

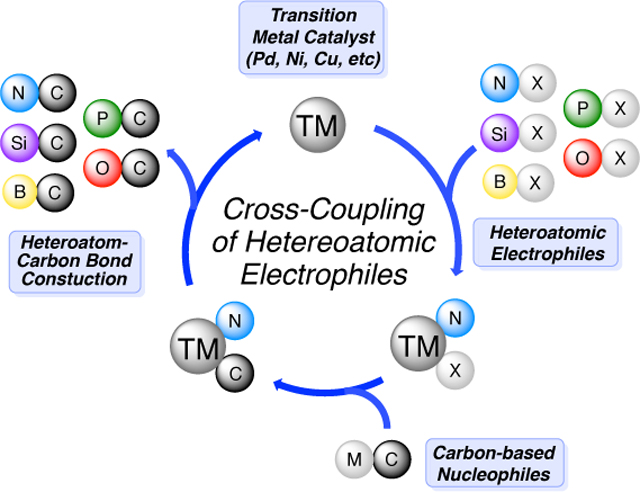
Abstract
At the advent of cross-coupling chemistry, carbon electrophiles based on halides or psuedohalides were the only suitable electrophilic coupling partners. Almost two decades passed before the first cross-coupling reaction of heteroatom-based electrophiles was reported. Early work by Murai and Tanaka initiated investigations into silicon electrophiles. Narasaka and Johnson pioneered the way in the use of nitrogen electrophiles; while Suginome began the exploration of boron electrophiles. The chemistry reviewed within provides prospective on the use of heteroatomic electrophiles specifically silicon-, nitrogen-, boron-, oxygen- and phosphorus-based electrophiles in transition-metal catalyzed cross-coupling. For the purposes of this review, a loose definition of cross-coupling is utilized; all reactions minimally proceed via an oxidative addition event. Although not cross-coupling in a traditional sense, we have also included catalyzed reactions that join a heteroatomic electrophile with an in situ generated nu-cleophile. However, for brevity, those involving hydroamination or C–H activation as a key step are largely excluded. This work includes primary references published up to and including October 2018.
TOC GRAPHIC:
1. Introduction
The importance of cross-coupling reaction is difficult to understate. Since early studies by Heck were disclosed the late 1960s, the development and application of cross-coupling chemistry has had an immense impact on the field of organic chemistry.1–7 In the nearly 50 years since those initial reports, an explosion of diversity in metals, ligands, and coupling partners has led to thousands of publications in this field, and many reviews and books have been published cataloging the advancements.8–44 This has led to cross-coupling reactions being a cornerstone technology in vastly diverse research industries, including pharmaceuticals,34–35 agrochemicals,37 polymers,45–47 and electronics48 – among others. Indeed, the importance of this general area was recognized by the award of the 2010 Nobel Prize in Chemistry to Professors Heck, Suzuki, and Negishi for their development of palladium-catalyzed cross-coupling.
As outlined below, the vast majority of cross-coupling reactions have focused on the use of carbon-based electrophiles. And, until recently, most of those have focused exclusively on sp2-hybridized carbon (C-sp2) electrophiles. The reasons for this stem both from the mechanistic aspects of early catalytic cross-coupling reactions, as well as a traditional focus on carboncarbon (C–C) bond formation in organic reaction method development. However, in parallel to the development of these “traditional” carbon-electrophile based cross-coupling reactions, non-traditional cross-coupling reactions involving non-carbon-based electrophiles have also been explored. Until recently, however, work in this area has progressed at a much slower pace. In the past few years, increased interest in transition-metal catalyzed reactions of these hetere-oatomic electrophiles has demonstrated that cross-couplings protocols cannot only utilize these reagents, but that interesting and useful chemistry can emerge from them. And in many cases, these “heteroatomic” cross-coupling reactions lead to more efficient access to hetereoatom-containing organic molecules than their “traditional” cross-coupling counterparts. Although certain classes of cross-coupling reactions have been described in highly-focused reviews,8, 10–14, 18–20, 26–27, 36, 38 to date there has been no comprehensive review of cross-coupling reactions that employ heteroatomic electrophiles. The chemistry reviewed within provides prospective on the use of heteroatomic electrophiles specifically silicon-, nitrogen-, boron-, oxygen- and phosphorus-based electrophiles in transition-metal catalyzed cross-coupling.
For the purposes of this review, a loose definition of cross-coupling is utilized to include all reactions that minimally proceed via an oxidative addition event of a transition-metal catalyst to a non-carbon-based electrophile. Because, in most cases, the details are not yet known, we have made no attempt to define or limit the mechanism by which this “oxidative addition event” occurs. Although not considered cross-couplings in a traditional sense, we have also included catalyzed reactions that join a heteroatomic electrophile with an in situ generated nucleophile. This work includes references published up to and including October 2018. Finally, we also note that heteroatomic electrophiles (particularly those of nitrogen) have been used in a range of other important reactions, including hydroamination and C-H functionalization. We have not included those here, as they are beyond the scope of cross-coupling and are expansive in their own right, but it is important to note the key contributions in these closely related areas.49–77
1.1. Traditional Cross-Coupling Reactions
To contrast cross-couplings of heteroatomic electrophiles, it is important to briefly discuss the chemistry of more “traditional” cross-coupling reactions. Going back to the work of Heck, the earliest examples of catalytic cross-coupling reactions (Heck,1–5 Kumada-Corriu,78–79 Sonogashira,80 Negishi,81 Stille,82–83 Suzuki,84–85 etc.) focused on the formation of carboncarbon bonds. Over the years, a vast number of related reactions have been developed. While a comprehensive cataloging of these transformation is not possible here, rough mechanistic outlines for several key reactions are shown in Figure 1.86 Prototypically, all of these reactions involve the cross-coupling of a C-sp2 electrophile (typically an aryl or vinyl halide or pseudohalide) to a variety of carbon-based nucleophiles. All of the reactions involve the oxidative addition of a low-valent metal complex into the electrophilic center. The resultant electrophilic organometallic complex, featuring a now oxidized metal center, then reacts with the nucleophile to generate an organometallic complex bearing two carbon centers. In the case of the Heck reaction, the nucleophilic component is an alkene that reacts via migratory insertion.87 In most other cases, the nucleophile is a metalated organic compound that undergoes transmetalation. Although the mechanistic details of these later steps vary considerably by reaction, in all cases, the formation of a new carbon-carbon (C–C) bond ultimately results, and the metal center is returned to the catalytic cycle via a reductive process (often concurrently with C–C bond formation). Reactions of this type have been employed countless times in in-dustrial applications, and have been instrumental in the construction of unsaturated organic molecules.
Figure 1.
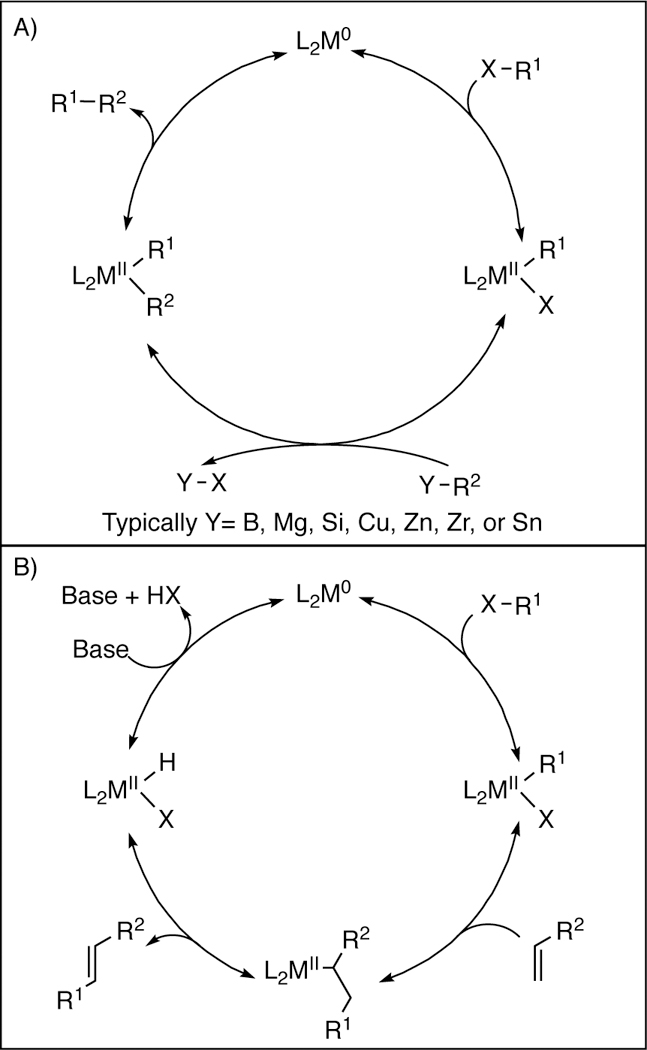
a) General Mechanism of Traditional Cross-Coupling Reactions (Suzuki Y=B, Kumada Y=Mg, Hiyama Y=Si, Sonogashira Y=Cu, Negishi Y=Zn, Stille Y=Sn) b) Mechanism of the Heck Reaction.
Historically, cross-coupling reactions had been limited to the use of C-sp2 electrophiles both because of the relative ease of oxidative addition and relative stability of the organometallic intermediated that resulted. For example, for many years, reactions involving C-sp3 electrophiles suffered both from slow oxidative addition and rapid β-hydride elimination from the resulting organometallic intermediates. However, more than two decades ago, early examples from Tamura88–90 and Miyaura91 paved the way for Knochel,92–94 Fu,95–97 and others25, 98–100 to understand how to control reactions involving C-sp3 electrophiles, and pioneered their development as useful agents in cross coupling reactions (see Figure 2 for an example). Today, a wide variety of C-sp3 electrophiles are broadly useful in Csp3–C cross-couplings to make saturated organic compounds, and more recently have been shown to also be compatible in related reductive cross-couplings processes.101–102
Figure 2.
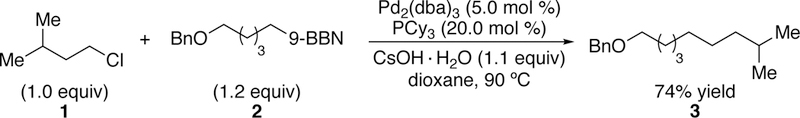
Example of C-sp3 Cross-Coupling of a Primary Alkyl Chloride with a Primary Alkyl 9-BBN Reagent from Fu.97
At about the same time C-sp3 electrophiles began to be explored in cross-coupling reactions, Buchwald and Hartwig (following earlier independent work from Migita103 and Boger104) recognized that heteroatomic nucleophiles could also be used in cross-coupling processes. The resultant amination reaction, now known as Buchwald-Hartwig amination, successfully coupled C-sp2 electrophiles with a range of nitrogen nucleophiles, and has become a critical reaction in the development of pharmaceutical agents and other amine containing materials. Over the intervening years, a number of related heteroatomic nucleophiles, and ranges of catalysts have also been shown to be compatible with this general process.8, 105–116 Mechanistically, studies have shown that these processes are closely related to the more classical C–C bond forming reactions, and proceed via initial oxidative addition of the low-valent metal catalyst to the C-sp2 electrophile, which undergoes exchange of anionic ligands, and ultimately reductive elimination provides the observed products (Figure 3).117–118 Additionally, during this time, catalytic protocols for cross-coupling aryl halides to heteroatomic nucleophiles based on classical Goldberg reactions were developed,119–120 as well as oxidative protocols to cross-couple nucleophilic C-sp2 nucleophiles (e.g. aryl boronic acids) to heteroatomic nucleophiles.121–124 Not only are all of these protocols important means of accessing heteroatom-containing organic molecules, they also highlight the compatibility of heteroatoms in cross-coupling reactions.
Figure 3.
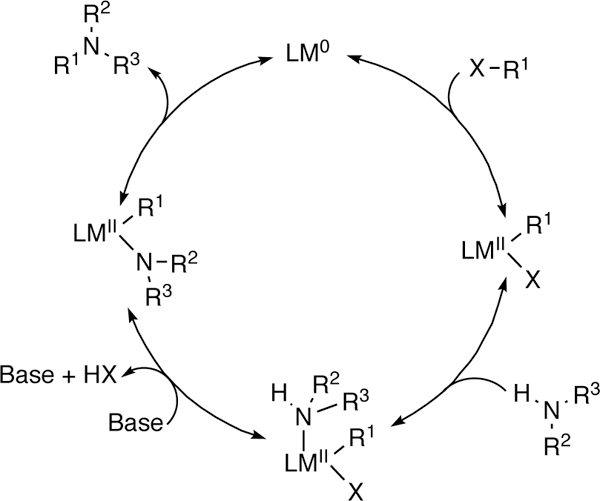
Mechanism of the Buchwald-Hartwig Reaction.
Common to all of the examples outlined above is the use of carbon-based electrophiles in the cross-coupling reaction.
1.2. Introduction to Heteroatomic Electrophiles
Beyond carbon, electrophilic species of many of the main group elements exist, and provide interesting opportunities to explore their use in cross-coupling reactions. From a fundamental standpoint, understanding which types of electrophiles are compatible with cross-coupling reactions and what types of nucleophiles they can be combined with in catalyst-controlled reactions provides an exciting intellectual arena for discovery. From a practical standpoint, as elaborated on below, the controlled introduction of heteroatoms into organic structures remains a key challenge in organic synthesis, as heteroatoms impart function and reactivity to most organic molecules. Cross-coupling of heteroatomic electrophiles presents an underexplored, yet highly promising means to accomplish those goals. And, in principle, by rendering compounds with different types of heteroatoms electrophilic, similar synthetic strategies can emerge for introducing disparate types of heteroatomic structures.
Through the lens of “traditional” cross-coupling reactions involving carbon electrophiles, there are two main classes of heteroatomic electrophiles to be considered for cross-coupling; those with electrophilic elements less electronegative than carbon (i.e. Si, B, P, etc), and those elements more electronegative than carbon (i.e. N and O). Each class presents unique challenges and opportunities.
Elements that are less electronegative than carbon are naturally electropositive and easily rendered into electrophiles. In contrast, in most cases, preparing nucleophiles of electropositive elements - such as would be required in “traditional” cross-couplings - is often challenging and frequently requires highly reducing conditions, which limits the scope of the reagents that can be prepared. The ready availability of these classes of electrophiles make exploration of their cross-coupling reactions particularly attractive.
In contrast, for elements more electronegative than carbon, preparation of electrophilic species is often less straightforward than for their nucleophilic counterparts. As detailed below, however, in many cases, access to these heteroatomic electrophiles is still readily accomplished, and in many cases, the reagents are remarkably easy to handle once prepared. The slight synthetic cost of preparing these reagents, however, is vastly off-set by the opportunities these reagents provide for developing new classes of heteroatom-installing reactions. Indeed, because the polarity of disconnections is reversed (umpoled) compared to traditional approaches, new coupling partners, new mechanistic paradigms, and new opportunities for control of reactivity present themselves.
As is cataloged in this review, regardless of the electronegativity of the heteroatomic electrophile, new, exciting, and efficient synthetic pathways have emerged by leveraging the reactivity of these reagents in cross-coupling reactions.
As mentioned above, in addition to the new strategies and efficiencies that can be gained by the use of heteroatomic electrophiles in cross-coupling reactions, all of these methods lead directly to the installation of heteroatoms into organic molecules. And the construction of carbon-heteroatom bonds (particularly carbon-silicon, carbon-boron, carbon-nitrogen, carbon-oxygen and carbon-phosphorus bonds, which are the focuses of this review) are of significant importance.
For example, organic compounds bearing compounds bearing silicon125–126 have increasing been used not only as coupling partners in allylation, Hiyama, and related cross-couplings,127–133 but also as masked alcohols or carbonyls through the Fleming-Tamao oxidation.134–135 Organosilicon compounds have also found extensive use in industrial and aerospace applications as lubricants or cryogenic fluids.136 Silicon atoms are also found in the structures of a number of small-molecule agrochemicals including the silylmethyltriazole fungicide Flusilazole (DPX-H6573) (4, Figure 4).137–140 Organosilicon compounds also appear in medicinal chemistry applications as bioisosteres because of their subtle structural and electronic differences compared to carbon and their non-toxicity.141–143
Figure 4.
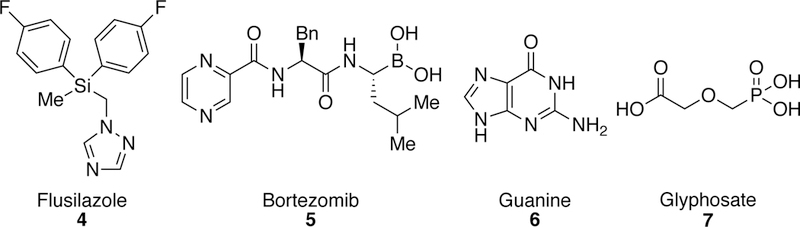
Relevant Structures Containing Silicon, Boron, Nitrogen, or Phosphorus Atoms.
Like silicon, boron has also found use as an atypical element in pharmaceutical agents,144 most notably with boronic acid-bearing chemotherapeutic agent bortezomib (VELACDE®) (5, Figure 4) being approved by the FDA in 2003.145 More notably, boronic acids, boronates, boroximes, and trifluoroborates can serve as functional group handles for further elaboration. The Suzuki-Miyaura coupling, arguably the most commonly employed cross-coupling reaction, pairs these organoboron species with carbon electrophiles to generate new C–C bonds.22, 146–149 Organoboronic acids or boronates are also employed in Chan-Lam-Evans coupling reactions110, 150–151 and Liebeskind-Srogl couplings.152 Also, boronic acids are employed in a variety of other transformations including homologations, conjugate additions, and allyl transfer reactions.153
The importance of nitrogen- and oxygen-containing molecules is hard to overstate given the prevalence of these atoms in natural products, pharmaceutical agents, agrochemicals, and the fundamental building blocks of life, proteins and DNA. Both the construction of C–N bonds and the use of C–N bonds as cross-coupling handles in metal-catalyzed processes has become a flourishing area of research.154 Key metal-mediated processes for the construction of C–N bonds will be discussed in section 3.1.155–156 As for the activation of carbon-nitrogen bonds in cross-coupling, a number of prominent examples and reviews can be found in the following references.157–161 There are a myriad of ways to install oxygen atoms in organic frameworks ranging from simple oxidations with peroxides or ozone to asymmetric processes like using Davis oxaziridine.162 Consequently, the number of cross-coupling-type methods for the introduction of oxygen is dwarfed in comparison with only a few examples of electrophilic oxygen cross-coupling detailed in Section 5.
Although C–P bonds are less prevalent in biologically relevant organic compounds, organophosphorus compounds serve important roles. Phosphorus atoms are commonly found in agrochemicals, with glyphosate (7, Figure 4), a phosphonate sold under the name Roundup, being one of the most widely used herbicides.163 Organophosphorus compounds also serve as synthetic intermediates in the commonly employed Wittig,164–165 Horner- Wadsworth-Emmons,166–167 and Seyferth-Gilbert reactions.168 The use of organophosphines, phosphites, phosphonites, and phosphoramidites as ligands is critical in many cross-coupling transformations,169–170 and they can even serves as organocatalysts in Morita-Baylis-Hilman171–172 and related Rauhut-Currier reactions.173
2. Silicon Cross-Coupling
Chronologically, the earliest studies related to the cross-coupling of heteroatomic electrophiles focused on silicon electrophiles. Initial studies focused on examining the stoichiometric addition of transition metals to silyl halides, which eventually gave way to the development of catalytic processes to prepare unsaturated organosilanes.
2.1. Early Stoichiometric Studies of Silicon-Halide Oxidative Addition
Silicon-halide bonds have been activated by transition metals beginning in the late 1970’s. The first report comes from Kuyper.174 This initial report explored the synthesis of platinum complexes derived from the activation of germanium-chloride bonds. Analogous reactions using chlorosilanes were attempted, although direct oxidative addition complexes could not be isolated or observed by NMR. However, platinum chloride complexes observed in the reaction were hypothesized to come from a transient silyl–platinum intermediate.
In 1988, Tanaka reported the first transition-metal complex from the oxidative addition of a trimethylsilylbromide (9) and the platinum(0) complex Pt(PEt3)3 (8, Figure 5).175 The resulting trans-Me3SiPtBr(PEt3)3 complex (10) was isolated as pale yellow needles and was characterized by single-crystal X-ray diffraction. Tanaka also found that the similar platinum(0) complex Pt(PEt3)4 could also be reacted with trimethylsilylbromide to give 10, albeit at a slower rate. Trimethylsilyliodide could also react with Pt(PEt3)4, but the analogous silylchloride was unreactive, likely due to the higher bond strength of the Si–Cl bond (98 kcal/mol) compared to that of Si–Br (76 kcal/mol).
Figure 5.
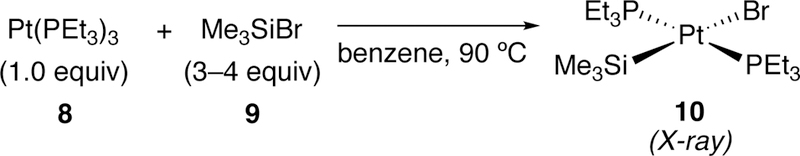
Tanaka’s Formation of a Platinum Oxidative Addition Complex from a Si–Br Bond.
This same platinum(0) precursor 8 was used again by Tanaka in 1990 for studies on the reactivity of halodisilanes at either the silylhalide bond or the disilyl bond in metal complex formation.176 Tanaka also reported on halogen-exchange reactions of rhodium and iridium complexes where the halide source is a trimethylsilylhalide.177 He suggests that two mechanisms may be possible. One of his proposed mechanisms involves the oxidative addition of the silylhalide followed by reductive elimination to produce the observed metal complex; however, his observed trends in reactivity disfavor this mechanism. Typically iridium complexes are more reactive than rhodium complexes in oxidative addition reactions but the opposite trend was observed in his system. The second proposed mechanism involves a four center σ-bond metathesis interaction, however, he could not conclusively determine a mechanism.
In 1989, Milstein synthesized and obtained a crystal structure for the oxidative addition product of methyltrichlorosilane (11) with iridium complex 12 (Figure 6).178 Reaction of the same iridium complex 12 with trimethylsilylchloride (14) did not provide the oxidative addition complex; rather, he proposes that it generates silene 16 and hydridocomplex 15, most likely arising from a β-hydride elimination.
Figure 6.
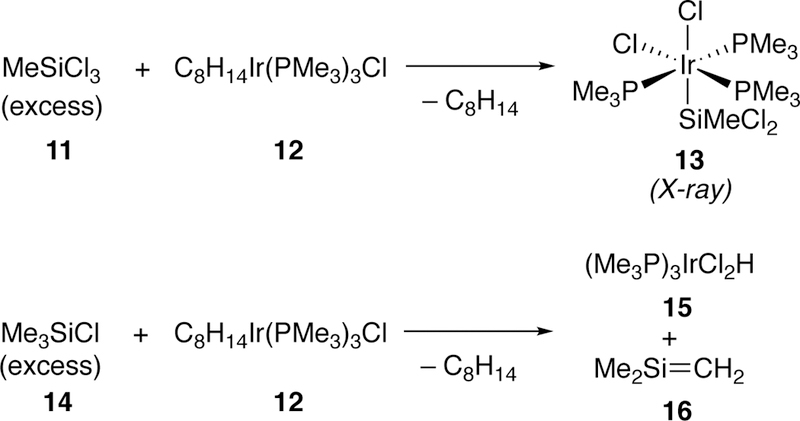
Milstein’s Iridium Oxidative Addition Complexes with Chlorosilanes.
Tanaka also investigated iridium complexes in the oxidative addition of halo(methyl)silanes including trichloromethylsilane, dichlorodimethylsilane, and trimethylsilyliodide.179 He also investigated the β-hydride elimination proposed by Milstein, and found NMR evidence to support the proposed pathway. When subjecting complexes of type 17 (Figure 7) to neohexene and silyl halides at 120 °C in a sealed NMR tube, he observed signals corresponding to the dihydrido complex, which would reasonably arise from β-hydride elimination, appear, and starting material peaks disappear. However, he could not completely rule out a disproportion reaction mechanism. More recent work on the activation of Si–Cl, Ge-Cl, and Ge–F bonds by iridium using computations has been completed by Kameo.180–181
Figure 7.
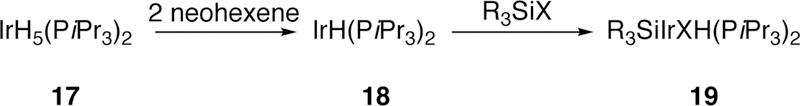
Tanaka’s Synthesis of Complexes to Probe Silene Formation by β-Hydride Elimination.
Use of platinum in the activation of silyl–halide bonds was revisited by Puddephatt in 1994 and 1996.182–183 His initial investigation studied the enthalpy differences between chloro-, bromo-, and iodotrimethylsilane in the oxidative addition step with a platinum(II) complex. It was found that the softer, larger iodide gave the most facile oxidative addition. He was able to isolate and crystallize the oxidative addition complex 21 of trimethylsilyliodide and platinum(II) precursor 20 (Figure 8).
Figure 8.
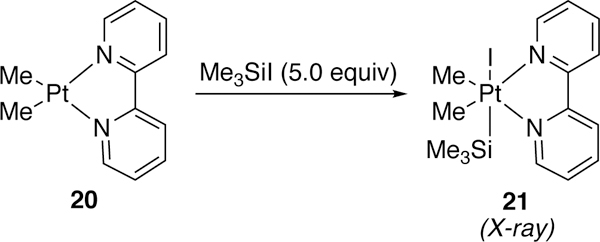
Puddephatt’s Platinum Oxidative Addition Complex with Trimethylsilyliodide.
The syntheses of additional platinum complexes were explored in his 1996 publication while platinum in the activation of silicon-chloride bonds was explored by Turculet.184 More recently, Ozerov has found that rhodium bearing a pincer ligand can been utilized in the activation of silicon-halide bonds, including less reactive silicon-chloride bonds (Figure 9).185 To our knowledge, this is the only trialkyl chlorosilane oxidative addition complex that has been characterized, and the authors argue that the reaction is driven by irreversible reductive elimination of the alkane from 22. In addition, NHC-ligated palladium complexes have been investigated for their silyliodide bond activation.186
Figure 9.
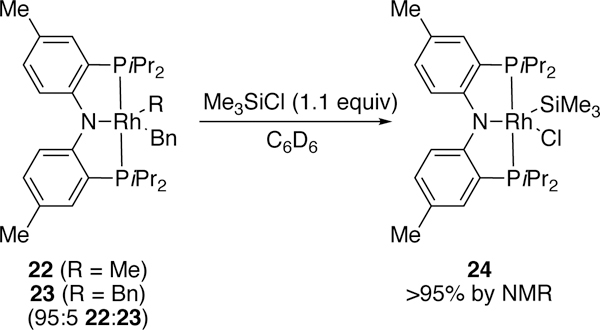
Ozerov’s Me3SiCl Oxidative Addition Complex with Rhodium.
2.2. Cross-Coupling of Silicon Electrophiles
2.2.1. Palladium-Catalyzed Silyl-Heck Reactions
In a seminal study on the cross-coupling of heteroatomic electrophiles, Tanaka also explored the formation of silicon-carbon bonds via a cross-coupling catalytic cycle initiated by his well-studied oxidative addition. In 1991, he reported a Heck-type reaction of trimethylsilyliodide with styrene derivatives in the presence of triethylamine base and catalytic PdCl2(PEt3)2.187 Only three simple styrene derivatives were examined, and modest yields ranging from 42–45% were observed. Although limited in scope and efficiency, this constitutes the first report of a silyl-Heck reaction. The trans product 26 was favored over the cis (>100:1) (Figure 10). A cumulative publication on Tanaka’s work with platinum(O) and silyl-halide bond activation including an example of platinum-mediated silyl-Heck reaction (providing only trace yield) was published in 1997.188
Figure 10.
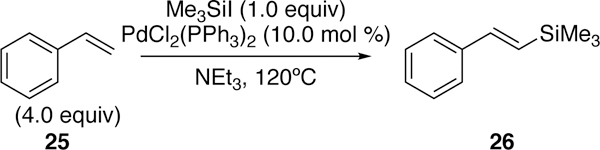
First Report of a Silyl-Heck Reaction of Styrenes and Silyliodides by Tanaka.
In 2012, an improved palladium-catalyzed silyl-Heck reaction of styrenes and isolated terminal alkenes with silyliodides to generate silyl styrenes and allyl silanes respectively was reported by our group.189–190 The key to the improved catalytic system was the use of the ligand tert-butyl diphenylphosphine. This ligand was rationalized to provide the optimal balance of steric and electronic factors while being both electron rich to support low-valent palladium and small enough to accommodate the bulky trimethylsilyl moiety. A variety of styrenes including those bearing heterocycles were coupled with iodotrimethylsilane giving exclusively the E-products (e.g. 29–31, Figure 11).
Figure 11.
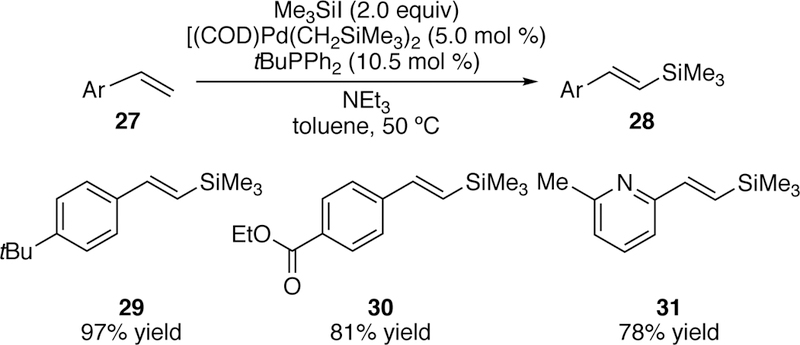
Scope of Watson’s Initial Silyl-Heck Reaction of Styrenes.
A slight adjustment in the reaction conditions - lowering the catalyst loading and temperature as well as switching the solvent from toluene to trifluorotoluene - allowed for the synthesis of allyl silanes from isolated terminal alkenes. Yields, however, were modest due to starting material isomerization. Good selectivity for the E-product was observed for many of the substrates although some substrates did exhibit minor amounts of the vinylsilane product (Figure 12). The synthetic use of these silanes by subsequent functionalization of these products including their participation in allyl transfer and crotylation reactions was also reported.
Figure 12.
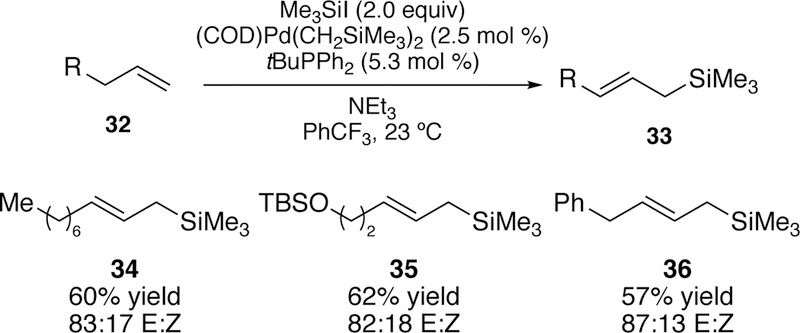
Scope of Watson’s Inital Silyl-Heck Reaction of Terminal Alkenes.
This reaction was further improved by the development of a second-generation ligand for the catalytic system.191 In the pursuit of improving the yields of the allyl silane products, additional phosphine ligands were synthesized and examined in the silyl-Heck reaction. Ligand 38 was designed with electron-donating groups on the aryl rings which leads to a more electron-rich phosphine center than the previous ligand. A more electron rich ligand promotes oxidative addition and consequently improves the overall reactivity of the catalyst. With the use of ligand 38, greatly improved yields were seen with an expanded allyl silane scope although the E:Z selectivity was comparable to the original ligand (Figure 13).
Figure 13.
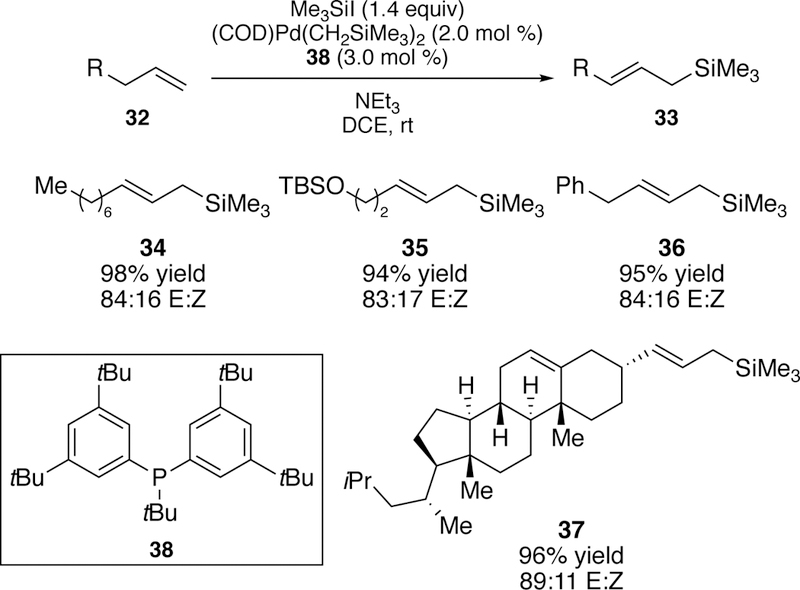
Scope of Watson’s Second-Generation Silyl-Heck Reaction of Terminal Alkenes.
In a second publication exploring the utility of ligand 38, the necessity of using a thermally sensitive palladium precatalyst and the limited scope in regards to the silane was addressed.192 It was found that ligand 38 accommodated the use of Pd2(dba)3 as the precatalyst and, with slight modifications to the catalyst loading and solvent, comparable yields of the vinylsilane products could be obtained. Also, for the first time, bulkier silanes could participate in the reaction, and also provided high yields for vinyl silanes. However, iodotri-ethylsilane failed to give more than traces of the desired product (Figure 14). In addition, only limited yields for allyl silanes were observed with the use of 38 and Pd2(dba)3, making the earlier protocol involving (COD)Pd(CH2SiMe3)2 still more advantageous for their synthesis.
Figure 14.
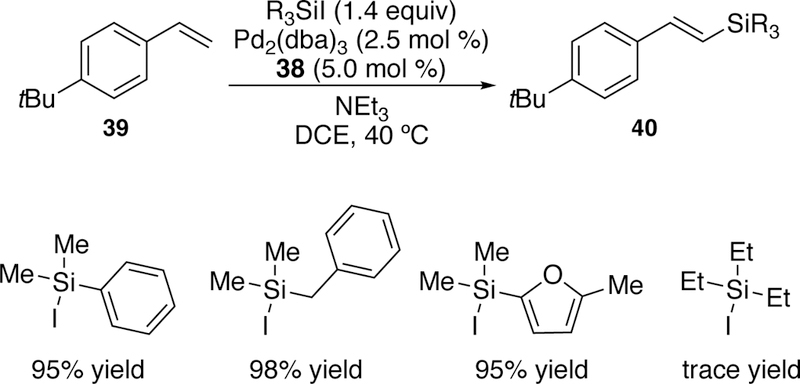
Watson’s Improved Silane Scope with Second-Generation Catalytic System for Silyl-Heck of Styrenes.
A third variation on this silyl-Heck reaction was reported in 2011.193 A single-component, bench-stable palladium precatalyst [(38)Pdl2]2 (Figure 15) was synthesized in one step from ligand 38 and palladium(II) iodide. The advantage of a single component catalyst is ease in reaction setup, the fact that the precatalyst is stable to long-term storage under ambient conditions, and it provides the allyl and vinylsilane products in uniformly high yield (Figure 16).
Figure 15.
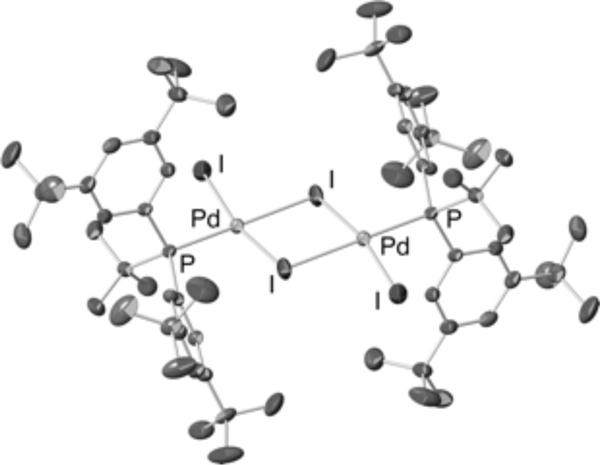
Crystal Structure of Watson’s Single-Component Precatalyst [(38)Pdl2]2 for Palladium-Catalyzed Silyl-Heck Reaction.
Figure 16.
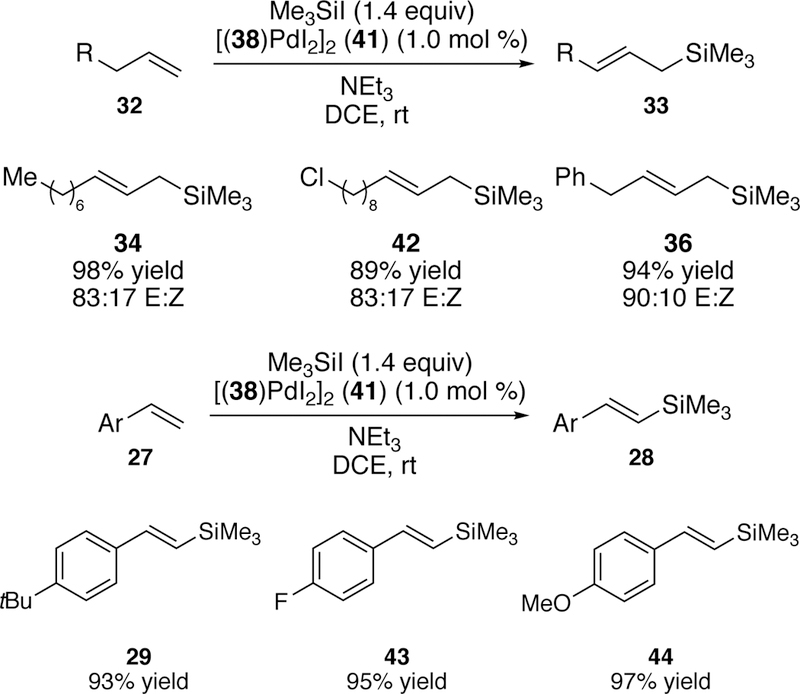
Scope of Watson’s Silyl-Heck Reaction of Terminal Alkenes and Styrenes Using Single-Component Precatalyst 35.
In addition to targeting trialkylsilylhalides as coupling partners, we have also explored the use of silyl ditriflates to generate vinylsilyl ethers (45) and disiloxanes (46) from styrenes (Figure 17).194 Using the same strategy as was developed for coupling trimethylsilylchlorides,189 we found that the addition of an iodide additive allowed the in situ generation of si-lyliodide, which readily undergoes oxidative addition. Only catalytic amounts of sodium iodide were required to achieve quantitative yield of the coupled product. Quenching the reaction with various alcohols led to a variety of substituted vinylsilylethers (e.g. 47–53) and quenching with water provided the disiloxane (e.g. 54). The product vinylsilylethers were also shown to be excellent substrates for Hiyama-Denmark coupling providing isomerically pure product over two steps in superior yields to the related direct Heck approach.
Figure 17.
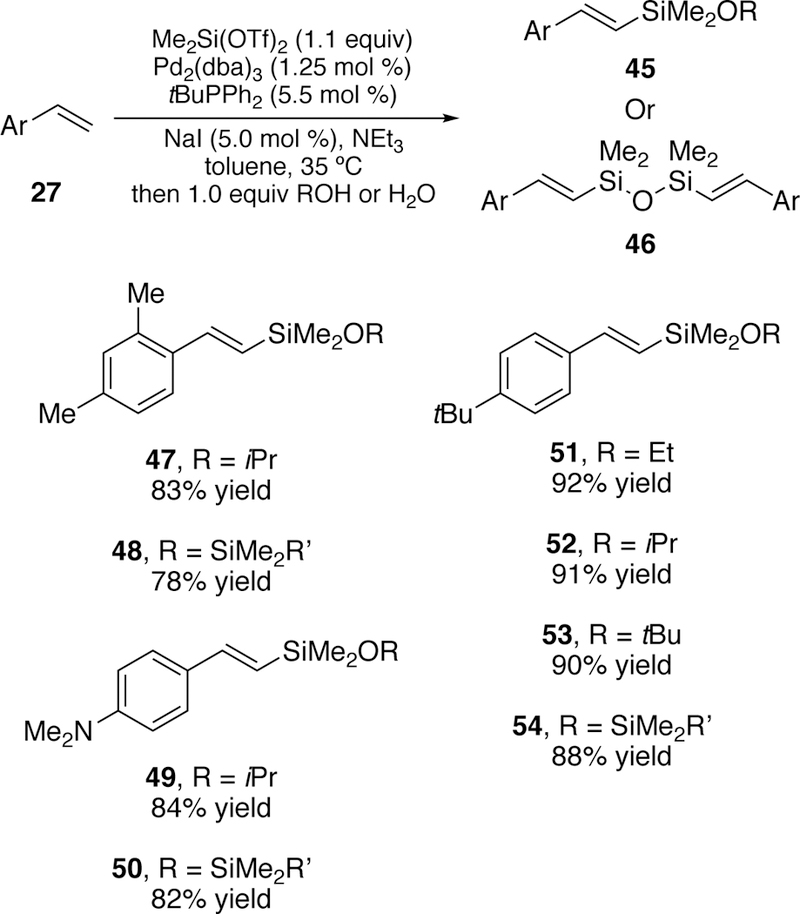
Scope of Watson’s Silyl-Heck Reaction of Silylditriflates with Styrenes Yielding Vinylsilylethers or Disiloxanes.
2.2.2. Nickel-Catalyzed Silyl-Heck Reactions
In addition to our interest in palladium-catalyzed silyl-Heck reactions, we have also developed a nickel-catalyzed variant on this reaction for the direct activation of silyltriflates.195 We found that we were able to couple trimethylsilyltriflate with styrenes without the addition of an iodide additive using Ni(COD)2 as the precatalyst. A number of ligands were investigated, and unlike with palladium, large trialkyl phosphines provided the best yields. The nickel variant was also able to tolerate bulkier silanes such as triethylsilane (Figure 18) which proved problematic in the second-generation palladium conditions with iodotriethylsilane (see Figure 10). The yields in many of these cases are modest due to steric bulk, and the reaction fails with triisopropylsilyltriflate. One downside is that a larger excess of the silyl halide is required, 3.0 equivalents compared to 1.4 equivalents used in most palladium catalyzed processes.
Figure 18.
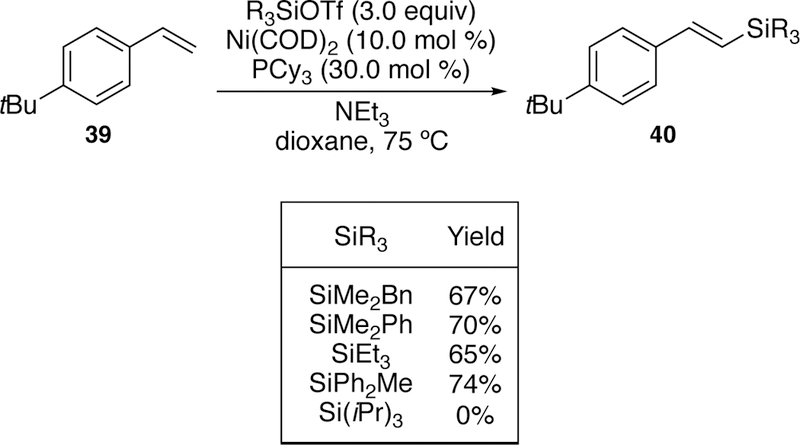
Scope of Watson’s Nickel-Catalyzed Silyl-Heck Reaction Using Silyltriflates.
In 2018, in a major advance in silyl-Heck reactions, Shimada and Nakajima reported on the use of nickel catalysis in the direct activation of Si–Cl bonds of mono-, di-, and tri-chlorosilanes and subsequent silyl-Heck reaction (Figure 19).196 By use of an electron-rich nickel catalyst and an added Lewis acid (AlMe3), styrenal-derived alkenes could easily be converted to the silylated alkene. Monochlorosilanes were sluggish with their initially optimized conditions, but by increasing the equivalents of silylchloride, catalyst loading, and temperature while also changing the base, high yields were observed.
Figure 19.
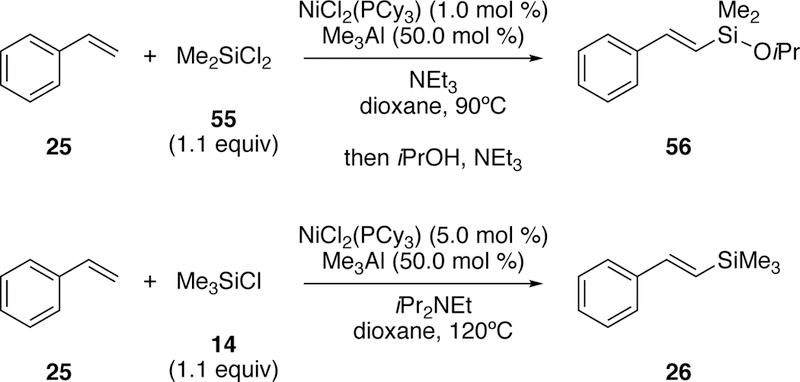
Shimada and Nakajima’s Silyl-Heck Reaction of Styrenes by Direct Si–Cl Bond Activation with Nickel.
2.2.3. Multicomponent Couplings of Silyl Electrophiles
Murai was also interested in cross-coupling reactions initiated by the oxidative addition of palladium across silyl-halide bonds. Contemporaneously to the catalytic reaction reported by Tanaka, his initial report in 1991 detailed a multicomponent coupling of silyliodides, terminal alkynes and tin reagents to generate vinylsilane products.197 The proposed mechanism (Figure 20) begins with the oxidative addition of palladium(0) across the Si–I bond generating intermediate 58 which then inserts into acetylene 59 to give vinylpalladium 60. This vinylpalladium undergoes transmetalation with the tin reagent 61 and then releases the product 62 by reductive elimination.
Figure 20.
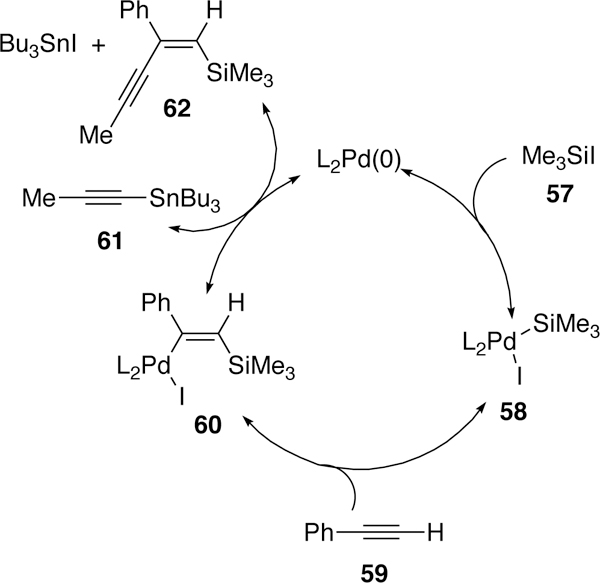
Proposed Catalytic Cycle of Murai’s Multicomponent Coupling of Trimethylsilyliodides.
Murai followed up this report in 1995 with a cross-coupling similar to his previous report, but utilizing organozinc reagents instead of alkynyl stannanes.198 This multicomponent reaction featured an expanded substrate scope in regards to the terminal alkyne and the dialkyl organozinc reagent. Additional silyl groups could be introduced both on alkyne or zinc reagent. However, the reaction was still limited to terminal alkynes and arylzinc reagents were not explored in this multicomponent coupling.
2.2.4. Silyl-Negishi and Silyl-Kumada–Tamao–Corriu Reactions
Our group has also applied palladium catalysis to the cross-coupling of silyl halides in other classes of cross-coupling reactions. In 2017, we reported the palladium-catalyzed silyl-Negishi reaction of alkylsilyliodide electrophiles and alkyl zinc halides.199 An initial ligand screen identified triarylphosphine ligand 65 as the optimal ligand for this reaction. Given success using single-component precatalysts in electrophilic silicon coupling (see section 2.2.1), (65)2PdI2 was prepared and used as catalyst, and showed excellent performance at even 1 mol % palladium loading. A variety of primary and secondary zinc bromides or iodides were competent coupling partners in the reaction and several trialkylsilyliodides could be utilized although the bulkier triethylsilyliodide provided diminished yield (Figure 21). One attractive feature of this reaction is that it can be run without rigorous exclusion of oxygen or water. A reaction run on the benchtop in a capped vial provided the product in 88% yield compared to 98% yield when run under rigorous anaerobic conditions.
Figure 21.
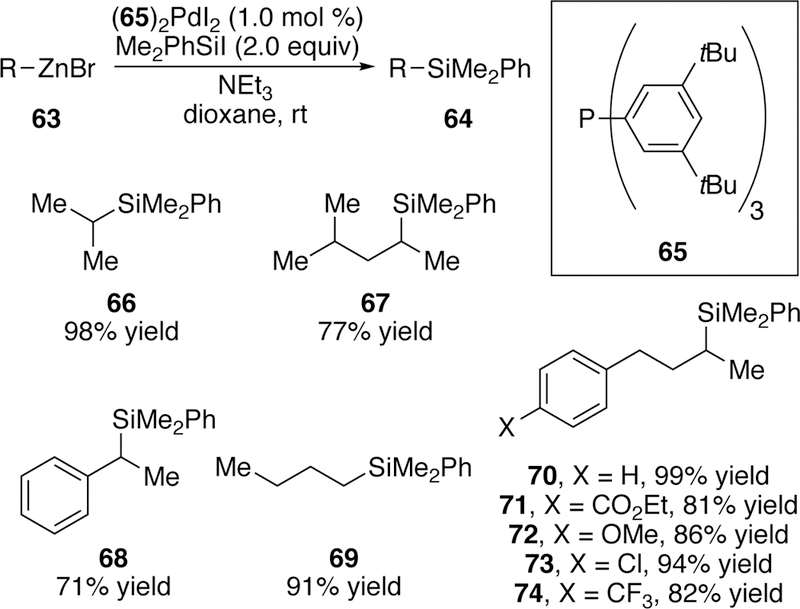
Scope of Watson’s Silyl-Negishi Reaction.
Later in 2017, we reported a silyl-Kumada–Tamao–Corriu reaction of silylchloride electrophiles and organomagnesium reagents.200 Although both this and the silyl-Negishi reaction use the same catalyst ((65)2Pdl2) and can provide identical products, the developed silyl-Kumada–Tamao–Corriu reaction utilizes less reactive chlorosilanes, which are much more tolerant of air and moisture than their iodide counterparts. More importantly, an immense array of silylchloride coupling partners are commercially available, yielding a diversified product library (Figure 22). This constituted the first time that chlorosilanes were used in a cross-coupling reaction. These conditions are also tolerant of bulky silylchlorides with triethylsi-lylchloride providing a near quantitative yield of 78. Again, these reactions can be run at room temperature but the optimal solvent is diethyl ether. Although in the standard reaction conditions heavier halides are present, the reaction can also run with (65)2PdCl2 and iPrMgCl with the silylchloride, suggesting that direct activation of the silicon chloride bond may be at play in the reactions.
Figure 22.
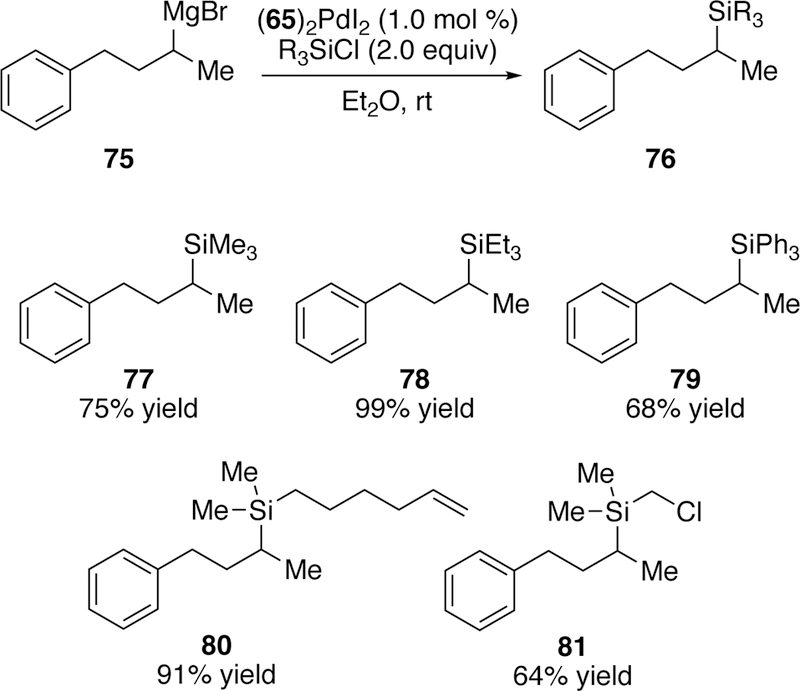
Scope of Watson’s Silyl-Kumada–Tamao–Corriu Reaction.
3. Nitrogen Cross-Coupling
3.1. Introduction
New methods for the formation of nitrogen-carbon bonds have been of interest since the advent of synthetic organic chemistry due to the prevalence of nitrogen atoms in biologically relevant molecules. Traditional methods for the generation of carbon–nitrogen bonds include the Gabriel amine synthesis (18 87),201 the Ritter reaction (1948),202–203 and reductive amination.204 Metal-mediated or metal-catalyzed methods have more recently been developed to complement traditional approaches and greatly extend the accessible chemical space.205 Examples of these metal-mediated reactions include the related Ullmann coupling,206 Goldberg reaction,120, 207–209 and Chan–Lam–Evans coupling,110, 150, 210–212 as well as the prototypical cross-coupling reaction for the generation of sp2C–N bonds, the Buchwald–Hartwig amination.213–218 Another metal-mediated reaction, the aza-Wacker reaction, reported by Hegedus beginning in 1976,219–222 generates sp3C–N bonds. Asymmetric variants223–224 have been developed and detailed mechanistic studies of the aza-Wacker reaction have been conducted.225–227
One feature common to the mechanisms of all the aforementioned metal-mediated C–N bond forming reactions is that the nitrogen coupling partner acts as the nucleophile. In general, reactions in which nitrogen acts as a nucleophile are very common; reactions involving electro- philic amination are less common and have been atopic of recent interest.228–233 Reactions combining both metals and electrophilic nitrogen in cross-coupling234–235 will be detailed in the following sections.
3.2. Cross-Coupling of Nitrogen Electrophiles
3.2.1. Oxime Ester Electrophiles with Palladium
Early studies by Narasaka investigated the activation of oxime N–O sulfonyl bonds with copper236 and in 1999 he found that a stoichiometric reaction of palladium(0) with O-sulfonyloxime 82, followed by aqueous quench, resulted in the formation of imine 84 (Figure 23). This result suggests the reaction proceeds through oxidative addition of palladium into the N–O bond followed by protodepalladation.237
Figure 23.
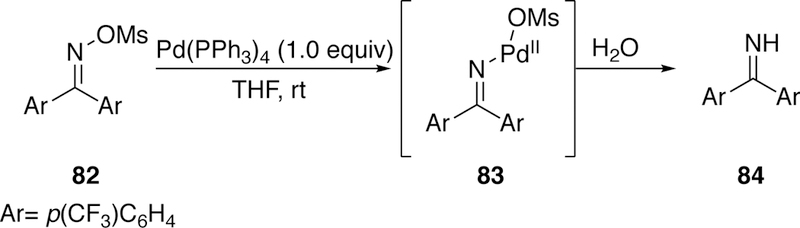
Narasaka’s Initial Discovery of Palladium Oxidative Addition into N–O Bonds.
Further support for the proposed oxidative addition of palladium across N–O bonds of oximes to generate species of type 83 was disclosed by Hartwig twenty years after Naraska’s initial investigation. Hartwig isolated and characterized by single X-ray diffraction complex 86, the direct oxidative addition complex of 85 and palladium(0) (Figure 24).238
Figure 24.
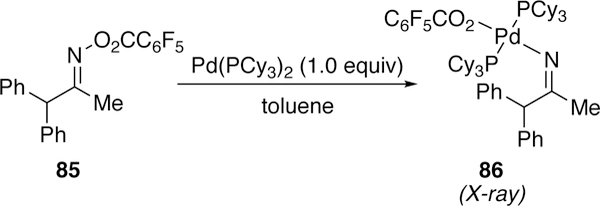
Hartwig’s Oxidative Addition Complex Derived from O-pentafluorobenzoyloxime and Palladium(0).
Following his initial report, in 2001, Narasaka exploited this reactivity in the development of novel N–C bond forming reactions, specifically to the cyclization of alkene-tethered O-pentafluorobenzoyloxime 87. He attributed this to a Heck-like pathway. The product 88, which under the reaction conditions or with the addition of trimethylsilylchlorides, isomerizes to pyrrole 89 (Figure 25). This work was further extended to sequential Heck reactions resulting in spiro-imines.239–240 During this study he also examined aldoximes as potential precursors. However, the cyclized product was not isolated but rather the nitrile, which he suggested results from Beckmann fragmentation of the aldoxime.
Figure 25.
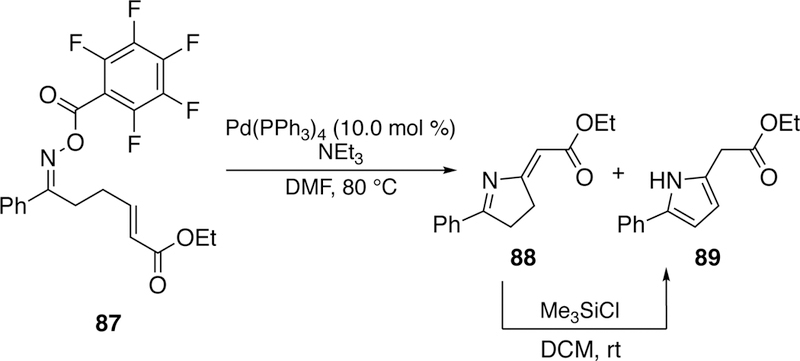
Narasaka’s Palladium-Catalyzed Intramolecular Heck Reaction of Alkene-Tethered O-pentafluorobenzoyloximes to Pyrroles.
The framework of other complex aza-heterocycles could be synthesized via this Heck reaction.241 5-Exo cyclization and subsequent isomerization allowed access to substituted pyrroles (Figure 25).242 Pyridines could also be accessed via a 6-endo cyclization of 90243 while isoquinolines 93 could be accessed following isomerization of a 6-exo cyclized product derived from 92 (Figure 26).244 Sequential Heck cyclization followed by chemical oxidation allowed access to azaazulenes.245–246 This methodology, specifically the use of oxime esters in Heck cyclizations to generate nitrogen heterocycles, is now commonly referred to as a Narasaka- Heck reation.247
Figure 26.
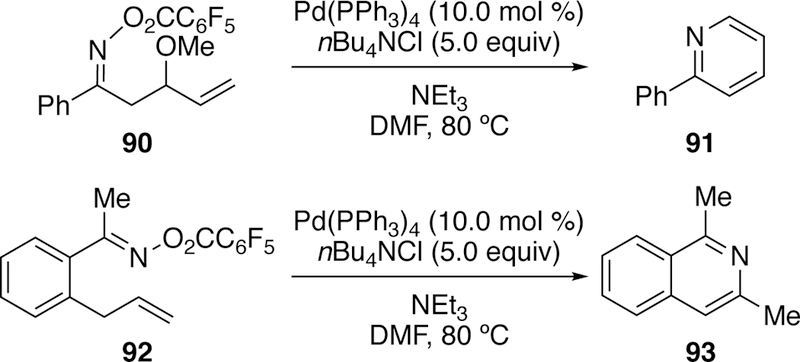
Narasaka’s Construction of Complex Aza-Heterocycles via Heck-type Cyclization.
O-Pentafluorobenzoyloximes were also utilized in a 5-endo Heck cyclization by Ichikawa to synthesize difluormethylene-substituted pyrrolines or fluorinated pyrroles after isomerization from appropriate substrates.248–249 In a related reaction, Abell found use of O-pentafluoroben-zoylamidoximes such as 94 in the synthesis of imidazoles (Figure 27).250
Figure 27.
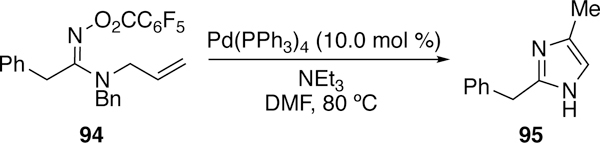
Abell’s Use of O-Pentafluorobenzoylamidoximes in the Synthesis of Imidazoles by Heck-type Cyclization.
Notably, a Narasaka-Heck reaction was utilized by Fürstner in his total synthesis of butylcy-cloheptylprodigiosin 98 (Figure 28).251–252
Figure 28.
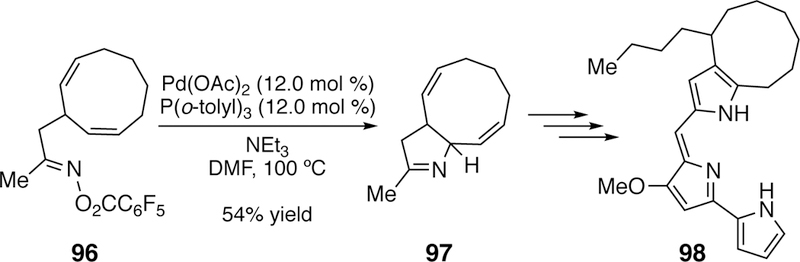
Fürstner’s Use of a Narasaka–Heck Reaction in the Total Synthesis of Butyl-cycloheptylprodigiosin (98).
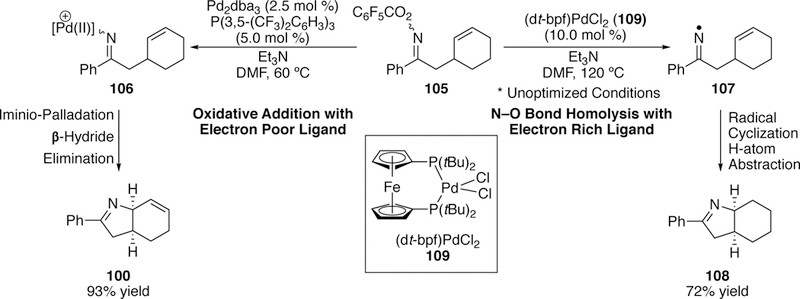
Bower has done extensive work extending the Narasaka–Heck reaction to the formation of stereogenic heterocycles. In 2012, he reported the formation of heterobicyclic structures via the 5-exo Narasaka–Heck cyclization from cyclohexene-tethered pentafluorobenzoyloxime esters (e.g. 99, Figure 29).253 His optimized conditions utilized a catalytic palladium(0) source (loadings as low as 2.5 mol %) with the electron-deficient phosphine ligand P(3,5-(CF3)2C6H3)3. He hypothesize that the electron-deficient ligand serves to improve the stability of the iminio–Pd(II) intermediate by reducing the electron density around palladium and consequently increasing the strength of the iminio–Pd bond, which limits protodepalladation. Electron-deficient phosphine ligands have been shown by Hartwig to accelerate the migratory insertion of alkenes into Pd–N bonds.254
Figure 29.
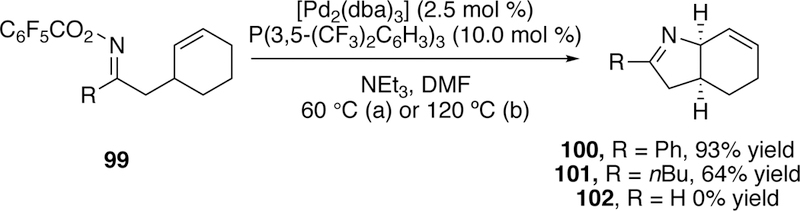
Bower’s Narasaka-Heck Reaction To Generate Stereogenic Bicyclic Imines From Pentafluorobenzoyloxime Esters.
Good yields were seen for aryl-substituted products such as 100, and many could proceed efficiently at 60 C; conversion could even be seen at ambient temperature. Alkyl substituted products such as 101 were more challenging to access as appreciable amounts of hydrolyzed ketone byproduct were formed in addition to the desired cyclized product. The alkyl-substituted substrates also required higher temperatures (up to 120 °C) to proceed efficiently. Ald- oximes failed to give the desired cyclized product 102, and instead underwent Beckman rearrangement255 to give the nitrile. This undesired reactivity of aldoximes was previously noted by Narasaka. For products bearing more than one stereocenter such as 103, good diastereose- lectivities were observed, and Bower attributes this to base-mediated equilibrium post-cyclization, since he observed that poor diastereomeric product mixtures could be subjected to the 120 °C conditions to improve the diastereomeric ratio (Figure 30).
Figure 30.
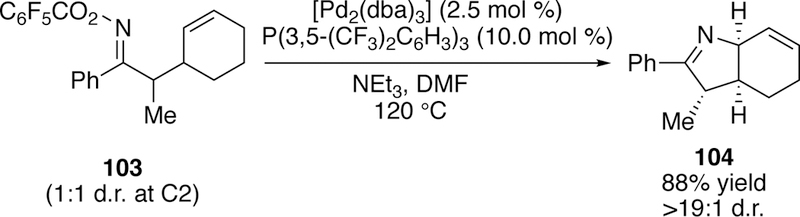
Bower’s Diastereoselective Narasaka-Heck Reaction of Cyclohexene-Tethered Pentafluorobenzoyloxime Esters.
Bower followed up this work a year later with a report of the Narasaka–Heck reaction of pentafluorobenzoyloxime esters with tethered 1,2-disubstituted alkenes to give unconjugated, stereogenic dihydropyrroles bearing an E-alkene (Figure 31a).256 The yields of some of the substrates such as 111, were reduced due to isomerization to the pyrrole. Bower suggests that formation of the pyrrole is a kinetic phenomenon, as resubjection of the desired product 111 to the reaction conditions did not result in isomerization to the pyrrole. He also utilized oximetethered 1,1-disubstituted alkenes to construct fully substituted nitrogen-bearing stereogenic centers which cannot undergo isomerization to the pyrrole (Figure 31b).257 In a subsequent report Bower further explores the mechanism of these reactions and notes that two mechanistic pathways are possible depending on the choice of ligand.258
Figure 31.
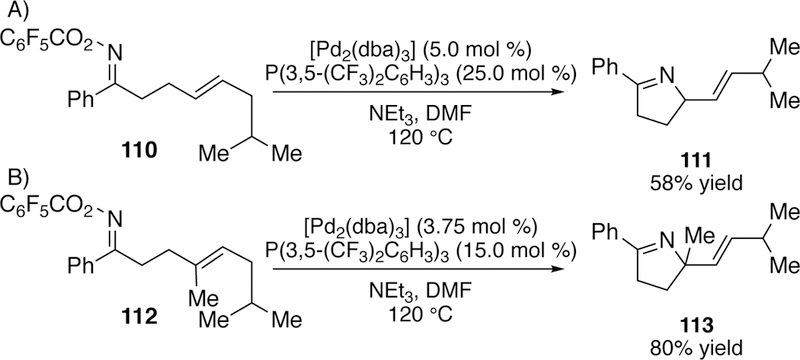
a) Bower’s Narasaka–Heck Reaction of 1,2-Disubstituted Alkenes b) Bower’s Narasaka–Heck Reaction of 1,1-Disubstituted Alkenes.
Electron-rich phosphines promote the generation of iminyl radicals through an interrupted one electron oxidative addition, whereas electron-poor phosphines promote a two-electron pathway more similar to that of conventional cross-coupling reactions (Figure 32).
Figure 32.
Bower’s Proposed Divergent Mechanism for Narasaka-Heck Cyclization.
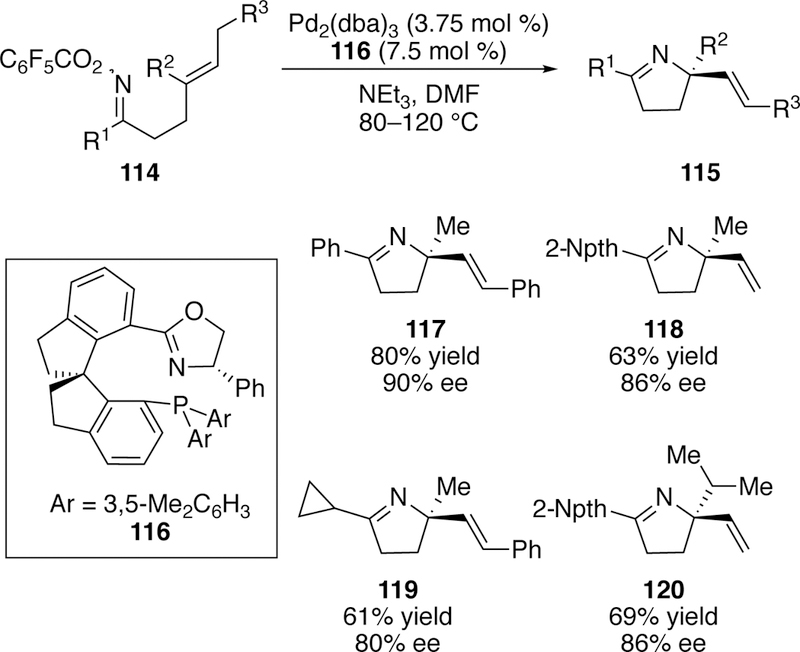
In 2017, Bower reported the first asymmetric Narasaka–Heck cyclization to form fully-substituted nitrogen-bearing stereogenic centers in a selective fashion.259 The key to his asymmetric induction was the use of a SIPHOX ligand (116) along with a palladium(0) precatalyst and triethylamine in DMF at elevated temperatures. A range of alkyl and aryl substitutions were well tolerated (Figure 33). Bower has also published a review on the use of aza-Heck reactions in the asymmetric synthesis of nitrogen-containing heterocycles.260
Figure 33.
Scope of Bower’s Asymmetric Narasaka-Heck Reaction.
The Narasaka–Heck reaction has also been extended beyond alkenes to alkyne-tethered pentafluorobenzoyloxime esters as reported by Abell in 2007.261 Following migratory insertion of the iminio-palladium(II) species 122 into the alkyne, reaction of the resulting vinylpalladium species 123 with carbon monoxide occurs yielding an acylpalladium(II) intermediate 124. This further reacts with ethanol to furnish the ethyl ester product 125 (Figure 34). Subsequent isomerization of the unconjugated diene yields the ester-substituted pyrrole 126. Unfortunately, this domino reaction results in mixtures of the desired ethyl ester pyrrole 126 and the unsubstituted pyrrole 129 which results due to the generation of vinylpalladium hydride 127.
Figure 34.
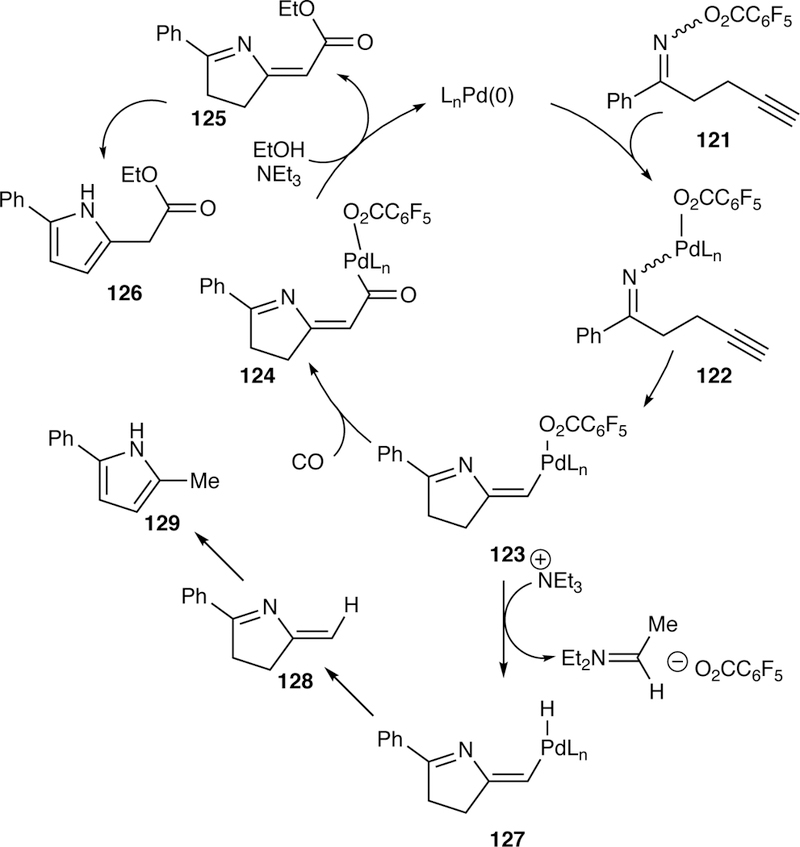
Proposed Catalytic Cycle of Abell’s Domino Narasaka–Heck Reaction and Carbonylation.
Kitamura took advantage of a similar domino reaction of alkyne-tethered pentafluorobenzo-yloximes (130) in the creation of substituted isoindoles (131).262 In this domino reaction the vinylpalladium species undergoes transmetalation with an arylboronic acid followed by reductive elimination of the arylated product (131, Figure 35).
Figure 35.
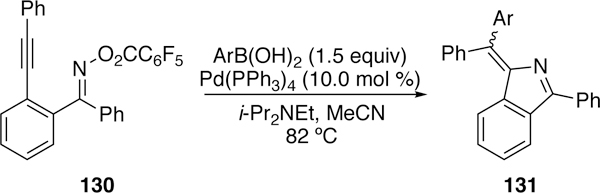
Kitamura’s Domino Reaction of Alkyne-Tethered Pentafluorobenzoyloximes to Form Isoindole Products.
Several other groups have also taken advantage of domino reactions involving interrupted Narasaka–Heck reactions. Bower has reported a carboamination reaction utilizing his Narasaka–Heck conditions in which the migratory insertion intermediate undergoes further reaction to generate one or more C–C bonds.263 Due to deliberate substrate design, there are no β-hydrogens present, prohibiting β-hydride elimination from occurring. Several variants were explored including the transmetalation of the migratory insertion product with an aryl-, alkenyl-, or alkynylboronate or stannane followed by reductive elimination to generate a Csp3–Csp2 or a Csp3–Csp bond. The authors also successfully intercepted the migratory insertion intermediate with carbon monoxide followed by a transmetalation with a boronate or tetraarylborate, and then reductive elimination to generate the ketone product (Figure 36).
Figure 36.
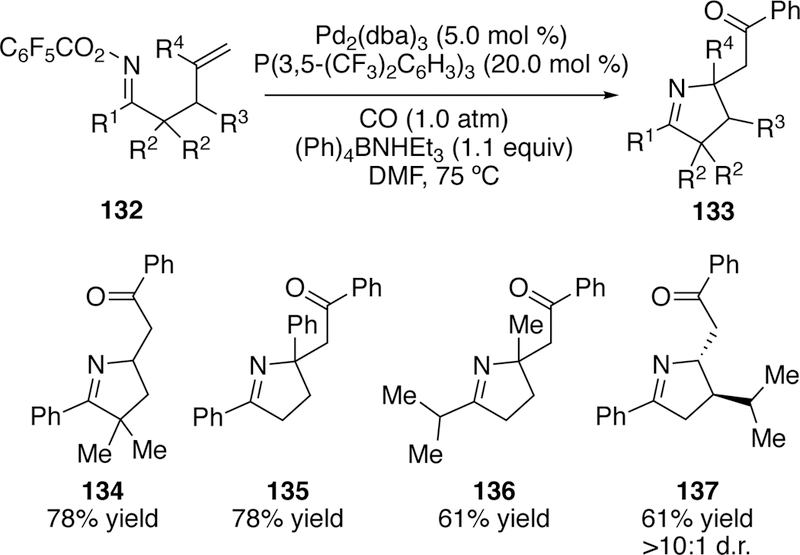
Scope of Bower’s Tandem Narasaka-Heck and Carbonylation.
The formation of carbon-halogen bonds via intercepted Narasaka–Heck cyclization was developed by Tong in 20 1 5.264 This iminohalogenation requires the use of four equivalents of either Nal, LiBr, or LiCl as the halide source, along with a palladium(II) precatalyst and an electron-deficient arylphosphine ligand. Mechanistic investigation ruled out a radical pathway as TEMPO did not interfere in the chlorination and bromination reactions. Deuteration studies found no erosion or scrambling when the vinyldeuteride 138 was subjected to the reaction, suggesting a stereospecific migratory insertion. Additional stereochemical data (see Figure 37) also suggests that the carbon-halogen bond formation occurs via an SN2-type reductive elimination mechanism.
Figure 37.
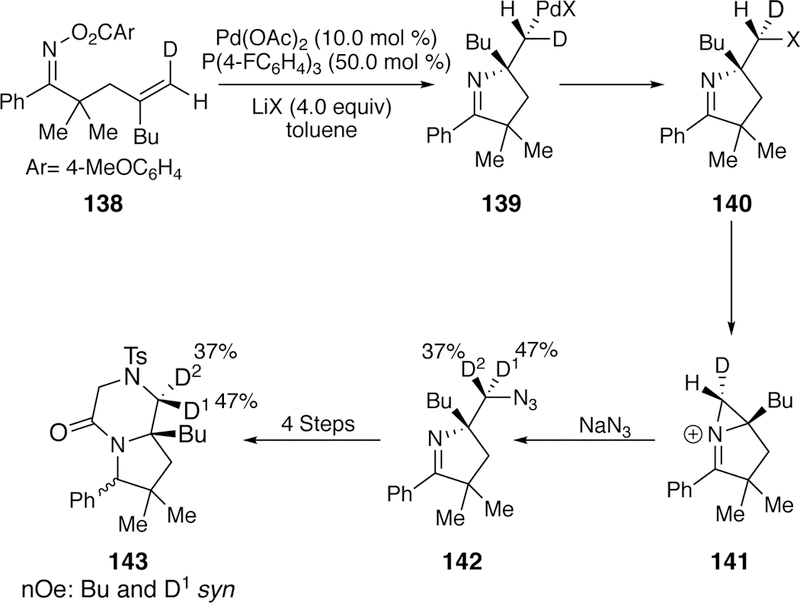
Tong’s Deuterium Studies of the Iminiohalogenation Reaction that Suggest an SN2-Type Reductive Elimination of the Product.
An enantioselective variant of an intercepted Narasaka–Heck cyclization was more recently reported by Zhu (Figure 38).265 He found that with the commercially available bidentate phosphine ligand SYNPHOS, a palladium(II) precatalyst, and base at elevated temperatures the cyclized and coupled products could be obtained in modest to good enantioselectivities (as high as 83% ee) when both substituents (R1 and R2) are aryl groups. Alkyl substitution on the oxadiazole (R2) resulted in diminished enantioselectivity, typically around 75% ee.
Figure 38.
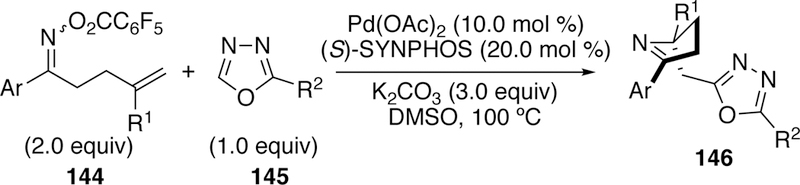
Zhu’s Tandem Asymmetric Narasaka–Heck Reaction and Arylation with Oxadiazole.
Initial studies found that the solvent can play an immense role in the reaction mechanism. A reaction run in toluene in the presence of TEMPO yielded the TEMPO-trapped adduct suggesting a radical mechanism. Switching the solvent to DMF yielded the product with no formation of the TEMPO-trapped adduct. The optimal conditions utilize DMSO as the solvent, and TEMPO experiments resulted in the formation of small amounts of TEMPO adduct, however, the high levels of enantioselectivity observed seems to suggest against a solely radical mechanism. Two competing mechanisms were proposed (Figure 39).
Figure 39.
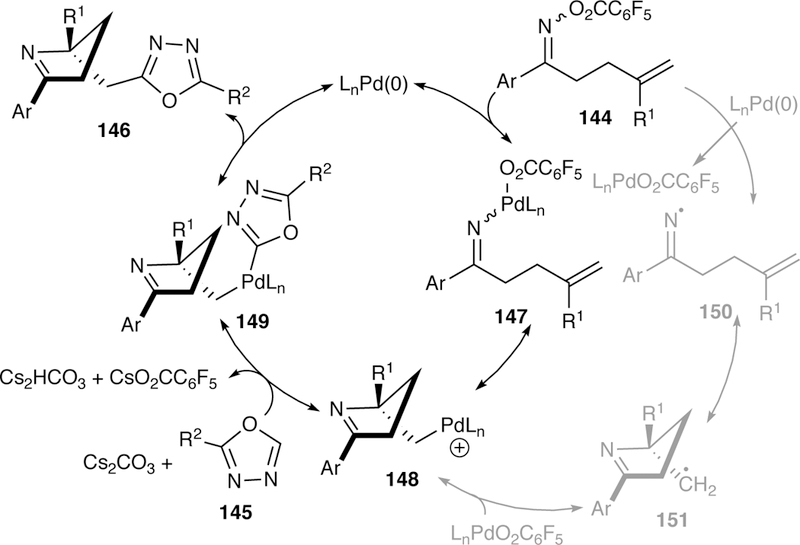
Proposed Two Electron and Single Electron Pathways for Zhu’s Enantioselective Interrupted Narasaka-Heck Reaction.
In the first two electron process mechanism, oxidative addition of palladium into the N–O bond followed by migratory insertion generates the alkylpalladium intermediate 148, which is intercepted by transmetalation with an oxadiazole anion generated in situ. The palladium(II) species 149 then undergoes reductive elimination releasing the product 146 and regenerating the catalyst. The second mechanism is a single-electron process in which iminyl radical 150 is generated and undergoes cyclization. The alkyl radical 151 recombines with the Pd(I), generating the same alkylpalladium 148 as described in the two-electron mechanism.
3.2.2. Oxime Ester Electrophiles with Copper
Copper has also been found competent in the activation of N–O bonds of oximes. In 2007, Liebeskind reported the N-imination of boronic acids and organostannanes using copper and O-acylketoximes (Figure 40).266 The proposed mechanism suggests oxidative addition of copper(I) into the N–O bond to generate a copper(III) intermediate which then undergoes transmetalation with the boronic acid or organostannane. Reductive elimination of the product and regeneration of the copper(I) catalyst then follows. When switching from the ketoximes to the aldoximes (152 R1 = H), the desired product was not isolated. Rather, as seen in Narasaka’s work with palladium (section 3.2.1), the nitrile was formed, which he suggests results from β-elimination of the oxidative addition intermediate. This work was extended to the synthesis of substituted pyridines via coupling of O-pentafluorobenzoylketoximes (160) with vinylboronic acids (161) followed by air oxidation (Figure 41).267
Figure 40.
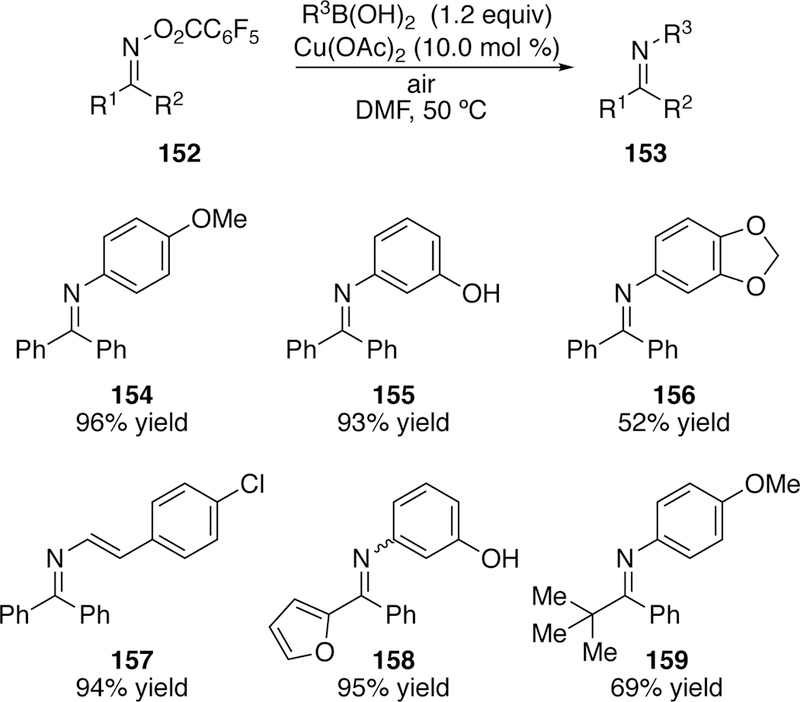
Scope of Liebeskind’s N-Imination of Boronic Acids.
Figure 41.
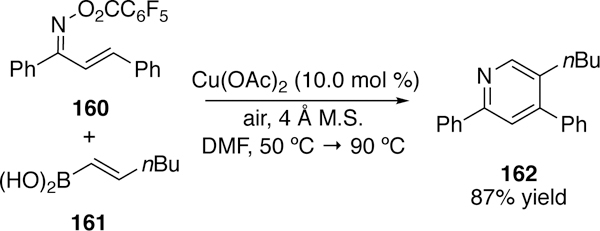
Liebeskind’s Synthesis of Substituted Pyridines via Coupling of Vinylboronic Acids and O-Pentafluoroenzoylketoximes.
Liebeskind also extended his copper-mediated coupling of boronic acids or organostannanes beyond oximes to O-acetylhydroxamic acids (163) yielding N-amidyl products with the use of stoichiometric or superstoichiometric copper (Figure 42).268 Different conditions were required to couple either the boronic acid or the stannane. Boronic acid coupling utilized one equivalent of copper(I) thiophene-2-carboxylate (CuTC), while the stannanes required two equivalents of copper(I) diphenylphosphinate (CuDPP) (Figure 42). He hypothesized that stoichiometric copper was required in the coupling of O-acetylhydroxamic acids (as opposed to catalytic copper with O-acyloximes) because the oxidative addition complex can form bridging amido-dimers, which are less susceptible to transmetalation.
Figure 42.
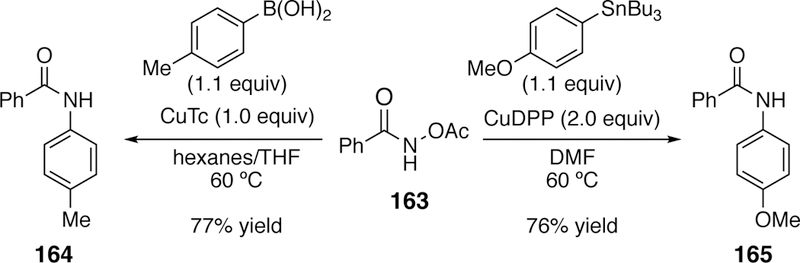
Liebeskind’s Copper-Mediated Coupling of O-Acyl Hydroxamic Acids with Boronic Acids and Stannanes.
Liu and Xi also used copper in their stoichiometric cross-coupling of O-benzoylketoximes and aldoximes with alkenylzirconocenes to generate 2-azadienes.269 This is the first example of a successful coupling of an aldoxime, as previous attempts by Narasaka, Liebeskind, and others resulted in undesired nitrile formation. In Liu and Xi’s proposed mechanism (Figure 43), an initial transmetalation from zirconium (166) to copper generates an alkenylcopper intermediate (167) which undergoes oxidative addition into the N–O bond of oxime 168. This is followed by reductive elimination of the product (170). One limitation of this reaction is the necessity to pre-generate the alkenylzirconocene from alkynes, resulting in diminished functional group tolerance.
Figure 43.
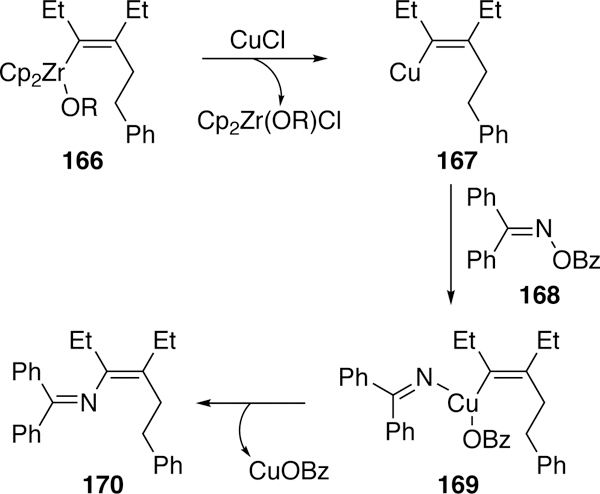
Proposed Mechanism of Liu and Xi’s Copper-Mediated Coupling of Alkenylzirconocenes and O-Benzoyloximes.
In 2014, Bower reported a copper-catalyzed cyclization of O-pivaloyl oximes to yield un-saturated pyrrolidines in a transformation similar to that of Narasaka.270 However, mechanistic studies revealed that the copper-catalyzed reaction does not proceed via migratory insertion of the M–N bond like in Heck reactions, but rather proceeds through addition of an iminyl radical or iminio–Cu(III) species. A similar transformation using stoichiometric copper was reported by Zard using alkene-tethered O-acetylketoximes.271
3.2.3. Other Oxime-Derived Electrophiles
O-Phosphinyloximes (e.g. 171) have also been utilized in aza-Heck cyclizations by Zhu.272–273 Under differing sets of conditions, either the 5-exo isomerized pyrrole or the 6-endo dehydrogenated pyridine (e.g. 172) could be obtained in modest yields from the linear substrate. The authors also examined fused substrates in a subsequent publication. They found that the trans-disposed substrates as shown in Figure 44 could cyclize and dehydrogenate under the reaction conditions to form the fused pyridine product.274
Figure 44.
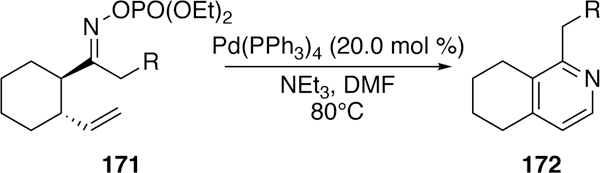
Zhu’s Aza-Heck Cyclization of O-Phosphinyloximes and Subsequent Dehydrogenation to Fused Pyridines.
3.2.4. Aza-Heck Reactions of Other Electrophiles
More recently, research efforts have extended the aza-Heck reaction beyond oxime substrates to electrophilic nitrogen centers bearing three different substituents. Bower, who made many advancements to the Narasaka–Heck reaction, has investigated (pentafluorobenzoyloxy)sulfonamide substrates allowing access to stereogenic sulfonamide-containing heterocycles in a 5-exo cyclization.275 The proposed mechanism follows similarly to that of the Narasaka–Heck reaction (Figure 45). First, an oxidative addition of palladium(0) into the N–O bond followed by a proposed dissociation of the pentafluorobenzoate gives cationic intermediate 175 which undergoes syn-migratory insertion254, 276–277 to give alkyl palladium intermediate 176. To complete the cycle, β-hydride elimination releases the product 177 and generates palladium hydride, which undergoes base-induced reductive elimination to regenerate the palladium(0) catalyst. Bower proposes that the catalytic cycle is driven by protodecarboxylation (corroborated by 19F NMR analysis) of the pentafluorobenzoate leaving group.
Figure 45.
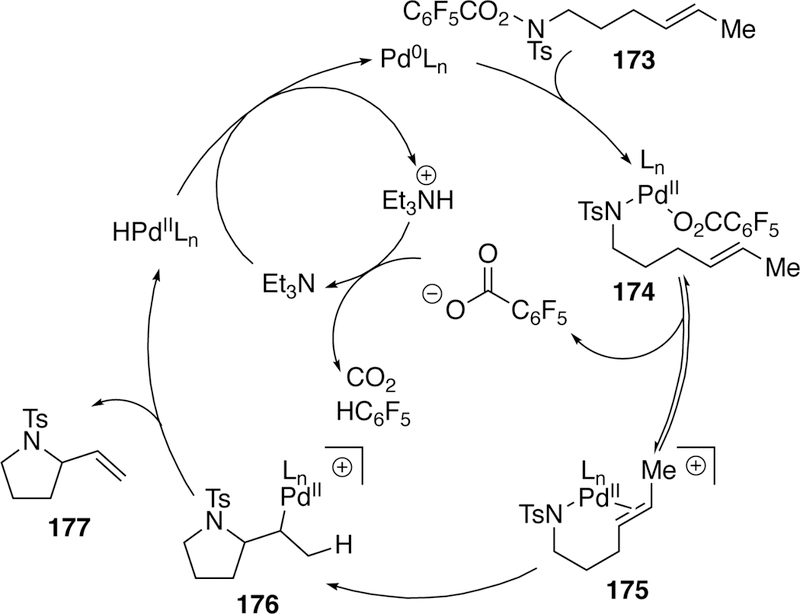
Proposed Catalytic Cycle for Bower’s Sulfonamide Aza-Heck Cyclization.
A variety of alkyl-substituted substrates could be cyclized to give the pyrrolidene products in modest to good yields; however, unified conditions were not utilized. Bower notes that solvent effects are pronounced, and combinations of THF, DMF, nBuCN, or toluene were utilized for optimal yields of many of the substrates. The sulfonamide protecting group on the nitrogen was also varied, some bearing tosyl while others, mesyl.
In addition to the 5-exo cyclization that was a hallmark of Narasaka–Heck cyclizations, he also briefly investigated a 6-exo cyclization with his catalytic conditions. The only 6-exo substrate that is reported (178) features a conformationally-restricting phenyl group and cyclizes to 179 in 42% yield (Figure 46).
Figure 46.
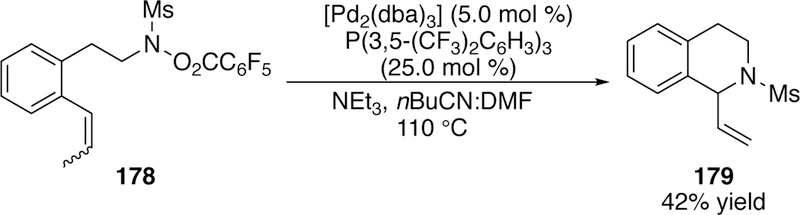
Bower’s 6-exo Sulfonamide Aza-Heck Cyclization.
Although this reaction produces products similar to that of the aza-Wacker reaction, the advantage of the aza-Heck approach is that it exhibits a wider tolerance of functional groups. Aza-Wacker reactions do not tolerate large α-substituents and electron-deficient alkenes susceptible to conjugate addition. Bower’s aza-Heck approach can accommodate both of these limitations and provides the additional advantage of potential domino processes.
Another unique solution to the aza-Heck reaction was developed by our group in 2016.278 This aza-Heck reaction eliminates the need to use the large pentafluorobenzoyl group, and like the Bower chemistry described above, moves beyond the oxime substrates of the Narasaka–Heck reaction.279 Like the Liebeskind work described in section 3.2.2, this approach uses hydroxamate derivatives. However, unlike the earlier chemistry, O-acyl activation proved incompatibile, and O-phenylhydroxamate ethers were identified as the key to success. These reactions proceed via 5-exo cyclization to form γ-lactams (Figure 47). For the first time, protection or full substitution on the nitrogen is not required and free N–H products are isolated directly from an aza-Heck reaction. It is noteworthy that the N–O bonds of O-arylhy-droxylamines have previously been broken in metal-free electrophilic amination of aryl boronic acids.280
Figure 47.
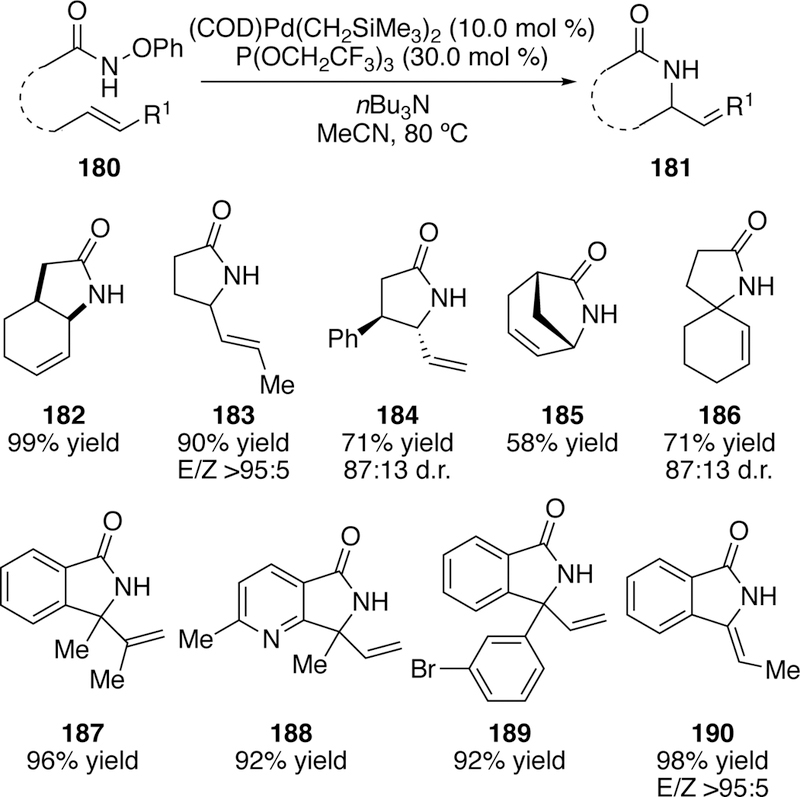
Scope of Watson’s 5-exo Aza-Heck Reaction of O-Phenylhydroxamates.
Although this may seem like a significant departure from the previously discussed Narasaka–Heck and aza-Heck reactions, preliminary mechanistic data suggests a palladium-catalyzed heteroatomic Heck reaction mechanism rather than an aza-Wacker reaction. An aza-Wacker mechanism would involve the activation of the N–H bond by Pd(II), whereas the aza-Heck mechanism would involve direct activation of the N–O bond by Pd(0). Optimized reaction conditions involved the use of a Pd(II) precatalyst (COD)Pd(CH2SiMe3)2281 which typically reduces in situ to Pd(0).282 To probe whether or not Pd(II) is the active catalyst (suggesting an aza-Wacker mechanism), Pd[P(OEt)3]4283 was synthesized and used as a single-component catalyst in the reaction. The reaction was high yielding of the desired product indicating that the Pd(0) catalyst is sufficient and Pd(II) is not the active catalyst, suggesting against an aza-Wacker mechanism.
We demonstrated a wide substrate scope including compatibility with other heteroatoms, aryl bromides (189), and a pyridine-derived substrate (188, Figure 47). The reaction provides good diastereoselectivity for vicinal stereocenter-containing compounds 184. Distal stereocenters fail to impart any selectivity. Interestingly, even tetrasubstituted alkenes could participate in the reaction resulting in 187, which remains a rare class of alkenes in aza-Heck reactions. It is also noteworthy that the products (183 and 190) of this aza-Heck reaction are isolated as single olefin isomers. It is hypothesized that phenoxide produced during this reaction mitigates the formation of palladium-hydride, which has plagued other related transformations with unwanted olefin scrambling.
Similar to Bower, we were also interested in expanding the scope of the reaction to include 6-exo cyclizations. We found that in this reaction, conformationally restricted substrates (191) are required for 6-exo cyclization to proceed in good yield (192, Figure 48).
Figure 48.
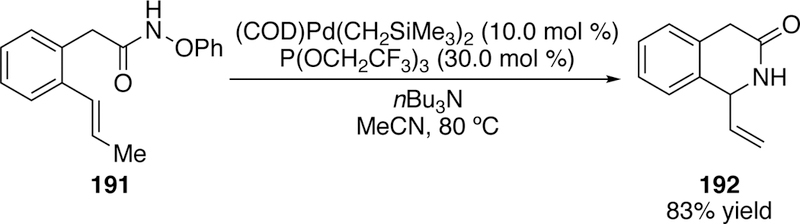
Watson’s 6-exo Aza-Heck Reaction of an O-Phenylhydroxamate.
In 2018, Bower reported an aza-Heck cyclization of N-pentafluorobenzoyl carbamates that accommodates both 5-exo and 6-exo cyclizations.284 This reaction takes advantage of a Pd(0) precatalyst, bulky phosphaadamantyl ligands, and elevated temperatures. For more challenging substrates the electronic properties of the ligand could be tuned by varying the aryl substituent on the phosphorus with ligands 196 and 197 preforming the best in both challenging 5-exo cyclizations and in the 6-exo cyclizations (Figure 43). Even a 7-exo cyclization could be accomplished to provide 203 using this method, although in modest yield. Bower found that the substituent on the carbamate (R1 in Figure 49) could be varied with the 5-exo substrates (e.g. 198–200). However, for the 6-exo and 7-exo substrates (201–203) only the tert-butyl substituent on the carbamate provided the optimal yields.
Figure 49.
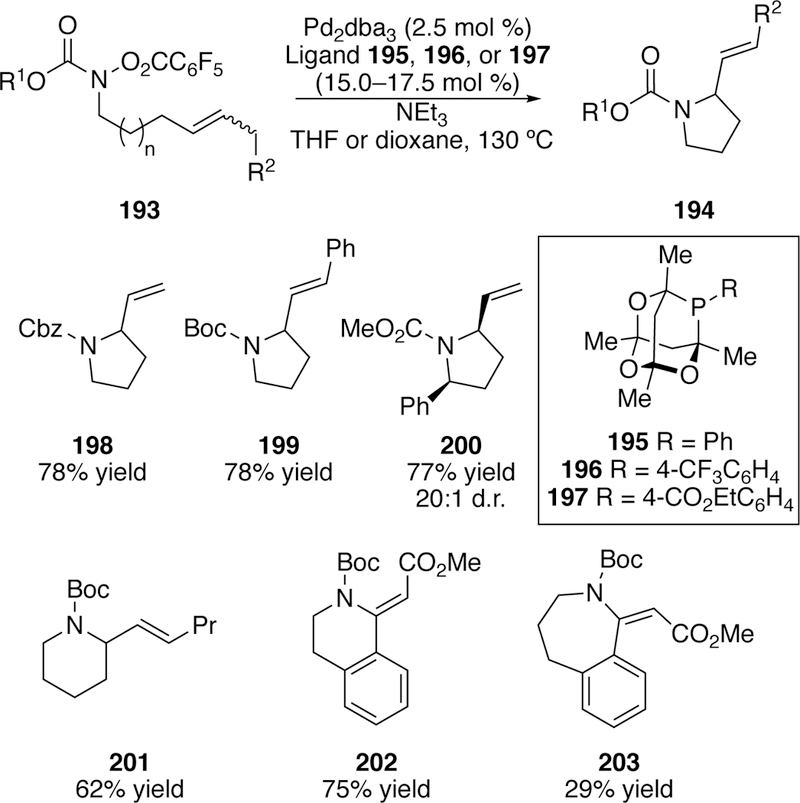
Scope of Bower’s Aza-Heck 5-exo and 6-exo Cyclizations of Carbamates.
Another recent development in aza-Heck chemistry reported by our group is the expansion of the hydroxamate-type electrophiles to more complex urea-derived electrophiles (204) generating imidazolidinones (205, Figure 50).285 To move away from (COD)Pd(CH2SiMe3)2, we found that [(cinnamyl)PdCl]2 could be used as the precatalyst with PhMgBr as a cocatalytic reductant. Like our earlier study, P(OCH2CF3)3 remained the optimal ligand. In some cases, addition of silver(I) tosylate boosted the yield of product by suppressing undesired alkene isomerization. In contrast, this alkene isomerization could be harnessed for the selective formation of dihydroimidazolones (206) by addition of iodide additives. A variety of substitution was well tolerated including when R1 = H (Figure 50), allowing for the synthesis of doubly unprotected products, a challenge not previously met in other metal-mediated methods for the synthesis of imidazolidinones.
Figure 50.
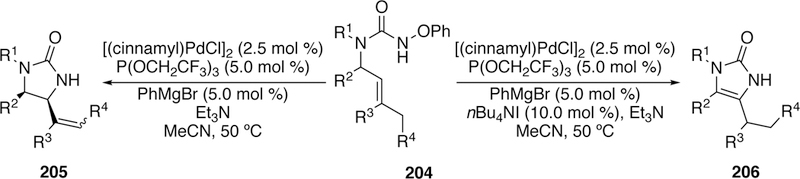
Watson’s Aza-Heck reaction of Urea-derived Electrophiles to Generate Imidazolidinones or Dihydroimidazolones.
In addition to the activation of N–O bonds in aza-Heck type cyclizations, N–Cl bonds have also been utilized. In 2000, Gottlich reported the activation of N–Cl bonds by copper in a radical cyclization reaction.286 In 2002, he followed up with the development of a palladium-catalyzed hetero-Heck-type reaction of alkene-tethered chloramines (207) to access a limited scope of cyclized alkyl chlorides (208) in modest to good yields (Figure 51).287
Figure 51.
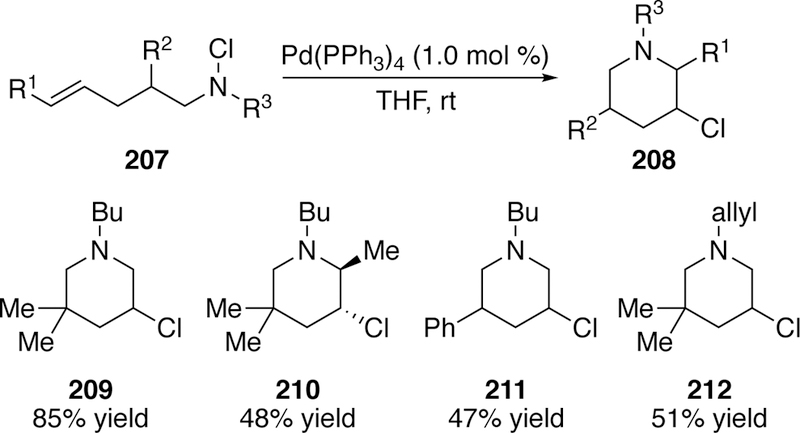
Scope of Gottlich’s Hetero-Heck-Type Reaction of Chloramines.
The authors propose that following the oxidative addition and migratory insertion, reductive elimination forming a C–Cl bond proceeds rather than the typical β-hydride elimination observed in the Heck reaction (Figure 52). Although a radical mechanism seems possible, Gottlich argues that the addition of acid or base, which normally affects aminyl-radical reactions, has no effect on this reaction. He also suggests that the diastereoselectivity seen with 210 would not be possible in a radical mechanism.
Figure 52.
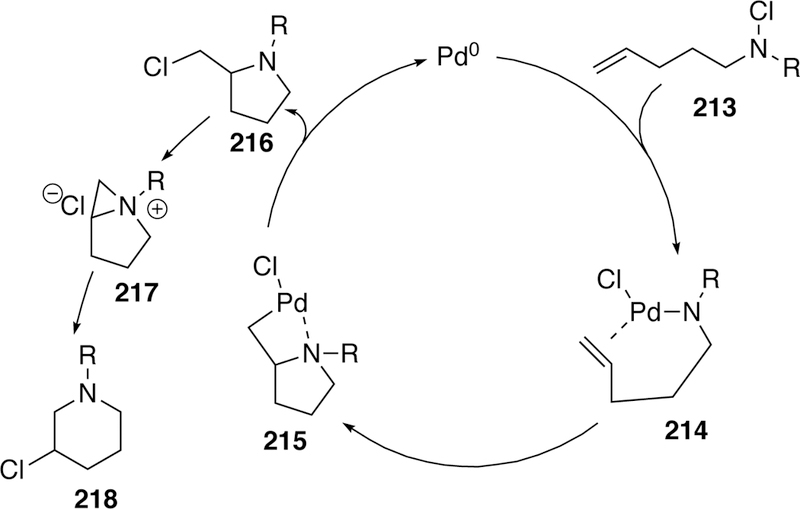
Proposed Catalytic Cycle of Gottlich’s Chloramine Heck-Type Cyclization.
3.2.5. Nitrogen Electrophiles in Arylation and Alkylation
3.2.5.1. N-Acyl Electrophiles in Arylation and Alkylation
In addition to Naraska, one of the early pioneers in the activation of N–O bonds using metals was Johnson. In 2004, he reported the copper-catalyzed coupling of diorganozinc reagents and O-benzoylhydroxylamine derivatives (Figure 53), an aza-version of the Negishi reaction.288 These O-benzoylhydroxylamines have become a very commonly used source of electrophilic nitrogen, which has recently been reviewed.235 In Johnson’s initial report a variety of O-benzoylhydroxylamine derivatives bearing N,N-dialkyl substitution could be efficiently coupled with dialkyl- or diarylzinc reagents at room temperature. Several copper sources were competent in the reaction. Even ortho-substitution on the diarylzinc reagent or diheteroarylzinc reagents could be accommodated with only slight drop in yield (222 and 223 respectively). The scope of diarylzinc reagents was expanded in a subsequent full paper.289
Figure 53.
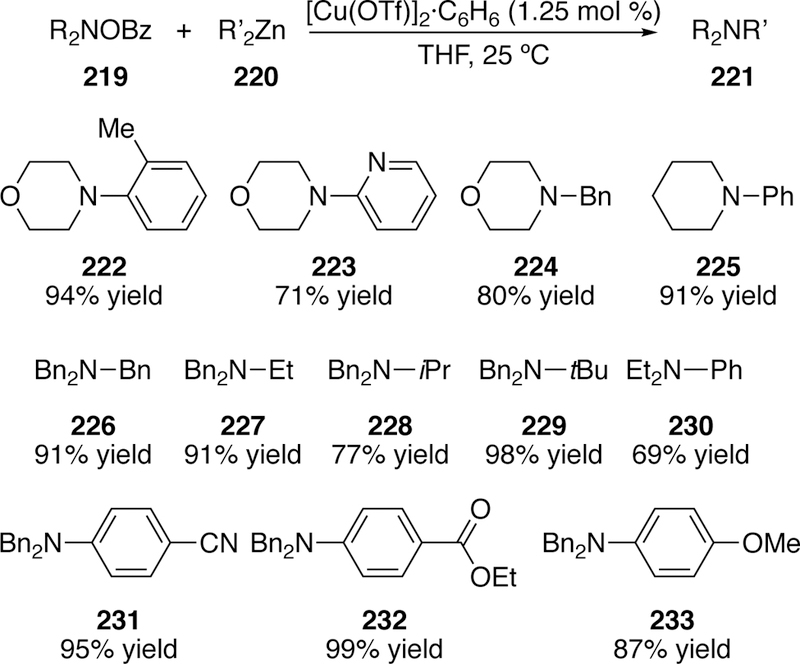
Scope of Johnson’s Copper-Catalyzed Coupling of Diorganozincs and O-Benzoylhydroxylamines.
In addition to using copper to activate N–O bonds, Johnson concurrently reported a nickelcatalyzed variant of the C–N cross-coupling reaction of O-benzoylhydroxylamines with arylzinc halides (Figure 54).290 In the previous copper-catalyzed method, arylzinc halides (234) were not well tolerated, however this complementary method uses aryl- or alkylzinc halides that are generated in situ from transmetalation of the corresponding Grignard reagent. The reaction also takes advantage of the commercially available and air stable bis(tri-phenylphosphine)-nickel(II) dichloride as the catalyst. The substrate scope in regards to the organozinc species was limited to aryl-(such as 222) or primary alkyl zincs (such as 224); secondary and tertiary alkylzinc halides failed to give the cross coupled product. Only N,N-dialkyl O-benzoylhydroxylamines were tested, but varied substitution on the arylzinc was well tolerated. For comparable products synthesized via the two methods, the yields using the copper-catalyzed method were typically higher and later investigations further explored the copper chemistry.
Figure 54.
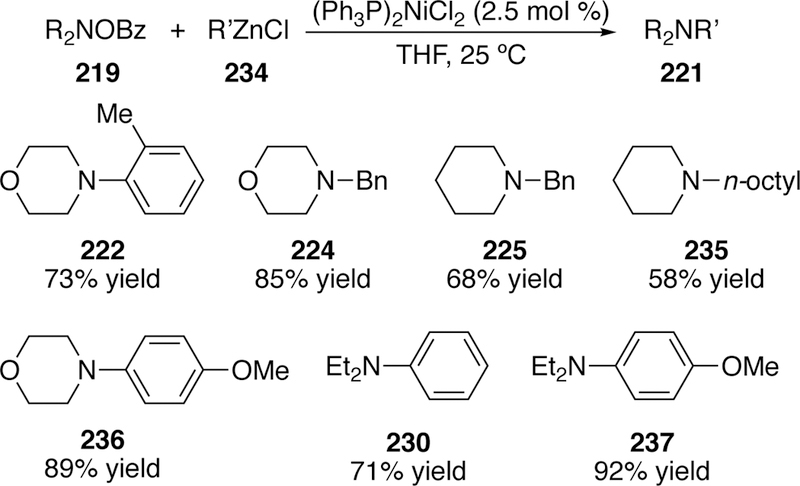
Scope of Johnson’s Nickel-Catalyzed Variant of Coupling of O-Benzoylhydroxylamines and Zinc Reagents.
In 2006, Johnson greatly expanded the scope of his copper-catalyzed cross-coupling method.291 This work highlights a simple method to prepare the O-benzoylhydroxylamines that improves upon the previously utilized method developed by Ganem.292 Shorter reaction times, lower reaction temperature, and limited side product formation could be achieved via a simple oxidation of the amine using a combination of benzoyl peroxide and an inorganic base in DMF. This method allowed expedient syntheses of a host of mono- and disubstituted O-benzoylhydroxylamines (219). Good to excellent yields were obtained when coupling with organozinc reagents for over twenty different coupled products including the sterically demanding coupling to form 245, albeit in modest yield (Figure 55). Even ortho-substituted diarylzincs, obtained from directed ortho-lithiation and metalation, could be coupled to morpholine-derived O-benzoylhydroxylamine.
Figure 55.
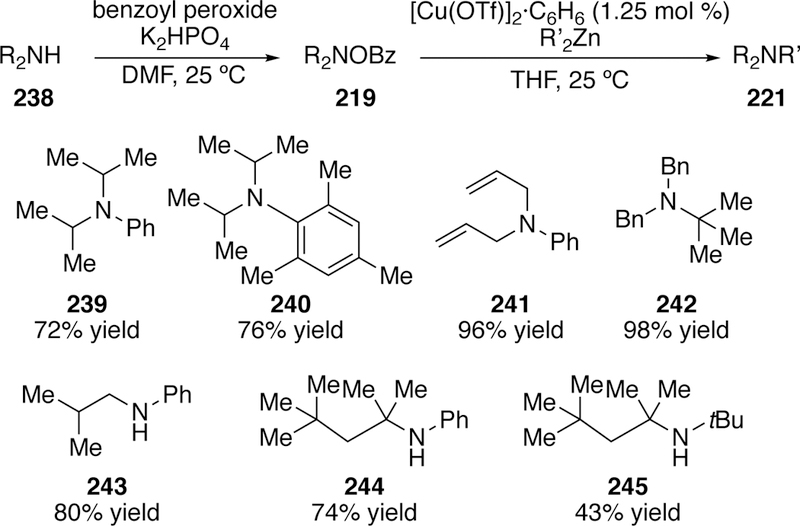
Scope of Johnson’s Copper-Catalyzed Coupling Utilizing an Improved O-Benzoylhydroxylamine Synthesis.
Mechanistic investigations by Johnson eliminated a radical pathway due to retention of stereochemistry in endo-and exonorbornyl substrates (Figure 56).293
Figure 56.
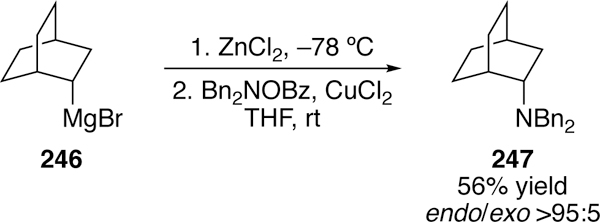
Retention of Endo-Stereochemistry In Johnson’s Copper-Catalyzed Cross-Coupling of Organozincs and O-Benzoylhydroxylamines.
By using an isotope labeling cross-over study with conformationally restricted substrate 248 (Figure 57), Johnson was able to gain insight into the mechanism of the reaction. In this experiment, a pathway involving oxidative addition to the N-O bond, followed by transmetalation and reductive elimination (traditional cross-coupling) would be expected to proceed via an intramolecular pathway. In this case, no cross-over of isotopic labels should be observed. In contrast, because of the constrained geometry, an intramolecular mechanism involving transmetalation followed by SN2 displacement at nitrogen would have to proceed through a strain-forbidden 6-endo-tet cyclization. In such a case, a similar intermolecular pathway would be expected to dominate, and cross-over of the isotope labels would be expected. In the event, subjecting a mixture of 248 and 248-d14 to the reaction conditions resulted in a statistical mixture of all four possible isotopic products (d0, d4, d10, and d14 Figure 57). This result supports a N-O cleavage occurring via an SN2-like pathway.
Figure 57.
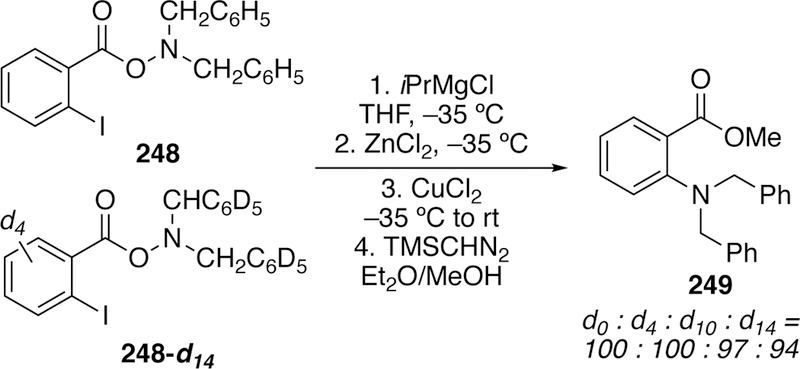
Johnson’s Deuteration Study Resulting In Statistical Mixture of Crossover And Non-Crossover Products Supporting An Intermolecular Reaction Mechanism.
In 2012, Miura utilized O-benzoyl hydroxyl am i nes in a cross-coupling with arylboronates.294 The optimized conditions utilized arylboronic acid neopentylglycol esters (250), monohydrated copper(II) acetate catalyst, l,2-bis(diphenylphosphino)-benzene ligand (252), and super-stoichiometric lithium tert-butoxide in tetrahydrofuran, along with N,N-dialkyl O-benzoylhydoxylamines (219, Figure 58). A variety of aryl groups could be coupled to the N,N-dibenzyl-O-benzoyl hydroxyl amine in yields ranging from 53–93%. Diminished yields were observed upon changing the alkyl substituents on the O-benzoyl hydroxyl ami ne from benzyl, as noted by 257–259.
Figure 58.
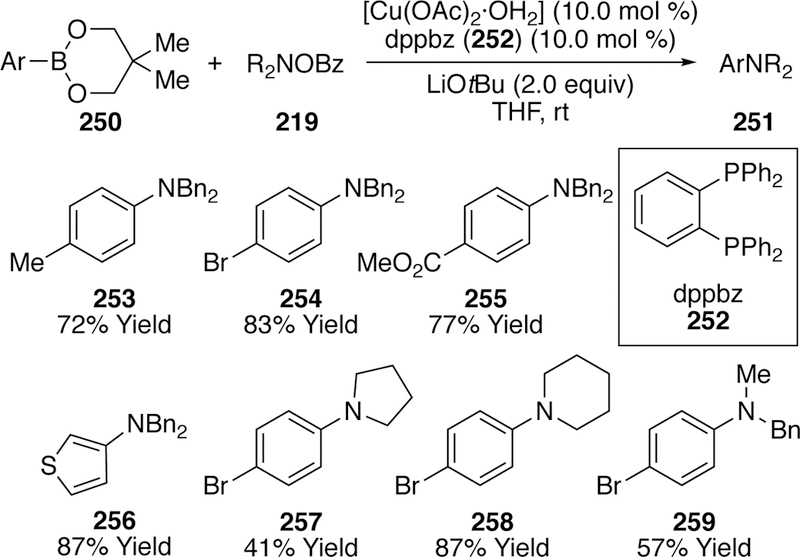
Scope of Miura’s Copper-Catalyzed Coupling of Arylboronates with O-Benzoylhydroxylamines.
Concurrently, efforts by Lalic expanded the substrate scope of the coupling of arylboronates and O-benzoyl hydroxyl ami nes.295 His optimized conditions also utilized arylboronic acid neopentylglycol esters (250), but the catalyst was derived from copper(I) tert-butoxide and Xantphos (Figure 59). An impressive array of highly substituted N,N-dialkyl O-benzoylhydroxylamines could be coupled, including even the highly sterically congested 2,2,6,6-tetramethylpiperidine with an ortho-substituted arylboronic ester to give 268 in 60% yield.
Figure 59.
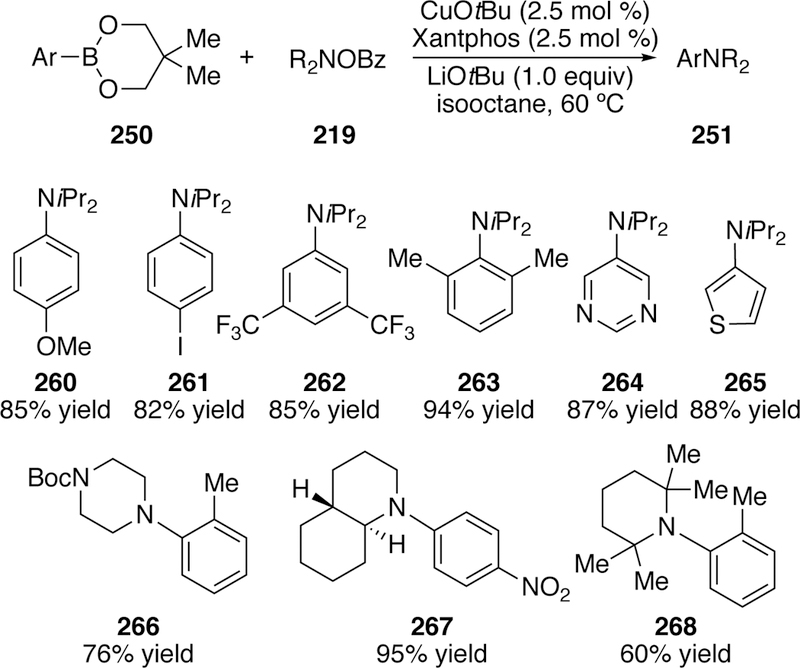
Scope of Lalic’s Coupling of Arylboronates and O-Benzoylhydroxylamines.
In 2013, Miura published a copper-catalyzed amination of arylsilanes using O-benzoylhy- droxylamines (219) to generate anilines.296 The optimized arylsilane was [(2-hydroxymethyl)-phenyl]dimethylphenylsilane (269), originally developed by Nakao and Hiyama, which forms the pentacoordinated arylsilicate in situ.297 A screen of ligands found that the combination of JohnPhos (270) and copper(I) iodide gave the best yields of the cross-coupled product. A variety of N,N-dialkyl substituted O-benzoylhydroxylamines performed well in this chemistry, but the aryl group was limited due to the requirement of the silane substitution (Figure 60).
Figure 60.
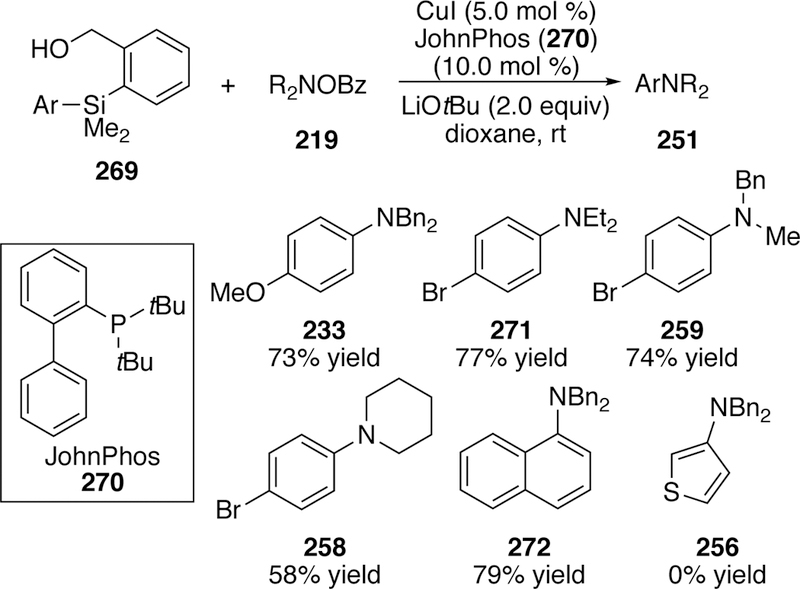
Scope of Miura’s Amination of Arylsilanes with O-Benzoylhydroxylamines.
A similar amination of O-benzoyl hydroxyl am i nes using silicon-based nucleophile was reported by Smith also in 20 1 3.298 He takes advantage of a siloxane transfer reagent 274 which in situ reacts with an aryllithium such as phenyllithium (273) generating a species 275 suited to transmetalate the aryl group (in this case phenyl) from silicon to copper giving 276 which can subsequently react with the electrophilic nitrogen species in the cross-coupling event (Figure 61).
Figure 61.
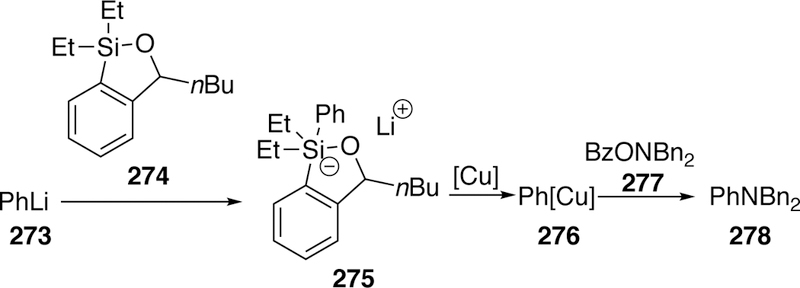
Proposed Mechanism of Smith’s Arylation Using Siloxane Transfer Reagents.
Another adaptation on the arylation of O-benzoyl hydroxyl am i nes was reported by Zhou and Wang in 2015.299 Similar to Johnson’s work with arylzincs and copper catalysis, this report details the use of aryl or heteroarylaluminum species with a copper catalyst (Figure 62). The high reactivity of organoalanes allows for full completion in an hour at room temperature.
Figure 62.
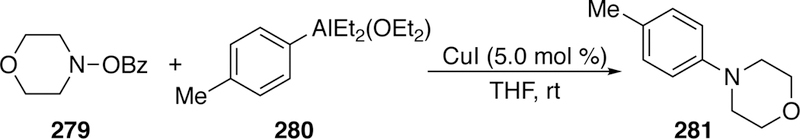
Zhou and Wang’s Cross-Coupling of Electrophilic Nitrogen with Organoalanes.
In addition to organoalanes, other organometallic nucleophiles have been coupled with O-benzoylhydroxylamines. Similar to the work of Liu and Xi who cross-coupled O-benzoylketoximes and aldoximes with alkenylzirconocenes (see section 3.2.2), Xi reports a cross-coupling of O-benzoylhydroxylamines with alkenylzirconocenes.300 Sterically demanding amines could be coupled giving products such as 286. Though in general, the scope in regards to the alkenylzirconocene (R2, R3, R4 in Figure 63) is limited to those with simple alkyl or aryl substituents.
Figure 63.
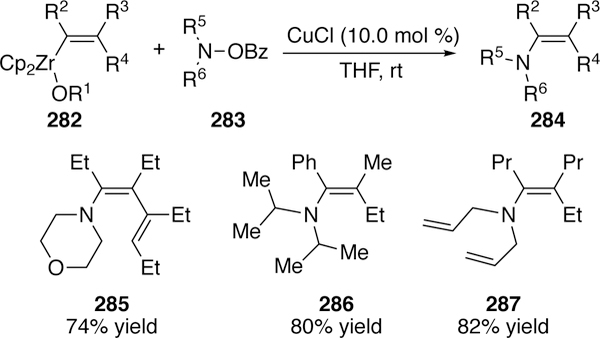
Scope of Xi’s Copper-Catalyzed Electrophilic Amination of Alkenylzirconecenes.
Knochel has built upon Gosmini’s precedence (see section 3.2.5.2) with a cobalt-catalyzed coupling of arylzincs with O-benzoylhydroxylamines as the electrophilic nitrogen source instead of chloramines as used by Gosmini.301 Using aryl zinc pivalates 289, accessed through initial halide-magnesium exchange, he is able to couple an assortment of functionalized arenes (both carbocyclic and heterocyclic) with a variety of both N,N-dialkyl O-benzoylhydoxylamines and N-alkyl,N-aryl O-benzoylhydoxylamines (Figure 64). This allows access to diarylalkylamines 290 which could not be accessed in the previously described copper- and nickel-catalyzed methods.
Figure 64.
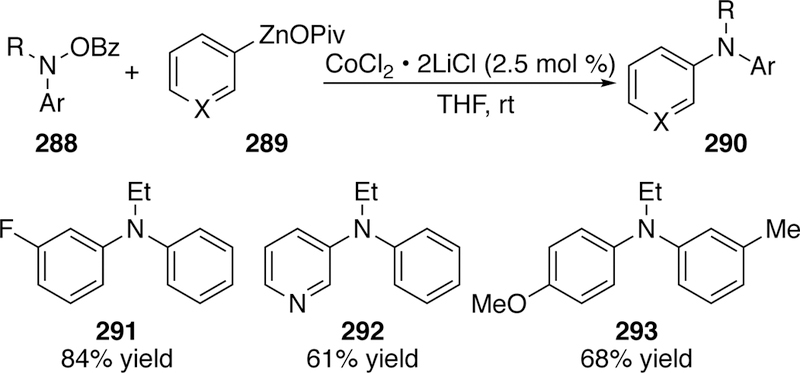
Scope of Knochel’s Cobalt-Catalyzed Coupling of Aryl-Substituted Hydroxylamine Electrophiles.
3.2.5.2. N-Acyl Electrophiles in Diaminations and Oxyaminations
O-benzoylhydroxylamines have also found use as one nitrogen component in diamination and oxyamination reactions. Wang first reported a copper-catalyzed diamination of alkenes with tethered N-methoxyamides (X = C in Figure 65a) or N-methoxyureas (X = C in Figure 65b) and O-benzoylhydroxylamines.302 This report was followed more recently by Wolfe who used palladium catalysis in the diamination of alkenes with tethered N-allylguanidines (X = N in Figure 65b) or N-allylureas (X = O in Figure 65b) and O-benzoylhydroxylamines.303 Variations of interrupted Wacker-type mechanism were proposed in both accounts.
Figure 65.
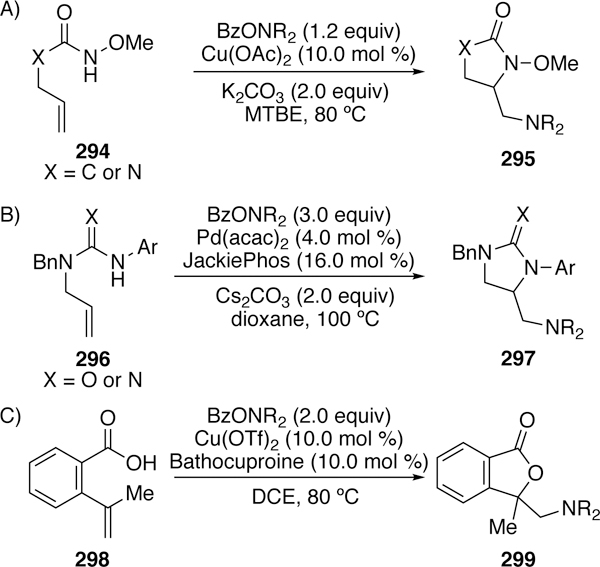
. Wang’s (A) and Wolfe’s (B) Diamination Reactions of O-Benzoylhydroxylamines and Wang’s (C) Aminolactonization Reaction.
Wang has also studied the use of O-benzoylhydroxylamines in the oxyamination of alkenes. Her two reports detail both three-component coupling and two-component couplings in which the alkene is tethered to the carboxylic acid (Figure 65c).304–305 There have been many examples of oxyaminations utilizing O-benzoylhydroxylamines or other N–O bond-bearing species; however, the mechanisms invoked are often radical-based or involve iron-nitrenoid species rather than an oxidative addition event, and they will not be described in detail here.306
3.2.5.3. Chloramines in Arylation
In 2008, Lei investigated the coupling of boronic acids with aniline-derived chloramines using copper catalysis (Figure 66).307 The author was able to rule out a radical mechanism because a reaction containing radical scavenger 1,1-diphenylethylene proceeded without inhibition. He also found that preforming the [PhCu] species resulted in no formation of coupled product. With this information, Lei’s proposed mechanism involves an initial oxidative addition of copper into the N–Cl bond. However, the details of the reaction mechanism post oxidative addition, including the key N–C bond forming step with the boronic acid remain unclear.
Figure 66.
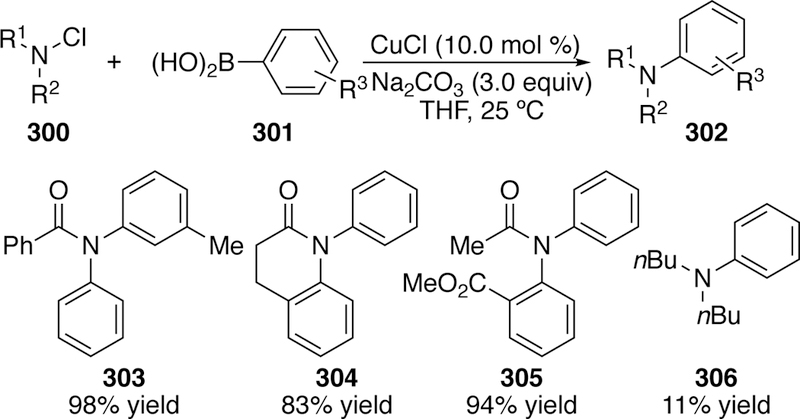
Scope of Lei’s Coupling of Chloramines with Aryl Boronic Acids.
Significant work in the field of N–Cl bond activation by metal catalysts has been done by Jarvo beginning with her publication in 2009. She reports a nickel-catalyzed amination of diorganozinc reagents using preformed N,N-dialkylchloramines (300).308
A variety of chloramines were coupled in good yields, including several bearing additional heteroatomic substitution (310), and in one case an unprotected primary amide (311) (Figure 67). However, the reaction is sensitive to substitution directly adjacent to the amine, as evidenced by α-branched amine product 312. Jarvo also demonstrated a one-pot chlorination/arylation protocol, which greatly improves the utility of this synthetic method by eliminating the need to isolate sensitive chloramine intermediates.
Figure 67.
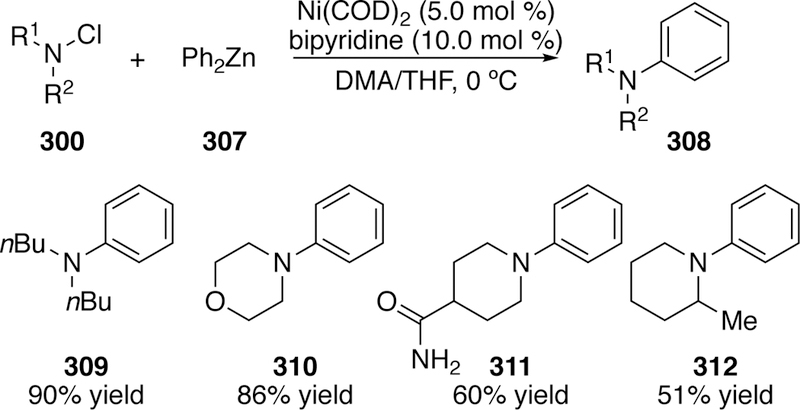
Scope of Jarvo’s First-Generation Nickel-Catalyzed Coupling of Chloramines and Diphenylzinc.
The one-pot chlorination and cross-coupling is featured again in her titanium-mediated reaction of primary chloramines and Grignard reagents (Figure 68).309 While she found that a nickel catalyst could direct the coupling with Grignard reagents, the yields were modest. Switching to titanium boosted the yields, and a ligand was not required. Unlike the previous method, substitution next to the nitrogen was well tolerated (e.g. 316). It is likely that this does not proceed through a traditional oxidative addition mechanism.
Figure 68.
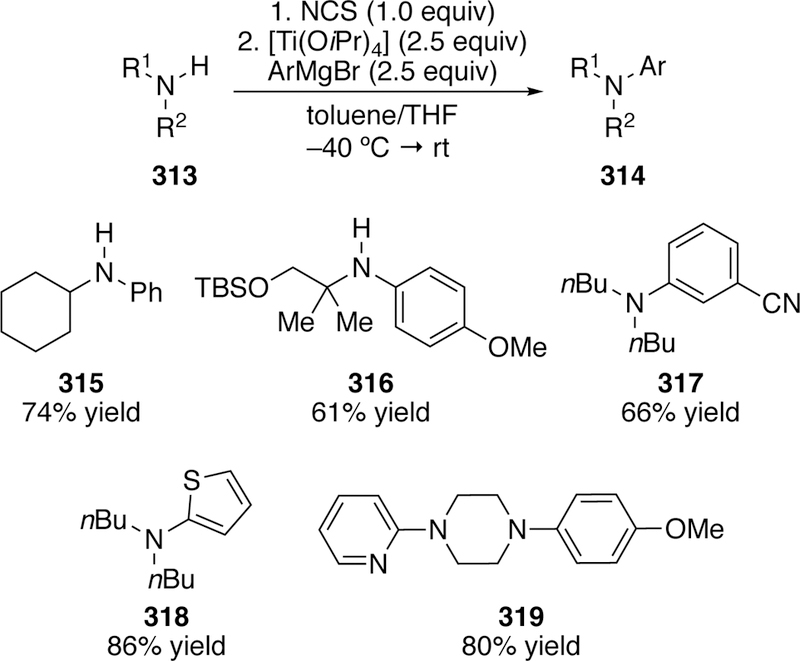
Scope of Jarvo’s Titanium-Mediated Coupling of in situ Generated Chloramines and Aryl Grignard Reagents.
The cross-coupling of chloramines and arylzincs has also been investigated by Gosmini using a cobalt catalyst (Figure 69).310 This two-step procedure utilizes cobalt(II) bromide and zinc powder to generate arylzinc species 321. Upon full conversion of the arylbromide, the zinc dust is filtered off and the solution is added directly to freshly prepared chloramine 283, bearing one or two alkyl substituents (R1 and R2 in Figure 69), and triethylamine in acetonitrile at 0 °C. A variety of aryl groups could be incorporated, including those bearing carbonates, tri-fluoromethyl groups, chlorides (322), fluorides (324), nitriles, thiols (323), and methoxy groups (325). Sterically encumbered chloramines could also be coupled but with slightly diminished yields as noted by 324.
Figure 69.
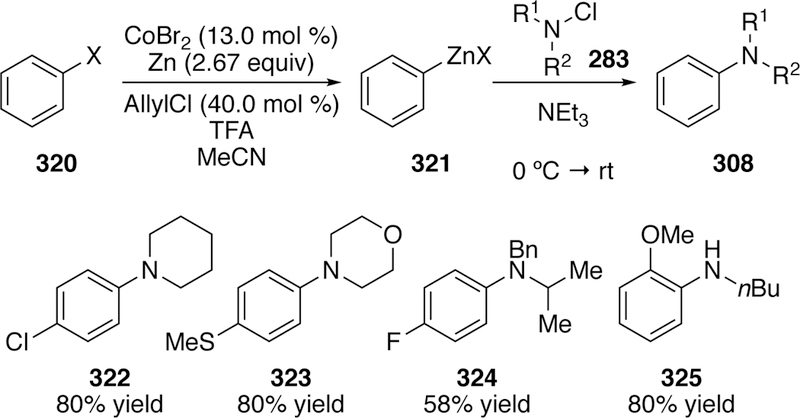
Scope of Gosmini’s Cobalt-Catalyzed Amination of Arylzincs with Chloramines.
3.2.5.4. N-Alkoxy Electrophiles in Arylation
In 2016, an interesting arylation of electrophilic nitrogen was reported by Sato.311 He utilized a novel source of electrophilic nitrogen, N-methoxyamines. He notes that, due to the poor leaving group ability of methoxy compared to the more commonly used benzoyl, the reaction requires additional activation of the methoxy by a Lewis acidic group. Modest coupling was seen between 326 and aryl boroxines using a copper catalyst (Figure 70).
Figure 70.
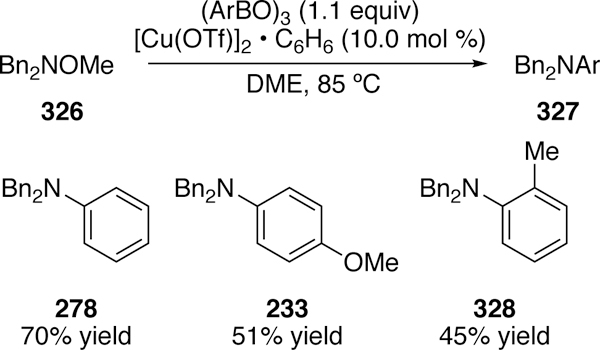
Scope of Sato’s Arylation of N-Methoxyamines with Copper Catalysis.
To access aminated arenes bearing primary amines several strategies have been employed involving different nitrogen electrophiles. In 2010, oxime derived electrophiles were employed by Daşkapan which upon hydrolysis gave rise to primary amines.312–313 Kürti has employed the use of unsubstituted oxaziridines.314 Oxaziridines had been used previously by Vidal in transition-metal free electrophilic amination.315 This unsubstituted oxaziridine 330 in the presence of stoichiometric copper could aminate aryl lithiums, aryl Grignards, and aryl zinc reagents which, upon aqueous workup, yields the primary arylamine (e.g. 331) products in modest to good yields (Figure 71). He also can access secondary amine products when using N-alkyl-O-benzoyl hydroxylamines (BzONHR) as the electrophilic nitrogen source. Two mechanistic pathways for this oxaziridine chemistry were proposed. In one proposal there is direct nucleophilic attack of the aryl cuprate on the oxaziridine. However, the mechanism favored involves oxidative addition of the arylcopper(I) across the N–O bond forming a metal-lacycle which reductively eliminates, and upon workup, hydrolyses to give the observed product.
Figure 71.
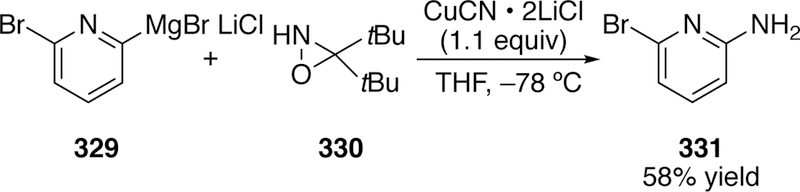
Example of Kürti’s Amination Using Oxaziridines to get Primary Arylamines.
3.2.6. Amination of Silyl Ketene Acetals with Nitrogen Electrophiles
T. Miura and Murakami have utilized chloramines to aminate silyl ketene acetals (332) and generate amino acid derivatives (334) using copper (Figure 72).316 Mechanistically the authors invokes oxidative addition of copper(I) to the chloramine to generate an amino copper(III) intermediate. Subsequent transmetalation with the silyl ketene acetal generates a copper(III) enolate, which reductively eliminates to form the new C–N bond and regenerate the catalyst. The substrate scope in regards to the chloramine is diverse, however the silyl ketene acetal was limited to those derived from triisopropylsilylchloride (TIPSC1) and methyl arylacetates.
Figure 72.
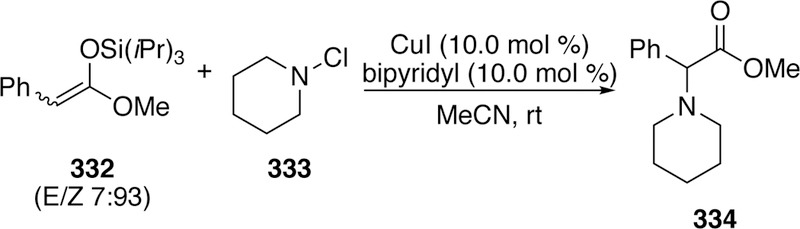
T. Miura and Murakami’s Amination of Silyl Ketene Acetals Using Chloramines.
Similar to T. Miura and Murakami’s work using chloramines to aminate silyl ketene acetals using copper,316 M. Miura developed a method to aminate silyl ketene acetals using O-benzoylhydroxylamines and a copper catalyst.317 The new method boasted a greatly expanded sub-strate scope in regards to the silyl ketene acetal. The method using chloramines was largely limited to aryl acetate-derived substrates, whereas a variety of alkyl and even unsubstituted acetates could be utilized as the silyl ketene acetal with O-benzoyl hydroxyl am i nes in good to excellent yields (Figure 73). Sterically hindered amines (343) could be coupled as well. Though he did not have enough data to propose an exact mechanism, Miura was able to rule out a radical pathway by conducting a TEMPO addition study and finding no inhibition.
Figure 73.
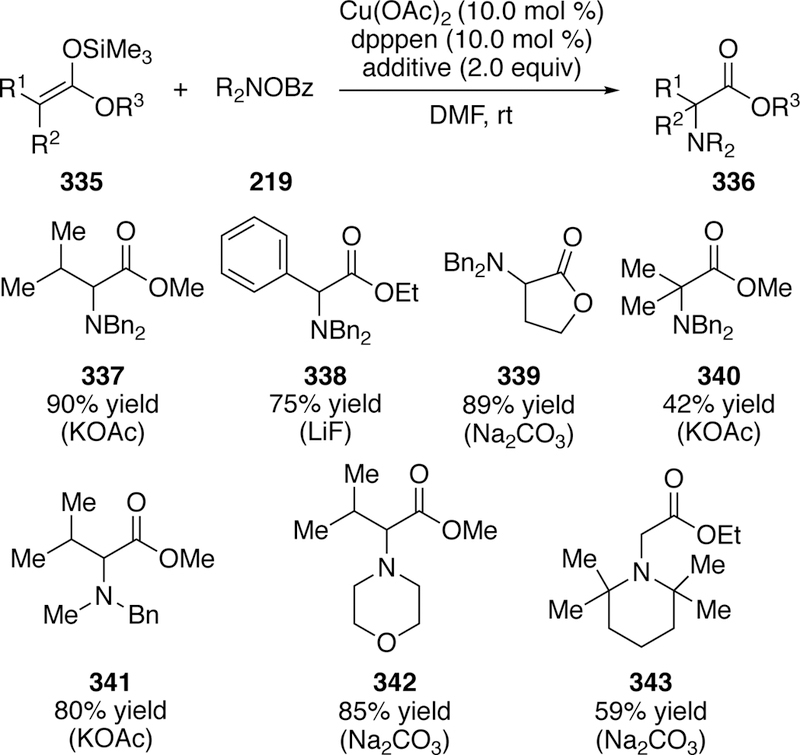
Scope of M. Miura’s Amination of Silyl Ketene Acetals Using O-Benzoylhydroxylamines.
Similar products as those described above could be obtained by Wang directly from the ester or amide using in situ generated zinc enolates rather than preformed silyl ketene acetals.318 In the presence of copper(II) chloride and bipyridine ligand the zinc enolate 345 could be converted to the aminated product 346 using O-benzoylhydroxylamine (Figure 74). In the absence of copper and ligand and at elevated temperatures, the zinc enolate reacts with the O-benzoylhydroxylamine to preferentially give the acylated product 347, suggesting that an oxidative addition event between copper and O-benzoylhydroxylamine is necessary to achieve the aminated product. Good yields were seen for a variety of ester and amide substrates as well as for a variety of differentially substituted hydroxylamines including one bearing a free N–H bond such as R2 = H in Figure 74. Wang reported another variant on this amination chemistry from phosphonates or phosphine oxides instead of esters.319
Figure 74.
Wang’s Coupling of in situ Generated Organozincs with Electrophilic Nitrogen.
3.2.7. Nitrogen Electrophiles in Nontraditional Alkylations
Lalic also developed a one-pot sequential hydroboration to generate primary alkyl boronates followed by transmetalation and a copper-catalyzed electrophilic amination to generate anti-Markovnikov tertiary amine products in a formal hydroamination via in situ generated boronates.320 The optimized conditions involve treating the terminal alkene substrate to a stoichiometric copper NHC complex in toluene along 9-borabicyclo(3.3.l)nonane, and an N,N-dialkyl-substituted O-benzoy1hydroxylamine. The reaction affords access to sterically demanding amines, including adamantyl-substituted 351 and 2,2,6,6-tetramethylpiperidine-derived 353 (Figure 75).
Figure 75.
Scope of Lalic’s One-Pot Hydroboration and Copper-Catalyzed Electrophilic Amination.
3.2.8. Nontraditional Arylations of N-Acyl Electrophiles
Another approach to the formation of aryl–N bonds utilizes in situ generated organometallic reagents as the nucleophile. One example comes from Wang. His net C–H amination of arenes, an adaptation of his work on the amination of organozinc reagents derived from esters and amides (see section 3.2.6), proceeds through in situ generation of aryl zinc species.321 Initial formation of the metalated arene (e.g. 355) is followed by addition of a symmetrical or unsymmetrical N,N-dialkyl-substituted O-benzoyl hydroxyl amine along with a copper(II) acetate catalyst, generating the coupled product (e.g. 356, Figure 76).
Figure 76.
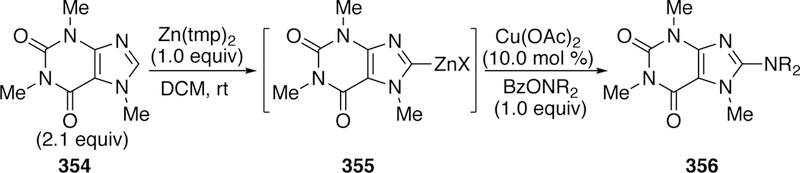
Wang’s C–H Zincation Followed by Electrophilic Amination with Copper.
A similar strategy was reported by Lee in 2017 on the coupling of in situ generated aryl alanes and N,N-dialkyl O-benzoyl hydroxyl am i nes using copper catalysis.322 The aryl ring is initially deprotonated, and the resulting anion is converted to the aryl alane (358). The copper catalyst and electrophile then are added (Figure 77). Good yields were obtained for the coupling of a variety of different heteroaryl starting materials and other electronically-biased arenes. The reaction could even be run effectively on gram scale.
Figure 77.
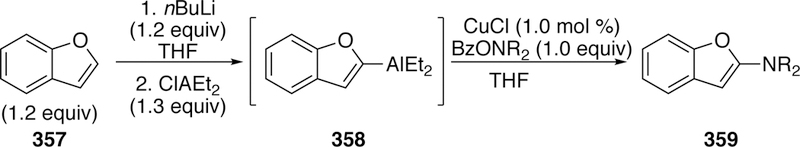
Lee’s in situ Generation of Organoalane for Cross-Coupling with Electrophilic nitrogen.
Another related transformation was reported by Wang.323 In this reaction the nucleophile, an arylzinc (361), is generated in situ from an electronically-biased arene or with the assistance of a directing group. This nucleophile couples to the nitrogen electrophile with a copper catalyst (Figure 78).
Figure 78.
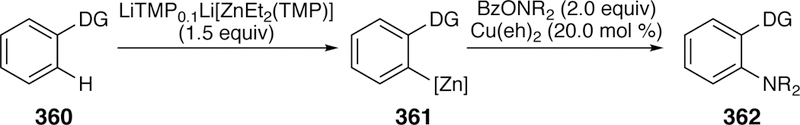
Strategy of Wang’s Directed Zincation of Arenes Followed by Copper-Catalyzed Electrophilic Amination.
A number of ortho-directing groups were suitable for phenyl rings, while biased heteroarenes were found to not require a directing group (Figure 79). A diverse array of aminated products were obtained using this method, and naturally occurring compounds like nicotine could be aminated giving 368 under the reaction conditions. This method was successfully extended by Wang to include azidation using azidoiodinane as the electrophilic nitrogen source in the same publication.
Figure 79.
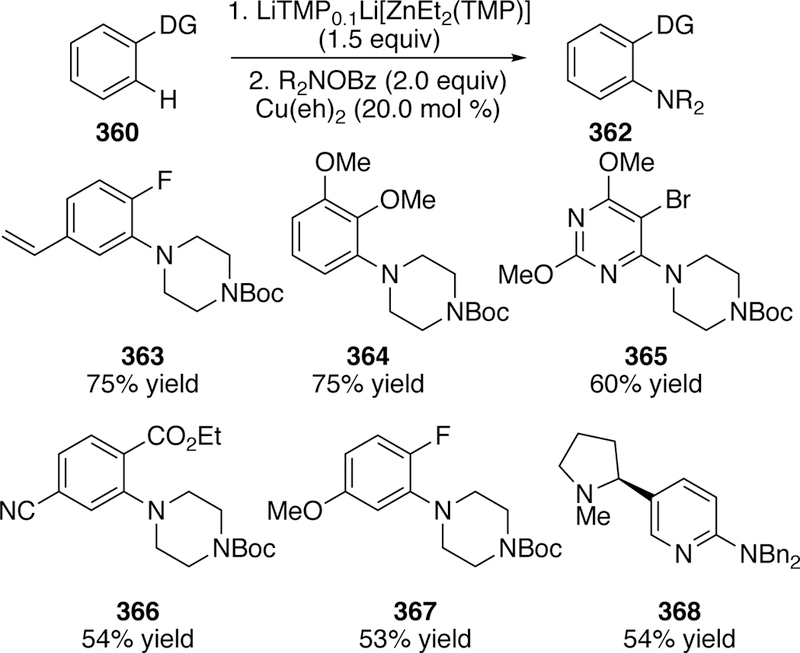
Scope of Wang’s Amination of in situ Generated Arylzincs with Electrophilic Nitrogen.
3.2.9. N–N Bond Electrophiles
3.2.9.1. Hydrazonium Electrophiles
In additional to the traditional pentafluorobenzoyloxime ester substrates that are a hallmark of the Narasaka–Heck reaction (see section 3.2.1), Narasaka also found that N,N,N-trialkylhy-drazonium salts can also react with palladium(0) in a similar transformation.324 When the benzophenone derived hydrazonium 369 was subjected to a Pd(0) source in deuterated DMF, the trimethylamine signal of the starting material disappeared and a new signal appeared up-field. He suggests that the shift of this upfield signal is more indicative of either free trimethylamine, the trimethylamine hydrochloride salt, or a trimethylamine bound to a palladium(II) species rather than a hydrazine species. Either of these three possibilities would result from the cleavage of the N–N bond supporting the proposed oxidative addition of palladium into the N–N bond (Figure 80).
Figure 80.
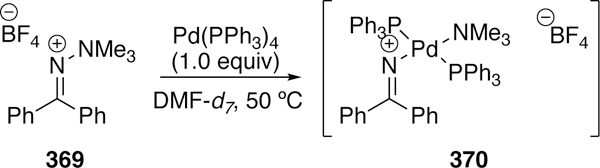
Narasaka’s NMR Studies of Proposed Oxidative Addition Intermediate of N,N,N-Trialkylhydrazonium Salts.
When Naraska subjected N,N,N-trialkylhydrazoniums bearing terminal or internal pendant alkenes he noted the formation of both 5-exo cyclization products (the unconjugated pyrrolidine and pyrrole (372) in varying amounts depending on the conditions used (Figure 81).
Figure 81.
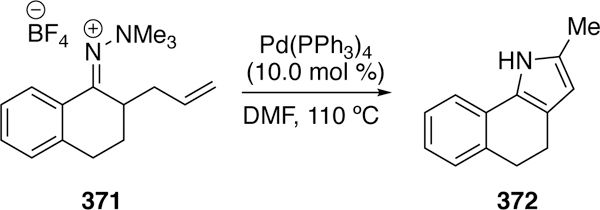
Narasaka’s 5-exo Cyclization of N,N,N-Trialkylhydrazoniums Bearing Pendant Alkene.
3.2.9.2. Diaziridinone Electrophiles
Shi has also activated the N–N bonds of diaziridinones and the related thiadiaziridines325–327 in a host of copper- or palladium-catalyzed diaminations of mono-, di-, and trienes as well as esters and ketones.328–342 From his work in palladium-catalyzed asymmetric diamination, Shi found that Pd-NHC complex 376 was found to be the optimal catalyst, but only diaziridinone 374 was utilized in the substrate scope in which moderate enantioselectivities were obtained (Figure 82). The mechanism of this transformation has been studied 343 and this chemistry was detailed in an account in 2014.344
Figure 82.
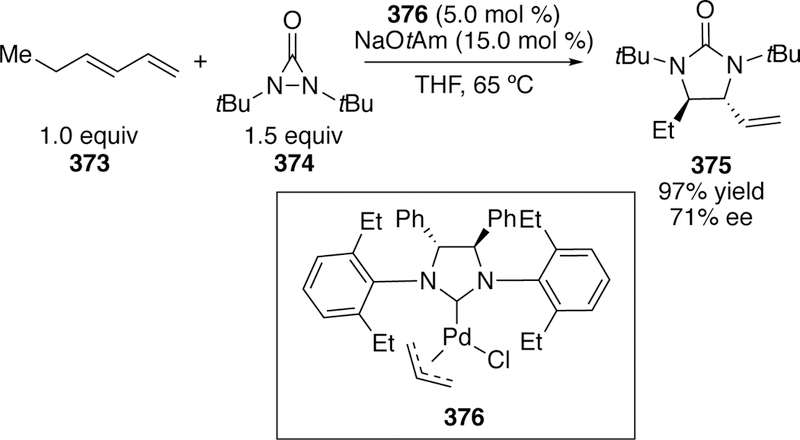
Shi’s Diamination using Diaziridinones and a Palladium-NHC Catalyst.
Shi has also applied the use of diaziridinone 374 in the synthesis of spirocyclic indolines. In 2013, he reported a polyamination of α-methylstyrenes with diaziridinone 374 via an oxidative addition of palladium across the N–N bond followed by sequential C–H animations.345 He built on this methodology in 2014 to synthesize mono-aminated norbornane-fused in-dolines 382 (Figure 83).346 In the reaction, a cascade Heck reaction with norbornene (378) was followed by C–H activation of the pendant arene. This generates palladium(II) palladacycle 380 which adds oxidatively across the N–N bond of the diaziridinone 374 to form palladium (IV) species 381 which releases the indoline product 382 and tert-butyl isocyanate (383).
Figure 83.
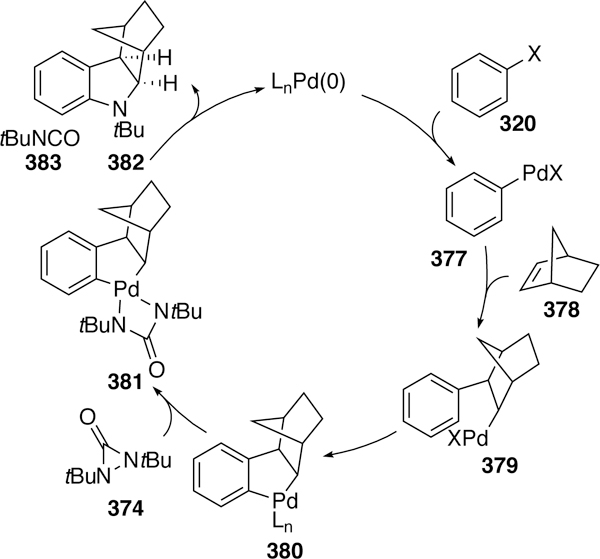
Proposed Catalytic Cycle of Shi’s Indoline Synthesis via C–H Activation and Electrophilic Amination.
3.2.10. N–CN Electrophiles
Recent work in the field of electrophilic nitrogen cross-coupling by Nakao has identified a novel class of compounds suitable for oxidative addition by a metal catalyst. Instead of activating a N–O bond or an N–N bond he is able to activate the N–CN bond of cyanamides (e.g. 384).347 A review detailing the construction of cyanamides has been reported.348 The initial report details the palladium-catalyzed aminocyanation of alkenes in the presence of an addition Lewis acid catalyst which likely serves to coordinate the nitrile nitrogen, further activating the N–C bond for oxidative addition by palladium(0). This is corroborated by the isolation and crystal structure of an oxidative addition complex. A variety of 5-exo cyclized and cyanated products of type 385 could be obtained, and modest enantioselectivity could be seen when using SKP ligands (e.g. 390, Figure 84). Similar to efforts in the field of aza-Heck chemistry, 6-exo cyclization was possible with conformationally restricted substrate giving product 388 in modest yield, though more challenging substrates were not reported.
Figure 84.
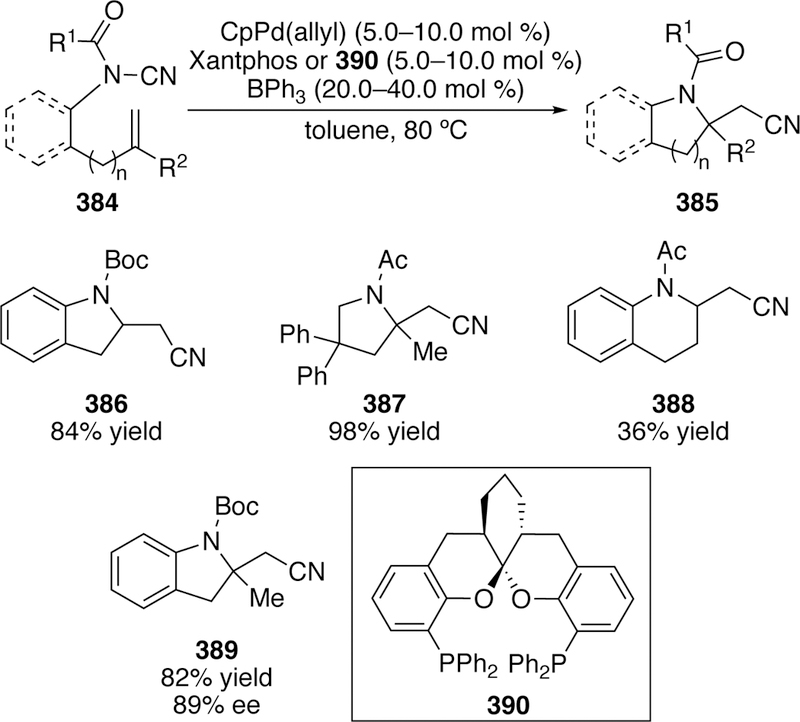
Scope of Nakao’s Aminocyanation of Alkene-Tethered Cyanamides.
Concurrently to this publication, a similar reaction was reported by Douglas in which the same transformation could be accomplished in the absence of a metal catalyst, albeit with sulfonamide-derived cyanamides instead of the carbamate-derived cyanamides (384) used by Nakao.349 Computational work investigating this metal-free variant have been conducted.350 An interesting variant on Nakao’s chemistry was reported by Shi in 2016 in which methylenecyclopropanes were oxycyanated by carbamate-derived cyanamides to give structurally complex products (Figure 85).351
Figure 85.
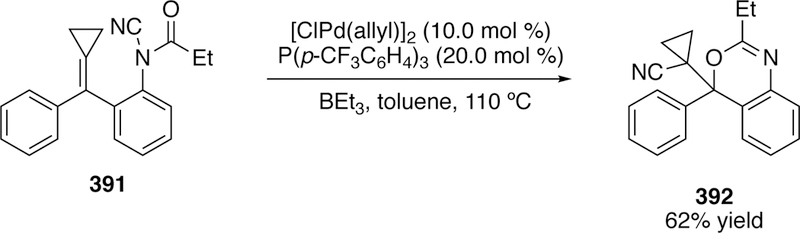
Shi’s Cyclopropyl Variant on Nakao’s Chemistry
In 2018, Douglas reported another similar aminocyanation of alkenes using cooperative palladium and Lewis-acid catalysis that allowed access to a diversified product library. He also explored downstream functionalization of the products and further explored the mechanism of this transformation, finding evidence to support an interrupted aza-Heck-type mechanism (Figure 86).352 First, the Lewis acid B (usually a BR3 species) coordinates to the terminal nitrogen and palladium(0) undergoes oxidative addition across the N–CN bond. Co-ordination of the alkene to the Pd(II) intermediate gives rise to 394 (Figure 86). Then syn-migratory insertion occurs giving cyclized 395, which then undergoes reductive elimination with the nitrile to yield the product 396 and regenerate the Pd(0) catalyst.
Figure 86.
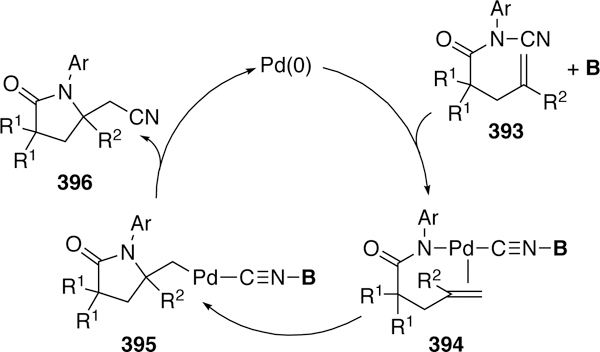
Proposed Mechanism of Douglas’ Aminocyanation Reaction.
4. Boron Cross-Coupling
Organoboron compounds have long been utilized as nucleophiles in traditional crosscoupling especially with palladium catalysis, and they have been lauded for their stability, low toxicity, and ready availability.22,353 In addition to metal catalyzed cross-coupling of organoborons, there has been a long standing interest in metal catalyzed hydroboration.354 Early interest in metal-boryl complexes stemming from hydroboration by Marder, Norman, Merola, Hartwig, Suzuki, and Miyaura in the 1990s found that metals can add oxidatively across B–H and B–B bonds.355–360 Typically, these reagents are associated with reductive borylation processes. In contrast, the more electrophilic boryl halides, which do not typically provide reductive byproducts in reactions with organic molecules, have also recently begun to be explored as a means to introduce boron centers via cross-coupling reactions.
4.1. Introduction
In 1998, Marder and Norman began to investigate the oxidative addition of boron-chlorine and boron-bromine bonds with platinum.361 In 2001, Tanaka reported the construction of a palladium complex derived from the oxidative addition of a B–Cl bond (see section 4.2).362 In the late 2000s, Braunschweig again investigated the oxidative addition of palladium into boron-halide bonds. First, complexes derived from the oxidative addition of palladium across B–Br were crystallized (Figure 87).363
Figure 87.
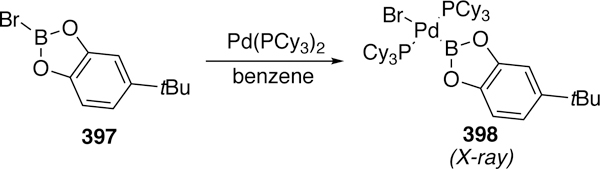
Braunschweig’s Substituted Catecholborylbromide Oxidative Addition Complex.
The oxidative addition of B–Br bonds would later be studied computationally by Sakaki.364 Then in 2009 Braunschweig investigated the oxidative addition of boryl chlorides with platinum.365 And lastly in 2011, Braunschweig also showed that platinum was able to activate B–F bonds.366
In 2001, Marder proposed a boron variant of a Heck reaction to give the same products as dehydrogenative borylation.367 However, in order for this reaction to be successful, he needed to determine if the chosen electrophilic boron source, chlorocatechol borane (catBCl, 400), was compatible with the phosphine ligand and base that are necessary in the Heck reaction and many other cross-couplings. In the presence of triethylamine or phosphines, adducts (399 and 401) were formed at ambient temperature according to NMR analysis (11B shift information in Figure 88) and crystal structures were obtained for catBCl adducts including for the trimethylphosphine adduct 399 and the triethylamine adduct 401. He concluded that undesireable side reactivity of the catBCl, in which ring opening and dimerization of the catBCl can occur, would be prohibitive of the use of labile phosphine ligands. Also, he concluded that the oxidative addition of the B–Cl bond in amine adducts by transition metals is unprecedented and would need to be explored.
Figure 88.
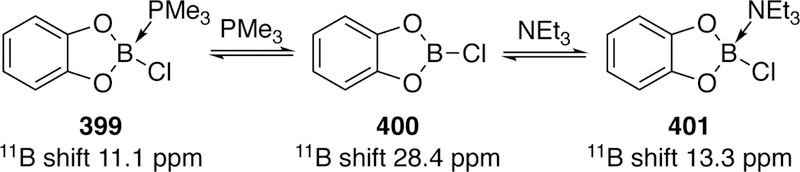
Marder’s NMR Studies and 11B Shift Information for Phosphine and Triethylamine Adducts in CDCl3.
4.2. Preliminary Efforts Towards Cross-Coupling
The first reported metal-mediated reaction of a boron electrophile and an alkyne nucleophile was published in 2001 by Tanaka.362 He previously had an interest in the activation of other chloro-heteroatom bonds specifically silyl–chloride bonds (see section 2.2.1). In his 2001 report, Tanaka found that oxidative addition of palladium across the B–Cl bond of 403 generates complex 404 which can insert into an alkyne 405 to give products of type 406 (Figure 89).
Figure 89.
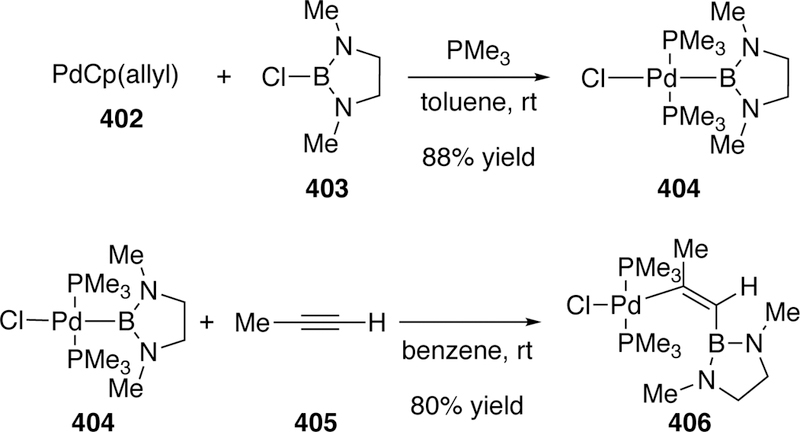
Tanaka’s Preliminary Efforts Towards Cross-Coupling of Boron Electrophiles.
4.3. Intramolecular Cross-Coupling of Boron Electrophiles
The first report of a catalytic coupling of a B–Cl species, (diisopropylamino)chloroboryl ether, and an alkyne was reported in 2005 by Suginome,368 who has subsequently done extensive work on the intramolecular coupling of boron electrophiles. The initial reaction was intramolecular, forming the 5-exo product, and utilized a nickel catalyst and triphenylphosphine ligand. By using the amine-substituted borylchloride (e.g. 407), which is more electron-rich than ClBcat, he was evidently able to avoid the undesireable phosphine- boron adduct formation that was reported by Marder (section 4.1).
Following oxidative addition across the B–Cl bond and insertion into the alkyne, the generated intermediate vinylnickel species 409 undergoes cis-to-trans isomerization to the sterically favored trans isomer 410 (Figure 90). This then undergoes transmetalation with an alkynylstannane 411 to yield 412. Reductive elimination yields the direct product of the coupling 413, which - due to its sensitivity to moisture - is transformed to the acetate-protected pinacol boronate 414 before isolation. Suginome was able to isolate and obtain a crystal structure of the cyclized vinylnickel intermediate of type 410, supporting the proposed cis-to-trans isomerization step.
Figure 90.
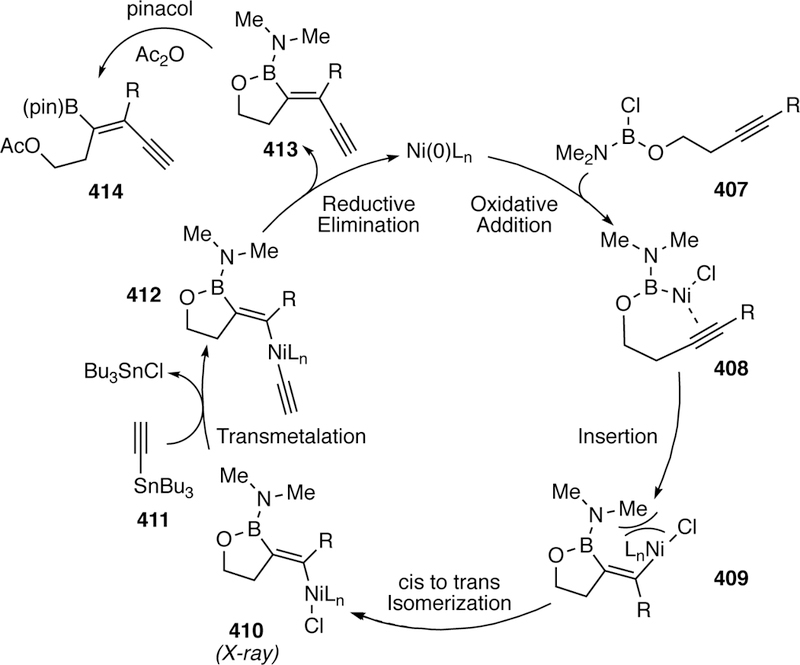
Proposed Catalytic Cycle of Suginome’s Nickel-Catalyzed Intramolecular Borylation/Stille Coupling.
In 2008, Suginome reported another intramolecular borylation reaction using similar starting materials. In this case, palladium was used instead of nickel, and an organozirconium reagent was used in place of the stannane.369 What is remarkable about this transformation is that the cis/trans selectivity could be controlled by the choice of phosphine ligand (Figure 91). When using trimethylphosphine as the ligand, the cis product 416 was obtained in greater than 99:1 selectivity for most substrates tested. Switching to a bulkier phosphine ligand such as tri-tert-butylphosphine or tricyclohexylphosphine inverted the selectivity to favor the trans product 417 again in greater than 99:1 selectivity for most substrates tested. The proposed mechanism is nearly identical to that of Figure 90. Suginome attributes the observed selectivity to the influence the ligand has on the cis-to-trans isomerization step. With a small ligand, the steric clash with the dialkylamino group is less significant and transmétalation proceeds faster than cis-to-trans isomerization resulting in the cis product 416. With a large ligand, the steric clash with the dialkylamino group is significant and cis-to-trans isomerization proceeds faster than transmétalation resulting in the trans product 417.
Figure 91.
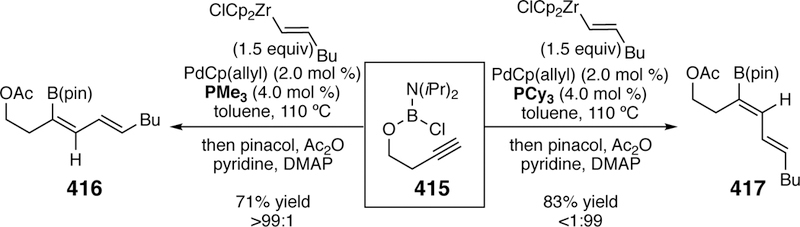
Suginome’s Observed Reversal of Selectivity in the Intramolecular Borylation and Coupling with Organozirconium Species.
Suginome and Daini later extended this method from alkynes to tethered alkenes generating products with a stereogenic center (Figure 92).370 The use of trimethylphosphine in a 2:1 ratio with palladium was crucial to obtaining a high yield of the trans product as was found in their previous work. However, unlike their previous work, only alkenylzirconiums were reactive; arylzirconiums and alkylzirconiums did not participate in the coupling reaction. 1,2-Disubstituted alkenes also failed to participate in the reaction and only 5-exo cyclizations were possible.
Figure 92.
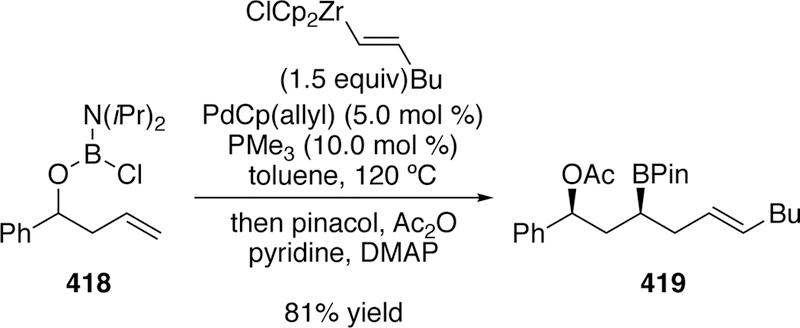
Suginome’s Borylation of Alkenes and Subsequent Cross-Coupling with Organozirconium Species.
In 2013, Suginome further extended this methodology to a tandem boration of an internal alkyne followed by a Heck reaction.371 Again PdCp(allyl) was found to be the optimal palladium source, but triphenylphosphine was the optimal ligand and triethylamine base was needed to turn over the palladium catalyst. The formation of unreactive triethylamine adducts as described by Marder (Section 4.1) was inconsequential - perhaps due to the use of an electron-rich aminoborylchloride (e.g. 420). Higher yields could be obtained in acetonitrile than toluene; however, this limited the temperature to 80 °C and many of the substrates were sluggish. Aryl- and heteroaryl-substituted alkenes gave modest yields while alkyl-substituted alkenes provided diminished yields of coupled product (Figure 93). Yields could be increased by the addition of geminal substitution and taking advantage of the Thorpe-Ingold effect.372–374
Figure 93.
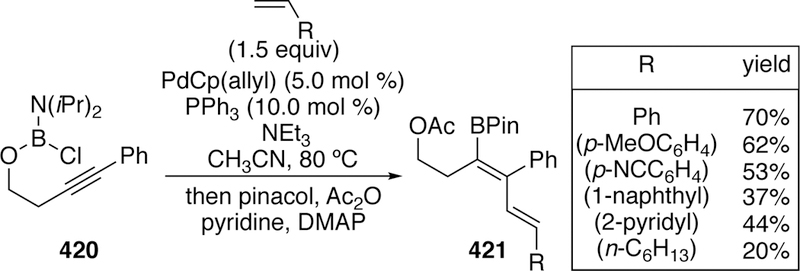
Suginome’s Tandem Electrophilic Borylation Followed by Heck Reaction.
4.4. Intermolecular Cross-Coupling of Boron Electrophiles
In addition to doing extensive work on intramolecular borylation reactions using electrophilic boron, Suginome also developed an intermolecular multicomponent coupling of alkynes, chloroborane 403, and vinylzirconocenes (423) or arylzirconocenes.375 Like his previous work his optimal conditions were a catalyst derived from PdCp(allyl) and trimethylphosphine and toluene solvent at elevated temperatures. As before, the direct products of the coupling were converted to the Bpin esters prior to isolation. Symmetrical, unsymmetrical, and terminal alkenes were all competent coupling partners with R1 (Figure 94) being the smaller of the two alkyne substituents.
Figure 94.
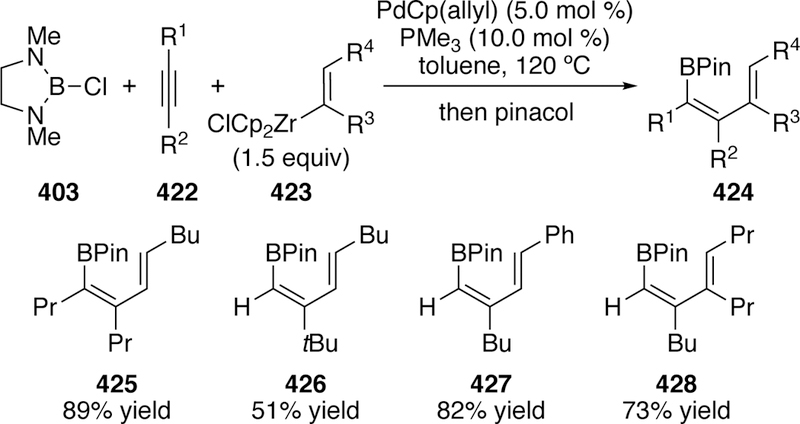
Scope of Suginome’s Intramolecular Carboboration Reaction.
In 2016, our group reported an intermolecular boryl-Heck reaction of catBCl and terminal alkenes.376 To overcome the issue of triethylamine sequestering the chloroborane (see section 4.1), a number of bases were investigated and it was found that large, sterically encumbered amine bases such as N,N-dicyclohexylmethylamine (Cy2NMe) provided the desired coupled product 429 with minimal starting material isomerization. 11B NMR experiments revealed that Cy2NMe binds less strongly to catBCl than does triethylamine. In a competition experiment, the triethylamine adduct was formed preferentially over the Cy2NMe adduct (Figure 95c). The broader, further downfield peak seen for the Cy2NMe adduct (Figure 95b) is also indicative that the exchange between free Cy2NMe and its adduct is more fluxional than that with triethylamine. This suggests that the oxidative addition of catBCl occurs via the equilibrium concentration of free catBCl.
Figure 95.
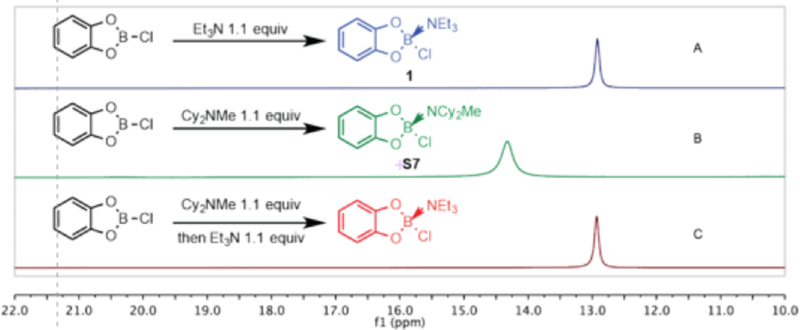
Watson’s 11B NMR Studies on Amine Adduct Formation with catBCl in CDCI3.
Reprinted with permission from Ref [346]. Copyright 2016 American Chemical Society
Further optimization of the reaction explored alternative catalysts and we found that a single-component catalyst derived from the optimal phosphine ligand 38 simplified reaction setup and gave better and more consistent yields of the vinylboronate products. Styrenyl substrates could also be coupled with slightly modified conditions. In these cases, catalytic lithium iodide is added which accelerates the reaction rate which allowed the reaction temperature to be decreased from 90 °C to a milder 70 °C and improved yields. A variety of boron acid derivatives could be obtained if alternative reagents were added at the end of the reaction (Figure 96).
Figure 96.
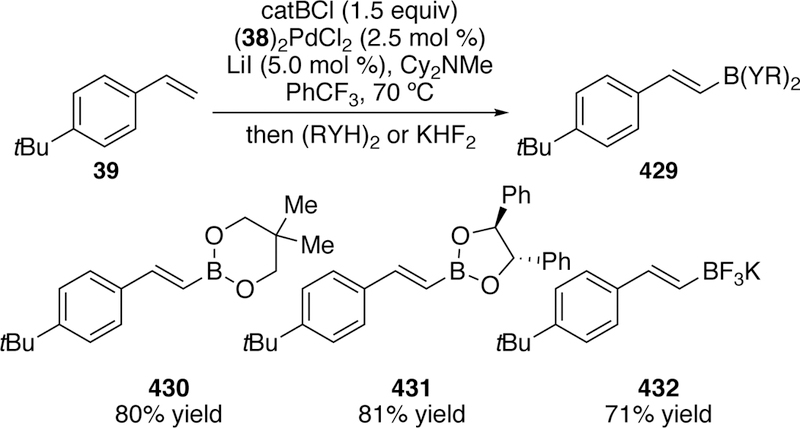
Scope of Boronic Derivatives Derived from Watson’s Boryl-Heck Reaction.
In the case of linear α-olefins, alkene isomerization proved problematic in initial studies. The addition of lithium iodide was not helpful here, and instead only increased starting material isomerization. Instead, it was found that the addition of stoichiometric lithium triflate (LiOTf) suppressed the starting material isomerization (Figure 97). The exact role of LiOTf is not well understood, though we hypothesize that the insolubility of LiCl in the reaction solvent may shift equilibria involving the palladium hydrides - which are likely the cause of starting material isomerization. The direct products of the coupling bearing catechol can be converted to the pinacol ester for ease of isolation simply by the addition of pinacol at the end of the reaction. A variety of linear products could be obtained (Figure 97) using the LiOTf conditions.
Figure 97.
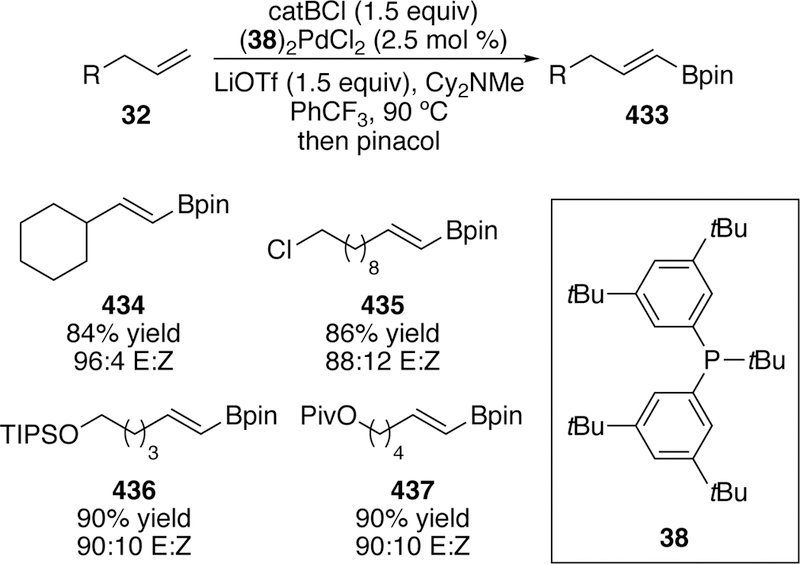
Scope of Linear α-Olefin Products in Watson’s Boryl-Heck Reaction.
More recent work in the field of boryl-Heck chemistry from our group has identified modified conditions for the coupling of 1,1-disubstituted and 1,2-disubstituted alkenes (Figure 98).377 These second generation conditions utilize a different electrophilic boron source B-bromocatecholborane (catBBr), and a more active catalyst, [(38)PdI2]2. Coupling with 1,1-disubstituted alkenes results in the trisubstituted vinyl boronate with high selectivity for the E-isomer (e.g. 439 in Figure 98). For styrenyl-derived 1,2-disubstituted alkenes, high selectivity for the Z-isomer (e.g. 441) was obtained when starting from the E-starting material (e.g. 440).
Figure 98.
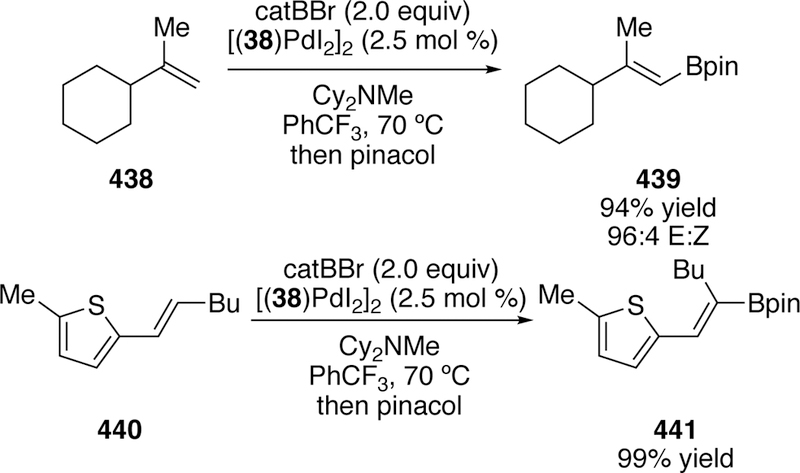
Watson’s Boryl-Heck Reaction of Disubstituted Alkenes with catBBr.
5. Cross-Coupling of Oxygen Electrophiles
Given the breadth of uncatalyzed ways to install oxygen in organic compounds, there have been comparatively few examples of the use of oxygen electrophiles in cross-coupling. The known examples, reviewed below, arise from the activation of C–O bonds in esters or oxycy-anated species, and result in intermediates that have the character of electrophilic oxygen centers.378 These reactions are distinct from a much larger class of reactions in which C–O bonds are activated to generate electrophilic carbon species in cross-coupling reaction,154, 379–381 which fall into mold of “traditional” cross-couplings and are not reviewed here.
5.1. Activation of C–O Bonds of Esters
The first report comes from Ohe in 2009 in which he proposes oxidative addition of palladium across the C–O bond of an ester (facilitated by the addition of zinc powder) followed by addition into a pendant nitrile ultimately leading to the formation of an aminobenzofuran (443, Figure 99).382
Figure 99.
Example of Ohe’s Formation of Aminobenzofurans.
This work was followed up by Douglas in 2011 who used rhodium to activate the C–O bond of an ester (444).383 A strategically placed quinoline group both directs the metal center to the acyl C–O bond and serves as a chelating ligand to support complex 445. Following oxidative addition, complex 445 undergoes C–O bond forming migratory insertion and reductive elimination to form product 447 (Figure 100). Both five- and six-membered rings could be formed and the reaction was tolerant of substitution on the backbone aryl ring. Douglas returned to his initial rhodium-catalyzed oxyacylation conditions in 2017 and further explored the mechanism and expanded the substrate scope.384 Additional DFT studies on the mechanism were completed by Cavallo in 2015.385
Figure 100.
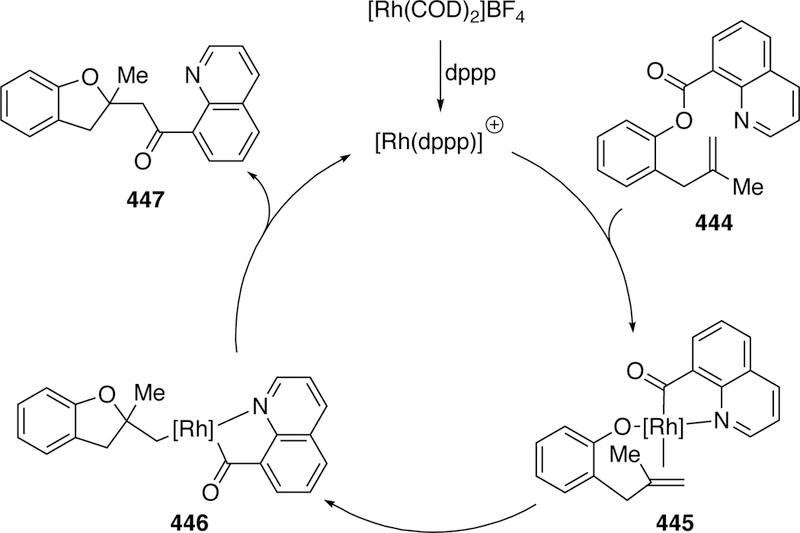
Proposed Mechanism of Douglas’ Intramolecular Oxyacylation Reaction.
Douglas followed this initial report up with another in 2014.386 Instead of using rhodium to activate the C–O bond, he uses iridium which allows the use of a simpler directing group handle, an easily functionalized phenol. Use of an iridium precatalyst with added MOP ligand (452) at elevated temperatures allowed for the formation of five- (451) or six- membered (450) ring products in good to modest yields (Figure 101). Although a chiral ligand 452 was used, no enantioselectivity was observed. A similar mechanism for the cyclization as to the prior rhodium report was proposed.
Figure 101.
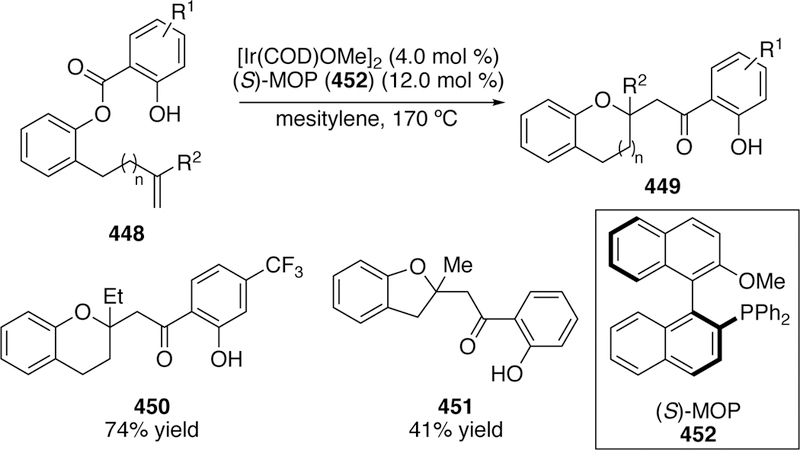
Substrate Scope of Douglas’ Iridium-Catalyzed Oxyacylation.
5.2. Activation of NC–O Bonds
While the activation of NC–N bonds in cross-coupling was discussed in 3.2.10, analogous chemistry on the activation of NC–O bonds was described concurrently.387 Nakao, similar to his reported work on cyanamides (section 3.2.10), reported a palladium-catalyzed oxycyanation reaction to form both five- and six-membered rings. Using a Pd(0) precatslyst and Xantphos ligand in conjunction with triphenylborane Lewis acid cocatalyst, a variety of substituted products could be synthesized (Figure 102).
Figure 102.
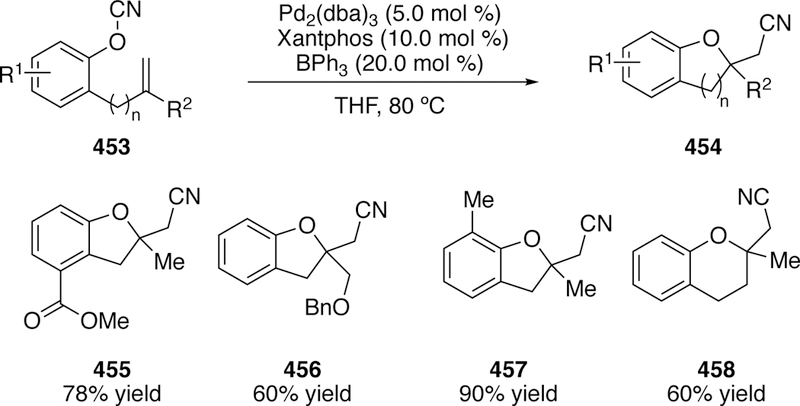
Products Synthesized by Nakao’s Oxycyanation Reaction.
6. Cross-Coupling of Phosphorus Electrophiles
The sole example of a cross-coupling involving a phosphorus electrophile was reported by Beletskaya in 2003. The reaction, a heteroanalog of a Sonogashira reaction, couples a terminal alkyne to a diaryl- or dialkylchlorophosphane using a nickel catalyst and triethylamine base.388 The proposed mechanism follows that of a traditional Sonogashira reaction (Figure 103).
Figure 103.
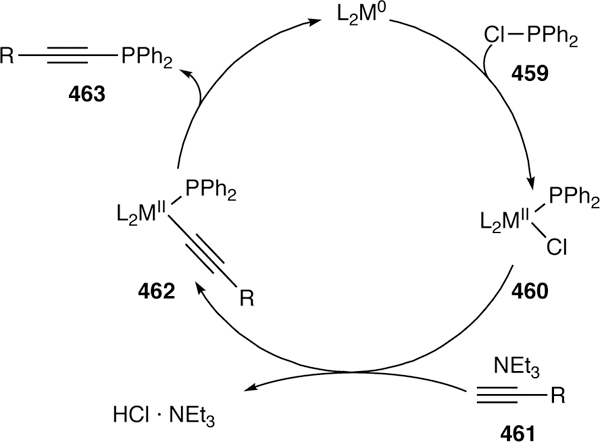
Proposed Mechanism of Belatskaya’s Coupling of Terminal Alkynes and Chlorophosphanes.
In addition to nickel, palladium could also catalyze the reaction but was found to be less active than the nickel catalysts. A number of solvents were compatible in the reaction ranging from nonpolar solvents like benzene and toluene to more polar solvents such as dichloromethane and acetonitrile. The reaction is very fast at 80 °C, reaching completion in many cases in under thirty minutes. Bulky substituents like tert-butyl groups on the chlorophosphane greatly slowed the reaction to the point of impractability with 471 being obtained in only 25% NMR yield after fourteen days at 120 °C. However, the bis-isoproylchlorophosphane reacted to give 468 in only ten minutes (Figure 104). Substitution on the phenyl rings of the diarylchlorophospane was not tolerated, resulting in complex mixtures.
Figure 104.

Scope of Beletskaya’s Cross-coupling of Chlorophosphanes and Terminal Alkynes.
7. Conclusion
Although couplings utilizing carbon electrophiles have been the gold standard in cross-coupling chemistry, the development of couplings utilizing heteroatomic electrophiles is an ever-expanding area of research. Since Tanaka’s seminal initial report of the use of silicon electrophiles in coupling, the overall field has expanded to the creation of C–Si, C–N, C–B, C–O and C–P bonds. The umpolung strategy of enlisting these heteroatoms as electrophiles, rather than more traditionally as nucleophiles, has allowed access to complementary structural diversity that would be hard to access using traditional cross-coupling. Although the past 10–15 years has seen an immense expansion of the cross-coupling reactions using heteroatomic electrophiles, the field remains in its infancy. Of the main group elements, only a hand full have been explored as electrophilic centers in these reactions. Likewise, the classes of reaction that have been investigated, particularly outside of nitrogen electrophiles, is very limited. Many opportunities for the development of new reactions, and in particular asymmetric reactions, using these general strategies remain.
Finally, as compared to traditional cross-coupling reactions using carbon electrophiles, very little is known about the mechanistic details of many of the reactions in this arena. Deeper understanding of the mechanism and pathways of these reactions will do doubt lead to further expansions of the scope and importance of this class of reactions. Future directions in the field will likely further explore the utility of the existing sources of electrophilic silicon, boron, oxygen, and nitrogen in cross-coupling chemistry with a variety of nucleophiles and metal catalysts, as well as identify new ways to activate heteroatoms as electrophiles. Likewise, as the field matures, heteroatomic electrophile cross-couplings will continue to find applications in more complex settings. Use of heteroatomic electrophile cross-coupling for the installation of heteroatoms has been scarcely utilized in total synthesis and industrial applications, though many opportunities exist given the host of high-yielding, robust processes in these contexts. We expect to see many developments in this area over the next decade and look forward to the exciting ways in which this field will advance.
Acknowledegement
The University of Delaware and the NIH NIGMS (P20 GM104316) are gratefully acknowledged for support in writing this review. Results described in this review from the DAW lab were supported by the University of Delaware, the Research Corp. Cottrell Scholars Program, NIH NIGMS (P20 GM104316), and NSF (CAREER CHE1254360).
The University of Delaware is thanked for support. Portions of the reviewed work were supported by the National Science Foundation (CAREER CHE-1254360), the National Institute of General Medical Science (P20GM104316), the Delaware Economic Development Office (Grant 16A00384), and the Research Corporation Cottrell Scholars Program.
Biographies:
Katerina M. Korch obtained a B.S. degree in chemistry, graduating Summa Cum Laude from Juniata College (Huntingdon, PA) in 2012. She then joined the group of Professor Brian M. Stoltz at Caltech as an NSF Graduate Research Fellow, completing her M.S. degree in chemistry in 2015. After a stint as a synthetic organic chemist at a small contract research organization in New Castle, DE, she returned to her Ph.D. studies at the University of Delaware. As a member of Professor Donald A. Watson’s research group she currently works on the development of aza-Heck chemistry and total synthesis.
Donald A. Watson received his B.S. in chemistry from UC San Diego in 1998, and his Ph.D. in chemistry from UC Irvine in 2004. After completing postdoctoral studies at UC Berkeley and MIT, he was appointed as an Assistant Professor in the Department of Chemistry and Biochemistry at the University of Delaware in 2009. He was promoted to Associate Professor with tenure in 2015, and to Professor in 2018. His laboratory is focused on the invention of new chemical transformations and the synthesis of complex molecules.
References
- (1).Heck RF The Addition of Alkyl- and Arylpalladium Chlorides to Conjugated Dienes. J. Am. Chem. Soc. 1968, 90, 5542–5546. [Google Scholar]
- (2).Heck RF The Palladium-Catalyzed Arylation of Enol Esters, Ethers, and Halides. A New Synthesis of 2-Aryl Aldehydes and Ketones. J. Am. Chem. Soc. 1968, 90, 5535–5538. [Google Scholar]
- (3).Heck RF Allylation of Aromatic Compounds with Organopalladium Salts. J. Am. Chem. Soc. 1968, 90, 5531–5534. [Google Scholar]
- (4).Heck RF The Arylation of Allylic Alcohols with Organopalladium Compounds. A New Synthesis of 2-Aryl Aldehydes and Ketones. J. Am. Chem. Soc. 1968, 90, 5526–5531. [Google Scholar]
- (5).Heck RF Acylation, Methylation, and Carboxyalkylation of Olefins by Group Viii Metal Derivatives. J. Am. Chem. Soc. 1968, 90, 5518–5526. [Google Scholar]
- (6).Heck RF Mechanism of Arylation and Carbomethoxylation of Olefins with Organopalladium Compounds. J. Am. Chem. Soc. 1969, 91, 6707–6714. [Google Scholar]
- (7).Heck RF; Nolley JP Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar]
- (8).Colacot TJ; Nolan S; Hardacre C; Stradiotto M; Ismagilov Z; Ozkan U; Lautens M; Spivey J; Lloyd-Jones G; Wu XF New Trends in Cross-Coupling: Theory and Applications. Royal Society of Chemistry: 2014. [Google Scholar]
- (9).Gildner PG; Colacot TJ Reactions of the 21st Century: Two Decades of Innovative Catalyst Design for Palladium-Catalyzed Cross-Couplings. Organometallics 2015, 34, 5497–5508. [Google Scholar]
- (10).Biscoe M; Bronger RPJ; Carboni B; Carreaux F; Chang TWT Science of Synthesis: Cross Coupling and Heck-Type Reactions Vol. 1: C-C Cross Coupling Using Organometallic Partners. Thieme: 2014. [Google Scholar]
- (11).Ahmad OK; McMurtrey KB; Chen H; Dong W; Driver TG Science of Synthesis: Cross Coupling and Heck-Type Reactions Vol. 2: C-C Cross Coupling of Acidic C-H Nucleophiles. Thieme: 2014. [Google Scholar]
- (12).Andersson CM; Andersson M; Borduas N; Borsini E; Broggini G Science of Synthesis: Cross Coupling and Heck-Type Reactions Vol. 3: C-C Cross Coupling Via C-H Activation. Thieme: 2014. [Google Scholar]
- (13).Molander GA; Wolfe JP; Larhed M Cross Coupling and Heck-Type Reactions: Carbon-Heteroatom Cross Coupling and C-C Cross Coupling of Acidic C-H Nucleophiles. Georg Thieme: 2013. [Google Scholar]
- (14).Molnár Á Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments. Wiley: 2013. [Google Scholar]
- (15).Melchor MG A Theoretical Study of Pd-Catalyzed C-C Cross-Coupling Reactions. Springer International Publishing: 2013. [Google Scholar]
- (16).Han FS Transition-Metal-Catalyzed Suzuki-Miyaura Cross-Coupling Reactions: A Remarkable Advance from Palladium to Nickel Catalysts. Chem. Soc. Rev. 2013, 42, 5270–5298. [DOI] [PubMed] [Google Scholar]
- (17).Evano G; Blanchard N Copper-Mediated Cross-Coupling Reactions. Wiley: 2013. [Google Scholar]
- (18).de Meijere A; Diederich F Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH: 2004. [Google Scholar]
- (19).de Meijere A; Bräse S; Oestreich M Metal Catalyzed Cross-Coupling Reactions and More, 3 Volume Set. Wiley: 2013. [Google Scholar]
- (20).Applied Cross-Coupling Reactions. Springer-Verlag Berlin Heidelberg: 2013.
- (21).Paul F; Patt J; Hartwig JF Palladium-Catalyzed Formation of Carbon-Nitrogen Bonds. Reaction Intermediates and Catalyst Improvements in the Hetero Cross-Coupling of Aryl Halides and Tin Amides. J. Am. Chem. Soc. 1994, 116, 5969–5970. [Google Scholar]
- (22).Miyaura N; Suzuki A Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- (23).Farina V New Perspectives in the Cross-Coupling Reactions of Organostannanes. Pure Appl. Chem. 1996, 68, 73–78. [Google Scholar]
- (24).Hartwig JF Recent Advances in Palladium- and Nickel-Catalyzed Chemistry Provide New Ways to Construct C–N and C–O Bonds. Angew. Chem. Int. Ed. 1998, 37, 2046–2067. [Google Scholar]
- (25).Cardenas DJ Advances in Functional-Group-Tolerant Metal-Catalyzed Alkyl-Alkyl Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2003, 42, 384–387. [DOI] [PubMed] [Google Scholar]
- (26).Miyaura N; Buchwald SL; Fugami K; Hiyama T; Kosugi M; Miura M; Miyaura N; Muci AR; Nomura M; Shirakawa E Cross-Coupling Reactions: A Practical Guide. Springer Berlin; Heidelberg: 2003. [Google Scholar]
- (27).Cross-Coupling Reactions. 1 ed.; Springer-Verlag Berlin Heidelberg: 2002.
- (28).Correa A Ni- and Fe-Based Cross-Coupling Reactions. Springer International Publishing: 2017. [Google Scholar]
- (29).Hartwig JF Carbon-Heteroatom Bond-Forming Reductive Eliminations of Amines, Ethers, and Sulfides. Acc. Chem. Res. 1998, 31, 852–860. [Google Scholar]
- (30).Johansson Seechurn C. C.; Kitching MO; Colacot TJ; Snieckus V Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [DOI] [PubMed] [Google Scholar]
- (31).Oestreich M The Mizoroki-Heck Reaction. Wiley: 2009. [Google Scholar]
- (32).Swift EC; Jarvo ER Asymmetric Transition Metal-Catalyzed Cross-Coupling Reactions for the Construction of Tertiary Stereocenters. Tetrahedron 2013, 69, 5799–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yamaguchi J; Muto K; Itami K Recent Progress in Nickel-Catalyzed Biaryl Coupling. Eur. J. Org. Chem. 2013, 2013, 19–30. [Google Scholar]
- (34).Prashad M, Palladium-Catalyzed Heck Arylations in the Synthesis of Active Pharmaceutical Ingredients. In Organometallics in Process Chemistry, Larsen RD, Ed. Springer: Berlin, Heidelberg: 2004; pp 181–203. [Google Scholar]
- (35).King AO; Yasuda N, Palladium-Catalyzed Cross-Coupling Reactions in the Synthesis of Pharmaceuticals. In Organometallics in Process Chemistry, Larsen RD, Ed. Springer: Berlin, Heidelberg, 2004; pp 205–245. [Google Scholar]
- (36).Diederich F; Stang PJ Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH: New York, 1998. [Google Scholar]
- (37).Beller M; Blaser HU Organometallics as Catalysts in the Fine Chemical Industry. Springer: Berlin; New York, 2012; Vol. 42. [Google Scholar]
- (38).Roberts SM Metal Catalysed Carbon-Carbon Bond-Forming Reactions. John Wiley: Chichester, West Sussex, England; Hoboken, N.J, 2004; Vol. 3. [Google Scholar]
- (39).Shaughnessy KH, Metal-Catalyzed Cross-Couplings of Aryl Halides to Form C-C Bonds in Aqueous Media In Metal-Catalyzed Reactions in Water, Dixneuf PH; Cadierno V, Eds. Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp 1–46. [Google Scholar]
- (40).Jana R; Pathak TP; Sigman MS Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross¬Coupling Reactions Using Alkyl-Organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Beletskaya IP; Averin AD New Trends in the Cross-Coupling and Other Catalytic Reactions. Pure Appl. Chem. 2017, 89, 1413–1428. [Google Scholar]
- (42).Cordovilla C; Bartolomé C; Martínez-Ilarduya JM; Espinet P The Stille Reaction, 38 Years Later. ACS Catal. 2015, 5, 3040–3053. [Google Scholar]
- (43).Chinchilla R; Nájera C The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev. 2007, 107, 874–922. [DOI] [PubMed] [Google Scholar]
- (44).Biffis A; Centomo P; Del Zotto A; Zecca M Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295. [DOI] [PubMed] [Google Scholar]
- (45).Lightowler S; Hird M Palladium-Catalyzed Cross-Coupling Reactions in the Synthesis of Novel Aromatic Polymers. Chem. Mater. 2004, 16, 3963–3971. [Google Scholar]
- (46).Baker MA; Tsai C-H; Noonan KJT Diversifying Cross-Coupling Strategies, Catalysts and Monomers for the Controlled Synthesis of Conjugated Polymers. Chem. Eur. J. 2018, 24, 13078–13088. [DOI] [PubMed] [Google Scholar]
- (47).Krishna A; Lunchev AV; Grimsdale AC Suzuki Polycondensation. Synth. Methods Conjugated Polym. Carbon Mater. 2017. [Google Scholar]
- (48).Xu S; Kim EH; Wei A; Negishi EI Pd- and Ni-Catalyzed Cross-Coupling Reactions in the Synthesis of Organic Electronic Materials. Sci. Technol. Adv. Mater. 2014, 15, 044201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Park Y; Kim Y; Chang S Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]
- (50).Huang L; Arndt M; Gooßen K; Heydt H; Gooßen LJ Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [DOI] [PubMed] [Google Scholar]
- (51).Ichikawa S; Zhu S; Buchwald SL A Modified System for the Synthesis of Enantioenriched N- Arylamines through Copper-Catalyzed Hydroamination. Angew. Chem. Int. Ed. 2018, 57, 8714–8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhou Y; Engl OD; Bandar JS; Chant ED; Buchwald SL Cuh-Catalyzed Asymmetric Hydroamidation of Vinylarenes. Angew. Chem. Int. Ed. 2018, 57, 6672–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Takata T; Nishikawa D; Hirano K; Miura M Synthesis of A-Aminophosphines by Copper-Catalyzed Regioselective Hydroamination of Vinylphosphines. Chem. Eur. J. 2018, 24, 10975–10978. [DOI] [PubMed] [Google Scholar]
- (54).Guo S; Yang JC; Buchwald SL A Practical Electrophilic Nitrogen Source for the Synthesis of Chiral Primary Amines by Copper-Catalyzed Hydroamination. J. Am. Chem. Soc. 2018, 140, 15976–15984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kato K; Hirano K; Miura M Copper/Bisphosphine Catalysts in the Internally Borylative Aminoboration of Unactivated Terminal Alkenes with Bis(Pinacolato)Diboron. J. Org. Chem. 2017, 82, 10418–10424. [DOI] [PubMed] [Google Scholar]
- (56).Kato K; Hirano K; Miura M Synthesis of B-Boryl-A-Aminosilanes by Copper-Catalyzed Aminoboration of Vinylsilanes. Angew. Chem. Int. Ed. 2016, 55, 14400–14404. [DOI] [PubMed] [Google Scholar]
- (57).Nishikawa D; Hirano K; Miura M Copper-Catalyzed Regio- and Stereoselective Aminoboration of Alkenylboronates. Org. Lett. 2016, 18, 4856–4859. [DOI] [PubMed] [Google Scholar]
- (58).Paudyal MP; Adebesin AM; Burt SR; Ess DH; Ma Z; Kürti L; Falck JR Dirhodium- Catalyzed C-H Arene Amination Using Hydroxylamines. Science 2016, 353, 1144–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Sakae R; Hirano K; Miura M Ligand-Controlled Regiodivergent Cu-Catalyzed Aminoboration of Unactivated Terminal Alkenes. J. Am. Chem. Soc. 2015, 137, 6460–6463. [DOI] [PubMed] [Google Scholar]
- (60).Nishikawa D; Hirano K; Miura M Asymmetric Synthesis of A-Aminoboronic Acid Derivatives by Copper-Catalyzed Enantioselective Hydroamination. J. Am. Chem. Soc. 2015, 137, 15620–15623. [DOI] [PubMed] [Google Scholar]
- (61).Niljianskul N; Zhu S; Buchwald SL Enantioselective Synthesis of Alpha-Aminosilanes by Copper-Catalyzed Hydroamination of Vinylsilanes. Angew. Chem. Int. Ed. 2015, 54, 1638–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Bandar JS; Pirnot MT; Buchwald SL Mechanistic Studies Lead to Dramatically Improved Reaction Conditions for the Cu-Catalyzed Asymmetric Hydroamination of Olefins. J. Am. Chem. Soc. 2015, 137, 14812–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Pirnot MT; Wang Y-M; Buchwald SL Copper Hydride Catalyzed Hydroamination of Alkenes and Alkynes. Angew. Chem. Int. Ed. 2015, 55, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).He J; Shigenari T; Yu J-Q Palladium(0)/Par3-Catalyzed Intermolecular Amination of C(Sp3)-H Bonds: Synthesis of B-Amino Acids. Angew. Chem. Int. Ed. 2015, 54, 6545–6549. [DOI] [PubMed] [Google Scholar]
- (65).Sakae R; Matsuda N; Hirano K; Satoh T; Miura M Highly Stereoselective Synthesis of (Borylmethyl)Cyclopropylamines by Copper-Catalyzed Aminoboration of Methylenecyclopropanes. Org. Lett. 2014, 16, 1228–1231. [DOI] [PubMed] [Google Scholar]
- (66).Miki Y; Hirano K; Satoh T; Miura M Copper-Catalyzed Enantioselective Formal Hydroamination of Oxa-and Azabicyclic Alkenes with Hydrosilanes and Hydroxylamines. Org. Lett. 2014, 16, 1498–1501. [DOI] [PubMed] [Google Scholar]
- (67).Huehls CB; Lin A; Yang J Iron-Catalyzed Intermolecular Hydroamination of Styrenes. Org. Lett. 2014, 16, 3620–3623. [DOI] [PubMed] [Google Scholar]
- (68).Sakae R; Hirano K; Satoh T; Miura M Copper-Catalyzed Stereoselective Aminoboration of Bicyclic Alkenes. Angew. Chem. Int. Ed. 2014, 54, 613–617. [DOI] [PubMed] [Google Scholar]
- (69).Zhu S; Niljianskul N; Buchwald SL Enantio- and Regioselective Cuh-Catalyzed Hydroamination of Alkenes. J. Am. Chem. Soc. 2013, 135, 15746–15749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Shang M; Zeng S-H; Sun S-Z; Dai H-X; Yu J-Q Ru(Ii)-Catalyzed Ortho-C-H Amination of Arenes and Heteroarenes at Room Temperature. Org. Lett. 2013, 15, 5286–5289. [DOI] [PubMed] [Google Scholar]
- (71).Matsuda N; Hirano K; Satoh T; Miura M Regioselective and Stereospecific Copper-Catalyzed Aminoboration of Styrenes with Bis(Pinacolato)Diboron and O-Benzoyl-N,N-Dialkylhydroxylamines. J. Am. Chem. Soc. 2013, 135, 4934–4937. [DOI] [PubMed] [Google Scholar]
- (72).Miki Y; Hirano K; Satoh T; Miura M Copper-Catalyzed Intermolecular Regioselective Hydroamination of Styrenes with Polymethylhydrosiloxane and Hydroxylamines. Angew. Chem. Int. Ed. 2013, 52, 10830–10834. [DOI] [PubMed] [Google Scholar]
- (73).Sakae R; Hirano K; Satoh T; Miura M Formal Anti-Markovnikov Hydroamination of Terminal Aryl Alkynes with Pinacolborane and Hydroxylamines Via Zr/Cu Sequential Catalysis. Chem. Lett. 2013, 42, 1128–1130. [Google Scholar]
- (74).Matsuda N; Hirano K; Satoh T; Miura M Copper-Catalyzed Direct Amination of Polyfluoroarenes and Azoles with Hydroxylamines and Its Application to the Synthesis of 3-Aminobenzoheteroles. Synthesis 2012, 44, 1792–1797. [Google Scholar]
- (75).Yoo EJ; Ma S; Mei T-S; Chan KSL; Yu J-Q Pd-Catalyzed Intermolecular C–H Amination with Alkylamines. J. Am. Chem. Soc. 2011, 133, 7652–7655. [DOI] [PubMed] [Google Scholar]
- (76).Matsuda N; Hirano K; Satoh T; Miura M Copper-Catalyzed Direct Amination of Electron-Deficient Arenes with Hydroxylamines. Org. Lett. 2011, 13, 2860–2863. [DOI] [PubMed] [Google Scholar]
- (77).Kawano T; Hirano K; Satoh T; Miura M A New Entry of Amination Reagents for Heteroaromatic C–H Bonds: Copper-Catalyzed Direct Amination of Azoles with Chloroamines at Room Temperature. J. Am. Chem. Soc. 2010, 132, 6900–6901. [DOI] [PubMed] [Google Scholar]
- (78).Tamao K; Sumitani K; Kumada M Selective Carbon-Carbon Bond Formation by Cross-Coupling of Grignard Reagents with Organic Halides. Catalysis by Nickel-Phosphine Complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar]
- (79).Corriu RJP; Masse JP Activation of Grignard Reagents by Transition-Metal Complexes. A New and Simple Synthesis of Trans-Stilbenes and Polyphenyls. J. Chem. Soc, Chem. Commun. 1972, 0, 144a–144a. [Google Scholar]
- (80).Sonogashira K; Tohda Y; Hagihara N A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes and Bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar]
- (81).Negishi E; King AO; Okukado N Selective Carbon-Carbon Bond Formation Via Transition Metal Catalysis. 3. A Highly Selective Synthesis of Unsymmetrical Biaryls and Diarylmethanes by the Nickel- or Palladium-Catalyzed Reaction of Aryl- and Benzylzinc Derivatives with Aryl Halides. J. Org. Chem. 1977, 42, 1821–1823. [Google Scholar]
- (82).Milstein D; Stille JK Palladium-Catalyzed Coupling of Tetraorganotin Compounds with Aryl and Benzyl Halides. Synthetic Utility and Mechanism. J. Am. Chem. Soc. 1979, 101, 4992–4998. [Google Scholar]
- (83).Milstein D; Stille JK A General, Selective, and Facile Method for Ketone Synthesis from Acid Chlorides and Organotin Compounds Catalyzed by Palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar]
- (84).Miyaura N; Suzuki A Stereoselective Synthesis of Arylated (E)-Alkenes by the Reaction of Alk-1-Enylboranes with Aryl Halides in the Presence of Palladium Catalyst. J. Chem. Soc., Chem. Commun. 1979, 866–867. [Google Scholar]
- (85).Miyaura N; Y amada K; Suzuki A A New Stereospecific Cross-Coupling by the Palladium-Catalyzed Reaction of 1-Alkenylboranes with 1-Alkenyl or 1-Alkynyl Halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar]
- (86).Iwasaki M; Nishihara Y, Mechanisms and Fundamental Reactions In Applied Cross-Coupling Reactions, Nishihara Y, Ed. Springer; Berlin, Heidelberg, 2013; Vol. 80, pp 17–39. [Google Scholar]
- (87).Jutand A, Mechanisms of the Mizoroki-Heck Reaction In The Mizoroki–HeckReaction, Oestreich M, Ed. John Wiley & Sons, Ltd: Chichester, UK, 2009; pp 1–50. [Google Scholar]
- (88).Kochi JK; Tamura M Alkylcopper(I) in the Coupling of Grignard Reagents with Alkyl Halides. J. Am. Chem. Soc. 1971, 93, 1485–1487. [Google Scholar]
- (89).Tamura M; Kochi JK Copper-Catalyzed Coupling of Grignard Reagents and Akyl Halides in Tetrahydrofuran Solutions. J. Organomet. Chem. 1972, 42, 205–228. [Google Scholar]
- (90).Kochi JK; Tamura M Mechanism of the Silver-Catalyzed Reaction of Grignard Reagents with Alkyl Halides. J. Am. Chem. Soc. 1971, 93, 1483–1485. [Google Scholar]
- (91).Ishiyama T; Abe S; Miyaura N; Suzuki A Palladium-Catalyzed Alkyl-Alkyl Cross-Coupling Reaction of 9-Alkyl-9-Bbn Derivatives with Iodoalkanes Possessing B-Hydrogens. Chem. Lett. 1992, 21, 691–694. [Google Scholar]
- (92).Tucker CE; Knochel P Cross-Coupling between Functionalized Alkylcopper Reagents and Functionalized Alkyl Halides. J. Org. Chem. 1993, 58, 4781–4782. [Google Scholar]
- (93).Giovannini R; Studemann T; Dussin G; Knochel P An Efficient Nickel-Catalyzed Cross-Coupling between Sp3 Carbon Centers. Angew. Chem. Int. Ed. 1998, 37, 2387–2390. [DOI] [PubMed] [Google Scholar]
- (94).Jensen AE; Knochel P Nickel-Catalyzed Cross-Coupling between Functionalized Primary or Secondary Alkylzinc Halides and Primary Alkyl Halides. J. Org. Chem. 2002, 67, 79–85. [DOI] [PubMed] [Google Scholar]
- (95).Zhou J; Fu GC Cross-Couplings of Unactivated Secondary Alkyl Halides: Room-Temperature Nickel-Catalyzed Negishi Reactions of Alkyl Bromides and Iodides. J. Am. Chem. Soc. 2003, 125, 14726–14727. [DOI] [PubMed] [Google Scholar]
- (96).Netherton MR; Dai C; Neuschutz K; Fu GC Room-Temperature Alkyl-Alkyl Suzuki Cross-Coupling of Alkyl Bromides That Possess B Hydrogens. J. Am. Chem. Soc. 2001, 123, 10099–10100. [DOI] [PubMed] [Google Scholar]
- (97).Kirchhoff JH; Dai C; Fu GC A Method for Palladium-Catalyzed Cross-Couplings of Simple Alkyl Chlorides: Suzuki Reactions Catalyzed by [Pd2(Dba)3]/Pcy3. Angew. Chem. Int. Ed. 2002, 41, 1945–1947. [PubMed] [Google Scholar]
- (98).Luh T-Y; Leung M-K; Wong K-T Transition Metal-Catalyzed Activation of Aliphatic C–X Bonds in Carbon-Carbon Bond Formation. Chem. Rev. 2000, 100, 3187–3204. [DOI] [PubMed] [Google Scholar]
- (99).Cardenas DJ Towards Efficient and Wide-Scope Metal-Catalyzed Alkyl-Alkyl Cross-Coupling Reactions. Angew. Chem. Int. Ed. 1999, 38, 3018–3020. [DOI] [PubMed] [Google Scholar]
- (100).Frisch AC; Beller M Catalysts for Cross-Coupling Reactions with Non-Activated Alkyl Halides. Angew. Chem. Int. Ed. 2005, 44, 674–688. [DOI] [PubMed] [Google Scholar]
- (101).Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem. 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Knappke CE; Grupe S; Gartner D; Corpet M; Gosmini C; von Wangelin Jacobi A. Reductive Cross-Coupling Reactions between Two Electrophiles. Chemistry 2014, 20, 6828–6842. [DOI] [PubMed] [Google Scholar]
- (103).Kosugi M; Kameyama M; Migita T Palladium-Catalyzed Aromatic Amination of Aryl Bromides with N,N-Diethylamino-Tributyltin. Chem. Lett. 1983, 12, 927–928. [Google Scholar]
- (104).Boger DL; Panek JS Palladium (O) Mediated B-Carboline Synthesis: Preparation of the Cde Ring System of Lavendamycin. Tetrahedron Lett. 1984, 25, 3175–3178. [Google Scholar]
- (105).Kannan M; Sengoden M; Punniyamurthy T, Transition Metal-Mediated Carbon-Heteroatom Cross-Coupling (C—N, C—O, C—S, C—Se, C—Te, C—P, C—as, C—Sb, and C—B Bond Forming Reactions) In Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, Mortier J, Ed. John Wiley & Sons, Inc.: Hoboken, NJ, 2015; pp 547–586. [Google Scholar]
- (106).Beletskaya IP; Ananikov VP Transition-Metal-Catalyzed C-S, C-Se, and C-Te Bond Formation Via Cross-Coupling and Atom-Economic Addition Reactions. Chem. Rev. 2011, 111, 1596–1636. [DOI] [PubMed] [Google Scholar]
- (107).Hartwig JF Carbon-Heteroatom Bond Formation Catalysed by Organometallic Complexes. Nature 2008, 455, 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Muci AR; Buchwald SL, Practical Palladium Catalysts for C-N and C-O Bond Formation In Cross-Coupling Reactions: A Practical Guide, Miyaura N, Ed. Springer Berlin Heidelberg: Berlin, Heidelberg, 2002; pp 131–209. [Google Scholar]
- (109).Beletskaya IP; Cheprakov AV The Complementary Competitors: Palladium and Copper in C–N Cross-Coupling Reactions. Organometallics 2012, 31, 7753–7808. [Google Scholar]
- (110).Qiao JX; Lam PYS Copper-Promoted Carbon-Heteroatom Bond Cross-Coupling with Boronic Acids and Derivatives. Synthesis 2011, 829–856. [Google Scholar]
- (111).Hartwig JF, Palladium-Catalyzed Synthesis of Aryl Ethers and Related Compounds Containing S and Se In Handbook of Organopalladium Chemistry for Organic Synthesis, Negishi E, Ed. Wiley-Interscience: New York, 2002; pp 1097–1106. [Google Scholar]
- (112).Kondo T; Mitsudo T. -a. Metal-Catalyzed Carbon–Sulfur Bond Formation. Chem. Rev. 2000, 100, 3205–3220. [DOI] [PubMed] [Google Scholar]
- (113).Schwan AL Palladium Catalyzed Cross-Coupling Reactions for Phosphorus–Carbon Bond Formation. Chem. Soc. Rev. 2004, 33, 218–224. [DOI] [PubMed] [Google Scholar]
- (114).Tappe FMJ; Trepohl VT; Oestreich M Transition-Metal-Catalyzed C–P Cross-Coupling Reactions. Synthesis 2010, 2010, 3037–3062. [Google Scholar]
- (115).Xue W; Oestreich M Silicon Grignard Reagents as Nucleophiles in Transition-Metal-Catalyzed Allylic Substitution. Synthesis 2019, 51, 233–239. [Google Scholar]
- (116).Bähr S; Xue W; Oestreich M C(Sp3)–Si Cross-Coupling. ACS Catal. 2019, 9, 16–24. [Google Scholar]
- (117).Singh UK; Strieter ER; Blackmond DG; Buchwald SL Mechanistic Insights into the Pd(Binap)-Catalyzed Amination of Aryl Bromides: Kinetic Studies under Synthetically Relevant Conditions. J. Am. Chem. Soc. 2002, 124, 14104–14114. [DOI] [PubMed] [Google Scholar]
- (118).Shekhar S; Ryberg P; Hartwig JF; Mathew JS; Blackmond DG; Strieter ER; Buchwald SL Reevaluation of the Mechanism of the Amination of Aryl Halides Catalyzed by Binap-Ligated Palladium Complexes. J. Am. Chem. Soc. 2006, 128, 3584–3591. [DOI] [PubMed] [Google Scholar]
- (119).Altman RA; Buchwald SL Cu-Catalyzed Goldberg and Ullmann Reactions of Aryl Halides Using Chelating N- and O-Based Ligands. Nat. Protocols 2007, 2, 2474–2479. [DOI] [PubMed] [Google Scholar]
- (120).Kunz K; Scholz U; Ganzer D Renaissance of Ullmann and Goldberg Reactions - Progress in Copper Catalyzed C-N-, C-O- and C-S-Coupling. Synlett 2003, 2428–2439. [Google Scholar]
- (121).Lei A; Shi W; Liu C; Liu W; Zhang H; He C Oxidative Cross-Coupling. John Wiley & Sons: 2016. [Google Scholar]
- (122).Liu C; Zhang H; Shi W; Lei A Bond Formations between Two Nucleophiles: Transition Metal Catalyzed Oxidative Cross-Coupling Reactions. Chem. Rev. 2011, 111, 1780–1824. [DOI] [PubMed] [Google Scholar]
- (123).Huang Z; Tang S; Lei A Oxidative Cross-Coupling: An Alternative Way for C–C Bond Formations. Sci. Bull. 2015, 60, 1391–1394. [Google Scholar]
- (124).Shi W; Liu C; Lei A Transition-Metal Catalyzed Oxidative Cross-Coupling Reactions to Form C–C Bonds Involving Organometallic Reagents as Nucleophiles. Chem. Soc. Rev. 2011, 40, 2761–2776. [DOI] [PubMed] [Google Scholar]
- (125).Brook MA Silicon in Organic, Organometallic, and Polymer Chemistry. Wiley: Chichester, UK, 2000. [Google Scholar]
- (126).Fleming I, Organic Silicon Chemistry In Comprehensive Organic Chemistry, Barton DH,R; Ollis WD, Eds. Pergamon Press: Oxford, UK, 1979; Vol. 3, pp 541–686. [Google Scholar]
- (127).Nakao Y; Hiyama T Silicon-Based Cross-Coupling Reaction: An Environmentally Benign Version. Chem. Soc. Rev. 2011, 40, 4893–4901. [DOI] [PubMed] [Google Scholar]
- (128).Sore HF; Galloway WRJD; Spring DR Palladium-Catalysed Cross-Coupling of Organosilicon Reagents. Chem. Soc. Rev. 2012, 41, 1845–1866. [DOI] [PubMed] [Google Scholar]
- (129).Komiyama T; Minami Y; Hiyama T Recent Advances in Transition-Metal-Catalyzed Synthetic Transformations of Organosilicon Reagents. ACS Catal. 2017, 7, 631–651. [Google Scholar]
- (130).Minami Y; Hiyama T Designing Cross-Coupling Reactions Using Aryl(Trialkyl)Silanes. Chemistry 2019, 25, 391–399. [DOI] [PubMed] [Google Scholar]
- (131).Denmark SE; Liu JHC Silicon-Based Cross-Coupling Reactions in the Total Synthesis of Natural Products. Angew. Chem. Int. Ed. 2010, 49, 2978–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Masse CE; Panek JS Diastereoselective Reactions of Chiral Allyl and Allenyl Silanes with Activated C:X.Pi.-Bonds. Chem. Rev. 1995, 95, 1293–1316. [Google Scholar]
- (133).Fleming I; Barbero A; Walter D Stereochemical Control in Organic Synthesis Using Silicon-Containing Compounds. Chem. Rev. 1997, 97, 2063–2192. [DOI] [PubMed] [Google Scholar]
- (134).Colvin EW, Oxidation of Carbon-Silicon Bonds In Comprehensive Organic Synthesis, Trost BM; Fleming I, Eds. Pergamon: Oxford, UK, 1991; pp 641–651. [Google Scholar]
- (135).Jones GR; Landais Y The Oxidation of the Carbon-Silicon Bond. Tetrahedron 1996, 52, 7599–7662. [Google Scholar]
- (136).Ando E; Goto Y; Morimoto K; Ariga K; Okahata Y Frictional Properties of Monomolecular Layers of Silane Compounds. Thin Solid Films 1989, 180, 287–291. [Google Scholar]
- (137).Sieburth SM, Isosteric Replacement of Carbon with Silicon in the Design of Safer Chemicals. In Designing Safer Chemicals, American Chemical Society: 1996; Vol. 640, pp 74–83. [Google Scholar]
- (138).Moberg WK; Basarab GS; Cuomo J; Liang PH, Biologically Active Organosilicon Compounds. In Synthesis and Chemistry of Agrochemicals, American Chemical Society: 1987; Vol. 355, pp 288–301. [Google Scholar]
- (139).Koyanagi T; Haga T, Bioisosterism in Agrochemicals. In Synthesis and Chemistry of Agrochemicals Iv, American Chemical Society: 1995; Vol. 584, pp 15–24. [Google Scholar]
- (140).Liebner F; Bankwitz U; Rühlmann K Synthese Von Fungicid Wirksamen (1h-1,2,4-Triazol-1-Yl- Methyl)Silanen Und -Siloxanen. Liebigs Ann. Chem. 1994, 1994, 145–150. [Google Scholar]
- (141).Franz AK; Wilson SO Organosilicon Molecules with Medicinal Applications. J. Med. Chem. 2013, 56, 388–405. [DOI] [PubMed] [Google Scholar]
- (142).Rèmond E; Martin C; Martinez J; Cavelier F Silicon-Containing Amino Acids: Synthetic Aspects, Conformational Studies, and Applications to Bioactive Peptides. Chem. Rev. 2016, 116, 11654–11684. [DOI] [PubMed] [Google Scholar]
- (143).Tacke R; Dörrich S, Drug Design Based on the Carbon/Silicon Switch Strategy In Atypical Elements in Drug Design, Schwarz J, Ed. Springer International Publishing: Cham, 2016; pp 29–60. [Google Scholar]
- (144).Yang W; Gao X; Wang B Boronic Acid Compounds as Potential Pharmaceutical Agents. Med. Res. Rev. 2003, 23, 346–368. [DOI] [PubMed] [Google Scholar]
- (145).Adams J; Kauffman M Development of the Proteasome Inhibitor Velcade™ (Bortezomib). Cancer Invest. 2004, 22, 304–311. [DOI] [PubMed] [Google Scholar]
- (146).Lennox AJJ; Lloyd-Jones GC Selection of Boron Reagents for Suzuki–Miyaura Coupling. Chem. Soc. Rev. 2014, 43, 412–443. [DOI] [PubMed] [Google Scholar]
- (147).Doucet H Suzuki–Miyaura Cross-Coupling Reactions of Alkylboronic Acid Derivatives or Alkyltrifluoroborates with Aryl, Alkenyl or Alkyl Halides and Triflates. Eur. J. Org. Chem. 2008, 2008, 2013–2030. [Google Scholar]
- (148).Rossi R; Bellina F; Carpita A Palladium Catalysts for the Suzuki Cross-Coupling Reaction: An Overview of Recent Advances. Synthesis 2004, 2004, 2419–2440. [Google Scholar]
- (149).Martin R; Buchwald SL Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands. Acc. Chem. Res. 2008, 41, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Qiao JX; Lam PYS, Recent Advances in Chan-Lam Coupling Reaction: Copper-Promoted C-Heteroatom Bond Cross-Coupling Reactions with Boronic Acids and Derivatives In Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, Second ed.; Hall DG, Ed. Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; Vol. 1 and 2. [Google Scholar]
- (151).Lam PYS, Chapter 7 Chan-Lam Coupling Reaction: Copper-Promoted C-Element Bond Oxidative Coupling Reaction with Boronic Acids In Synthetic Methods in Drug Discovery: Volume 1, The Royal Society of Chemistry: 2016; Vol. 1, pp 242–273. [Google Scholar]
- (152).Cheng H-G; Chen H; Liu Y; Zhou Q The Liebeskind-Srogl Cross-Coupling Reaction and Its Synthetic Applications. Asian J. Org. Chem. 2018, 7, 490–508. [Google Scholar]
- (153).Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials. 1&2 ed.; Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar]
- (154).Desnoyer AN; Love JA Recent Advances in Well-Defined, Late Transition Metal Complexes That Make and/or Break C–N, C–O and C-S Bonds. Chem. Soc. Rev. 2017, 46, 197–238. [DOI] [PubMed] [Google Scholar]
- (155).Bariwal J; Van der Eycken E C-N Bond Forming Cross-Coupling Reactions: An Overview. Chem. Soc. Rev. 2013, 42, 9283–9303. [DOI] [PubMed] [Google Scholar]
- (156).Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Basch CH; Liao J; Xu J; Piane JJ; Watson MP Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings Via C-N Bond Activation. J. Am. Chem. Soc. 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Dander JE; Garg NK Breaking Amides Using Nickel Catalysis. ACS Catal. 2017, 7, 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Liu L; Chen P; Sun Y; Wu Y; Chen S; Zhu J; Zhao Y Mechanism of Nickel-Catalyzed Selective C–N Bond Activation in Suzuki-Miyaura Cross-Coupling of Amides: A Theoretical Investigation. J. Org. Chem. 2016, 81, 11686–11696. [DOI] [PubMed] [Google Scholar]
- (160).Ouyang K; Hao W; Zhang W-X; Xi Z Transition-Metal-Catalyzed Cleavage of C–N Single Bonds. Chem. Rev. 2015, 115, 12045–12090. [DOI] [PubMed] [Google Scholar]
- (161).Wang Q; Su Y; Li L; Huang H Transition-Metal Catalysed C-N Bond Activation. Chem. Soc. Rev. 2016, 45, 1257–1272. [DOI] [PubMed] [Google Scholar]
- (162).Williamson KS; Michaelis DJ; Yoon TP Advances in the Chemistry of Oxaziridines. Chem. Rev. 2014, 114, 8016–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Franz JE; Mao MK; Sikorski JA Glyphosate: A Unique Global Herbicide. American Chemical Society: Washington, 1997. [Google Scholar]
- (164).Nicolaou KC; Härter MW; Gunzner JL; Nadin A The Wittig and Related Reactions in Natural Product Synthesis. Liebigs Ann. 1997, 1997, 1283–1301. [Google Scholar]
- (165).Wittig Reaction. Comprehensive Organic Name Reactions and Reagents 2010. [Google Scholar]
- (166).Boutagy J; Thomas R Olefin Synthesis with Organic Phosphonate Carbanions. Chem. Rev. 1974, 74, 87–99. [Google Scholar]
- (167).Horner-Wadsworth-Emmons Olefination. Comprehensive Organic Name Reactions and Reagents 2010. [Google Scholar]
- (168).Seyferth-Gilbert Homologation. Comprehensive Organic Name Reactions and Reagents 2010. [Google Scholar]
- (169).Allen DW, Phosphines and Related P-C-Bonded Compounds In Organophosphorus Chemistry, The Royal Society of Chemistry: Cambridge, UK, 2014; Vol. 43, pp 1–51. [Google Scholar]
- (170).Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Applications. Wiley-VCH: Weinheim, Germany, 2008; Vol. 1.
- (171).Wei Y; Shi M Recent Advances in Organocatalytic Asymmetric Morita-Baylis-Hillman/Aza-Morita–Baylis–Hillman Reactions. Chem. Rev. 2013, 113, 6659–6690. [DOI] [PubMed] [Google Scholar]
- (172).Basavaiah D; Rao AJ; Satyanarayana T Recent Advances in the Baylis-Hillman Reaction and Applications. Chem. Rev. 2003, 103, 811–892. [DOI] [PubMed] [Google Scholar]
- (173).Aroyan CE; Dermenci A; Miller SJ The Rauhut–Currier Reaction: A History and Its Synthetic Application. Tetrahedron 2009, 65, 4069–4084. [Google Scholar]
- (174).Kuyper J Dimethyl Compounds of Platinum(Ii). 2. Reactions Involving Carbon, Silicon, and Germanium Halogen Bonds. Inorg. Chem. 1978, 17, 77–81. [Google Scholar]
- (175).Yamashita H; Hayashi T; Kobayashi T; Tanaka M; Goto M Oxidative Addition of Halosilanes to Zero-Valent Platinum Complexes. J. Am. Chem. Soc. 1988, 110, 4417–4419. [Google Scholar]
- (176).Yamashita H; Kobayashi T; Hayashi T; Tanaka M Dichotomous Reactivity of Halodisilanes in Oxidative Addition with Pt(Pet3)3. Formation of Bis(Silyl)- or (Disilanyl)Platinum Complexes. Chem. Lett. 1990, 19, 1447–1450. [Google Scholar]
- (177).Yamashita H; Kobayashi T; Hayashi T; Tanaka M Novel Halogen Exchange Reactions between Halosilanes and Rh(I) or Ir(I) Complexes. Chem. Lett. 1989, 18, 471–474. [Google Scholar]
- (178).Zlota AA; Frolow F; Milstein D Oxidative Addition of Si–Cl Bonds to Electron-Rich Iri Complexes. J. Chem. Soc, Chem. Commun. 1989, 1826–1827. [Google Scholar]
- (179).Yamashita H; Kawamoto AM; Tanaka M; Goto M Oxidative Addition of Halo(Methyl)Silanes to an in Situ Generated Ir(I) Complex and B-Hydride Elimination Reaction of the Resulting (Methylsilyl)Iridium Complexes. Chem. Lett. 1990, 19, 2107–2110. [Google Scholar]
- (180).Kameo H; Ikeda K; Sakaki S; Takemoto S; Nakazawa H; Matsuzaka H Experimental and Theoretical Studies of Si–Cl and Ge–Cl Ʃ-Bond Activation Reactions by Iridium-Hydride. Dalton Trans. 2016, 45, 7570–7580. [DOI] [PubMed] [Google Scholar]
- (181).Kameo H; Ikeda K; Bourissou D; Sakaki S; Takemoto S; Nakazawa H; Matsuzaka H Transition-Metal-Mediated Germanium–Fluorine Activation: Inverse Electron Flow in Ʃ-Bond Metathesis. Organometallics 2016, 35, 713–719. [Google Scholar]
- (182).Levy CJ; Puddephatt RJ; Vittal JJ The Halogen Effect in Oxidative Addition of Trimethylsilyl and Trimethyltin Halides to Platinum(Ii)- Synthesis and Characterization of Silylplatinum(Iv) Complexes. Organometallics 1994, 13, 1559–1560. [Google Scholar]
- (183).Levy CJ; Vittal JJ; Puddephatt RJ Synthesis and Characterization of Group 14–Platinum(Iv) Complexes. Organometallics 1996, 15, 2108–2117. [Google Scholar]
- (184).Mitton SJ; McDonald R; Turculet L Synthesis and Characterization of Neutral and Cationic Platinum(Ii) Complexes Featuring Pincer-Like Bis(Phosphino)Silyl Ligands: Si–H and Si–Cl Bond Activation Chemistry. Organometallics 2009, 28, 5122–5136. [Google Scholar]
- (185).Gatard S; Chen CH; Foxman BM; Ozerov OV Oxidative Addition Reactions of Silyl Halides with the (Pnp)Rh Fragment. Organometallics 2008, 27, 6257–6263. [Google Scholar]
- (186).Esposito O; Roberts DE; Cloke FGN; Caddick S; Green JC; Hazari N; Hitchcock PB Carbon-Silicon Bond Activation by [Pd(Itbu)2] - the Molecular Structures of [Pd(Me3si)(Itbu)(M-I)]2and [Pd(Ch2itbu)I2]. Eur. J. Inorg. Chem. 2009, 2009, 1844–1850. [Google Scholar]
- (187).Yamashita H; Kobayashi T; Hayashi T; Tanaka M Heck-Type Reaction of Lodotrimethylsilane with Olefins Affording Alkenyltrimethylsilanes. Chem. Lett. 1991, 20, 761–762. [Google Scholar]
- (188).Yamashita H; Tanaka M; Goto M Oxidative Addition of Silicon–Halogen Bonds to Platinum(0) Complexes and Reactivities of the Resulting Silylplatinum Species. Organometallics 1997, 16, 4696–4704. [Google Scholar]
- (189).McAtee JR; Martin SE; Ahneman DT; Johnson KA; Watson DA Preparation of Allyl and Vinyl Silanes by the Palladium-Catalyzed Silylation of Terminal Olefins: A Silyl-Heck Reaction. Angew. Chem. Int. Ed. 2012, 51, 3663–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Watson D; Martin S Silyl-Heck Reactions for the Preparation of Unsaturated Organosilanes. Synlett 2013, 24, 2177–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).McAtee JR; Yap GPA; Watson DA Rational Design of a Second Generation Catalyst for Preparation of Allylsilanes Using the Silyl-Heck Reaction. J. Am. Chem. Soc. 2014, 136, 10166–10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).McAtee JR; Krause SB; Watson DA Simplified Preparation of Trialkylvinylsilanes Via the Silyl-Heck Reaction Utilizing a Second Generation Catalyst. Adv. Synth. Catal. 2015, 357, 2317–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Krause SB; McAtee JR; Yap GPA; Watson DA A Bench-Stable, Single-Component Precatalyst for Silyl-Heck Reactions. Org. Lett. 2017, 19, 5641–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Martin SE; Watson DA Preparation of Vinyl Silyl Ethers and Disiloxanes Via the Silyl-Heck Reaction of Silyl Ditriflates. J. Am. Chem. Soc. 2013, 135, 13330–13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).McAtee JR; Martin SE; Cinderella AP; Reid WB; Johnson KA; Watson DA The First Example of Nickel-Catalyzed Silyl-Heck Reactions: Direct Activation of Silyl Triflates without Iodide Additives. Tetrahedron 2014, 70, 4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Matsumoto K; Huang J; Naganawa Y; Guo H; Beppu T; Sato K; Shimada S; Nakajima Y Direct Silyl-Heck Reaction of Chlorosilanes. Org. Lett. 2018, 20, 2481–2484. [DOI] [PubMed] [Google Scholar]
- (197).Chatani N; Amishiro N; Murai S A New Catalytic Reaction Involving Oxidative Addition of Iodotrimethylsilane (Me3sii) to Pd(0). Synthesis of Stereodefined Enynes by the Coupling of Me3sii, Acetylenes, and Acetylenic Tin Reagents. J. Am. Chem. Soc. 1991, 113, 7778–7780. [Google Scholar]
- (198).Chatani N; Amishiro N; Morii T; Yamashita T; Murai S Pd-Catalyzed Coupling Reaction of Acetylenes, Iodotrimethylsilane, and Organozinc Reagents for the Stereoselective Synthesis of Vinylsilanes. J. Org. Chem. 1995, 60, 1834–1840. [Google Scholar]
- (199).Cinderella AP; Vulovic B; Watson DA Palladium-Catalyzed Cross-Coupling of Silyl Electrophiles with Alkylzinc Halides: A Silyl-Negishi Reaction. J. Am. Chem. Soc. 2017, 139, 7741–7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Vulovic B; Cinderella AP; Watson DA Palladium-Catalyzed Cross-Coupling of Monochlorosilanes and Grignard Reagents. ACS Catal. 2017, 8113–8117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Gabriel S Ueber Eine Darstellungsweise Primarer Amine Aus Den Entsprechenden Halogenverbidungen. Ber. Dtsch. Chem. Ges. 1887, 20, 2224–2236. [Google Scholar]
- (202).Ritter JJ; Kalish J A New Reaction of Nitriles. Ii. Synthesis of T-Carbinamines. J. Am. Chem. Soc. 1948, 70, 4048–4050. [DOI] [PubMed] [Google Scholar]
- (203).Ritter JJ; Minieri PP A New Reaction of Nitriles. I. Amides from Alkenes and Mononitriles1. J. Am. Chem. Soc. 1948, 70, 4045–4048. [DOI] [PubMed] [Google Scholar]
- (204).Gomez S; Peters JA; Maschmeyer T The Reductive Amination of Aldehydes and Ketones and the Hydrogenation of Nitriles: Mechanistic Aspects and Selectivity Control. Adv. Synth. Catal. 2002, 344, 1037–1057. [Google Scholar]
- (205).Ricci A Modern Amination Methods. Wiley: 2008. [Google Scholar]
- (206).Ullmann F Ueber Eine Neue Bildungsweise Von Diphenylaminderivaten. Ber. Dtsch. Chem. Ges. 1903, 36, 2382–2384. [Google Scholar]
- (207).Goldberg I Ueber Phenylirungen Bei Gegenwart Von Kupfer Als Katalysator. Ber. Dtsch. Chem. Ges. 1906, 39, 1691–1692. [Google Scholar]
- (208).Lindley J Copper Assisted Nucleophilic Substitution of Aryl Halogen. Tetrahedron 1984, 40, 1433–1456. [Google Scholar]
- (209).Sambiagio C; Marsden SP; Blacker AJ; McGowan PC Copper Catalysed Ullmann Type Chemistry: From Mechanistic Aspects to Modern Development. Chem. Soc. Rev. 2014, 43, 3525–3550. [DOI] [PubMed] [Google Scholar]
- (210).Chan DMT; Monaco KL; Wang R-P; Winters MP New N- and O-Arylations with Phenylboronic Acids and Cupric Acetate. Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar]
- (211).Lam PYS; Clark CG; Saubern S; Adams J; Winters MP; Chan DMT; Combs A New Aryl/Heteroaryl C-N Bond Cross-Coupling Reactions Via Arylboronic Acid/Cupric Acetate Arylation. Tetrahedron Lett. 1998, 39, 2941–2944. [Google Scholar]
- (212).Evans DA; Katz JL; West TR Synthesis of Diaryl Ethers through the Copper-Promoted Arylation of Phenols with Arylboronic Acids. An Expedient Synthesis of Thyroxine. Tetrahedron Lett. 1998, 39, 2937–2940. [Google Scholar]
- (213).Wolfe JP; Wagaw S; Marcoux J-F; Buchwald SL Rational Development of Practical Catalysts for Aromatic Carbon-Nitrogen Bond Formation. Acc. Chem. Res. 1998, 31, 805–818. [Google Scholar]
- (214).Hartwig JF Approaches to Catalyst Discovery. New Carbon–Heteroatom and Carbon–Carbon Bond Formation. Pure Appl. Chem. 1999, 71, 1417–1423. [Google Scholar]
- (215).Buchwald-Hartwig Amination. In Comprehensive Organic Name Reactions and Reagents, Wang Z, Ed. Wiley: 2009; pp 575–581. [Google Scholar]
- (216).Louie J; Hartwig JF Palladium-Catalyzed Synthesis of Arylamines from Aryl Halides. Mechanistic Studies Lead to Coupling in the Absence of Tin Reagents. Tetrahedron Lett. 1995, 36, 3609–3612. [Google Scholar]
- (217).Guram AS; Rennels RA; Buchwald SL A Simple Catalytic Method for the Conversion of Aryl Bromides to Arylamines. Angew. Chem. Int. Ed. 1995, 34, 1348–1350. [Google Scholar]
- (218).Guram AS; Buchwald SL Palladium-Catalyzed Aromatic Aminations with in Situ Generated Aminostannanes. J. Am. Chem. Soc. 1994, 116, 7901–7902. [Google Scholar]
- (219).Hegedus LS; Allen GF; Waterman EL Palladium Assisted Intramolecular Amination of Olefins. A New Synthesis of Indoles. J. Am. Chem. Soc. 1976, 98, 2674–2676. [Google Scholar]
- (220).Hegedus LS; Allen GF; Bozell JJ; Waterman EL Palladium-Assisted Intramolecular Amination of Olefins. Synthesis of Nitrogen Heterocycles. J. Am. Chem. Soc. 1978, 100, 5800–5807. [Google Scholar]
- (221).Hegedus LS; Allen GF; Olsen DJ Palladium-Assisted Cyclization-Insertion Reactions. Synthesis of Functionalized Heterocycles. J. Am. Chem. Soc. 1980, 102, 3583–3587. [Google Scholar]
- (222).Hegedus LS; McKearin JM Palladium-Catalyzed Cyclization Of.Omega.-Olefinic Tosamides. Synthesis of Nonaromatic Nitrogen Heterocycles. J. Am. Chem. Soc. 1982, 104, 2444–2451. [Google Scholar]
- (223).Jiang F; Wu Z; Zhang W Pd-Catalyzed Asymmetric Aza-Wacker-Type Cyclization Reaction of Olefinic Tosylamides. Tetrahedron Lett. 2010, 51, 5124–5126. [Google Scholar]
- (224).Yang G; Shen C; Zhang W An Asymmetric Aerobic Aza-Wacker-Type Cyclization: Synthesis of Isoindolinones Bearing Tetrasubstituted Carbon Stereocenters. Angew. Chem. Int. Ed. 2012, 51, 9141–9145. [DOI] [PubMed] [Google Scholar]
- (225).Liu G; Stahl SS Two-Faced Reactivity of Alkenes: Cis- Versus Trans-Aminopalladation in Aerobic Pd-Catalyzed Intramolecular Aza-Wacker Reactions. J. Am. Chem. Soc. 2007, 129, 6328–6335. [DOI] [PubMed] [Google Scholar]
- (226).Ye X; Liu G; Popp BV; Stahl SS Mechanistic Studies of Wacker-Type Intramolecular Aerobic Oxidative Amination of Alkenes Catalyzed by Pd(Oac)2/Pyridine. J. Org. Chem. 2011, 76, 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Wang D; Weinstein AB; White PB; Stahl SS Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions. Chem. Rev. 2018, 118, 2636–2679. [DOI] [PubMed] [Google Scholar]
- (228).Corpet M; Gosmini C Recent Advances in Electrophilic Amination Reactions. Synthesis 2014, 46, 2258–2271. [Google Scholar]
- (229).Greck C; Genet JP Electrophilic Amination- New Synthetic Applications. Synlett 1997, 1997, 741–748. [Google Scholar]
- (230).Dembech P; Seconi G; Ricci A The Electrophilic Amination of Carbanions- an Unconventional New Entry to C–N Bond Formation. Chem. Eur. J. 2000, 6, 1281–1286. [DOI] [PubMed] [Google Scholar]
- (231).Erdik E; Ay M Electrophilic Amination of Carbanions. Chem. Rev. 1989, 89, 1947–1980. [Google Scholar]
- (232).Hendrick CE; Wang Q Emerging Developments Using Nitrogen-Heteroatom Bonds as Amination Reagents in the Synthesis of Aminoarenes. J. Org. Chem. 2017, 82, 839–847. [DOI] [PubMed] [Google Scholar]
- (233).Yan X; Yang X; Xi C Recent Progress in Copper-Catalyzed Electrophilic Amination. Catal. Sci. Technol. 2014, 4, 4169–4177. [Google Scholar]
- (234).Jarvo E; Barker T Developments in Transition-Metal-Catalyzed Reactions Using Electrophilic Nitrogen Sources. Synthesis 2011, 2011, 3954–3964. [Google Scholar]
- (235).Dong X; Liu Q; Dong Y; Liu H Transition-Metal-Catalyzed Electrophilic Amination: Application of O-Benzoylhydroxylamines in the Construction of the C-N Bond. Chemistry 2017, 23, 2481–2511. [DOI] [PubMed] [Google Scholar]
- (236).Tsutsui H; Hayashi Y; Narasaka K Preparation of Primary Amines by the Copper(I) Catalyzed Reaction of 4,4'-Bis(Trifluoromethyl)Benzophenone O-Methylsulfonyloxime and Alkyl Grignard Reagents. Chem. Lett. 1997, 26, 317–318. [Google Scholar]
- (237).Tsutsui H; Narasaka K Synthesis of Pyrrole Derivatives by the Heck-Type Cyclization of Γ,Δ-Unsaturated Ketone O-Pentafluorobenzoyloximes. Chem. Lett. 1999, 28, 45–46. [Google Scholar]
- (238).Tan Y; Hartwig JF Palladium-Catalyzed Amination of Aromatic C–H Bonds with Oxime Esters. J. Am. Chem. Soc. 2010, 132, 3676–3677. [DOI] [PubMed] [Google Scholar]
- (239).Kitamura M; Zaman S; Narasaka K Synthesis of Spiro Imines from Oximes by Palladium-Catalyzed Cascade Reaction. Synlett 2001, 974–976. [Google Scholar]
- (240).Zaman S; Kitamura M; Narasaka K Synthesis of Polycyclic Imines by Palladium-Catalyzed Domino Cyclization of Di- and Trienyl Ketone O-Pentafluorobenzoyloximes. Bull. Chem. Soc. Jpn. 2003, 76, 1955–1062. [Google Scholar]
- (241).Kitamura M; Narasaka K Synthesis of Aza-Heterocycles from Oximes by Amino-Heck Reaction. Chem. Rec. 2002, 2, 268–277. [DOI] [PubMed] [Google Scholar]
- (242).Tsutsui H; Kitamura M; Narasaka K Synthesis of Pyrrole Derivatives by Palladium-Catalyzed Cyclization of Γ,Δ-Unsaturated Ketone O-Pentafluorobenzoyloximes. Bull. Chem. Soc. Jpn. 2002, 75, 1451–1460. [Google Scholar]
- (243).Kitamura M; Kudo D; Narasaka K Palladium(0)-Catalyzed Synthesis of Pyridines from B-Acetoxy-Γ,Δ-Unsaturated Ketone Oximes. ARKIVOC 2006, 2006, 148–162. [Google Scholar]
- (244).sutsui TH; Narasaka K Synthesis of Pyridine and Isoquinoline Derivatives by the Palladium-Catalyzed Cyclization of Olefinic Ketone O-Pentafluorobenzoyloximes. Chem. Lett. 2001, 30, 526–527. [Google Scholar]
- (245).Kitamura M; Chiba S; Saku O; Narasaka K Palladium-Catalyzed Synthesis of 1-Azaazulenes from Cycloheptatrienylmethyl Ketone O-Pentafluorobenzoyl Oximes. Chem. Lett. 2002, 31, 606–607. [Google Scholar]
- (246).Chiba S; Kitamura M; Saku O; Narasaka K Synthesis of 1-Azaazulenes from Cycloheptatrienylmethyl Ketoneo-Pentafluorobenzoyloximes by Palladium-Catalyzed Cyclization and Oxidation. Bull. Chem. Soc. Jpn. 2004, 77, 785–796. [Google Scholar]
- (247).Narasaka K; Kitamura M Amination with Oximes. Eur. J. Org. Chem. 2005, 2005, 4505–4519. [Google Scholar]
- (248).Sakoda K; Mihara J; Ichikawa J Heck-Type 5-Endo-Trig Cyclization Promoted by Vinylic Fluorines: Synthesis of 5-Fluoro-3h- Pyrroles. Chem. Commun. 2005, 4684–4686. [DOI] [PubMed] [Google Scholar]
- (249).Ichikawa J; Nadano R; Ito N 5-Endo Heck-Type Cyclization of 2-(Trifluoromethyl)Allyl Ketone Oximes-Synthesis of 4-Difluoromethylene-Substituted 1-Pyrrolines. Chem. Commun. 2006, 4425–4427. [DOI] [PubMed] [Google Scholar]
- (250).Zaman S; Kitamura M; Abell AD Synthesis of Trisubstituted Imidazoles by Palladium-Catalyzed Cyclization of O-Pentafluorobenzoylamidoximes- Application to Amino Acid Mimetics with a C-Terminal Imidazole. Org. Lett. 2005, 7, 609–611. [DOI] [PubMed] [Google Scholar]
- (251).Furstner A; Radkowski K; Peters H Chasing a Phantom by Total Synthesis: The Butylcycloheptylprodigiosin Case. Angew. Chem. Int. Ed. 2005, 44, 2777–2781. [DOI] [PubMed] [Google Scholar]
- (252).Furstner A; Radkowski K; Peters H; Seidel G; Wirtz C; Mynott R; Lehmann CW Total Synthesis, Molecular Editing and Evaluation of a Tripyrrolic Natural Product: The Case of "Butylcycloheptylprodigiosin". Chemistry 2007, 13, 1929–1945. [DOI] [PubMed] [Google Scholar]
- (253).Faulkner A; Bower JF Highly Efficient Narasaka-Heck Cyclizations Mediated by P(3,5- (Cf3)2c6h3)3: Facile Access to N-Heterobicyclic Scaffolds. Angew. Chem. Int. Ed. 2012, 51, 1675–1679. [DOI] [PubMed] [Google Scholar]
- (254).Hanley PS; Hartwig JF Intermolecular Migratory Insertion of Unactivated Olefins into Palladium Nitrogen Bonds. Steric and Electronic Effects on the Rate of Migratory Insertion. J. Am. Chem. Soc. 2011, 133, 15661–15673. [DOI] [PubMed] [Google Scholar]
- (255).Gawley RE, The Beckmann Reactions: Rearrangements, Elimination-Additions, Fragmentations, and Rearrangement-Cyclizations In Organic Reactions, John Wiley & Sons, Inc: Chichester, UK, 1988; Vol. 35. [Google Scholar]
- (256).Race NJ; Bower JF Palladium Catalyzed Cyclizations of Oxime Esters with 1,2-Disubstituted Alkenes-Synthesis of Dihydropyrroles. Org. Lett. 2013, 15, 4616–4619. [DOI] [PubMed] [Google Scholar]
- (257).Faulkner A; Scott JS; Bower JF Palladium Catalyzed Cyclizations of Oxime Esters with 1,1-Disubstituted Alkenes: Synthesis of Alpha,Alpha-Disubstituted Dihydropyrroles and Studies Towards an Asymmetric Protocol. Chem. Commun. 2013, 49, 1521–1523. [DOI] [PubMed] [Google Scholar]
- (258).Race NJ; Faulkner A; Shaw MH; Bower JF Dichotomous Mechanistic Behavior in Narasaka- Heck Cyclizations: Electron Rich Pd-Catalysts Generate Iminyl Radicals. Chem. Sci. 2016, 7, 1508–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Race NJ; Faulkner A; Fumagalli G; Yamauchi T; Scott JS; Ryden-Landergren M; Sparkes HA; Bower JF Enantioselective Narasaka-Heck Cyclizations: Synthesis of Tetrasubstituted Nitrogen-Bearing Stereocenters. Chem. Sci. 2017, 8, 1981–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Race NJ; Hazelden IR; Faulkner A; Bower JF Recent Developments in the Use of Aza-Heck Cyclizations for the Synthesis of Chiral N-Heterocycles. Chem. Sci. 2017, 8, 5248–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Zaman S; Kitamura M; Abell AD Synthesis of 5-Phenyl 2-Functionalized Pyrroles by Amino Heck and Tandem Amino Heck Carbonylation Reactions. Aust. J. Chem. 2007, 60, 624–626. [Google Scholar]
- (262).Kitamura M; Moriyasu Y; Okauchi T Synthesis of Isoindole Derivatives by Palladium-Catalyzed Domino Reaction of (2-Alkynyl)Phenylketone O-Pentafluorobenzoyloximes. Synlett 2011, 2011, 643–646. [Google Scholar]
- (263).Faulkner A; Scott JS; Bower JF An Umpolung Approach to Alkene Carboamination-Palladium Catalyzed 1,2-Amino-Acylation, -Carboxylation, -Arylation, -Vinylation, and -Alkynylation. J. Am. Chem. Soc. 2015, 137, 7224–7230. [DOI] [PubMed] [Google Scholar]
- (264).Chen C; Hou L; Cheng M; Su J; Tong X Palladium(0)-Catalyzed Iminohalogenation of Alkenes: Synthesis of 2-Halomethyl Dihydropyrroles and Mechanistic Insights into the Alkyl Halide Bond Formation. Angew. Chem. Int. Ed. 2015, 54, 3092–3096. [DOI] [PubMed] [Google Scholar]
- (265).Bao X; Wang Q; Zhu J Palladium-Catalyzed Enantioselective Narasaka-Heck Reaction/Direct C-H Alkylation of Arenes: Iminoarylation of Alkenes. Angew. Chem. Int. Ed. 2017, 56, 9577–9581. [DOI] [PubMed] [Google Scholar]
- (266).Liu S; Yu Y; Liebeskind LS N-Substituted Imines by the Copper-Catalyzed N-Imination of Boronic Acids and Organostannanes with O-Acyl Ketoximes. Org. Lett. 2007, 9, 1947–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Liu S; Liebeskind LS A Simple, Modular Synthesis of Substituted Pyridines. J. Am. Chem. Soc. 2008, 130, 6918–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Zhang Z; Yu Y; Liebeskind LS N-Amidation by Copper-Mediated Cross-Coupling of Organostannanes or Boronic Acids with O-Acetyl Hydroxamic Acids. Org. Lett. 2008, 10, 3005–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Liu H; Yan X; Chen C; Liu Q; Xi C Copper-Mediated Electrophilic Imination of Alkenylzirconocenes with O-Benzoyl Ketoximes and Aldoximes. Chem. Commun. 2013, 49, 5513–5515. [DOI] [PubMed] [Google Scholar]
- (270).Faulkner A; Race NJ; Scott JS; Bower JF Copper Catalyzed Heck-Like Cyclizations of Oxime Esters. Chem. Sci. 2014, 5, 2416–2421. [Google Scholar]
- (271).Zard ZS; Bingham M; Moutrille C A Simple Route to Functionalized Δl-Pyrrolenines. Heterocycles 2014, 88, 953–960. [Google Scholar]
- (272).Zhu J-L; Su Y-L; Chang Y-H; Chen IC; Liao C-C Studies on the Amino-Heck Reactions of Unsaturated Ketones O-Phosphinyloximes for the Preparation of Substituted Pyridines. Heterocycles 2009, 78, 369–387. [Google Scholar]
- (273).Zhu J-L; Chan Y-H Palladium-Catalyzed Amino-Heck Reaction of r,A-Unsaturated Ketone O-Diethylphosphinyloximes: A New Synthesis of Substituted Pyrroles and Indoles. Synlett 2008, 2008, 1250–1254. [Google Scholar]
- (274).Zhu J-L; Tsao S-W Investigation on Amino-Heck Cyclization of 1-(2-Vinylcyclohexyl)Ketone Diethyl Phosphinyloximes. Heterocycles 2012, 85, 383–401. [Google Scholar]
- (275).Hazelden IR; Ma X; Langer T; Bower JF Diverse N-Heterocyclic Ring Systems Via Aza-Heck Cyclizations of N-(Pentafluorobenzoyloxy)Sulfonamides. Angew. Chem. Int. Ed. 2016, 55, 11198–11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Hanley PS; Markovic D; Hartwig JF Intermolecular Insertion of Ethylene and Octene into a Palladium-Amide Bond. Spectroscopic Evidence for an Ethylene Amido Intermediate. J. Am. Chem. Soc. 2010, 132, 6302–6303. [DOI] [PubMed] [Google Scholar]
- (277).Hanley PS; Hartwig JF Migratory Insertion of Alkenes into Metal-Oxygen and Metal-Nitrogen Bonds. Angew. Chem. Int. Ed. 2013, 52, 8510–8525. [DOI] [PubMed] [Google Scholar]
- (278).Vulovic B; Watson DA Heck-Like Reactions Involving Heteroatomic Electrophiles. Eur. J. Org. Chem. 2017, 2017, 4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Shuler SA; Yin G; Krause SB; Vesper CM; Watson DA Synthesis of Secondary Unsaturated Lactams Via an Aza-Heck Reaction. J. Am. Chem. Soc. 2016, 13830–13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Zhu C; Li G; Ess DH; Falck JR; Kürti L Elusive Metal-Free Primary Amination of Arylboronic Acids: Synthetic Studies and Mechanism by Density Functional Theory. J. Am. Chem. Soc. 2012, 134, 18253–18256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Pan Y; Young GB Syntheses and Spectroscopic Characteristics of Dialkylpalladium(Ii) Complexes; Pdr2(Cod) as Precursors for Derivatives with N- or P-Donor Ligands. J. Organomet. Chem. 1999, 577, 257–264. [Google Scholar]
- (282).Lee HG; Milner PJ; Colvin MT; Andreas L; Buchwald SL Structure and Reactivity of [(LPd)N(1,5-Cyclooctadiene)] (N=1–2) Complexes Bearing Biaryl Phosphine Ligands. Inorg. Chim. Acta 2014, 422, 188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Meier M; Basolo F; Kroll WR; Moy D; Romanelli MG, Tetrakis(Triethyl Phosphite)Nickel(0), Palladium(0), and Platinum(0) Complexes In Inorganic Syntheses: Reagents for Transition Metal Complex and Organometallic Syntheses, Angelici RJ, Ed. John Wiley & Sons, Inc: Hoboken, NJ, USA, 1990; Vol. 28, pp 104–107. [Google Scholar]
- (284).Hazelden IR; Carmona RC; Langer T; Pringle PG; Bower JF Pyrrolidines and Piperidines by Ligand-Enabled Aza-Heck Cyclizations and Cascades of N-(Pentafluorobenzoyloxy)Carbamates. Angew. Chem. Int. Ed. 2018, 57, 5124–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Xu F; Shuler SA; Watson DA Synthesis of N-H Bearing Imidazolidinones and Dihydroimidazolones Using Aza-Heck Cyclizations. Angew. Chem. Int. Ed. 2018, 57, 12081–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Gottlich R Copper(I)-Catalyzed Intramolecular Addition of N-Chloroamines to Double Bonds under Aprotic Conditions. Towards a Stereoselective Catalytic Radical Reaction. Synthesis 2000, 2000, 1561–1564. [Google Scholar]
- (287).Helaja J; Gottlich R A New Catalytic Hetero-Heck Type Reaction. Chem. Commun. 2002, 720–721. [DOI] [PubMed] [Google Scholar]
- (288).Berman AM; Johnson JS Copper-Catalyzed Electrophilic Amination of Diorganozinc Reagents. J. Am. Chem. Soc. 2004, 126, 5680–5681. [DOI] [PubMed] [Google Scholar]
- (289).Berman AM; Johnson JS Copper-Catalyzed Electrophilic Amination of Functionalized Diarylzinc Reagents. J. Org. Chem. 2005, 70, 364–366. [DOI] [PubMed] [Google Scholar]
- (290).Johnson JS; Berman AM Nickel-Catalyzed Electrophilic Amination of Organozinc Halides. Synlett 2005, 1799–1801. [Google Scholar]
- (291).Berman AM; Johnson JS Copper-Catalyzed Electrophilic Amination of Organozinc Nucleophiles-Documentation of O-Benzoyl Hydroxylamines as Broadly Useful R2n(+) and Rhn(+) Synthons. J. Org. Chem. 2006, 71, 219–224. [DOI] [PubMed] [Google Scholar]
- (292).Biloski AJ; Ganem B Improved Oxidation of Amines with Dibenzoyl Peroxide. Synthesis 1983, 537–538. [Google Scholar]
- (293).Campbell MJ; Johnson JS Mechanistic Studies of the Copper-Catalyzed Electrophilic Amination of Diorganozinc Reagents and Development of a Zinc-Free Protocol. Org. Lett. 2007, 9, 1521–1524. [DOI] [PubMed] [Google Scholar]
- (294).Matsuda N; Hirano K; Satoh T; Miura M Copper-Catalyzed Amination of Arylboronates with N,N- Dialkylhydroxylamines. Angew. Chem. Int. Ed. 2012, 51, 3642–3645. [DOI] [PubMed] [Google Scholar]
- (295).Rucker RP; Whittaker AM; Dang H; Lalic G Synthesis of Hindered Anilines: Copper-Catalyzed Electrophilic Amination of Aryl Boronic Esters. Angew. Chem. Int. Ed. 2012, 51, 3953–3956. [DOI] [PubMed] [Google Scholar]
- (296).Miki Y; Hirano K; Satoh T; Miura M Copper-Catalyzed Electrophilic Amination of Arylsilanes with Hydroxylamines. Org. Lett. 2013, 15, 172–175. [DOI] [PubMed] [Google Scholar]
- (297).Nakao Y; Imanaka H; Sahoo AK; Yada A; Hiyama T Alkenyl- and Aryl[2-(Hydroxymethyl)Phenyl]Dimethylsilanes: An Entry to Tetraorganosilicon Reagents for the Silicon-Based Cross-Coupling Reaction. J. Am. Chem. Soc. 2005, 127, 6952–6953. [DOI] [PubMed] [Google Scholar]
- (298).Nguyen MH; Smith AB Copper-Catalyzed Electrophilic Amination of Organolithiums Mediated by Recoverable Siloxane Transfer Agents. Org. Lett. 2013, 15, 4872–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Zhou S; Yang Z; Chen X; Li Y; Zhang L; Fang H; Wang W; Zhu X; Wang S Copper-Catalyzed Electrophilic Amination of Organoaluminum Nucleophiles with O-Benzoyl Hydroxylamines. J. Org. Chem. 2015, 80, 6323–6328. [DOI] [PubMed] [Google Scholar]
- (300).Yan X; Chen C; Zhou Y; Xi C Copper-Catalyzed Electrophilic Amination of Alkenylzirconocenes with O-Benzoylhydroxylamines: An Efficient Method for Synthesis of Enamines. Org. Lett. 2012, 14, 4750–4753. [DOI] [PubMed] [Google Scholar]
- (301).Chen Y-H; Graßl S; Knochel P Cobalt-Catalyzed Electrophilic Amination of Aryl- and Heteroarylzinc Pivalates with N-Hydroxylamine Benzoates. Angew. Chem. Int. Ed. 2018, 57,, 1108–1111. [DOI] [PubMed] [Google Scholar]
- (302).Shen K; Wang Q Copper-Catalyzed Diamination of Unactivated Alkenes with Hydroxylamines. Chem. Sci. 2015, 6,4279–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Peterson LJ; Kirsch JK; Wolfe JP Pd-Catalyzed Alkene Diamination Reactions of Nitrogen Electrophiles: Synthesis of Cyclic Guanidines and Ureas Bearing Dialkylaminomethyl Groups. Org. Lett. 2018, 20, 3513–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Hemric BN; Shen K; Wang Q Copper-Catalyzed Amino Lactonization and Amino Oxygenation of Alkenes Using O-Benzoylhydroxylamines. J. Am. Chem. Soc. 2016, 138, 5813–5816. [DOI] [PubMed] [Google Scholar]
- (305).Hemric BN; Wang Q Copper-Catalyzed Intermolecular Oxyamination of Olefins Using Carboxylic Acids and O-Benzoylhydroxylamines. Beilstein J. Org. Chem. 2016, 12, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Donohoe TJ; Callens CKA; Flores A; Lacy AR; Rathi AH Recent Developments in Methodology for the Direct Oxyamination of Olefins. Chem. Eur. J. 2011, 17, 58–76. [DOI] [PubMed] [Google Scholar]
- (307).He C; Chen C; Cheng J; Liu C; Liu W; Li Q; Lei A Aryl Halide Tolerated Electrophilic Amination of Arylboronic Acids with N-Chloroamides Catalyzed by Cucl at Room Temperature. Angew. Chem. Int. Ed. 2008, 47, 6414–6417. [DOI] [PubMed] [Google Scholar]
- (308).Barker TJ; Jarvo ER Umpolung Amination-Nickel-Catalyzed Coupling Reactions of N,N-Dialkyl-N-Chloroamines with Diorganozinc Reagents. J. Am. Chem. Soc. 2009, 131, 15598–15599. [DOI] [PubMed] [Google Scholar]
- (309).Barker TJ; Jarvo ER Titanium-Mediated Amination of Grignard Reagents Using Primary and Secondary Amines. Angew. Chem. Int. Ed. 2011, 50, 8325–8328. [DOI] [PubMed] [Google Scholar]
- (310).Qian X; Yu Z; Auffrant A; Gosmini C Cobalt-Catalyzed Electrophilic Amination of Arylzincs with N-Chloroamines. Chemistry 2013, 19, 6225–6229. [DOI] [PubMed] [Google Scholar]
- (311).Fukami Y; Wada T; Meguro T; Chida N; Sato T Copper-Catalyzed Electrophilic Amination Using N-Methoxyamines. Org. Biomol. Chem. 2016, 14, 5486–5489. [DOI] [PubMed] [Google Scholar]
- (312).DaSkapan T; Koca S Highly Efficient Catalytic System for Electrophilic Amination of Arylzinc Reagents. Appl. Organomet. Chem. 2009, 24, 12–16. [Google Scholar]
- (313).Daskapan T Preparation of Primary Arylamines Via Arylzinc Chlorides in Good Yields. Tetrahedron Lett. 2006, 47, 2879–2881. [Google Scholar]
- (314).Zhou Z; Ma Z; Behnke NE; Gao H; Kürti L Non-Deprotonative Primary and Secondary Amination of (Hetero)Arylmetals. J. Am. Chem. Soc. 2017, 139, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Ghoraf M; Vidal J Electrophilic Amination of Diorganozinc Reagents by Oxaziridines. Tetrahedron Lett. 2008, 49, 7383–7385. [Google Scholar]
- (316).Miura T; Morimoto M; Murakami M Copper-Catalyzed Amination of Silyl Ketene Acetals with N-Chloroamines. Org. Lett. 2012, 14, 5214–5217. [DOI] [PubMed] [Google Scholar]
- (317).Matsuda N; Hirano K; Satoh T; Miura M Copper-Catalyzed Amination of Ketene Silyl Acetals with Hydroxylamines: Electrophilic Amination Approach to Alpha-Amino Acids. Angew. Chem. Int. Ed. 2012, 51, 11827–11831. [DOI] [PubMed] [Google Scholar]
- (318).McDonald SL; Wang Q Selective A-Amination and A-Acylation of Esters and Amides Via Dual Reactivity of O-Acylhydroxylamines toward Zinc Enolates. Chem. Commun. 2014, 50, 2535–2538. [DOI] [PubMed] [Google Scholar]
- (319).McDonald SL; Wang Q Copper-Catalyzed A-Amination of Phosphonates and Phosphine Oxides: A Direct Approach to A-Amino Phosphonic Acids and Derivatives. Angew. Chem. 2014, 126, 1898–1902. [DOI] [PubMed] [Google Scholar]
- (320).Rucker RP; Whittaker AM; Dang H; Lalic G Synthesis of Tertiary Alkyl Amines from Terminal Alkenes: Copper-Catalyzed Amination of Alkyl Boranes. J. Am. Chem. Soc. 2012, 134, 6571–6574. [DOI] [PubMed] [Google Scholar]
- (321).McDonald SL; Hendrick CE; Wang Q Copper-Catalyzed Electrophilic Amination of Heteroarenes and Arenes by C-H Zincation. Angew. Chem. Int. Ed. 2014, 53, 4667–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Yoon H; Lee Y Copper-Catalyzed Electrophilic Amination of Heteroarenes Via C-H Alumination. J. Org. Chem. 2015, 80, 10244–10251. [DOI] [PubMed] [Google Scholar]
- (323).Hendrick CE; Bitting KJ; Cho S; Wang Q Site-Selective Copper-Catalyzed Amination and Azidation of Arenes and Heteroarenes Via Deprotonative Zincation. J. Am. Chem. Soc. 2017, 139, 11622–11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Kitamura M; Yanagisawa H; Yamane M; Narasaka K Palladium(0)-Catalyzed Amino-Heck Reaction of Γ,Δ-Unsaturated Ketone N,N,N-Trimethylhydrazonium Salts. Heterocycles 2005, 65, 273–277. [Google Scholar]
- (325).Zhao B; Yuan W; Du H; Shi Y Cu(I)-Catalyzed Intermolecular Diamination of Activated Terminal Olefins. Org. Lett. 2007, 9, 4943–4945. [DOI] [PubMed] [Google Scholar]
- (326).Cornwall RG; Zhao B; Shi Y Complementary Regioselectivity in the Cu(I)-Catalyzed Diamination of Conjugated Dienes to Form Cyclic Sulfamides. Org. Lett. 2011, 13, 434–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Cornwall RG; Zhao B; Shi Y Catalytic Asymmetric Synthesis of Cyclic Sulfamides from Conjugated Dienes. Org. Lett. 2013, 15, 796–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Xu L; Shi Y Chiral N-Heterocyclic Carbene-Pd(0)-Catalyzed Asymmetric Diamination of Conjugated Dienes and Triene. J. Org. Chem. 2008, 73, 749–751. [DOI] [PubMed] [Google Scholar]
- (329).Du H; Yuan W; Zhao B; Shi Y Catalytic Asymmetric Diamination of Conjugated Dienes and Triene. J. Am. Chem. Soc. 2007, 129, 11688–11689. [DOI] [PubMed] [Google Scholar]
- (330).Du H; Yuan W; Zhao B; Shi Y A Pd(0)-Catalyzed Diamination of Terminal Olefins at Allylic and Homoallylic Carbons Via Formal C-H Activation under Solvent-Free Conditions. J. Am. Chem. Soc. 2007, 129, 7496–7497. [DOI] [PubMed] [Google Scholar]
- (331).Du H; Zhao B; Shi Y Catalytic Asymmetric Allylic and Homoallylic Diamination of Terminal Olefins Via Formal C-H Activation. J. Am. Chem. Soc. 2008, 130, 8590–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Yuan W; Du H; Zhao B; Shi Y A Mild Cu(I)-Catalyzed Regioselective Diamination of Conjugated Dienes. Org. Lett. 2007, 9, 2589–2591. [DOI] [PubMed] [Google Scholar]
- (333).Xu L; Du H; Shi Y Diamination of Conjugated Dienes and Trienes Catalyzed by N-Heterocyclic Carbene-Pd(0) Complexes. J. Org. Chem. 2007, 72, 7038–7041. [DOI] [PubMed] [Google Scholar]
- (334).Zhao B; Du H; Shi Y Cu(I)-Catalyzed Cycloguanidination of Olefins. Org. Lett. 2008, 10, 1087–1090. [DOI] [PubMed] [Google Scholar]
- (335).Zhao B; Du H; Shi Y A Cu(I)-Catalyzed C–H A-Amination of Esters. Direct Synthesis of Hydantoins. J. Am. Chem. Soc. 2008, 130, 7220–7221. [DOI] [PubMed] [Google Scholar]
- (336).Du H; Zhao B; Yuan W; Shi Y Cu(I)-Catalyzed Asymmetric Diamination of Conjugated Dienes. Org. Lett. 2008, 10, 4231–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Zhao B; Du H; Shi Y Cu(I)-Catalyzed C-H A-Amination of Aryl Ketones: Direct Synthesis of Imidazolinones. J. Org. Chem. 2009, 74, 4411–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Wen Y; Zhao B; Shi Y Cu(I)-Catalyzed Diamination of Disubstituted Terminal Olefins: An Approach to Potent Nk1 Antagonist. Org. Lett. 2009, 11, 2365–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (339).Zhao B; Du H; Shi Y Cu(I)-Catalyzed Diamination of Conjugated Olefins with Tunable Anionic Counterions. A Possible Approach to Asymmetric Diamination. J. Org. Chem. 2009, 74, 8392–8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (340).Zhao B; Peng X; Cui S; Shi Y Cu(I)-Catalyzed Regioselective Diamination of Conjugated Dienes Via Dual Mechanistic Pathways. J. Am. Chem. Soc. 2010, 132, 11009–11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Zhao B; Peng X; Zhu Y; Ramirez TA; Cornwall RG; Shi Y Cu(I)-Catalyzed Diamination of Conjugated Dienes. Complementary Regioselectivity from Two Distinct Mechanistic Pathways Involving Cu(Ii) and Cu(Iii) Species. J. Am. Chem. Soc. 2011, 133, 20890–20900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Zhu Y; Shi Y Cui-Catalyzed Sequential Diamination and Dehydrogenation of Terminal Olefins: A Facile Approach to Imidazolinones. Chem. Eur. J. 2014, 20, 13901–13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Zhao B; Du H; Cui S; Shi Y Synthetic and Mechanistic Studies on Pd(0)-Catalyzed Diamination of Conjugated Dienes. J. Am. Chem. Soc. 2010, 132, 3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Zhu Y; Cornwall RG; Du H; Zhao B; Shi Y Catalytic Diamination of Olefins Via N-N Bond Activation. Acc. Chem. Res. 2014, 47, 3665–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Ramirez TA; Wang Q; Zhu Y; Zheng H; Peng X; Cornwall RG; Shi Y Pd(0)-Catalyzed Sequential C-N Bond Formation Via Allylic and Aromatic C-H Amination of A-Methylstyrenes with Diaziridinone. Org. Lett. 2013, 15, 4210–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Zheng H; Zhu Y; Shi Y Palladium(0)-Catalyzed Heck Reaction/C–H Activation/Amination Sequence with Diaziridinone: A Facile Approach to Indolines. Angew. Chem. Int. Ed. 2014, 53, 11280–11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Miyazaki Y; Ohta N; Semba K; Nakao Y Intramolecular Aminocyanation of Alkenes by Cooperative Palladium/Boron Catalysis. J. Am. Chem. Soc. 2014, 136, 3732–3735. [DOI] [PubMed] [Google Scholar]
- (348).Yu J-T; Teng F; Cheng J The Construction of X–Cn (X=N, S, O) Bonds. Adv. Synth. Catal. 2017, 359, 26–38. [Google Scholar]
- (349).Pan Z; Pound SM; Rondla NR; Douglas CJ Intramolecular Aminocyanation of Alkenes by N–Cn Bond Cleavage. Angew. Chem. Int. Ed. 2014, 53, 5170–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Zhao J; Wang G; Li S An Insight into the Lewis Acid-Catalyzed Intramolecular Aminocyanation and Oxycyanation of Alkenes: A Concerted or Stepwise Mechanism. Chem. Commun. 2015, 51, 15450–15453. [DOI] [PubMed] [Google Scholar]
- (351).Yuan Y-C; Yang H-B; Tang X-Y; Wei Y; Shi M Unprecedented Oxycyanation of Methylenecyclopropanes for the Facile Synthesis of Benzoxazine Compounds Containing a Cyano Group. Chem. Eur. J. 2016, 22, 5146–5150. [DOI] [PubMed] [Google Scholar]
- (352).Pan Z; Wang S; Brethorst JT; Douglas CJ Palladium and Lewis-Acid-Catalyzed Intramolecular Aminocyanation of Alkenes: Scope, Mechanism, and Stereoselective Alkene Difunctionalizations. J. Am. Chem. Soc. 2018, 140, 3331–3338. [DOI] [PubMed] [Google Scholar]
- (353).Suzuki A Cross-Coupling Reactions Via Organoboranes. J. Organomet. Chem. 2002, 653, 83–90. [Google Scholar]
- (354).Burgess K; Ohlmeyer MJ Transition-Metal Promoted Hydroborations of Alkenes, Emerging Methodology for Organic Transformations. Chem. Rev. 1991, 91, 1179–1191. [Google Scholar]
- (355).Baker RT; Ovenall DW; Calabrese JC; Westcott SA; Taylor NJ; Williams ID; Marder TB Boryliridium and Boraethyliridium Complexes Fac-[Irh2(Pme3)3(Brr')] and Fac-[Irh(Pme3)3(.Eta.2-Ch2bhrr')]. J. Am. Chem. Soc. 1990, 112, 9399–9400. [Google Scholar]
- (356).Knorr JR; Merola JS Synthesis and Structure of a [(1,2-Phenylenedioxy)Boryl]Iridium Hydride Complex: A Model System for Studying Catalytic Hydroboration. Organometallics 1990, 9, 3008–3010. [Google Scholar]
- (357).Nguyen P; Lesley G; Taylor NJ; Marder TB; Pickett NL; Clegg W; Elsegood MRJ; Norman NC Oxidative Addition of B-B Bonds by Rhodium(I) Phosphine Complexes: Molecular Structures of B2cat2 (Cat = 1,2-O2c6h4) and Its 4-but and 3,5-But2 Analogs. Inorg. Chem. 1994, 33, 4623–4624. [Google Scholar]
- (358).Baker RT; Nguyen P; Marder TB; Westcott SA Transition Metal Catalyzed Diboration of Vinylarenes. Angew. Chem. Int. Ed. 1995, 34, 1336–1338. [Google Scholar]
- (359).Hartwig JF; He X Structural and Reaction Chemistry of Tungstenocene Boryl Complexes. Organometallics 1996, 15, 5350–5358. [Google Scholar]
- (360).Ishiyama T; Matsuda N; Murata M; Ozawa F; Suzuki A; Miyaura N Platinum(0)-Catalyzed Diboration of Alkynes with Tetrakis(Alkoxo)Diborons: An Efficient and Convenient Approach to Cis-Bis(Boryl)Alkenes. Organometallics 1996, 15, 713–720. [Google Scholar]
- (361).Clegg W; Lawlor FJ; Lesley G; Marder TB; Norman NC; Orpen AG; Quayle MJ; Rice CR; Scott AJ; Souza FES Oxidative Addition of Boron–Boron, Boron–Chlorine and Boron–Bromine Bonds to Platinum(0)1. J. Organomet. Chem. 1998, 550, 183–192. [Google Scholar]
- (362).Onozawa S; Tanaka M Oxidative Addition of the B–Cl Bond with Palladium Species and Insertion of Alkynes and a Vinyl Ketone into the Resulting B-Pd Bond. Organometallics 2001, 20, 2956–2958. [Google Scholar]
- (363).Braunschweig H; Gruss K; Radacki K; Uttinger K Oxidative Addition of B–Br Bonds to Pd0: Synthesis and Structure Oftrans-Bromo(Boryl)Palladium Complexes. Eur. J. Inorg. Chem. 2008, 2008, 1462–1466. [Google Scholar]
- (364).Zeng G; Sakaki S Noble Reaction Features of Bromoborane in Oxidative Addition of B–Br Ʃ-Bond to [M(Pme3)2] (M = Pt or Pd): Theoretical Study. Inorg. Chem. 2011, 50, 5290–5297. [DOI] [PubMed] [Google Scholar]
- (365).Braunschweig H; Fuß M; Radacki K; Uttinger K Synthesis of Chloro Boryl Complexes by Oxidative Addition of B-Cl Bonds. Z. Anorg. Allg. Chem. 2009, 635, 208–210. [Google Scholar]
- (366).Bauer J; Braunschweig H; Kraft K; Radacki K Oxidative Addition of Boron Trifluoride to a Transition Metal. Angew. Chem. Int. Ed. 2011, 50, 10457–10460. [DOI] [PubMed] [Google Scholar]
- (367).Coapes RB; Souza FES; Fox MA; Batsanov AS; Goeta AE; Yufit DS; Leech MA; Howard JAK; Scott AJ; Clegg W; Marder TB Phosphine Promoted Substituent Redistribution Reactions of B-Chlorocatechol Borane: Molecular Structures of Clbcat, Brbcat and LClbcat (Cat = 1,2-O2c6h4; L = Pme3, Pet3, Pbut3, Pcy3, Net3)†. J. Chem. Soc, Dalton Trans. 2001, 1201–1209. [Google Scholar]
- (368).Yamamoto A; Suginome M Nickel-Catalyzed Trans-Alkynylboration of Alkynes Via Activation of a Boron-Chlorine Bond. J. Am. Chem. Soc. 2005, 127, 15706–15707. [DOI] [PubMed] [Google Scholar]
- (369).Daini M; Yamamoto A; Suginome M Palladium-Catalyzed Trans- and Cis-Carboboration of Alkynes Tethered to Chloroborane with Organozirconium Reagents-Ligand-Dependent Complementary Stereoselectivity. J. Am. Chem. Soc. 2008, 130, 2918–2919. [DOI] [PubMed] [Google Scholar]
- (370).Daini M; Suginome M Palladium-Catalyzed, Stereoselective, Cyclizative Alkenylboration of Carbon–Carbon Double Bonds through Activation of a Boron-Chlorine Bond. J. Am. Chem. Soc. 2011, 133, 4758–4761. [DOI] [PubMed] [Google Scholar]
- (371).Nakada K; Daini M; Suginome M Palladium-Catalyzed Carboboration: Borylative Coupling of Alkynes with Alkenes through Activation of Boron-Chlorine Bonds. Chem. Lett. 2013, 42, 538–540. [Google Scholar]
- (372).Beesley RM; Ingold CK; Thorpe JF Cxix.-the Formation and Stability of Spiro-Compounds. Part I. Spiro-Compounds from Cyclohexane. J. Chem. Soc, Trans. 1915, 107, 1080–1106. [Google Scholar]
- (373).Jung ME; Piizzi G Gem-Disubstituent Effect: Theoretical Basis and Synthetic Applications. Chem. Rev. 2005, 105, 1735–1766. [DOI] [PubMed] [Google Scholar]
- (374).Bachrach SM The Gem-Dimethyl Effect Revisited. J. Org. Chem. 2008, 73, 2466–2468. [DOI] [PubMed] [Google Scholar]
- (375).Daini M; Suginome M Palladium-Catalyzed Carboboration of Alkynes Using Chloroborane and Organozirconium Reagents. Chem. Commun. 2008, 5224–5226. [DOI] [PubMed] [Google Scholar]
- (376).Reid WB; Spillane JJ; Krause SB; Watson DA Direct Synthesis of Alkenyl Boronic Esters from Unfunctionalized Alkenes: A Boryl-Heck Reaction. J. Am. Chem. Soc. 2016, 138, 5539–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Reid WB; Watson DA Synthesis of Trisubstituted Alkenyl Boronic Esters from Alkenes Using the Boryl-Heck Reaction. Org. Lett. 2018, 20, 6832–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Wolfe JP Intramolecular Alkoxycyanation and Alkoxyacylation Reactions: New Types of Alkene Difunctionalizations for the Construction of Oxygen Heterocycles. Angew. Chem. Int. Ed. 2012, 51, 10224–10225. [DOI] [PubMed] [Google Scholar]
- (379).Yu D-G; Luo S; Zhao F; Shi Z-J, Homogeneous Transition-Metal-Catalyzed C–O Bond Activation In Homogeneous Catal. Unreact. Bond Act, Shi Z-J, Ed. John Wiley & Sons, Inc: Hoboken, NJ, 2015; pp 347–440. [Google Scholar]
- (380).Rosen BM; Quasdorf KW; Wilson DA; Zhang N; Resmerita A-M; Garg NK; Percec V Nickel-Catalyzed Cross-Couplings Involving Carbon-Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Cornella J; Zarate C; Martin R Metal-Catalyzed Activation of Ethers Via C-O Bond Cleavage: A New Strategy for Molecular Diversity. Chem. Soc. Rev. 2014, 43, 8081–8097. [DOI] [PubMed] [Google Scholar]
- (382).Murai M; Miki K; Ohe K A New Route to 3-Acyl-2-Aminobenzofurans: Palladium-Catalysed Cycloisomerisation of 2-(Cyanomethyl)Phenyl Esters. Chem. Commun. 2009, 3466–3468. [DOI] [PubMed] [Google Scholar]
- (383).Hoang GT; Reddy VJ; Nguyen HHK; Douglas CJ Insertion of an Alkene into an Ester: Intramolecular Oxyacylation Reaction of Alkenes through Acyl C-O Bond Activation. Angew. Chem. Int. Ed. 2011, 50, 1882–1884. [DOI] [PubMed] [Google Scholar]
- (384).Hoang GT; Walsh DJ; McGarry KA; Anderson CB; Douglas CJ Development and Mechanistic Study of Quinoline-Directed Acyl C–O Bond Activation and Alkene Oxyacylation Reactions. J. Org. Chem. 2017, 82, 2972–2983. [DOI] [PubMed] [Google Scholar]
- (385).Vummaleti SVC; Al-Ghamdi M; Poater A; Falivene L; Scaranto J; Beetstra DJ; Morton JG; Cavallo L Mechanism of Intramolecular Rhodium- and Palladium-Catalyzed Alkene Alkoxyfunctionalizations. Organometallics 2015, 34, 5549–5554. [Google Scholar]
- (386).Hoang GT; Pan Z; Brethorst JT; Douglas CJ Intramolecular Oxyacylation of Alkenes Using a Hydroxyl Directing Group. J. Org. Chem. 2014, 79, 11383–11394. [DOI] [PubMed] [Google Scholar]
- (387).Wang R; Falck JR Transformations of X (C, O, N)–Cn Bonds: Cases of Selective X (C, O, N)–C Activation. RSC Adv. 2014, 4, 1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Beletskaya IP; Afanasiev VV; Kazankova MA; Efimova IV New Approach to Phosphinoalkynes Based on Pd- and Ni-Catalyzed Cross-Coupling of Terminal Alkynes with Chlorophosphanes. Org. Lett. 2003, 5, 4309–4311. [DOI] [PubMed] [Google Scholar]