

Abstract
G protein-coupled receptors (GPCRs) mediate the transduction of extracellular signals into complex intracellular responses. Despite their ubiquitous roles in physiological processes and as drug targets for a wide range of disorders, the precise mechanisms of GPCR function at the molecular, cellular, and systems levels remain partially understood. To dissect the function of individual receptor subtypes with high spatiotemporal precision, various optogenetic and photopharmacological approaches have been reported that use the power of light for receptor activation and deactivation. Here, we introduce a novel and, to date, most remote way of applying photoswitchable orthogonally remotely tethered ligands by using a SNAP-tag fused nanobody. Our nanobody–photoswitch conjugates can be used to target a green fluorescent protein-fused metabotropic glutamate receptor by either gene-free application of purified complexes or coexpression of genetically encoded nanobodies to yield robust, reversible control of agonist binding and subsequent downstream activation. By harboring and combining the selectivity and flexibility of both nanobodies and self-labeling proteins (or suicide enzymes), we set the stage for targeting endogenous receptors in vivo.
GRAPHICAL ABSTRACT
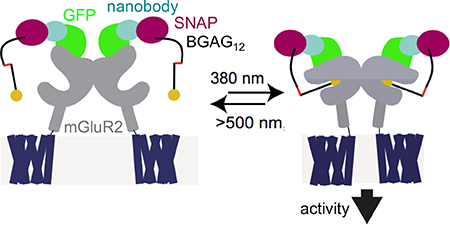
Optical control of proteins and their associated cellular functions has evolved into a powerful technique for gaining a deeper understanding of biological processes.1,2 In particular, the ability to optically control ion channels, transporters, and receptors has made it possible to move toward a more mechanistic understanding of the role of specific cells and synapses in the circuit-driven processes of the brain. While classical optogenetic tools, such as those based on opsins, allow for the general control of cellular excitability or broadly defined signaling pathways, the ability to control specific signaling molecules has the potential to extend such analysis to probe the molecular basis of physiological functions and disease pathophysiology. This circumvents the limitations of pharmacology, where subtype selectivity is difficult to achieve, and compounds cannot be applied or removed in a fast and efficient manner to defined locations due to diffusion.
Ideally, one would be able to control native receptors with subtype-specificity, high spatiotemporal precision, and genetic targeting to defined cell types. A number of previous strategies achieved a subset of these desired parameters (e.g., opsin-based receptors, DREADDs, drugs acutely restricted by tethering, caged compounds and photochromic ligands, and modulatory nanobodies (NBs)), but it remains a major challenge to attain all of them within the same toolset (see Supplementary Table 1 for a detailed outline and comparison).3 We previously developed approaches to optogenetically manipulate specific G protein-coupled metabotropic glutamate receptors (mGluRs) through the covalent attachment of photoswitchable tethered ligands (PTLs).4–6 Initially, these compounds were targeted directly to an engineered cysteine via maleimide conjugation (Figure 1A),7 but more recently, we improved this method through the use of an N-terminal self-labeling tag (Figure 1B). This strategy yielded enhanced labeling efficiency in culture and in vivo,9 simplified photoswitch design, enabled multiplexing of tags and, most importantly, is fully orthogonal to native chemistry.10 Based on the demonstrated utility of self-labeling proteins in this context and the key finding that tethered photoswitches can optically control ligand binding when attached to a distinct protein domain, we wondered if we could further move photoswitch attachment to an antibody or antibody fragment to add an extra layer of specificity and flexibility (Figure 1C). The ability to photoswitch a membrane receptor via immunochemistry would, in principle, produce an approach that could be used to target native receptors (Supplementary Table 1). To test if such a design is possible and provide a stepping stone toward the ultimate goal of endogenous control, we turned to single chain antibodies (i.e., nanobodies) that target green fluorescent protein (GFP), which proved to be a robust and reliable system in previous studies.11 Nanobodies are beneficial because of their small size, target specificity, and ability to be genetically encoded for expression in mammalian cells.12,13
Figure 1.

Optical control of ligand binding via distinct photoswitch attachment strategies. Photoswitchable tethered ligands may be presented to target ligand binding domains through previously established strategies of directly binding to the target protein (A), traditionally via cysteine-maleimide linkage, through binding to introduced self-labeling protein tags (B), such as SNAP, CLIP, or Halo-tag, or potentially through immunochemistry with labeled antibodies (C).
RESULTS
SNAP-Tagged Anti-GFP NBs Retain GFP Affinity and Efficient Labeling of BG-Conjugated Compounds
To test the ability of NBs to be used for gene-free optical control of target proteins, we utilized previously characterized NBs that target GFP11 to produce a nanobody–photoswitch conjugate (NPC). We genetically fused the self-labeling SNAP-tag14,15 protein to either the N-terminus (SNAP-NB) or C-terminus (NB-SNAP) of the anti-GFP nanobody GBP1, which enhances the fluorescence of wtGFP. The SNAP-fusion proteins remained reactive in this context and allowed us to covalently attach small molecules such as fluorophores and photoswitches to the NBs via O6-benzylguanine (BG)-linked compounds in vitro (Figure S1; Supplementary Table 2). We next tested if SNAP incorporation would alter the ability of the NB to efficiently target its antigen, GFP. Application of both SNAP-NB and NB-SNAP fusions to purified wtGFP led to a dose-dependent increase in fluorescence (Figure 2A), with an apparent affinity of ~3 nM. Together, this shows that incorporation of SNAP into the NB at either end does not affect its ability to target GFP, consistent with the localization of the N- and C-termini of the NB distant from the GFP binding site.11
Figure 2.

SNAP-tagged anti-GFP nanobodies enable stoichiometric labeling of GFP-mGluR2 in living cells. (A) In vitro dose–response curve for SNAP-NB, NB-SNAP, NB, and SNAP against wtGFP assessed by fluorescence enhancement upon binding indicates that attachment of N- or C-terminal SNAP domains does not alter NB affinity (error bars indicate SEM). (B) Representative images showing labeling of surface-expressed GFP-mGluR2 (green) by application of purified NB-SNAP labeled with Alexa-546 (red). Right, line scan analysis shows overlap in GFP and Alexa-546 fluorescence at the plasma membrane. (C) Representative images, left, and line scan, right, showing that no red surface fluorescence is observed following application of Alexa-546-labeled NB-SNAP to cells expressing mGluR2-GFP. Scale bars = 20 μm. (D) Left, schematic of single molecule pulldown (SiMPull) of GFP-mGluR2 and SNAP-fused NBs via an anti-mGluR2 antibody on PEG passivated coverslip. Right, representative images show colocalized GFP-mGluR2 (green circles) and NB-SNAP (Alexa-546; red circles) molecules; 74.6% of red spots were colocalized with a green spot (773/1034). Scale bars = 20 μm. (E and F) Counting photobleaching steps in the red channel (representative trace, E) allow for analysis of NB stoichiometry (F), confirming that 2 NBs can bind to a receptor dimer. Data comes from 2 separate transfections and 13 individual movies. Bars show the average proportion of 1-step and 2-step spots for each movie, and error bars show SEM.
SNAP-Tagged NBs Label GFP-mGluR2 in Living Cells with 2:2 Stoichiometry
We next asked if purified SNAP-tagged NBs could enable efficient labeling of a GFP-tagged membrane receptor to ultimately mediate optical control in living cells. We genetically fused GFP to either the extracellular N-terminus or the intracellular C-terminus of mGluR2 (see online methods). Application of purified SNAP-tagged NBs that had been prelabeled with Alexa-546 produced clear fluorescence on the surface of HEK 293T cells expressing GFP-mGluR2, but not mGluR2-GFP (Figures 2B and C; Figures S2A and B). Importantly, following cell surface labeling with NBs, washing with buffer for 30 min did not reduce the fluorescence (Figure S2C), indicating that the extracellular GFP–NB interaction is sufficiently stable for probing signaling on the physiological time scales relevant to GPCR signaling.
Many membrane receptors are oligomeric, raising the need for labeling of each subunit within a complex if one is going to use NBs for efficient optical control of ligand binding with a tethered agonist. Because mGluRs are dimers in living cells16,17 and two agonists are required for full activation,16,18 we sought to determine if two NBs can bind to an mGluR2 dimer. Compared to typical antibodies (~150 kDa), we hypothesized that the small size of NBs (~15 kDa) would decrease the chance of steric hindrance between NBs on adjacent subunits. To measure the stoichiometry of SNAP-tagged NBs bound to GFP-mGluR2 dimers, we performed a single-molecule pulldown (SiMPull) of lysate from cells expressing GFP-mGluR2 and labeled with NB-SNAP(Alexa-546). After immobilization of GFP-mGluR2 on a polyethylene-glycol (PEG) passivated coverslip using an anti-mGluR2 antibody (Figure 2D), we used photobleaching step analysis of spots in the red channel that were colocalized with GFP spots to determine the number of NBs per GFP-mGluR2 (Figures 2D and E). Approximately 45% of these colocalized red spots bleached in two steps, showing that two NBs can indeed bind to an mGluR2 dimer (Figure 2E). Importantly, very few spots were observed when lysate from cells expressing mGluR2-GFP was immobilized (Figure S2D).
Purified SNAP-Tagged NBs Allow Photoactivation of mGluR2 via BGAG Photoswitches
Given the efficient binding of SNAP-tagged NBs to GFP-mGluR2, we next tested if they could enable optical control of mGluR2 via attachment of BGAG photoswitches.8,10 As a prerequisite to optical control, we first assessed the ability of GFP-mGluR2 to retain normal function following NB conjugation. As G protein-gated inward rectifying potassium (GIRK) channels are well-characterized native downstream effectors of mGluR2 activity,19,20 we performed whole cell patch clamp recordings from HEK 293T cells coexpressing GIRK channels as a readout of activity (see Materials and Methods for details). Indeed, we observed no effect of NB attachment on apparent glutamate affinity or maximum current amplitude (Figure 3A). We next labeled GFP-mGluR2 expressing cells with either SNAP-NB or NB-SNAP along with BGAG variants of different PEG linker length (BGAG0, BGAG12, or BGAG28) (Figure 3B). Robust photocurrents were observed for most conditions with maximal photoactivation of up to 40% relative to saturating glutamate observed for NB-SNAP with BGAG12 (Figures 3C, E, and F; S3A–C). The directionality of photoswitching, with cis-BGAG serving as an agonist, was conserved for NB conjugation to all BGAG variants as was seen in previous studies with direct conjugation to an N-terminal SNAP domain.8,10 This supports a mechanism where relative efficacy of the cis and trans configurations are due to alterations in the inherent pharmacology of the azobenzene–glutamate moiety rather than the relative reach of the two forms. In the presence of saturating (1 mM) glutamate, no photoswitching was observed, indicating that BGAG12 does not serve as a partial agonist or antagonist in the NB-tethered context (Figure S3D). Subtle differences in the BGAG length dependence of SNAP-NB and NB-SNAP indicate differences in the relative orientation of the attachment domain to the glutamate binding site. SNAP-NB showed similar efficacy with BGAG0 and BGAG12, whereas NB-SNAP showed a twofold increase in efficacy for BGAG12 relative to BGAG0 (Figure 3F). This likely indicates that the SNAP domain of SNAP-NB is, on average, closer to the glutamate binding site than the SNAP domain of NB-SNAP. Both SNAP-NB and NB-SNAP showed negligible photoswitching with BGAG28, consistent with the notion that PEG chains of this length provide too low of an effective concentration due to the large volume of conformational space sampled. Structural models showing a possible arrangement of SNAP-tagged nanobodies relative to GFP-mGluR2 indicate distinct geometries of the two systems which could account for differences in photoswitch efficacy (Figure S3E). Protein flexibility likely also plays a major role in facilitating the ability of NB-tethered BGAGs to effectively bind and activate mGluR2. This is especially likely for BGAG0 which would be too short to reach the binding site without major flexibility introduced either from interdomain linkers (i.e., between GFP and mGluR2 or SNAP and NB) or the receptor itself. Importantly, photocurrents mediated by NB-SNAP were blocked by either an mGluR2/3 antagonist (Figure 3G) or a blocker of GIRK channels (Figure 3H). These controls confirm that photoactivation is indeed mediated by mGluR2-mediated activation of GIRK channels.
Figure 3.

BGAG conjugation to SNAP-tagged NBs permits robust photoactivation of GFP-mGluR2. (A) Glutamate dose–response curves from GIRK activation assays indicate that conjugation of SNAP-NB or NB-SNAP does not change the glutamate response of mGluR2. Current amplitudes are normalized to the response to saturating 1 mM glutamate. (B) Schematic showing arrangement of BGAG-labeled SNAP-tagged NBs attached to GFP-mGluR2 and light-mediated receptor activation via photoisomerization of the azobenzene moiety of BGAG. (C and D) Representative traces showing reversible photoactivation of GFP-mGluR2 by BGAG12 conjugated to (C) SNAP-NB or (D) NB-SNAP. 380 nm light (magenta) induces photoisomerization of the azobenzene from trans to cis, and 500 nm light (green) reverses the process. (E) Representative current trace for GFP-mGluR2 labeled with NB-SNAP + BGAG12 showing repeated photoactivations followed by application of saturating glutamate. (F) Summary bar graph showing relative photoswitch efficacy compared to saturating 1 mM glutamate for BGAG variants of different length conjugated to either SNAP-NB or NB-SNAP. The numbers of cells tested for each condition are shown in parentheses. (G and H) Photoactivation of GFP-mGluR2 with NB-SNAP conjugated to BGAG12 is blocked by either LY341495 (G), a competitive mGluR2/3 antagonist, or barium (H), a pore blocker of GIRK channels.
Coexpression of Genetically Encoded SNAP-Tagged NBs Enables Optical Control of mGluR2
While the ability to target proteins for optical control using purified nanobodies opens many experimental possibilities that do not require genetic manipulation, an alternative approach would take advantage of the relative ease at which NBs can be genetically encoded and expressed in mammalian systems.21,22 Genetically encoding NBs that can then target specific proteins and provide a tethering point for photoswitch attachment would allow for cell-type specific targeting, a crucial property for dissecting the role of specific signaling molecules in many physiological systems, especially within the circuitry of the nervous system. To see if NBs can be used to target extracellular protein domains, we added an mGluR signal sequence (ss) to NB-SNAP to promote trafficking through the endoplasmic reticulum (ER) via the same pathway as integral membrane proteins. This should allow NB-SNAP to remain in the same compartment (ER lumen, Golgi lumen, extracellular space) as the GFP of GFP-mGluR2 during the entire trafficking process (Figure 4A). Consistent with this, coexpression of ss-NB-SNAP and GFP-mGluR2, but not mGluR2-GFP, led to clear surface labeling with BG-Alexa-546 (Figure 4B; S4A). Coexpression of NB-SNAP, without the addition of the ss, was unable to label surface GFP-mGluR2 (Figure S4B). We next tested the ability of genetically encoded, coexpressed NB-SNAP to optically control GFP-mGluR2. Similar to what was observed with purified NB-SNAP, BGAG12 produced clear photocurrents that were ~20% in amplitude (20.4 ± 4.7%, n = 6 cells) relative to saturating 1 mM glutamate (Figure 4C). Together, these data demonstrate that anti-GFP NBs may be genetically encoded to efficiently target proteins for optical control. In addition to the advantage of permitting cell-type specific optical control in complex systems, this approach also provides the flexibility to either label a nanobody–GFP complex for optical control on the cell surface (using a membrane-impermeable photoswitch or fluorophore) or inside of a cell (using a membrane-permeable photoswitch or fluorophore) to probe the distinct roles and regulation of a signaling protein in different cellular locations, an increasingly appreciated aspect of GPCR function.23,24
Figure 4.

Genetic encoding of SNAP-tagged anti-GFP nanobodies allows targeting of extracellular GFP-tags for optical control. (A) Schematic showing cotrafficking and assembly of ss-NB-SNAP with GFP-mGluR2. (B) Representative images showing labeling of surface-expressed GFP-mGluR2 with coexpressed ss-NB-SNAP following labeling with Alexa-546. Scale bars = 20 μm. (C) Representative traces showing photoactivation of GFP-mGluR2 by BGAG12 conjugated to coexpressed ss-NB-SNAP.
DISCUSSION
In this study, we established a new, general approach to targeting signaling proteins for optical control using NPCs. In contrast to a recent study using an antibody to deliver an irreversible photosensitizer to inactivate AMPA receptors,25 this work establishes the suitability of immunochemistry for targeting a membrane protein for reversible optical control, opening the door to the manipulation of native proteins with high spatiotemporal precision. Similarly, Scholler et al.26 recently reported allosteric nanobodies to manipulate mGluR2, but these tools also lack reversibility and were not shown to permit genetic encoding for cell-type targeting, limiting their utility for precise, dynamic manipulation of receptors. In addition, the ability of NPCs to manipulate signaling via the orthosteric binding site via PORTLs permits a more physiological receptor perturbation through a defined, native mechanism.
Recent work has supported the idea that antibody-based approaches to manipulating GPCRs show great promise for treatment of a wide range of diseases, including various cancers and neurological disorders.27 Importantly, the proof-of-principle reported in this manuscript using GFP-targeting NBs indicates that NB-mediated optical control is possible and should be widely applicable via other types of antibodies and other receptor subtypes and may ultimately be useful clinically by adding reversible spatiotemporal control to traditional antibody–drug conjugates (ADCs).28 Crucially, we demonstrate that NPCs can either be applied as purified complexes, which are most applicable in a clinical context, or as genetically encoded tools that can be used to target specific cell types for maximal precision in mechanistic biological studies. While the ability to target NBs directly to wild-type, endogenous proteins of interest offers great potential, the targeting of proteins with incorporated GFP tags also offers many advantages. Strong antibodies or nanobodies with sufficient strength and specificity are not available for many antigens but GFP, for which high affinity nanobodies have been developed, extensively characterized and applied in vitro and in living cells11,29,30 has been successfully incorporated into many proteins, often in multiple different positions, that have been well-characterized functionally.31 In addition, recent genetic advances based on the CRISPR-Cas9 system have made it increasingly feasible to incorporate GFP or other tags into native proteins either at the transgenic level or even in postmitotic cells in the nervous system.32,33 Ultimately, the existing toolbox of photoswitchable ligands for SNAP and CLIP with different spectral properties, NBs and single chain antibodies for ubiquitous tags, along with the complement of genetic lines for targeting NB expression, should allow for sophisticated multiplexed experiments that can manipulate native signaling proteins to probe their roles in physiology with optimal precision.
MATERIALS AND METHODS
Chemical Synthesis
Chemical synthesis was performed as previously described.8,10
Cloning, Purification, and Characterization of Nanobodies
All fusion proteins were cloned using standard cloning techniques. SNAP NB-fusions with an N-terminal Strep-tag and 10xHis-tag were cloned into a pBAD expression vector for bacterial expression, wtGFP with an N-terminal Strep-tag and a C-terminal 10xHis-tag was cloned into pET51b(+) for bacterial expression, and complete amino acid sequences for constructs used can be found in the Supporting Information. ss-NB-SNAP contains the mGluR5 signal sequence (MVLLLILSVLLLKEDVRGSA) at the N-terminus. For purification, SNAP-fused NBs were expressed in the E. coli strain DH10B, and wtGFP was expressed in BL21(DE3) pLysS cell. All LB media contained ampicillin (100 μg/mL) for protein expression. A culture was grown at 37 °C until an OD600 of 0.6 was reached at which point cells were induced with L-arabinose (0.02%, (w/v)) or IPTG (1 mM). Protein constructs were expressed overnight at 25 °C. Cells were harvested by centrifugation and sonicated to produce cell lysates. The lysate was cleared by centrifugation and purified by Ni-NTA resin (Thermofisher) and Strep-Tactin II resin (IBA) according to the manufacturer’s protocols. Purified protein samples were stored in 50 mM Hepes, 50 mM NaCl (pH 7.3) and either flash frozen and stored at −80 °C or stored at 4 °C. SNAP-tag was labeled with fluorescent dyes or photoswitchable compounds at 2–10 μM protein with a 2–5 fold molar ratio of fluorophores in 50 mM Hepes, 50 mM NaCl (Ph 7.3). For in vitro experiments, excess label was removed by centrifugal filter devices with 10 kDa cutoff (Microcon YM-10, Millipore), and the labeled protein was stored in 50 mM Hepes, 50 mM NaCl (pH 7.3) at 4 °C.
Activity testing of purified nanobody–SNAP fusions against wtGFP was performed in 50 mM Hepes, 50 mM NaCl (pH 7.3), 0.05 mg mL−1 BSA, 0.05% Triton X-100 in black 384-well plates (Corning, 3820). Fluorescent intensities were recorded on a Spark 20 M (Tecan) with excitation at 480 nm, emission at 535 nm, and bandwidths of 5 nm for both excitation and emission. The dose–response was fit with eq 1:
| (1) |
with
| (2) |
where Fsb is defined in eq 2, I is the intensity of light at 535 nm, Rmax is the maximum light emitted at 535 nm, Rmin is the minimum light emitted at 535 nm, L is the concentration of wtGFP and fixed to 4 nM, x is the concentration of the nanobody–SNAP fusion protein, Kd is the calculated dissociation constant, and N accounts for nonspecific binding. The fit was performed in Prism (GraphPad).
Competitive labeling and in-gel imaging were used to assay the efficiency of BGAG12 labeling of SNAP-tag NB-fusions. SNAP-NB or NB-SNAP (5 μM) was treated with BGAG12 (15 μM, 3-fold equivalence) in 50 mM Hepes, 50 mM NaCl (pH 7.3). Aliquots were taken at indicated time points and labeled with BG-TMR (150 μM, 30 equiv) for 1 h. The reaction was then quenched with SDS-loading buffer, heated to 95 °C for 10 min, and stored at −20 °C until separated by SDS-PAGE. In-gel fluorescence of BG-TMR was measured on a ChemiDoc MP imager (Bio Rad) equipped with a CCD camera. Imaging was performed using green epifluorescent light and emission filters for Cy3 (605/50 nm). The gel was then labeled with coomasie. Image analysis was performed with ImageJ (W. Rasband, NIH). Fluorescence intensities in the Cy3 channel were background subtracted and normalized to the fluorescence with no BGAG12 present at time 0.
Mass Analysis
A Bruker maXis II ETD quadrupole-time-of-flight (QTOF) instrument with reverse phase liquid chromatography was used to measure accurate protein masses. All measurements were performed by injecting 5 μL of protein in 50 mM Hepes, 50 mM NaCl (pH 7.3) at a concentration of 5 μM. Proteins were first passed over a C8 wide pore column (100 mm) heated to 50 °C in a gradient of H2O + 1% formic acid (A)/MeCN + 1% formic acid (B) from 10 to 98% B over 6 min. The charge envelope of the protein within the corresponding spectrum was deconvoluted using Bruker’s Data-Analysis software with the Maximum Entropy deconvolution option enabled.
Molecular Biology, Cell Culture, and Transfection
GFP-mGluR2 in pcDNA3 was produced using standard PCR-based techniques. In brief, enhanced GFP (eGFP) and a 17 amino acid glycine-rich linker were added after the mGluR2 signal sequence. HEK293T cells were maintained in DMEM media supplemented with 5% fetal bovine serum. Cells were seeded on 18 mm coverslips and transfected 12 h later using Lipofectamine 3000 (Thermo Fisher). Then, 0.3–0.7 μg each of GFP-mGluR2 DNA, GIRK1-F137S DNA (for electrophysiology experiments), and NB-SNAP (for NB coexpression experiments) was added to each well. The GIRK1-F137S mutant was used to allow for channel homotetramerization, as previously reported,34 which minimizes the number of plasmids that need to be transfected without altering the properties of the GIRK channel. Experiments were performed 24–48 h after transfection.
Fluorescence Imaging
Imaging was performed on an IX-73 microscope (Olympus) with a cellTIRF laser illumination system and an sCMOS ORCA-Flash4 v3.0 camera (Hamamatsu). GFP was excited using a 488 nm laser diode and a 561 nm DPSS laser was used for Alexa-546. Cellular imaging was performed in wide field mode using a 60× objective (NA = 1.49). Data was acquired using cellSens software (Olympus) and analyzed using ImageJ. Prior to imaging, cells were incubated with purified, Alexa-546 labeled NBs (50 nM) for 45 min at 37 °C. For genetically encoded ss-NB-SNAP experiments, BG-Alexa-546 was applied at 1 μM for 45 min at 37 °C. Single molecule pulldown of mGluR2 was performed as previously described.16 Briefly, following immobilization using an anti-mGluR2 antibody, images were acquired with a 100× objective (NA = 1.49) at 20 Hz in TIRF mode, and bleaching steps for individual molecules were manually analyzed using a previously described custom software.35
Patch Clamp Electrophysiology
Whole cell patch clamp electrophysiology was performed as previously described.10 In brief, HEK 293T cells coexpressing GFP-mGluR2 along with GIRK1-F137S (homotetramerization mutant) were voltage clamped at −60 mV using an Axopatch 200B amplifier (Molecular Devices) attached to a 3–6 MΩ pipet filled with intracellular solution containing 140 mM KCl, 10 mM Hepes, 5 mM EGTA, and 3 mM MgCl2 (pH 7.4). Recordings were performed in high potassium extracellular solution containing 120 mM KCl, 25 mM NaCl, 1 mM MgCl2, 2 mM CaCl2, and 10 mM Hepes (pH 7.4). Cells were incubated with purified NBs (100 nM) and/or BGAG variants (10 μM) for 45 min at 37 °C. Prior to any recordings, cells were extensively washed with high potassium extracellular solution using a gravity-driven perfusion system, which was also used to control glutamate application. Light stimulation (~1 mW/mm2) was provided by a coolLED pE-4000 illumination system that was controlled via pClamp software via a Digidata 1550B digitizer. Data were analyzed with Clampfit (Molecular Devices) and Prism (GraphPad).
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Y. Isacoff (UC Berkeley), J. Dittman (Weill Cornell), and P. Leippe (LMU) for helpful discussion. We also thank S. Fabritz (MPImR) for assistance with mass spectrometry. D.T. is grateful to the Center for Integrated Protein Science, Munich, and the European Research Council (ERC Advanced Grant 268795) for financial support. K.J. is grateful to finding from the Swiss Science Foundation, the NCCR Chemical Biology, and EPFL. This work was supported by an R35 grant to J.L. from the National Institute of General Medicine (1 R35 GM124731). J.B. acknowledges support from the “EPFL Fellows” fellowship program cofunded by Marie Sklodowska-Curie, Horizon 2020 Grant 665667.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.8b00628.
Details on labeling (including mass spectrometry), binding properties, NB-mediated photoactivation, extracellular GFP targeting, protein sequences, and a summary for manipulation of GPCRs and other membrane proteins (PDF)
REFERENCES
- (1).Tischer D, and Weiner OD (2014) Illuminating cell signalling with optogenetic tools. Nat. Rev. Mol. Cell Biol 15, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Deisseroth K (2015) Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci 18, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Spangler SM, and Bruchas MR (2017) Optogenetic approaches for dissecting neuromodulation and GPCR signaling in neural circuits. Curr. Opin. Pharmacol 32, 56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Broichhagen J, Frank JA, and Trauner D (2015) A Roadmap to Success in Photopharmacology. Acc. Chem. Res 48, 1947–1960. [DOI] [PubMed] [Google Scholar]
- (5).Broichhagen J, and Trauner D (2014) The in vivo chemistry of photochromic tethered ligands. Curr. Opin. Chem. Biol 21, 121–127. [DOI] [PubMed] [Google Scholar]
- (6).Reiner A, Levitz J, and Isacoff EY (2015) Controlling ionotropic and metabotropic glutamate receptors with light: principles and potential. Curr. Opin. Pharmacol 20, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Levitz J, Pantoja C, Gaub B, Janovjak H, Reiner A, Hoagland A, Schoppik D, Kane B, Stawski P, Schier AF, Trauner D, and Isacoff EY (2013) Optical control of metabotropic glutamate receptors. Nat. Neurosci 16, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Broichhagen J, Damijonaitis A, Levitz J, Sokol KR, Leippe P, Konrad D, Isacoff EY, and Trauner D (2015) Orthogonal Optical Control of a G Protein-Coupled Receptor with a SNAP-Tethered Photochromic Ligand. ACS Cent. Sci 1, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Berry M, Holt A, Levitz J, Broichhagen J, Gaub B, Visel M, Stanley C, Aghi K, Kim JY, Trauner D, Flannery J, and Isacoff EY (2017) Restoration of patterned vision with an engineered G protein-coupled receptor. Nat. Commun 8, 1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Levitz J, Broichhagen J, Leippe P, Konrad D, Trauner D, and Isacoff EY (2017) Dual optical control and mechanistic insights into photoswitchable group II and III metabotropic glutamate receptors. Proc. Natl. Acad. Sci. U. S. A 114, E3546–E3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, Pellis M, Muyldermans S, Casas-Delucchi CS, Cardoso MC, Leonhardt H, Hopfner KP, and Rothbauer U (2010) Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol 17, 133–138. [DOI] [PubMed] [Google Scholar]
- (12).Beghein E, and Gettemans J (2017) Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Interaction Analysis, and Protein Function Exploration. Front. Immunol 8, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Manglik A, Kobilka BK, and Steyaert J (2017) Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu. Rev. Pharmacol. Toxicol 57, 19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mollwitz B, Brunk E, Schmitt S, Pojer F, Bannwarth M, Schiltz M, Rothlisberger U, and Johnsson K (2012) Directed evolution of the suicide protein O(6)-alkylguanine-DNA alkyltransferase for increased reactivity results in an alkylated protein with exceptional stability. Biochemistry 51, 986–994. [DOI] [PubMed] [Google Scholar]
- (15).Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, and Johnsson K (2003) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol 21, 86–89. [DOI] [PubMed] [Google Scholar]
- (16).Levitz J, Habrian C, Bharill S, Fu Z, Vafabakhsh R, and Isacoff EY (2016) Mechanism of Assembly and Cooperativity of Homomeric and Heteromeric Metabotropic Glutamate Receptors. Neuron 92, 143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Doumazane E, Scholler P, Zwier JM, Trinquet E, Rondard P, and Pin JP (2011) A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 25, 66–77. [DOI] [PubMed] [Google Scholar]
- (18).Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prezeau L, and Pin JP (2004) Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat. Struct. Mol. Biol 11, 706–713. [DOI] [PubMed] [Google Scholar]
- (19).Dutar P, Petrozzino JJ, Vu HM, Schmidt MF, and Perkel DJ (2000) Slow synaptic inhibition mediated by metabotropic glutamate receptor activation of GIRK channels. J. Neurophysiol 84, 2284–2290. [DOI] [PubMed] [Google Scholar]
- (20).Watanabe D, and Nakanishi S (2003) mGluR2 postsynaptically senses granule cell inputs at Golgi cell synapses. Neuron 39, 821–829. [DOI] [PubMed] [Google Scholar]
- (21).Tang JC, Szikra T, Kozorovitskiy Y, Teixiera M, Sabatini BL, Roska B, and Cepko CL (2013) A nanobody-based system using fluorescent proteins as scaffolds for cell-specific gene manipulation. Cell 154, 928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, and von Zastrow M (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Irannejad R, Tsvetanova NG, Lobingier BT, and von Zastrow M (2015) Effects of endocytosis on receptor-mediated signaling. Curr. Opin. Cell Biol 35, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Thomsen AR, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, and Lefkowitz RJ (2016) GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 166, 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Takemoto K, Iwanari H, Tada H, Suyama K, Sano A, Nagai T, Hamakubo T, and Takahashi T (2016) Optical inactivation of synaptic AMPA receptors erases fear memory. Nat. Biotechnol 35, 38–47. [DOI] [PubMed] [Google Scholar]
- (26).Scholler P, Nevoltris D, de Bundel D, Bossi S, Moreno-Delgado D, Rovira X, Moller TC, El Moustaine D, Mathieu M, Blanc E, McLean H, Dupuis E, Mathis G, Trinquet E, Daniel H, Valjent E, Baty D, Chames P, Rondard P, and Pin JP (2017) Allosteric nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat. Commun 8, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hutchings CJ, Koglin M, Olson WC, and Marshall FH (2017) Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat. Rev. Drug Discovery 16, 787–810. [DOI] [PubMed] [Google Scholar]
- (28).Beck A, Goetsch L, Dumontet C, and Corvaia N (2017) Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discovery 16, 315–337. [DOI] [PubMed] [Google Scholar]
- (29).Kubala MH, Kovtun O, Alexandrov K, and Collins BM (2010) Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 19, 2389–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sommese RF, Hariadi RF, Kim K, Liu M, Tyska MJ, and Sivaramakrishnan S (2016) Patterning protein complexes on DNA nanostructures using a GFP nanobody. Protein Sci. 25, 2089–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Giepmans BN, Adams SR, Ellisman MH, and Tsien RY (2006) The fluorescent toolbox for assessing protein location and function. Science 312, 217–224. [DOI] [PubMed] [Google Scholar]
- (32).Mikuni T, Nishiyama J, Sun Y, Kamasawa N, and Yasuda R (2016) High-Throughput, High-Resolution Mapping of Protein Localization in Mammalian Brain by In Vivo Genome Editing. Cell 165, 1803–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nishiyama J, Mikuni T, and Yasuda R (2017) Virus-Mediated Genome Editing via Homology-Directed Repair in Mitotic and Postmitotic Cells in Mammalian Brain. Neuron 96, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, and Logothetis DE (1997) Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J. Biol. Chem 272, 31553–31560. [DOI] [PubMed] [Google Scholar]
- (35).Ulbrich MH, and Isacoff EY (2007) Subunit counting in membrane-bound proteins. Nat. Methods 4, 319–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.