

Summary
Pulmonary hypertension (PH) is a common but dangerous complication in chronic obstructive pulmonary disease (COPD). We hypothesized that dysregulation in the T helper type 17 (Th17) compartment could contribute to the development of COPD‐associated PH (COPD‐PH). To investigate this hypothesis, patients with COPD‐PH and age‐ and sex‐matched healthy controls were recruited, and their circulating CD4+ T cells were activated using anti‐CD3/CD28 antibodies. The frequency of interleukin‐17 (IL‐17) ‐secreting cells was significantly higher in COPD‐PH patients than in healthy controls. The secretion of IL‐17 was significantly higher from COPD‐PH CD4+ T cells than from control CD4+ T cells, whereas the secretion of interferon‐γ and IL‐4 was not significantly different. The expression of transforming growth factor‐β, on the other hand, was significantly higher in healthy controls than in COPD‐PH patients. Activated CD4+ T cells from COPD‐PH patients also presented significantly lower forkhead box P3 (FOXP3) and higher retinoic acid receptor‐related orphan C2 (RORC2) expression than CD4+ T cells from healthy controls. In both controls and patients, a negative correlation between RORC2 and FOXP3 was found, ex vivo and after CD3/CD28 activation. The serum IL‐6 level was slightly higher in COPD‐PH patients than in controls, but the IL‐6 transcription by monocytes was comparable in COPD‐PH patients and controls. Interestingly, CD4+ T cells from COPD‐PH patients presented significantly higher levels of Kirsten rat sarcoma viral oncogene homolog and neuroblastoma RAS viral oncogene homolog than CD4+ T cells from healthy controls. Inhibiting Ras‐GTPases using farnesylthiosalicylic acid significantly reduced the ratio of RORC2/FOXP3 expression in CD4+ T cells. Overall, we demonstrated that an imbalance of Th17/regulatory T cells was a hallmark of COPD‐PH.
Keywords: chronic obstructive pulmonary disease, pulmonary hypertension, regulatory T cells, T helper type 17 cells
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by restrictions in the airflow caused by pathological airway inflammation and progressive alveolar destruction.1 One of the complications of COPD is pulmonary hypertension (PH), where a mild to severe increase in the mean pulmonary arterial pressure is observed over time. The severity of PH independently affects the prognosis of COPD patients with PH.2, 3 It is thought that alveolar hypoxia may cause vasoconstriction in the pulmonary artery, which then leads to mild to moderate PH.4 Remodeling of pulmonary vascular lesions may also contribute to PH.5 In addition, the level of interleukin‐6 (IL‐6), an inflammatory cytokine, is associated with the risk of PH in COPD patients.6
Dysregulations in T helper type 17 (Th17) activation and function have been implicated in the pathogenesis of a variety of inflammatory diseases, including COPD.7, 8 Th17 cells secrete a variety of inflammatory mediators, including its namesake IL‐17. Airway epithelial cells express IL‐17 receptor at the basolateral surface.9 Upon receiving IL‐17, cells can produce a series of chemokines and growth factors that act as chemoattractants and stimulants to neutrophils and macrophages.10, 11 Combined with tumor necrosis factor‐α, IL‐17 can increase the mRNA stability and further enhance the production of pro‐inflammatory cytokines such as IL‐6 and IL‐8.12, 13 These IL‐17‐mediated effects can contribute to airway inflammation and remodeling in COPD patients, thus increasing the risk of PH development.
The polarization of Th17 cells is thought to involve a complex network of cytokines, including transforming growth factor‐β (TGF‐β), IL‐1β, IL‐6 and IL‐23.14, 15, 16 TGF‐β could induce retinoic‐acid‐receptor‐related orphan nuclear receptor γt (RORγt), a signature transcription factor for the Th17 lineage. The production of IL‐17 requires the signal transducer and activator of transcription 3 signaling pathway, which can be activated by IL‐6 and IL‐23. Interestingly, RORγt is not only present in Th17 cells but also found in regulatory T (Treg) cells.17 TGF‐β alone was found to stimulate forkhead box P3 (FOXP3) expression and Treg cell polarization,18, 19 and Th17 may be induced in the absence of TGF‐β, if IL‐1β and IL‐6 were present.20 In addition, plasticity between Treg and Th17 cells has been demonstrated, as FOXP3+ CD4+ T cells can lose Foxp3 expression in the presence of IL‐6, and differentiate into Th17 cells.21, 22 A high Th17/Treg cell ratio has been shown to associate with the development of inflammatory disorders, such as autoimmune diseases and inflammatory bowel diseases.7, 23
Based on the current evidence, we hypothesize that the dysregulations in the Th17 compartment can contribute to the development of COPD‐induced PH (COPD‐PH). To investigate this hypothesis, patients with COPD‐PH and age‐ and sex‐matched healthy controls were recruited, and the characteristics of Th17 cells in these study participants were examined and compared.
Methods
Study participants
We included 20 COPD‐PH patients, including five women at aged 61–71 (median 66) years and 15 men aged 59–70 (median 65) years, with body mass indices ranging from 23·1 to 28·6 (median 26·4) kg/m2. The pulmonary artery pressure at rest in the patients was 33–55 (median 45) mmHg. Healthy control participants were recruited from people without past or present pulmonary diseases, cardiovascular diseases, autoimmune diseases, or malignancies, and were not experiencing acute or chronic infections at the time of recruitment. The healthy group comprised five women between 60 and 72 (median 65) years of age, and 15 men between 58 and 71 (median 64) years of age, with body mass indices ranging from 23·8 to 28·5 (median 26·1) kg/m2. Diagnosis and participant recruitment were performed at The Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University. Ethical approval was given by the ethics committee of The Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University. All COPD‐PH patients and healthy controls had given written informed consent of participation and sample donation.
Sample collection
Venous blood was collected from each participant in citrated tubes (Thermo Fisher, Waltham, MA). Peripheral blood mononuclear cells (PBMCs) were harvested using centrifugation at 300 g for 30 min across a Ficoll (Sigma, St Louis, MO) gradient. The PBMCs were then cryopreserved in fetal calf serum supplemented with 10% (volume/volume) dimethylsulfoxide (Sigma) at −150°. Before the experiments, cells were thawed at 37° and dimethylsulfoxide was removed by rigorous washing. Cells were then rested overnight inside a CO2 incubator before use.
Th17 frequency and cytokine production
The PBMCs were stimulated with 2 μg/ml anti‐CD3 antibody OKT3 and 4 μg/ml anti‐CD28 antibody CD28.2 (BioLegend, San Diego, CA) for 24 hr. When indicated, recombinant human IL‐6 (PeproTech, Rocky Hill NJ) was added. The next day, cells were transferred into a 96‐well round‐bottom plate and were centrifuged at 300 g for 5 min to separate the supernatant and the cells. Cytokine secretion in the supernatant was measured using a custom MILLIPLEX Human TH17 Magnetic Bead Panel (Millipore, Billerica, MA) through a Luminex assay. CD4+ T cells were purified using Human CD4+ T Cell Enrichment Kit (Stemcell Technologies, Vancouver, BC, Canada) according to the manufacturer's protocol. Live CD4+ T cells were counted using 0·4% Trypan Blue staining (Gibco, Grand Island, NY) and were placed in a Human IL‐17 Microplate (R&D Systems, Minneapolis, MN) at serially diluted concentrations. The ELISpot proceeded according to the Human IL‐17 ELISpot Kit (R&D Systems) protocol. Each condition was performed in triplicates.
IL‐6 enzyme‐linked immunosorbent assay
A portion of blood was centrifuged to collect the upper serum. The concentration of IL‐6 was measured using the Human IL‐6 Quantikine ELISA Kit (R&D Systems), which has an assay range of 3·1–300 pg/ml and a sensitivity of 0·7 pg/ml.
mRNA transcript analysis
CD4+ T cells were isolated from ex vivo PBMCs, or PBMCs that were stimulated with 2 μg/ml anti‐CD3 antibody OKT3 and 4 μg/ml anti‐CD28 antibody CD28.2 for 24 hr. Monocytes were isolated by placing PBMCs in a six‐well plate and collecting adherent cells. The monocytes were then stimulated with 1 μg/ml lipopolysaccharide for 24 hr. Afterwards, each cell type was lyzed, and the mRNA was obtained using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and transcribed into cDNA using QuantiTect Reverse Transcription Kit (Qiagen). The transcripts were then quantified using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA), with the following IDs: Hs00364284_g1 (Kirsten rat sarcoma viral oncogene homolog, KRAS), Hs00180035_g1 (neuroblastoma RAS viral oncogene homolog, NRAS), Hs00978050_g1 (Harvey rat sarcoma viral oncogene homolog, HRAS), Hs00174383_m1 (IL17A), Hs00174131_m1 (IL6), Hs01076112_m1 (RORC2) and Hs01085834_m1 (FOXP3).
Statistical analysis
Data between controls and patients were examined using unpaired t‐test with Welch's correction. Correlation between FOXP3 and RORC2 was examined using a Pearson correlation test. The ratio of RORC2/FOXP3 with different levels of farnesylthiosalicylic acid (FTS) treatment was examined using repeated measures one‐way analysis of variance followed by Dunnett's multiple comparison tests. P < 0·05 was considered significant.
Results
COPD‐PH patients presented greater Th17 response than controls
To evaluate the role of Th17 cells in COPD‐PH patients, we first examined the frequency of Th17 cells using the expression of IL‐17. Directly ex vivo and in the absence of stimulation, few IL‐17‐secreting circulating CD4+ T cells were found. Hence, to improve detection, circulating CD4+ T cells were activated using anti‐CD3 antibody OKT3 and anti‐CD28 antibody CD28.2. The frequency of IL‐17‐secreting cells was examined in each participant using ELISpot and was significantly higher in COPD‐PH patients than in healthy controls (Fig. 1a). The secretion of IL‐17 was significantly higher from COPD‐PH CD4+ T cells than from control CD4+ T cells (Fig. 1b). The secretion of interferon‐γ and IL‐4, on the other hand, was not significantly different between COPD‐PH CD4+ T cells and control CD4+ T cells (Fig. 1c), suggesting that the cytokine up‐regulation in COPD‐PH CD4+ T cells was restricted to IL‐17. The expression of TGF‐β was significantly higher in healthy controls than in COPD‐PH patients (Fig. 1c).
Figure 1.
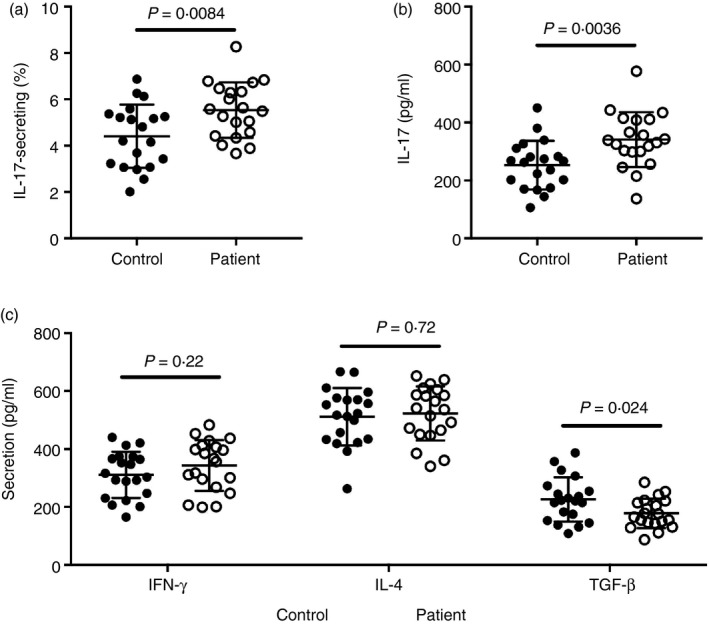
Expression of cytokines by CD4+ T cells from patients with chronic obstructive pulmonary disease‐associated pulmonary hypertension (COPD‐PH) and controls. (a) The frequency of interleukin‐17 (IL‐17) ‐secreting CD4+ T cells upon CD3/CD28 activation. (b) The amount of IL‐17 secretion by CD4+ T cells upon CD3/CD28 activation. (c) The amount of interferon‐γ (IFN‐γ), IL‐4 and transforming growth factor‐β (TGF‐β) secretion by CD4+ T cells upon CD3/CD28 activation. Unpaired t‐test with Welch's correction.
COPD‐PH patients presented an imbalance of RORC2 and FOXP3 expression
Having shown an up‐regulation of the Th17 response and a down‐regulation of the Treg cell responses, we investigated the expression of transcription factors, including the Th17‐lineage transcription factor RORC2 and the Treg‐lineage transcription factor FOXP3, in CD4+ T cells ex vivo and following CD3/CD28 activation. When ex vivo, the RORC2 expression in CD4+ T cells was borderline not significantly different between controls and COPD‐PH patients, whereas the FOXP3 expression was borderline significantly lower in patients compared with controls (Fig. 2a). After CD3/CD28 activation, CD4+ T cells from COPD‐PH patients presented significantly higher RORC2 and lower FOXP3 expression than CD4+ T cells from healthy controls (Fig. 2b). Because Treg and Th17 cells exhibit a certain degree of plasticity and may convert to each other under certain circumstances,24 we examined the correlation between RORC2 and FOXP3 expression. In both controls and patients, a negative correlation between RORC2 and FOXP3 was found, ex vivo and after CD3/CD28 activation (Fig. 2c,d).
Figure 2.
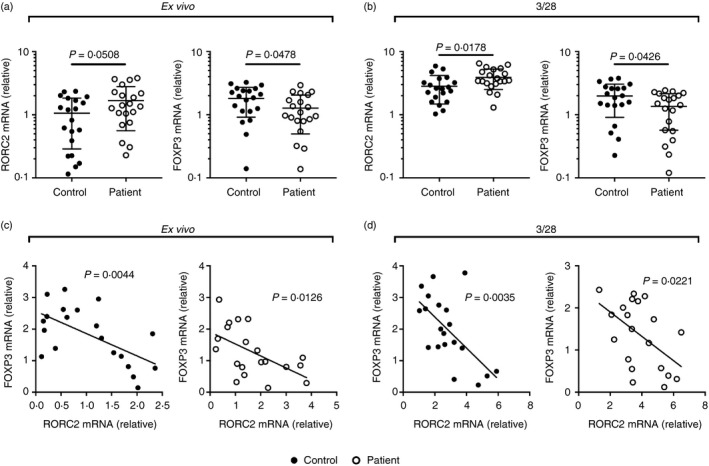
Expression of RORC2 and FOXP3 in CD4+ T cells from patients with chronic obstructive pulmonary disease‐associated pulmonary hypertension (COPD‐PH) and controls. (a) The expression of RORC2 and FOXP3 in CD4+ T cells ex vivo. (b) The expression of RORC2 and FOXP3 in CD4+ T cells upon CD3/CD28 activation. Unpaired t‐test with Welch's correction. (c) The correlation between ex vivo RORC2 and FOXP3 expression. (d) The correlation between RORC2 and FOXP3 expression upon CD3/CD28 stimulation. Pearson correlation test.
Serum but not monocyte IL‐6 was elevated in COPD‐PH patients
Previous studies have shown that IL‐6 is involved in switching between Treg and Th17 differentiation, and was involved in the development of pulmonary hypertension.7, 25 Inhibition of IL‐6 could alleviate the disease. Here, we investigated the effect of IL‐6 on the RORC2/FOXP3 ratio, by adding exogenous IL‐6 during CD3/CD28 activation. We found that in both controls and patients, the expression of RORC2 was significantly elevated with increasing levels of IL‐6 (Fig. 3a,b). Subsequently, serum IL‐6 concentration and monocyte IL‐6 production were examined in COPD‐PH patients and healthy controls. The serum IL‐6 level was slightly higher in COPD‐PH patients than in controls (Fig.3c). The IL‐6 transcription by monocytes, however, was comparable in COPD‐PH patients and controls, both ex vivo and after stimulation by lipopolysaccharide (Fig. 3d,e).
Figure 3.
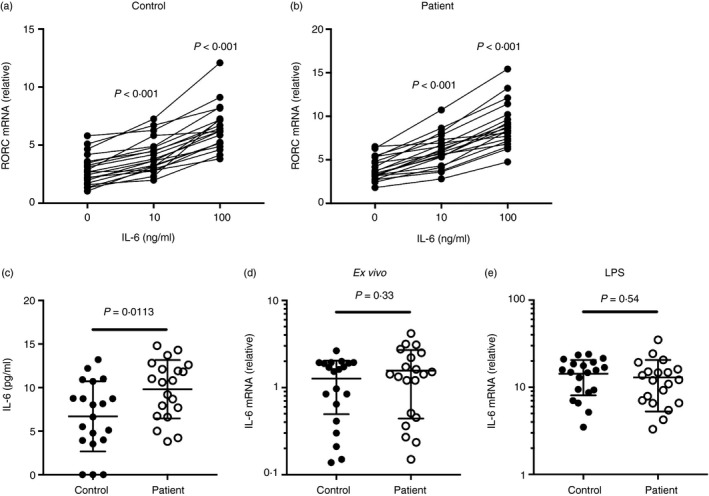
Interleukin‐6 (IL‐6) level in serum and monocytes from patients with chronic obstructive pulmonary disease‐associated pulmonary hypertension (COPD‐PH) and controls. (a) The expression of RORC2 in control CD4+ T cells upon CD3/CD28 activation and various levels of exogenous IL‐6. (b) The expression of RORC2 in patient CD4+ T cells upon CD3/CD28 activation and various levels of exogenous IL‐6. Repeated measures one‐way analysis of variance followed by Dunnett's multiple comparison tests. (c) The serum IL‐6 level in COPD‐PH patients and healthy controls. (d) IL‐6 mRNA expression in ex vivo monocytes. (e) IL‐6 mRNA expression in lipopolysaccharide‐stimulated monocytes. Unpaired t‐test with Welch's correction.
Ras‐GTPases demonstrated higher expression in COPD‐PH patients than in controls
Ras‐GTPases are key switches that act downstream of the CD3/CD28 signaling and have critical roles in many cellular processes, such as mobilization, differentiation, proliferation and apoptosis.26 Excessive Ras signaling was shown to associate with excessive Th17 responses in autoimmune diseases.27 Therefore, we examined the expression of Ras in CD4+ T cells from COPD‐PH patients and controls. CD4+ T cells from COPD‐PH patients presented significantly lower levels of KRAS and NRAS than CD4+ T cells from healthy controls both ex vivo and after CD3/CD28 activation (Fig. 4a,b). The HRAS expression, on the other hand, was not significantly different between patients and controls. In both the patient groups and the control group we split the participants into High‐KRAS and Low‐KRAS, and High‐NRAS and Low‐NRAS groups, using median KRAS and NRAS expression values as division standards, respectively. Under ex vivo conditions, no significant difference in terms of the RORC2/FOXP3 ratio was observed between High‐KRAS and Low‐KRAS individuals, and between High‐NRAS and Low‐NRAS individuals (data not shown). Following CD3/CD28 activation, High‐KRAS controls presented higher average RORC2/FOXP3 ratios than Low‐KRAS controls, but the difference was not statistically significant (Fig. 4c). However, the High‐KRAS patients presented significantly higher RORC2/FOXP3 ratio than Low‐KRAS patients (Fig. 4c). The High‐NRAS controls and patients also presented higher RORC2/FOXP3 ratio than Low‐NRAS controls (Fig. 4d).
Figure 4.
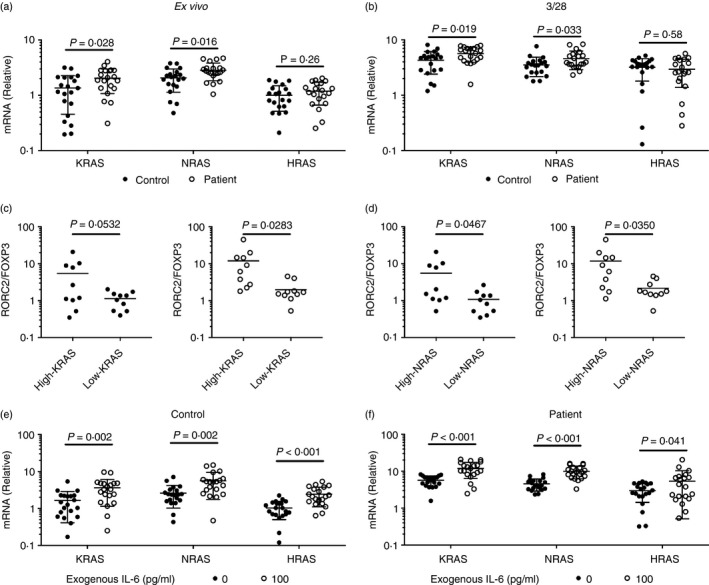
Expression of Ras‐GTPases in CD4+ T cells from patients with chronic obstructive pulmonary disease‐associated pulmonary hypertension (COPD‐PH) and controls. (a) The expression of KRAS, NRAS, and HRAS in CD4+ T cells ex vivo. (b) The expression of KRAS, NRAS and HRAS in CD4+ T cells upon CD3/CD28 activation. (c) The RORC3/FOXP3 ratio in High‐KRAS and Low‐KRAS controls (filled) and patients (open). (d) The RORC3/FOXP3 ratio in High‐NRAS and Low‐NRAS controls (filled) and patients (open). (e) The expression of KRAS, NRAS and HRAS in control CD4+ T cells upon CD3/CD28 activation and exogenous interleukin (IL‐6). (f) The expression of KRAS, NRAS and HRAS in patient CD4+ T cells upon CD3/CD28 activation and exogenous IL‐6. Unpaired t‐test with Welch's correction.
To investigate the effect of IL‐6 on the expression of Ras‐GTPases, we stimulated CD4+ T cells using CD3/CD28 and IL‐6. The expression of KRAS, NRAS and HRAS was then examined. In both controls and patients; IL‐6 had significantly increased the expression of KRAS, NRAS and HRAS (Fig. 4e,f).
Inhibition of Ras‐GTPases resulted in lower RORC2/FOXP3 expression ratio
Due to the association between RORC2/FOXP3 ratio and the KRAS and NRAS expression, we investigated whether Th17 responses could be reduced by inhibiting Ras‐GTPases. CD4+ T cells were activated by CD3/CD28, and FTS, a small molecule inhibitor of Ras‐GTPases, was added during the activation process at various concentrations. In both healthy controls and COPD‐PH patients, FTS at ≥ 10 ng/ml significantly reduced the ratio of RORC2/FOXP3 expression in CD4+ T cells (Fig. 5a). Subsequently, we examined the cytokine release, including IL‐17 and TGF‐β, in the absence of FTS or in the presence of 100 ng/ml FTS. In both controls and COPD‐PH patients, IL‐17 release was significantly reduced in the presence of FTS (Fig. 5b). The TGF‐β release, on the other hand, was unaffected by FTS (Fig. 5c).
Figure 5.
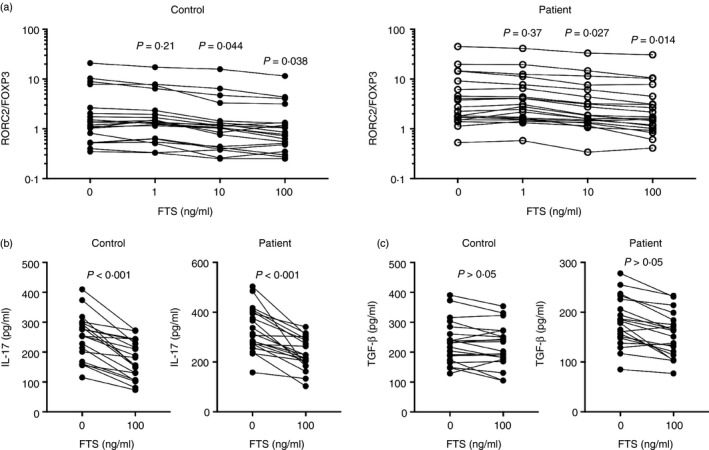
Inhibition of Ras‐GTPases on the ratio of RORC2/FOXP3 expression. (a) The RORC3/FOXP3 ratio in healthy controls and patients with chronic obstructive pulmonary disease‐associated pulmonary hypertension (COPD‐PH) upon CD3/CD28 activation and farnesylthiosalicylic acid (FTS) treatment. Repeated measures one‐way analysis of varaince followed by Dunnett's multiple comparison tests. (b) Interleukin‐17 (IL‐17) secretion in healthy controls and COPD‐PH patients upon CD3/CD28 activation and FTS treatment. (c) Transforming growth factor‐β (TGF‐β) secretion in healthy controls and COPD‐PH patients upon CD3/CD28 activation and FTS treatment. Paired t‐test.
Discussion
In this study, we showed that as a group, COPD‐PH patients presented more active Th17 responses than healthy controls, though individual variations also existed and varied greatly from participant to participant. The up‐regulation in Th17 responses, including the expression of IL‐17 and RORC2, was accompanied by a down‐regulation in Treg responses, including the expression of TGF‐β and FOXP3. No changes in the Th1 signature cytokine interferon‐γ or Th2 signature cytokine IL‐4 were observed in COPD‐PH patients, indicating that these patients did not present an overactive response across various CD4+ T cells. Overall, findings here demonstrated that an imbalance of Th17/Treg cell ratio was a hallmark of COPD‐PH.
Interleukin‐6, a pro‐inflammatory cytokine that favors the differentiation of Th17 responses,7, 23 was higher in COPD‐PH patients than in controls; however, the difference between the patient and the control group was small. It is unclear whether such a small difference in IL‐6 could result in Th17/Treg cell imbalance. Also, the level of IL‐6 secretion by circulating monocytes was comparable between COPD‐PH patients and healthy controls. The clinical significance of this small difference in the serum level of IL‐6 remains unclear. Potentially, chronic up‐regulation of IL‐6, even at low levels, may contribute to an increasingly imbalanced immune system, ultimately resulting in clinical manifestations. Here, we found that IL‐6 could significantly increase the expression of RORC2 and Ras‐GTPases, demonstrating that IL‐6 could have a direct effect on CD4+ T‐cell imbalance. Of note, IL‐6 can be produced in the lung by epithelial cells, fibroblasts and infiltrating immune cells.28 Potentially, the IL‐6 concentration in pulmonary infiltration might be significantly higher than that in the serum, but experimental verification is required. Also, whether the serum IL‐6 level is associated with the pulmonary production of IL‐6 should be investigated. Interestingly, mutations in the IL‐6 gene and dysregulation of IL‐6 expression have been associated with the development of multiple pulmonary diseases, including asthma, COPD and lung cancer.28, 29 Interleukin‐6 in the lung not only contributes to Th17 inflammation, but may also act on neutrophils and fibroblasts to increase inflammation and collagen deposition. Future studies may focus on suppressing the IL‐6 pathway to treat PH.
The Ras‐GTPase family of molecules acts downstream of many receptors, such as the T‐cell receptor CD3 complex and the growth factor receptors.26 Activation of upstream pathways transforms the inactive Ras‐GDP to the active Ras‐GTP, which is bound by Raf kinases. A series of downstream phosphorylation events then occur, ultimately leading to the activation of a series of transcription factors that dramatically alter the transcriptome of the cell. The Ras isoforms, including KRAS, NRAS and HRAS, are among the earliest known oncogenes in human cancers due to their capacity to mediate proliferative responses in cells.30 Here, we showed that KRAS and NRAS presented up‐regulated expression in COPD‐PH CD4+ T cells compared with control CD4+ T cells, and participants with high KRAS and NRAS also tended to present higher RORC2/FOXP3 ratios, which could be suppressed by the inhibition of Ras‐GTPases. In a murine lung cancer model where the oncogenic K‐RasG12D is expressed at the lung epithelium, accumulation of Th17 cells in the lung tumors was observed.31 The Ras‐GTPase inhibitor FTS suppressed the differentiation of Th17 cells and the expression of Th17‐associated IL‐17 and IL‐22.27 These studies, together with the current one, suggest that Ras signaling might be critical to the development of Th17 cells and the Th17/Treg cell imbalance.
Overall, our study indicated that COPD‐PH patients displayed a number of inflammatory features, such as high Th17/Treg cell ratio, high Ras‐GTPase expression and high IL‐6 expression. The precise cause of PH is still unclear and is likely a multifactorial process that involves both genetic and environmental components and is dependent on the individual medical history. Animal experiments have shown that several of the above features, such as high IL‐6 and low Treg cells, could significantly contribute to the pathogenesis of PH.25, 32 Our investigation has uncovered the underlying connections between these features, that the Th17/Treg cell imbalance was associated with Ras‐GTPase overexpression, and IL‐6, a cytokine slightly elevated in the serum of COPD‐PH patients, could enhance the expression of Ras‐GTPase. Future investigations should examine whether the above process could be stopped and reversed for the treatment of COPD‐PH.
Disclosures
The authors declare no conflict of interest.
References
- 1. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol Mech Dis 2009; 4:435–59. [DOI] [PubMed] [Google Scholar]
- 2. Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducoloń A et al Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 172:189–94. [DOI] [PubMed] [Google Scholar]
- 3. Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J 2008; 32:1371–85. [DOI] [PubMed] [Google Scholar]
- 4. Skjørten I, Hilde JM, Melsom MN, Hansteen V, Steine K, Humerfelt S. Pulmonary artery pressure and Pao 2 in chronic obstructive pulmonary disease. Respir Med 2013; 107:1271–9. [DOI] [PubMed] [Google Scholar]
- 5. Carlsen J, Hasseriis Andersen K, Boesgaard S, Iversen M, Steinbrüchel D, Bøgelund Andersen C. Pulmonary arterial lesions in explanted lungs after transplantation correlate with severity of pulmonary hypertension in chronic obstructive pulmonary disease. J Heart Lung Transplant 2013; 32:347–54. [DOI] [PubMed] [Google Scholar]
- 6. Chaouat A, Savale L, Chouaid C, Tu L, Sztrymf B, Canuet M et al Role for interleukin‐6 in COPD‐related pulmonary hypertension. Chest 2009; 136:678–87. [DOI] [PubMed] [Google Scholar]
- 7. Kimura A, Kishimoto T. IL‐6: regulator of Treg/Th17 balance. Eur J Immunol 2010; 40:1830–5. [DOI] [PubMed] [Google Scholar]
- 8. Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol 2010; 72:495–516. [DOI] [PubMed] [Google Scholar]
- 9. Huang F, Kao CY, Wachi S, Thai P, Ryu J, Wu R. Requirement for both JAK‐mediated PI3K signaling and ACT1/TRAF6/TAK1‐dependent NF‐κB activation by IL‐17A in enhancing cytokine expression in human airway epithelial cells. J Immunol 2007; 179:6504–13. [DOI] [PubMed] [Google Scholar]
- 10. McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C et al Role of IL‐17A, IL‐17F, and the IL‐17 receptor in regulating growth‐related oncogene‐ and granulocyte colony‐stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol 2005; 175:404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laan M, Cui Z‐H, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC et al Neutrophil recruitment by human IL‐17 via C‐X‐C chemokine release in the airways. J Immunol 1999; 162:2347–52. [PubMed] [Google Scholar]
- 12. Van Den Berg A, Kuiper M, Snoek M, Timens W, Postma DS, Jansen HM et al Interleukin‐17 induces hyperresponsive interleukin‐8 and interleukin‐6 production to tumor necrosis factor‐α in structural lung cells. Am J Respir Cell Mol Biol 2005; 33:97–104. [DOI] [PubMed] [Google Scholar]
- 13. Shen F, Gaffen SL. Structure–function relationships in the IL‐17 receptor: implications for signal transduction and therapy. Cytokine 2008; 41:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E et al A critical function for transforming growth factor‐β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH‐17 responses. Nat Immunol 2008; 9:650–7. [DOI] [PubMed] [Google Scholar]
- 15. Manel N, Unutmaz D, Littman DR. The differentiation of human TH‐17 cells requires transforming growth factor‐β and induction of the nuclear receptor RORγt. Nat Immunol 2008; 9:641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harri TJ, Gross JF, Ye H‐R, Xi H, Kortylewsk M, Albesian E et al Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17‐dependent autoimmunity. J Immunol 2007; 179:4313–7. [DOI] [PubMed] [Google Scholar]
- 17. Burgler S, Ouaked N, Bassin C, Basinski TM, Mantel PY, Siegmund K et al Differentiation and functional analysis of human TH17 cells. J Allergy Clin Immunol 2009; 123:588–95. [DOI] [PubMed] [Google Scholar]
- 18. Nakamur K, Kitan A, Strobe W. Cell contact‐dependent immunosuppression by CD4+ CD25+ regulatory T cells is mediated by cell surface‐bound transforming growth factor β . J Exp Med 2001; 194:629–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Das J, Ren G, Zhang L, Roberts AI, Zhao X, Bothwell ALM et al Transforming growth factor β is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med 2009; 206:2407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE et al Generation of pathogenic TH17 cells in the absence of TGF‐β2 signalling. Nature 2010; 467:967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh‐Hora M, Kodama T et al Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 2014; 20:62–8. [DOI] [PubMed] [Google Scholar]
- 22. Koenen HJPM, Smeets RL, Vink PM, Van Rijssen E, Boots AMH, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL‐17 producing cells. Blood 2008; 122:2340–52. [DOI] [PubMed] [Google Scholar]
- 23. Omenetti S, Pizarro TT. The Treg/Th17 axis: a dynamic balance regulated by the gut microbiome. Front Immunol 2015; 6:639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kleinewietfeld M, Hafler DA. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin Immunol 2013; 25:305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin‐6 overexpression induces pulmonary hypertension. Circ Res 2009; 104:236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lapinski PE, King PD. Regulation of Ras signal transduction during T cell development and activation. Am J Clin Exp Immunol 2012; 1:147–53. [PMC free article] [PubMed] [Google Scholar]
- 27. Zayoud M, Marcu‐Malina V, Vax E, Jacob‐Hirsch J, Elad‐Sfadia G, Barshack I et al Ras signaling inhibitors attenuate disease in adjuvant‐induced arthritis via targeting pathogenic antigen‐specific Th17‐type cells. Front Immunol 2017; 8:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rincon M, Irvin CG. Role of IL‐6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci 2012; 8:1281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Silva EM, Mariano VS, Pastrez PRA, Pinto MC, Castro AG, Syrjanen KJ et al High systemic IL‐6 is associated with worse prognosis in patients with non‐small cell lung cancer. PLoS ONE 2017; 12:e0181125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takashima A, Faller DV. Targeting the RAS oncogene. Expert Opin Ther Targets 2013; 17:507–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS et al T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci USA 2014; 111:5664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tamosiunien R, Manouvakhov O, Mesang P, Sait T, Qia J, Sanyal M et al Dominant role for regulatory T cells in protecting females against pulmonary hypertension. Circ Res 2018; 122:1689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]