Fig. 6D). This, together with the increased binding of tRNA by Elp123 in the absence of Elp456, strongly suggests that the role of Elp456 in eukaryotes is rather related to active site clearance and efficient cm5-modified tRNA removal than to tRNA delivery or contributing directly to the modification reaction. In addition, we and others (25, 27) have previously shown that Elp456 binding to tRNA is regulated by its intrinsic ATPase activity. Notably, we observed reduced tRNA binding to the Elongator complex in the presence of adenosine triphosphate (ATP), whereas addition of SAM and acetyl-CoA does not affect tRNA release (fig. S6C). Hence, it has to be analyzed in the future whether ATP hydrolysis triggers the dissociation of tRNA-bound Elp456 or releases the tRNA from Elongator after the modification reaction has been performed. In summary, our structural and functional analyses of Elp123 provide a comprehensive picture of the eukaryotic Elongator at the molecular level and clarify its mechanisms of substrate tRNA recognition.
MATERIALS AND METHODS
Sample preparation
Saccharomyces cerevisiae strain SCCM49 (MATa; his3Δ1; leu2Δ0; met15Δ0, ura3Δ0; YLR384C-3xFLAG::natNT2), the Elp1-3xFLAG strain, was generated by integrating a cassette of the 3xFLAG tag and a natNT2 marker into the genome of the parental strain BY4741. Yeast cells were grown as described previously (26). Cells (350 to 400 g) expressing C terminally FLAG-tagged Elp1 were suspended in buffer A [250 mM Hepes (pH 7.9), 125 mM NaCl, 0.1% NP-40, 10% glycerol, 50 mM NaF, 0.1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF)] supplemented with protease inhibitor cocktail (cOmplete EDTA-free, Roche) and lysed at 4°C with glass beads in BeadBeater (BioSpec). The soluble fraction obtained after centrifugation (1 hour at 30,000g in a Beckman JA-14 rotor) was incubated with 5 ml of preequilibrated ANTI-FLAG M2 affinity gel (Sigma-Aldrich) for 2 to 3 hours at 4°C. After washing the M2 resin with buffer B [10 mM Hepes (pH 7.9), 125 mM NaCl, 0.1% NP-40, 10% glycerol, 50 mM NaF, 0.1 mM Na3VO4, 1 mM PMSF supplemented with protease inhibitor cocktail] and buffer C [10 mM Hepes (pH 7.9), 125 mM NaCl, 10% glycerol, 50 mM NaF, 0.1 mM Na3VO4], bound Elp123 was eluted in buffer C containing 3× FLAG peptide (0.1 mg/ml; Sigma-Aldrich). A final concentration of 5 mM 2-mercaptoethanol was added to the eluate. The sample was concentrated to 7 to 20 mg/ml and subjected to a final purification step using a Superose 6 Increase 3.2/300 column (GE Healthcare) in buffer D [10 mM Hepes (pH 7.9), 125 mM NaCl, 50 mM NaF, 0.1 mM Na3VO4, 5 mM 2-mercaptoethanol] or buffer E [10 mM Hepes (pH 7.9), 125 mM NaCl, 50 mM NaF, 0.1 mM Na3VO4, 5 mM dithiothreitol (DTT), 5 mM MgCl2]. For the apoElp123 reconstruction, the gel filtration was performed in buffer D. Purified Elp123 was incubated with in vitro–transcribed S. cerevisiae tRNAAlaUGC at a 1:3 molar ratio for 30 min at 25°C in the presence of 1 mM MgCl2 and 1 mM acetyl-CoA. The sample was cross-linked with 0.01% glutaraldehyde (Electron Microscopy Sciences) for 1 hour at 4°C, quenched with 40 mM tris-HCl buffer (final concentration) for 10 min, and then plunged. Following tRNA binding, experiments showed that Elp123 binding to tRNA was abolished in the presence of 0.01% glutaraldehyde (fig. S4A), validating the observation of apoElp123 but not Elp123-tRNA complexes in the cross-linked sample. Hence, for the Elp123-tRNA complexes, pure Elp123 (from gel filtration in buffer E) was incubated with in vitro–transcribed tRNAAlaUGC at a 1:5 molar ratio for 30 min at 25°C (in the presence of 1 mM acetyl-CoA for the Elp123-tRNA lobe dataset and without acetyl-CoA for the Elp123-2tRNA dataset) and then immediately plunged.
Electron microscopy
Quantifoil grids (200 mesh 2/1 copper) were glow-discharged on a PELCO easiGlow glow discharger. A total of 2.5 μl of sample (0.37 to 0.4 mg/ml) was plunge-frozen using FEI Vitrobot Mark IV set to 100% humidity and 4°C (blotting parameters: wait time, 15 s; blot force, 5; blot time, 5 to 8 s). Micrographs were acquired at 300 kV using FEI Titan Krios equipped with a Gatan Quantum energy filter and a K2 Summit direct detector. The detector was operated in super-resolution mode at ×105,000 magnification, resulting in a pixel size of 0.675 Å on the object scale. A total of 4614 micrographs were collected for the apoElp123 reconstruction, 6990 for the Elp123-tRNA lobe reconstruction, and 5412 for the Elp123-2tRNAs reconstruction. We collected at under-focus varying between 1 and 2.5 μm a total of 40 frames accumulating to doses of 43 e−/Å2 (apoElp123), 44 e−/Å2 (Elp123-tRNA lobe), and 39.8 e−/Å2 (Elp123-2tRNAs).
Image processing
Frame alignment and dose weighting were performed with MotionCor2 (using Fourier space cropping by a factor of 2, resulting in a pixel size of 1.35 Å) (41), and contrast transfer function was determined with CTFIND4 (42). Initially, 8563 (apoElp123), 10888 (Elp123-tRNA lobe), and 9757 (Elp123-2tRNAs) particles were manually selected using EMAN2 boxer swarm tool (43). 2D classes obtained from these subsets in RELION2 (44) were used as templates in the autopicking procedure with the full datasets. Classification, refinement, post-processing, and local resolution estimation were performed with RELION2.
For the Elp123 structure, 1 million autopicked particles were extracted and binned four times to reduce computational costs (fig. S1G). These particles were subjected to reference-free classification, from which 490,000 particles (49%) were selected for subsequent processing. 3D classification was performed with the selected unbinned particles using as a starting model the available Elp123 map at a resolution of 27 Å (EMD-4151) low pass–filtered to 60 Å. Three classes (containing 32% of the particles) were selected on the basis of their recognizable Elp123-like shape and subjected to a second round of 3D classification, using either a mask that covered one lobe of the Elp123 subcomplex (for the Elp123 lobe map) or a mask covering the whole Elp123 subcomplex [for the Elp123 lobe DD map]. For the Elp123 lobe, the classification with the lobe mask yielded two classes that showed better resolution and gathered 54% of the particles, which were pooled and refined using a lobe mask. We obtained the Elp123 lobe map with a resolution of 3.4 Å and 84,135 particles, which after post-processing yielded the final 3.3 Å resolution map. For the Elp123 lobe DD, the classification using a full mask yielded two classes with both lobes nicely defined, so we pooled the 81,709 particles and then refined them using a mask covering the whole molecule that yielded the Elp123 lobe DD map at a resolution of 3.97 Å. The post-processing of this map yielded a final 3.7 Å resolution map that included well-resolved Elp1-DD. Alternatively, for the Elp123 full map, after the first round of 3D classification, we selected the best resolution class that showed an apparent twofold symmetry axis, although no symmetry was previously imposed. This class (with 45,236 particles) was refined with C2 symmetry, applying a mask that covered the whole molecule. This procedure yielded the Elp123 full map at a resolution of 7.5 Å, which was post-processed, yielding the final map at a resolution of 4.5 Å.
For the Elp123-tRNA lobe reconstruction (fig. S4F), 1.5 million particles were autopicked, extracted with bin 4, and reference-free–classified. A total of 751,000 particles (50%) were selected and subjected to 3D classification using the same starting model as for apoElp123, the available 27 Å map of Elp123 (EMD-4151) low pass–filtered to 60 Å as a reference. Two classes (that gathered 194,000 particles) were selected on the basis of their Elp123-like shape and the presence of an extra density on one or two lobes of the molecule.
The unbinned particles were used for a second round of 3D classification, yielding a good class of 65,000 particles with a conspicuous extra density on one lobe. A subsequent focused 3D classification step was performed after partial signal subtraction of the lobe (45). Only the signal coming from the extra density was considered, and masked 3D classification without alignment was performed yielding one class with a tRNA-shaped density in the lobe. This class with 23,020 particles was refined using a lobe mask, yielding an Elp123-tRNA lobe map at a resolution of 4.5 Å. The final map after post-processing was obtained at a resolution of 4.4 Å.
For the Elp123-2tRNA complex (fig. S6C), 798,000 autopicked particles were extracted, binned four times, and subjected to reference-free classification, from which 654,000 particles (82%) were selected. 3D classification was performed with the subset of binned particles using the Elp123 map (EMD-4151) low pass–filtered to 60 Å. Two classes were selected on the basis of their Elp123-like shape and the presence of an extra density on one or two lobes of the molecule. The unbinned 189,000 particles from the two classes were subjected to a second round of 3D classification. One class with 52,000 particles was selected on the basis of the presence of a strong extra density on each lobe. This subset of particles was subjected to partial signal subtraction of the two lobes. Then, masked 3D classification without alignment was performed, yielding two classes with extra densities in both lobes. The 12,921 particles from these classes were pooled and refined independently with and without C2 symmetry, using a mask covering the whole molecule and yielding two equivalent reconstructions at resolutions of 7.8 Å (without symmetry) and 6.9 Å (with C2 symmetry) (fig. S6A). The Elp123-2tRNA reconstructions were post-processed, yielding the final 7.6 Å resolution map (without symmetry) and the 6.7 Å resolution map (with C2 symmetry).
Model building
The building of initial models into the experimental density maps was guided by the available crystal structure of Elp2 [Protein Data Bank (PDB) ID, 5M2N], the crystal structure of the Elp1 CTD (PDB ID, 5CQS), as well as the homology models of Elongator subunits Elp1 and Elp3 described previously (26). For initial model building, the models were placed into the apoElp123 lobe map at a resolution of 3.3 Å by rigid body fitting in Chimera (46), and this initial model was used for the first round of LocScale (see below). The fitted structures of individual subunits were used to aid de novo model building and rebuilding reference models in the LocScale maps using Coot (47). In detail, the WD40 domains of Elp1 were only used for the generation of the first LocScale map, as they were regarded as low quality with the confident assignment of the fold but uncertain sequence register. Thus, both WD40 domains of Elp1 were built de novo, aided by secondary structure prediction and side-chain densities in this region of the apoElp123 lobe map (fig. S2). The fit of the Elp1 CTD was optimized by flexible fitting of the crystal structure (PDB 5CQS) using iMODfit (48) until convergence of cross-correlation below a threshold of 0.00001 over 5000 steps (with maximum 100,000 steps) and refined in Coot in regions with good enough map quality. Before the fitting, missing side chains were added with Modeller (49), and the stereochemistry was refined with energy minimization using GROMACS (50). To prevent overfitting during the flexible fitting, constraints on the secondary structure were applied. Moreover, overfitting was controlled by comparing the resulting models with the rigid body fragments of the starting crystal structure of the Elp1 CTD (PDB ID, 5CQS), ensuring that relative arrangements of secondary structure elements are locally preserved and that side chains are similar to the starting structure. Moreover, we extended the Elp1 structure with the “link loop” (residues 811 to 853), which was predicted on a secondary structure level and perfectly followed a prominent density. In addition, the Elp3 structure was extended with the helix (residues, 392 to 403) that was clearly visible in the cryo-EM density (apoElp123 lobe map) showing side-chain density and was also predicted on a secondary structure level. Our current model of apoElp123 accounts for most features observed in the experimental EM density maps (Elp123 lobe, Elp123 lobe DD, and full Elp123), including two strong densities located close to the active site that corresponds to the [4Fe4S] cluster and the 5′dA molecule.
tRNA-bound models were generated by flexible fitting of the apoElp123 model using iMODFit until convergence (with the convergence achieved in less than 500,000 steps). To identify the optimal fit of the tRNA and to select the template for comparative modeling of tRNAAlaUGC, fitting 802 tRNA chains available in the RNA Bricks database (51) to the segment of the EM map corresponding to the apparent tRNA density. For each structure, the fitting was performed using the University of California, San Francisco (UCSF) Chimera (46) global search with 10,000 random initial positions and normalized cross-correlation as the fitting metric. The statistical significance of the fits was assessed as a P value calculated from the normalized cross-correlation scores. To calculate the P values, the cross-correlation scores were first transformed to z scores (Fisher’s z transform) and centered, from which two-sided P values were computed using standard deviation derived from an empirical null distribution [derived from all obtained fits and fitted using fdrtool (52) from the R package] (53, 54). All P values were corrected for multiple testing using the Benjamini-Hochberg procedure, with the number of tests corresponding to all resulting fits from all 802 structures (i.e., treating each fitting attempt from a random initial position as an independent test). From the top fits, the structure of tRNA-Glu that bound to RlmN methyltransferase (PDB ID, 5HR6, chain D) was selected as the modeling template. This structure and the corresponding fit ranked first according to the normalized cross-correlation score (adjusted P = 6 × 10−7). The comparative model based on this template was built using ModeRNA (55). The G:U base pair 50–64 of the model was further refined using SimRNA (56) to recapitulate appropriate hydrogen bond interactions, and the final models were minimized using GROMACS (50). Note that because of limitations of comparative modeling and limited resolution of the tRNA density, the comparative model of the tRNA-Ala should be regarded with caution with deviations from the true structure expected, particularly in the regions of D- and T-loop and the acceptor stem. These deviations are not expected to affect the overall fit and the anticodon loop, where the quality of the density is sufficient for de novo tracing. The comparative model was fitted into the Elp123-tRNA density map at a resolution of 4.4 Å by superposition onto the template fitted as described above and local optimization using “Fit in Map” tool of UCSF Chimera. The anticodon loop was built de novo by manual building in Coot.
Local amplitude scaling
To generate the LocScale maps, unsharpened and unfiltered cryo-EM maps were scaled against simulated model maps using a rolling window of 20 voxels. The model maps were simulated from full-chain models refined against cryo-EM maps that were sharpened in RELION2. The dmin parameter of the LocScale procedure was set to the Nyquist frequency (2.7 Å), as recommended by the authors (32). Two rounds of LocScale were performed for the Elp123 lobe map, and one round was performed for the Elp123 lobe DD map.
Model refinement and validation
Models were refined against respective cryo-EM maps, which were B factor–sharpened with RELION2’s post-process tool. Automated real-space refinement cycles were alternated with manual model building in Coot. Manual model building was aided by experimental maps filtered at lower resolution and LocScale maps. Geometry statistics were calculated with MolProbity (57). Figures were prepared using PyMOL (58) and Chimera (46).
tRNA preparation
S. cerevisiae tRNAAlaUGC was in vitro–transcribed by T7 RNA polymerase from a linearized pUC19 vector harboring a corresponding tRNA sequence under the control of a T7 promoter. Transcription took place overnight at 37°C in 20 mM tris-HCl (pH 8.0), 150 mM NaCl, 8 mM MgCl2, 2 mM spermidine, template DNA (100 μg/ml), RNasin (25 mg/ml), pyrophosphatase (0.001 U/ml), and T7 RNA polymerase. To modify tRNA internally with Cy5 dye during transcription, 20% of cytidine triphosphate was substituted with 5-propargylamino-cytidine-5′-triphosphate labeled with Cy5 (Jena Bioscience). Deoxyribonuclease (3 to 10 U; Thermo Fisher Scientific) was added for 30 min at 37°C to remove template DNA followed by purification on HiTrap DEAE FF column (GE Healthcare). tRNA eluate was precipitated overnight at −20°C with 0.3 M sodium acetate (pH 5.5) and 75% ethanol. After centrifugation for 30 min at 4°C (20,000g), the tRNA pellet was washed with 75% ethanol; dried and suspended in 20 mM Hepes (pH 7.5), 50 mM NaCl, and 50 mM KCl; and annealed by gradual cooling from 80° to 40°C at a speed of 0.5°C/min. After annealing, tRNA was supplemented with 1 mM MgCl2 and 1 mM DTT and purified on a Superdex 75 Increase 10/300 GL column (GE Healthcare).
Wild-type bulk tRNA and bulk Δelp3 tRNA were purified from UMY2893 and UMY2916 strains, respectively. Cells were grown in yeast extract/peptone/dextrose (YPD) medium to an OD600 (optical density at 600 nm) of 1.0 to 1.5, collected by centrifugation, and suspended in equal volume of 10 mM tris-HCl (pH 7.5), 1 mM EDTA, 100 mM sodium acetate, and 1% SDS. After freezing in liquid nitrogen, cells were cryo-milled and thawed at 4°C with agitation. The lysate was mixed with equal volume of TRIzol (Thermo Fisher Scientific) and incubated at room temperature for 20 min. Next, 20% (v/v) of chloroform was added, mixed, and incubated at room temperature for 15 min. After centrifugation at 4°C with 3000g, the aqueous phase was collected and further centrifuged at 4°C for another 60 min at 12,000g. Large RNA from the supernatant was precipitated by incubation at −20°C for 3 hours with 33% volume of 8 M lithium chloride and centrifugation at 4°C with 4000g (30 min). tRNA was precipitated as described above and suspended in 4 ml of 10 mM sodium acetate (pH 5.2). tRNA was purified on HiLoad 26/600 Superdex 200 pg (GE Healthcare). Both transcribed and bulk tRNAs were concentrated using an Amicon Ultra-0.5 3k concentrator (Merck Millipore), aliquoted, and stored at −20°C.
Recombinant protein production and purification
S. cerevisiae ELP1 to ELP6 genes were amplified in polymerase chain reactions (PCRs) and cloned in pairs into vectors with opposite-oriented, inducible GAL1 and GAL10 promoters to allow simultaneous expression of two genes from one vector (59). Each vector harbored different selection marker cassettes (LEU2, TRP1, and URA3) to ensure the exclusive growth of transformants. For the purpose of later protein purification, the ELP3 or ELP1 gene was modified with FLAG and Twin-Strep-tag (IBA Lifesciences) coding sequences, while the C terminus of Elp6 was fused to Staphylococcus aureus protein A and a cleavage site for the protease of tobacco etch virus (His-TEV). Elp1, Elp2, and Elp3 mutant proteins were prepared by mutating respective genes in modified QuikChange reactions. DS1-2b yeast strain was cotransformed with vectors carrying ELP genes. Transformants were grown on selection medium agar plates. To overproduce Elongator, transformed cells were grown at 30°C with shaking in raffinose selection medium to an OD600 of 0.3 to 0.6 and induced for the following 21 hours with galactose at a 2% final concentration. Cells were next spun down, suspended in a lysis buffer [LB; 250 mM Hepes (pH 7.5), 100 mM NaCl, 10% glycerol, 0.1% Tween 20, 1 mM sodium orthovanadate, 20 mM sodium fluoride, aprotinin (2 μg/ml), leupeptin (5 μg/ml), 1 μM pepstatin A, and 1 mM PMSF; 3 ml of LB per 20 g of cell pellet], and frozen in liquid nitrogen in the form of small drops. Drops were lysed by cryo-milling to a powder on a Qiagen TissueLyser II instrument and stored at −80°C. All purification steps were performed at 4°C or on ice. To isolate recombinant wild-type and mutant forms of Elongator, 12 ml of LB was added per 24 to 26 g of yeast pellet and thawed for 1 hour with agitation. Lysates were clarified by centrifugation first at 7000g (30 min) followed by another round of centrifugation at 20,000g (20 min). Cell extracts were incubated with agitation for 30 min with 2 ml of immunoglobulin G Sepharose 6 Fast Flow affinity resin (GE Healthcare), followed by two wash steps (10 ml). The bound fraction was incubated with 1 mg of His-TEV protease in the presence of 1 mM DTT for 1 hour and eluted. Next, Elongator was bound to a 1-ml StrepTrap HP column (GE Healthcare) and eluted with 20 mM d-desthiobiotin. After concentrating on an Amicon Ultra-15 100k, Elongator was purified on a Superose 6 Increase 10/300 GL column (GE Healthcare) equilibrated with 20 mM Hepes (pH 7.5) buffer, 100 mM NaCl, and 3 mM DTT. Selected fractions were pooled and concentrated to ~1 mg/ml with an Amicon Ultra-0.5 100k concentrator. Aliquots were frozen in liquid nitrogen and stored at −80°C.
tRNA binding assays
The internally Cy5 labeled tRNAAlaUGC (0.1 μM) was incubated with endogenous yeast Elp123 subcomplex or recombinant wild-type or mutant forms of Elongator (0.1 to 0.9 μM) for 10 min on ice. The reaction was carried out in the buffer condition containing 20 mM Hepes (pH 7.5), 100 mM NaCl, 5 mM MgCl2, and 3 mM DTT. Next, samples were supplemented with 30% glycerol and resolved on a tris-boric acid EDTA–based 6% native polyacrylamide gel electrophoresis (PAGE) gels containing 15% sucrose using EMSA. Gels were run with 120 V for 1 hour and 40 min in 1× tris-glycine/DTT buffer at 4°C. Individual cofactors (1 mM), such as acetyl-CoA, SAM, and ATP, were added as indicated in figures. To quantify the tRNA binding affinity of Elp123 and Elongator, microscale thermophoresis assays were performed. The reactions (20 μl) were carried out as described above and applied to premium capillaries (NanoTemper). The interactions were determined using the MO.Control software (NanoTemper), and dissociation constants (Kd’s) were calculated based on triplicate measurement using the MO.Affinity software (NanoTemper).
Acetyl-CoA hydrolysis assays
Acetyl-CoA hydrolysis rates were measured using the Acetyl-CoA Assay Kit (MAK039, Sigma-Aldrich). Endogenous yeast Elp123 subcomplex or Elp3-compensated recombinant Elongator (108 nM) was mixed with 2 μM tRNA (in vitro–transcribed yeast tRNAAlaUGC or bulk tRNA) in the presence of 100 μM acetyl-CoA in 1× acetyl-CoA assay buffer. Next, samples were incubated in a thermocycler for 185 min at 30°C, and proteins were removed using an Amicon Ultra-0.5 column. The flow through was collected, and measurement of acetyl-CoA concentration was performed as indicated in the manual. The end-point assay was performed, and the fluorescence intensity (λExcitation, 535 nm; λEmission, 587 nm) was measured using a plate reader (Molecular Devices). Hydrolysis rates were calculated from at least three independent experiments.
Plasmid constructs
Plasmid carrying the c-myc3–tagged versions of wild-type and mutated alleles of the ScELP3 gene [ScELP3-(myc)3] are based on the centromeric plasmid p424TDH (60). The gene was amplified by PCR from genomic DNA using yeast strain FFY3t (ELP3-(myc)3::SpHIS5) as a template. The PCR fragment was cloned into p424TDH using Eco RI and Xho I to give pON34 (ELP3-(myc)3). pON34 served as a template to generate mutated elp3-(myc)3 alleles by using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) to construct the mutated versions elp3-H110A-(myc)3, elp3-C118A/C121A-(myc)3, elp3-V119A/Y120A-(myc)3, (elp3-Y136A-myc)3, elp3-R232A-(myc)3, elp3-R269A-(myc)3, elp3-H271A-(myc)3, and elp3-W341A-(myc)3. The other mutations were transferred from plasmids of the GAL-driven overexpression system. Plasmids carrying elp1_Δ811-853 or elp3_Δ1-93 were generated using the In-Fusion HD Cloning Kit (Takara Bio USA Inc.). For elp1_Δ811-853, two fragments were amplified by PCR from genomic DNA using yeast strain UMY2893. Fragment 1 was amplified using ELP1_pUC19-F (5′-CGACGGCCAGTGAATTCATGGTTGAACATGACAAGAGTG-3′) and ELP1_d811-R (5′-TATTTTGTTGACTTTATCTTCCGACAAACAGGAAA-3′). Fragment 2 was amplified with the following oligonucleotides: ELP1_d853-F (5′-AAAGTCAACAAAATATGCGATGCTGTGC-3′) and ELP1_pUC19-R (5′-ATTACGCCAAGCTTGCATGCCTGCTGCTCAAAAATCAACAATATGACTCTTAGGG-3′). For elp3_Δ1-93, the first fragment was amplified using Elp3-pUC19-F (5′-CGACGGCCAGTGAATTCATCACTTATAACGTTGTGTT-3′) and Elp3_d2-93_R (5′-CACTGCAATACCCGACATCTTTGTCAGGGTGTTCTTCGA-3′) from pRZ130 (pJet200 XbaI/XhoI + PELP3-elp3_E485A-3myc- SpHIS3-TermELP3) and the second fragment was amplified using Elp3_d2-93_F (5′-TCGGGTATTGCAGTGGTAGC-3′) and Elp3-pUC19-R (5′-CCAAGCTTGCATGCCTGCTGAATCTTCAAAATATATGTACGT-3′) from pRZ135 (pJet200 XbaI/XhoI + PELP3-elp3_K86A-K88A-3myc-SpHIS3-TermELP3). Both fragments were cloned in the linearized (with EcoRI and SphI, respectively) plasmid pUC19 (61).
Phenotypical analyses of Elongator function in vivo
To analyze the presence of the Elongator-dependent tRNA anticodon modification in elp1, elp2, or elp3 mutants, two established assays were used: the zymocin sensitivity assay and the SUP4 suppression assay (62). Zymocin, a trimeric toxin secreted by so-called killer strains of Kluyveromyces lactis strains, inhibits growth of sensitive yeasts by cleaving tRNAs that contain mcm5s2U34-modified tRNA anticodons. Growth inhibition by zymocin in the growth medium or intracellular expression of the catalytic γ-toxin subunit of zymocin, a tRNA endonuclease, is abolished by mutations inactivating Elongator function. The SUP4 suppression assay makes use of the SUP4-encoded suppressor tRNA, which has an mcm5U34-modified anticodon. Strains carrying the SUP4 suppressor allele plus the ochre stop codon containing ade2-1 or can1-100 alleles provide convenient readouts for Elongator activity on medium lacking adenine or containing canavanine. The latter is a toxic arginine analog taken up by the arginine permease encoded by CAN1. Elongator deficiency results in canavanine resistance or adenine auxotrophy correlating with the degree of mcm5 modification. Zymocin or γ-toxin resistance reflects the Elongator-dependent mcm5s2 modification status. For the γ-toxin assay, the γ-toxin gene was expressed from the galactose-inducible GAL1 promoter on plasmid pLF16 (63). Transformed strains were grown overnight in single dropout medium lacking leucine, and serial dilutions of the cell suspensions were spotted on either 2% dextrose–containing (repressing conditions) or 2% galactose–containing (inducing conditions) plates and incubated for 3 days at 30°C. In the zymocin or killer eclipse assay, the zymocin-producing K. lactis strain AWJ137 (64) was inoculated next to a drop of the S. cerevisiae strains to be tested (table S2). The plates were incubated at 30°C, and the growth inhibition zone around the killer strain was analyzed in the elp1, elp2, or elp3 mutants after 2 days. Alternatively, zymocin was provided by the supernatant of strain AWJ137 grown overnight in YPD.
Supplementary Material
Acknowledgments
We thank G. Chojnowski for help with the RNA Bricks database and A. Chramiec-Głąbik for tRNA samples. We thank C. Alfieri and J. Heddle for critical comments on the manuscript, E. Hurt and M. Gaik for sharing the bicistronic yeast overexpression system, W. Hagen for data acquisition, A. Jakobi for support with the LocScale procedure, and R. Wetzel and H. Groetsch for technical support. Funding: We acknowledge support by the MCB Structural Biology Core Facility (supported by the TEAM TECH CORE FACILITY/2017-4/6 grant from the Foundation for Polish Science; to S.G.). The computational part of this work was done on the High-Performance Cluster of the EMBL, supported by the EMBL Heidelberg IT Services. This work was further supported by OPUS10 grant UMO-2015/19/B/NZ1/00343 from the National Science Centre (to M.J., R.K., and S.G.), the Homing program (Homing/2016-2/14 to T.-Y.L. and N.E.H.A.) from the Foundation for Polish Science, grant 703432 from the EU Horizon 2020 program Marie Sklodowska-Curie (to H.K.), and grants BR921/9-1 and Mu3173/2-1 from the Deutsche Forschungsgemeinschaft (to M.I.D., C.K., K.D.B., and C.W.M.). Author contributions: M.I.D. and M.J. established protein purification procedures; M.I.D. performed EM characterization and analyses; F.W. performed data acquisitions and H.K. helped to optimize plunging conditions; M.J. performed biochemical and biophysical characterization; T.-Y.L. produced tRNAs and established acetyl-CoA hydrolysis assays; N.E.H.A. and R.K. purified the Elp3 N terminus and performed EMSAs; C.K. and K.D.B. performed site-directed mutagenesis and phenotypical analyses in yeast; J.K. conducted tRNA modeling and fitting of Elongator subunits; M.I.D. and S.G. performed de novo building and refinements; M.I.D., M.J., C.W.M., and S.G. designed experiments and analyzed the data; M.I.D., J.K., C.W.M., and S.G. wrote the manuscript with input from all other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Materials and methods are available as supplementary materials at the Science website. The atomic coordinates and EM densities for Elp123 lobe (PDB ID 6QK7 and EMD-4571) and the EM densities of full Elp123 (EMD-4573), tRNA-Elp123 lobe (EMD-4574), and 2tRNAs-Elp123 (EMD-4576) have been deposited in the EMData Bank (EMDB) and the European Protein Data Bank (PDBe). All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw2326/DC1
Fig. S1 Elp123 cryo-EM reconstruction.
Fig. S2. Atomic structure of the Elp1 N terminus and intersubunit contacts.
Fig. S3. Structural comparison of individual Elongator subunits.
Fig. S4. Cryo-EM reconstruction of Elp123-tRNA.
Fig. S5. Cryo-EM reconstruction of Elp123 bound to two tRNAs.
Fig. S6. In vivo and in vitro characterization of Elongator mutants.
Table S1. Collection and refinement statistics.
Table S2. Yeast strains used in this study.
Movie S1. Conformational changes of Elp123 upon tRNA binding.
REFERENCES AND NOTES
- 1.Vare V. Y. P., Eruysal E. R., Narendran A., Sarachan K. L., Agris P. F., Chemical and conformational diversity of modified nucleosides affects tRNA structure and function. Biomolecules 7, 29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phizicky E. M., Hopper A. K., tRNA biology charges to the front. Genes Dev. 24, 1832–1860 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tumaitis T. D., Lane B. G., Differential labelling of the carboxymethyl and methyl substituents of 5-carboxymethyluridine methyl ester, a trace nucleoside constituent of yeast transfer RNA. Biochim. Biophys. Acta 224, 391–403 (1970). [DOI] [PubMed] [Google Scholar]
- 4.Paraskevopoulou C., Fairhurst S. A., Lowe D. J., Brick P., Onesti S., The elongator subunit Elp3 contains a Fe4S4 cluster and binds S-adenosylmethionine. Mol. Microbiol. 59, 795–806 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Selvadurai K., Wang P., Seimetz J., Huang R. H., Archaeal Elp3 catalyzes tRNA wobble uridine modification at C5 via a radical mechanism. Nat. Chem. Biol. 10, 810–812 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johansson M. J. O., Xu F., Byström A. S., Elongator—A tRNA modifying complex that promotes efficient translational decoding. Biochim. Biophys. Acta 1861, 401–408 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Ranjan N., Rodnina M. V., Thio-modification of tRNA at the wobble position as regulator of the kinetics of decoding and translocation on the ribosome. J. Am. Chem. Soc. 139, 5857–5864 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Rezgui V. A. N., Tyagi K., Ranjan N., Konevega A. L., Mittelstaet J., Rodnina M. V., Peter M., Pedrioli P. G. A., tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc. Natl. Acad. Sci. U.S.A. 110, 12289–12294 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vendeix F. A. P., Murphy F. V. IV, Cantara W. A., Leszczyńska G., Gustilo E. M., Sproat B., Malkiewicz A., Agris P. F., Human tRNALys3 UUU is pre-structured by natural modifications for cognate and wobble codon binding through keto–enol tautomerism. J. Mol. Biol. 416, 467–485 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goffena J., Lefcort F., Zhang Y., Lehrmann E., Chaverra M., Felig J., Walters J., Buksch R., Becker K. G., George L., Elongator and codon bias regulate protein levels in mammalian peripheral neurons. Nat. Commun. 9, 889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodnina M. V., Fischer N., Maracci C., Stark H., Ribosome dynamics during decoding. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thommen M., Holtkamp W., Rodnina M. V., Co-translational protein folding: Progress and methods. Curr. Opin. Struct. Biol. 42, 83–89 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Dauden M. I., Jaciuk M., Müller C. W., Glatt S., Structural asymmetry in the eukaryotic Elongator complex. FEBS Lett. 592, 502–515 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Nedialkova D. D., Leidel S. A., Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161, 1606–1618 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zinshteyn B., Gilbert W. V., Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLOS Genet. 9, e1003675 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laguesse S., Creppe C., Nedialkova D. D., Prévot P.-P., Borgs L., Huysseune S., Franco B., Duysens G., Krusy N., Lee G., Thelen N., Thiry M., Close P., Chariot A., Malgrange B., Leidel S. A., Godin J. D., Nguyen L., A dynamic unfolded protein response contributes to the control of cortical neurogenesis. Dev. Cell 35, 553–567 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Cohen J. S., Srivastava S., Farwell K. D., Lu H.-M., Zeng W., Lu H., Chao E. C., Fatemi A., ELP2 is a novel gene implicated in neurodevelopmental disabilities. Am. J. Med. Genet. A 167, 1391–1395 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Close P., Gillard M., Ladang A., Jiang Z., Papuga J., Hawkes N., Nguyen L., Chapelle J.-P., Bouillenne F., Svejstrup J., Fillet M., Chariot A., DERP6 (ELP5) and C3ORF75 (ELP6) regulate tumorigenicity and migration of melanoma cells as subunits of elongator. J. Biol. Chem. 287, 32535–32545 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kojic M., Wainwright B., The many faces of elongator in neurodevelopment and disease. Front. Mol. Neurosci. 9, 115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rapino F., Delaunay S., Rambow F., Zhou Z., Tharun L., de Tullio P., Sin O., Shostak K., Schmitz S., Piepers J., Ghesquière B., Karim L., Charloteaux B., Jamart D., Florin A., Lambert C., Rorive A., Jerusalem G., Leucci E., Dewaele M., Vooijs M., Leidel S. A., Georges M., Voz M., Peers B., Büttner R., Marine J.-C., Chariot A., Close P., Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 558, 605–609 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Kojic M., Gaik M., Kiska B., Salerno-Kochan A., Hunt S., Tedoldi A., Mureev S., Jones A., Whittle B., Genovesi L. A., Adolphe C., Brown D. L., Stow J. L., Alexandrov K., Sah P., Glatt S., Wainwright B. J., Elongator mutation in mice induces neurodegeneration and ataxia-like behavior. Nat. Commun. 9, 3195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esberg A., Huang B., Johansson M. J. O., Byström A. S., Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell 24, 139–148 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Huang B., Johansson M. J. O., Bystrom A. S., An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 11, 424–436 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otero G., Fellows J., Li Y., de Bizemont T., Dirac A. M. G., Gustafsson C. M., Erdjument-Bromage H., Tempst P., Svejstrup J. Q., Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol. Cell 3, 109–118 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Glatt S., Létoquart J., Faux C., Taylor N. M. I., Séraphin B., Müller C. W., The elongator subcomplex Elp456 is a hexameric RecA-like ATPase. Nat. Struct. Mol. Biol. 19, 314–320 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Dauden M. I., Kosinski J., Kolaj-Robin O., Desfosses A., Ori A., Faux C., Hoffmann N. A., Onuma O. F., Breunig K. D., Beck M., Sachse C., Séraphin B., Glatt S., Müller C. W., Architecture of the yeast Elongator complex. EMBO Rep. 18, 264–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Setiaputra D. T., Cheng D. T. H., Lu S., Hansen J. M., Dalwadi U., Lam C. H. Y., To J. L., Dong M.-Q., Yip C. K., Molecular architecture of the yeast Elongator complex reveals an unexpected asymmetric subunit arrangement. EMBO Rep. 18, 280–291 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu H., Lin Z., Li F., Diao W., Dong C., Zhou H., Xie X., Wang Z., Shen Y., Long J., Dimerization of elongator protein 1 is essential for Elongator complex assembly. Proc. Natl. Acad. Sci. U.S.A. 112, 10697–10702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glatt S., Zabel R., Kolaj-Robin O., Onuma O. F., Baudin F., Graziadei A., Taverniti V., Lin T.-Y., Baymann F., Séraphin B., Breunig K. D., Müller C. W., Structural basis for tRNA modification by Elp3 from Dehalococcoides mccartyi. Nat. Struct. Mol. Biol. 23, 794–802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.T.-Y. Lin, S. Glatt, in Encyclopedia of Inorganic and Bioinorganic Chemistry, R. A. Scott, Ed. (Wiley Online Library, 2018), vol. eibc2623. [Google Scholar]
- 31.Lin T.-Y., Abbassi N. E. H., Zakrzewski K., Chramiec-Głąbik A., Jemioła-Rzemińska M., Różycki J., Glatt S., The Elongator subunit Elp3 is a non-canonical tRNA acetyltransferase. Nat. Commun. 10, 625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jakobi A. J., Wilmanns M., Sachse C., Model-based local density sharpening of cryo-EM maps. eLife 6, e27131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Santo R., Bandau S., Stark M. J. R., A conserved and essential basic region mediates tRNA binding to the Elp1 subunit of the Saccharomyces cerevisiae Elongator complex. Mol. Microbiol. 92, 1227–1242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong C., Lin Z., Diao W., Li D., Chu X., Wang Z., Zhou H., Xie Z., Shen Y., Long J., The Elp2 subunit is essential for elongator complex assembly and functional regulation. Structure 23, 1078–1086 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Benítez-Páez A., Villarroya M., Armengod M. E., The Escherichia coli RlmN methyltransferase is a dual-specificity enzyme that modifies both rRNA and tRNA and controls translational accuracy. RNA 18, 1783–1795 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwalm E. L., Grove T. L., Booker S. J., Boal A. K., Crystallographic capture of a radical S-adenosylmethionine enzyme in the act of modifying tRNA. Science 352, 309–312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boal A. K., Grove T. L., McLaughlin M. I., Yennawar N. H., Booker S. J., Rosenzweig A. C., Structural basis for methyl transfer by a radical SAM enzyme. Science 332, 1089–1092 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou C., Huang R. H., Crystallographic snapshots of eukaryotic dimethylallyltransferase acting on tRNA: Insight into tRNA recognition and reaction mechanism. Proc. Natl. Acad. Sci. U.S.A. 105, 16142–16147 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sokołowski M., Klassen R., Bruch A., Schaffrath R., Glatt S., Cooperativity between different tRNA modifications and their modification pathways. Biochim. Biophys. Acta 1861, 409–418 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Abdel-Fattah W., Jablonowski D., Di Santo R., Thüring K. L., Scheidt V., Hammermeister A., ten Have S., Helm M., Schaffrath R., Stark M. J. R., Phosphorylation of Elp1 by Hrr25 is required for elongator-dependent tRNA modification in yeast. PLOS Genet. 11, e1004931 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Kimanius D., Forsberg B. O., Scheres S. H. W., Lindahl E., Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, e18722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai X.-c., Rajendra E., Yang G., Shi Y., Scheres S. H. W., Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 4, e11182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Lopéz-Blanco J. R., Chacón P., iMODFIT: Efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J. Struct. Biol. 184, 261–270 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M.-y., Pieper U., Sali A., Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 15, 5.6.1–5.6.30 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E., GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chojnowski G., Waleń T., Bujnicki J. M., RNA Bricks—A database of RNA 3D motifs and their interactions. Nucleic Acids Res. 42, D123–D131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strimmer K., fdrtool: A versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Bui K. H., von Appen A., DiGuilio A. L., Ori A., Sparks L., Mackmull M.-T., Bock T., Hagen W., Andrés-Pons A., Glavy J. S., Beck M., Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155, 1233–1243 (2013). [DOI] [PubMed] [Google Scholar]
- 54.von Appen A., Kosinski J., Sparks L., Ori A., DiGuilio A. L., Vollmer B., Mackmull M.-T., Banterle N., Parca L., Kastritis P., Buczak K., Mosalaganti S., Hagen W., Andres-Pons A., Lemke E. A., Bork P., Antonin W., Glavy J. S., Bui K. H., Beck M., In situ structural analysis of the human nuclear pore complex. Nature 526, 140–143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rother M., Rother K., Puton T., Bujnicki J. M., ModeRNA: A tool for comparative modeling of RNA 3D structure. Nucleic Acids Res. 39, 4007–4022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boniecki M. J., Lach G., Dawson W. K., Tomala K., Lukasz P., Soltysinski T., Rother K. M., Bujnicki J. M., SimRNA: A coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 44, e63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., Snoeyink J., Richardson J. S., Richardson D. C., MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.W. L. DeLano, The PyMOL Molecular Graphics System (DeLano, 2002). [Google Scholar]
- 59.Gaik M., Flemming D., von Appen A., Kastritis P., Mücke N., Fischer J., Stelter P., Ori A., Bui K. H., Baßler J., Barbar E., Beck M., Hurt E., Structural basis for assembly and function of the Nup82 complex in the nuclear pore scaffold. J. Cell Biol. 208, 283–297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mumberg D., Müller R., Funk M., Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 (1995). [DOI] [PubMed] [Google Scholar]
- 61.Norrander J., Kempe T., Messing J., Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene 26, 101–106 (1983). [DOI] [PubMed] [Google Scholar]
- 62.Frohloff F., Fichtner L., Jablonowski D., Breunig K. D., Schaffrath R., Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J. 20, 1993–2003 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jablonowski D., Zink S., Mehlgarten C., Daum G., Schaffrath R., tRNAGlu wobble uridine methylation by Trm9 identifies Elongator's key role for zymocin-induced cell death in yeast. Mol. Microbiol. 59, 677–688 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Kamper J., Esser K., Gunge N., Meinhardt F., Heterologous gene expression on the linear DNA killer plasmid from Kluyveromyces lactis. Curr. Genet. 19, 109–118 (1991). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw2326/DC1
Fig. S1 Elp123 cryo-EM reconstruction.
Fig. S2. Atomic structure of the Elp1 N terminus and intersubunit contacts.
Fig. S3. Structural comparison of individual Elongator subunits.
Fig. S4. Cryo-EM reconstruction of Elp123-tRNA.
Fig. S5. Cryo-EM reconstruction of Elp123 bound to two tRNAs.
Fig. S6. In vivo and in vitro characterization of Elongator mutants.
Table S1. Collection and refinement statistics.
Table S2. Yeast strains used in this study.
Movie S1. Conformational changes of Elp123 upon tRNA binding.