

In Bacillus subtilis, biofilm formation is triggered in response to environmental and cellular signals. It was proposed that serine limitation acts as a proxy for nutrient status and triggers biofilm formation at the onset of biofilm entry through a novel signaling mechanism caused by global ribosome pausing on selective serine codons. In this study, we reveal that serine levels decrease at the biofilm entry due to catabolite control and a serine shunt mechanism. We also show that levels of five serine tRNA isoacceptors are differentially decreased in stationary phase compared with exponential phase; three isoacceptors recognizing UCN serine codons are reduced much more than the two recognizing AGC and AGU codons. This finding indicates a possible mechanism for selective ribosome pausing.
KEYWORDS: Bacillus subtilis, biofilms, catabolite control, serine, tRNA
ABSTRACT
Biofilm development in Bacillus subtilis is regulated at multiple levels. While a number of known signals that trigger biofilm formation do so through the activation of one or more sensory histidine kinases, it was discovered that biofilm activation is also coordinated by sensing intracellular metabolic signals, including serine starvation. Serine starvation causes ribosomes to pause on specific serine codons, leading to a decrease in the translation rate of sinR, which encodes a master repressor for biofilm matrix genes and ultimately triggers biofilm induction. How serine levels change in different growth stages, how B. subtilis regulates intracellular serine levels, and how serine starvation triggers ribosomes to pause on selective serine codons remain unknown. Here, we show that serine levels decrease as cells enter stationary phase and that unlike most other amino acid biosynthesis genes, expression of serine biosynthesis genes decreases upon the transition into stationary phase. The deletion of the gene for a serine deaminase responsible for converting serine to pyruvate led to a delay in biofilm formation, further supporting the idea that serine levels are a critical intracellular signal for biofilm activation. Finally, we show that levels of all five serine tRNA isoacceptors are decreased in stationary phase compared with exponential phase. However, the three isoacceptors recognizing UCN serine codons are reduced to a much greater extent than the two that recognize AGC and AGU serine codons. Our findings provide evidence for a link between serine homeostasis and biofilm development in B. subtilis.
IMPORTANCE In Bacillus subtilis, biofilm formation is triggered in response to environmental and cellular signals. It was proposed that serine limitation acts as a proxy for nutrient status and triggers biofilm formation at the onset of biofilm entry through a novel signaling mechanism caused by global ribosome pausing on selective serine codons. In this study, we reveal that serine levels decrease at the biofilm entry due to catabolite control and a serine shunt mechanism. We also show that levels of five serine tRNA isoacceptors are differentially decreased in stationary phase compared with exponential phase; three isoacceptors recognizing UCN serine codons are reduced much more than the two recognizing AGC and AGU codons. This finding indicates a possible mechanism for selective ribosome pausing.
INTRODUCTION
Bacteria in the natural environment are often found in surface-attached multicellular communities known as biofilms (1–3). Biofilms are a leading cause of hospital-acquired infections (4). Biofilms also pose problems in the environment and industrial settings. Bacillus subtilis is a Gram-positive, rod-shaped bacterium commonly used as a model system for studies of biofilm formation (5, 6). Under laboratory settings, B. subtilis is capable of forming two different types of biofilms, namely, pellicles, which form at the air-liquid interface, and colony biofilms, which form on solid surfaces (2, 5). B. subtilis is also able to form plant root-associated biofilms when living in the rhizosphere (7–9). The genetic circuitry governing biofilm formation in B. subtilis has been well studied (reviewed in reference 6), but less is known about the signals and signal transduction mechanisms that trigger biofilm formation.
Canonically, extracellular signals activate one or more sensory histidine kinases, which then trigger a phosphorelay, leading to the phosphorylation of Spo0A, a master regulator for cell development in B. subtilis (10–12). Upon phosphorylation, Spo0A∼P activates transcription of the sinI gene (13). sinI encodes an antagonist for the master biofilm repressor SinR (14, 15). SinI binds to SinR and sequesters it off its DNA targets, leading to expression of SinR-repressed genes responsible for production of the biofilm matrix, which include a 15-gene epsA-to-epsO operon and a three-gene tapA-sipW-tasA operon (6). Furthermore, small changes in SinR protein levels are shown to have dramatic effects on the expression of matrix genes (13, 16), primarily due to the ultrasensitivity of the SinI-SinR regulatory module, indicating that its levels need to be tightly regulated within the cell. Metabolic stimuli of biofilm formation have also been identified (16–18). In a previous study, we showed that serine starvation acts as a proxy for nutrient limitation at the onset of stationary phase and triggers biofilm formation in B. subtilis (16). Serine starvation results in decreased translation of sinR due to ribosomes preferentially pausing on the four UCN (N stands for A, G, U, and C) serine codons. These four UCN codons are overly abundant in the sinR gene (16). It remains to be confirmed if serine levels decrease during the transition from exponential- to stationary-phase growth. This also leads to other interesting questions, such as how B. subtilis cells regulate serine levels and how decreased serine levels cause ribosomes to pause on selective serine codons.
The amino acid serine is a centrally important biomolecule, not only as a building block for protein synthesis but also as a precursor of nucleotides, other amino acids (such as cysteine, tryptophan, and glycine), and phospholipids (19). While the biochemical reactions involved in serine metabolism are well known, how bacterial cells regulate serine homeostasis and maintain intracellular serine concentrations remains poorly understood. Serine biosynthesis is intimately linked to central metabolism (Fig. 1A). Glycerate-3-phosphate serves as both a glycolytic intermediate and a precursor to serine. The first, rate-limiting step in serine biosynthesis is the conversion of glycerate-3-phosphate to 3-phosphohydroxypyruvate, catalyzed by SerA (Fig. 1A) (20). The regulation of serA transcription has been elucidated in Escherichia coli. serA is positively regulated by two global metabolic regulators, cAMP receptor protein (CRP) and leucine rich repeat protein (Lrp) (21). CRP, also known as catabolite activator protein (CAP), activates genes responsible for secondary carbon source utilization in E. coli in a cAMP-dependent fashion (22, 23). Lrp responds to the amino acid leucine and is activated under leucine starvation (24). This indicates that under nutrient starvation, transcription of serA is activated in E. coli. In B. subtilis, regulation of serA on a genetic level remains unclear. Different from E. coli, in B. subtilis, carbon metabolism is chiefly regulated in a cAMP-independent manner by catabolite control protein A (CcpA) (25, 26). A link between carbon metabolism and biofilm formation has been investigated in several previous studies (27, 28). Biofilm formation is triggered when cells are starved for glucose and dependent on catabolite control. Deletion of the ccpA gene increases biofilm formation (27). This indicates that CcpA negatively regulates biofilm formation in B. subtilis (27). Another global metabolic regulator CodY is also shown to be involved in biofilm formation in B. subtilis (29). In both cases, the underlying mechanisms remain unknown.
FIG 1.

Supernatants of B. subtilis cells support growth of a serine auxotroph. (A) Serine metabolism is linked to central metabolism through glycerate-3-phosphate (G-3-P). G-3-P is a precursor for serine biosynthesis through the activities of SerA, SerC, and SerB. SerA carries the rate-limiting step in serine biosynthesis. The SdaA deaminase complex is involved in converting serine back to G-3-P. (B) Microbiological assays were used to assess serine levels in the supernatant. Supernatants from three different strains (WT, JG08 [serA overexpression], and JG59 [ΔsdaAA]) grown in MSgg medium were prepared during exponential phase (OD600, ∼0.6) and early stationary phase (OD600, ∼1.4) to mimic biofilm entry. Auxotroph cultures, ΔserA (YC913), were washed and subcultured for growth into the supernatants and then plated for CFU counts. Adjusted fold CFU was defined as fold changes of CFU and calculated by comparing the CFU of the serine auxotroph strain after growth in the supplemented supernatant to the CFU of the initial inoculum. As shown, exponential-phase supernatant from cells with serA overexpression (serA++, JG08) was able to support growth about 10 times better than stationary-phase supernatant (****, P ≤ 0.0001). Also, adding supernatant from significantly increased population growth during exponential phase compared with stationary phase from cells with a deleted sdaAA gene (JG59) (*, P ≤ 0.05). Supernatant did not show a significant decrease in cell growth from wild-type supernatant in stationary compared with exponential stage (ns, P > 0.05). Exponential-phase supernatants of both JG08 and JG59 supported much higher growth of the auxotroph than the exponential-phase supernatant of the wild type (****, P ≤ 0.0001), indicating the effectiveness of genetic manipulation in increasing cellular serine accumulation. Dots represent individual data points. Error bars represent standard deviations. (C) Standard curve of ΔserA strain (YC913) in MSgg growth medium supplemented with different concentrations of serine. After 24 h of incubation, YC913 cells were plated, and CFU were counted the next day. CFU count was adjusted to the CFU of initial inoculum to get the adjusted fold CFU per milliliter. The best-fit curve has equation y = 397.40x + 40.72 (R = 0.9626) and can be used to better estimate the concentration of serine in the harvested supernatant. Error bars represent standard deviations.
In bacteria, serine levels are also affected by a single enzymatic reaction, which converts serine to pyruvate and ammonia (Fig. 1A) (30). This reaction is carried out by a serine deaminase (also known as serine dehydratase) (30, 31). Serine deamination is clearly important because in E. coli there are three serine deaminases and at least one of the three serine deaminases is active under any given growth condition (32). In B. subtilis, only one serine deaminase has been identified (33). This two-subunit protein complex is encoded by the sdaAA and sdaAB genes (33). The metabolic benefits(s) of serine deamination has not been fully investigated and remains largely unclear. Previous studies have shown that serine is depleted far faster than other amino acids in E. coli upon nutrient limitation or when cells enter stationary phase (34–36). Presumably, serine is converted to pyruvate and used for central metabolism, energy generation, and even gluconeogenesis. This can be considered a shunt pathway for serine from being used as a building block for protein synthesis. It is unclear how B. subtilis cells would regulate serine homeostasis to ensure an adequate balance between carbon flow into amino acid biosynthesis and central metabolism.
In this study, we set out to elucidate the mechanism by which serine depletion triggers biofilm formation and how cells regulate intracellular serine levels in B. subtilis. We found that serine levels decreased in stationary phase relative to exponential phase and that expression of serine biosynthesis genes declined as cells enter stationary phase. The key serine biosynthetic gene serA was regulated in an indirect manner by catabolite control in B. subtilis. But unlike in E. coli, serA in B. subtilis appeared to be expressed when nutrients are plentiful. In addition to a decrease in serine levels, the abundance of serine tRNAs (a total of 5 isoacceptors) that recognize six synonymous serine codons (UCA, UCC, UCG, UCU, AGC, and AGU) also decreased in stationary phase compared with exponential phase, but the decrease differed significantly for different isoacceptors.
RESULTS
Serine levels decrease upon entry into stationary phase.
We previously showed that early-stationary-phase conditions (biofilm entry) or induced serine starvation in the exponential phase led to ribosome pausing on selective serine codons and reduced translation of sinR (16). Thus, we proposed that serine starvation occurred in early stationary phase and acted as a trigger for biofilm induction. It remained to be confirmed if serine levels were decreasing upon cells entering stationary phase under normal conditions. We initially applied biochemical approaches (primarily high-pressure liquid chromatography [HPLC] and mass spectrometry [MS]/spectrometry) to measure concentrations of free intracellular amino acids, but experienced an inconsistency of the results in both experimental and control samples. We then decided to develop a novel microbiological assay for the measurement (see Materials and Methods). Briefly, wild-type B. subtilis cells capable of synthesizing serine were grown in a minimal medium (MSgg) without exogenous serine. Supernatant from either the exponential- (optical density at 600 nm [OD600], 0.6) or early stationary-phase (OD600, 1.4) culture was prepared as described in the Materials and Methods and then added to the fresh minimal medium inoculated with a B. subtilis serine auxotroph strain (ΔserA) (16). ΔserA cells were cultured for 24 h and then plated. The relative growth of the auxotroph was calculated by comparing the final CFU to the initial inoculum. We also engineered two B. subtilis strains predicted to accumulate higher levels of serine (JG08 for serA overexpression or serA++ and JG59 for ΔsdaAA). We collected supernatants from those cultures accordingly and tested growth support of the serine auxotroph by those supernatants. As shown in Fig. 1B, supernatant from exponentially growing wild-type cells was able to support more than four times higher growth of the auxotroph strain than the stationary-phase supernatant (as calculated by CFU) (Fig. 1B). The exponential-phase supernatant of JG08 (serA++) was able to support even higher growth of the auxotroph (about 10 times) than the supernatant from the stationary-phase culture (P < 0.0001) (Fig. 1B). The exponential-phase supernatant from the ΔsdaAA strain (JG59) also demonstrated a significant increase in supporting auxotroph growth compared with supernatant from the stationary phase (P < 0.05) (Fig. 1B).
Compared to auxotroph growth in the media with known concentrations of serine (Fig. 1C), we estimated about 0.82 μg/ml and 0.08 μg/ml serine in the exponential and early stationary phase of the wild-type supernatants, 38.0 μg/ml and 3.65 μg/ml serine in the exponential and early stationary phase of JG08 (serA++) supernatants, and 80.99 μg/ml and 27.85 μg/ml serine in the exponential and early stationary phase of JG59 (ΔsdaAA) supernatants, respectively. Our results supported the note of significantly higher levels of free serine in the supernatant of the exponential-phase culture than the stationary-phase culture. Although we could not directly measure the intracellular serine levels, we infer that intracellular serine levels are likely proportionally higher in the exponential-phase cells than in the stationary culture.
Manipulation of intracellular serine levels alters biofilm formation.
We previously showed that adding exogenous serine to the media delayed biofilm formation by B. subtilis (16). This suggested that manipulation of serine levels might affect the timing of biofilm induction. To understand the physiological implications of changes in intracellular serine levels during growth transition, we decided to investigate if manipulation of the serine biosynthesis or shunt pathway would affect biofilm formation. As described above (Fig. 1A), two enzymatic pathways play key roles in serine homeostasis in B. subtilis, namely, de novo serine biosynthesis primarily from pyruvate mediated by SerA/SerB/SerC and deamination of serine back to pyruvate, mediated by the serine deaminase complex SdaAA/SdaAB. Based on the results of microbiological assays (Fig. 1B), we already observed that supernatants from the two engineered strains (JG08 and JG59) supported substantially higher serine auxotroph cell growth than that of the wild type (P < 0.0001 for both JG08 and JG59), indicating that both genetic manipulations successfully allowed increased serine accumulation.
Next, we investigated how the genetic manipulations could affect biofilm formation by performing biofilm assays for the two engineered strains. We observed that deletion of the sdaAA gene severely delayed biofilm formation. The ΔsdaAA mutant formed a very weak pellicle biofilm at 24 and 48 h, and only at 72 h, the pellicle biofilm of the mutant showed up to a level comparable to the wild type (Fig. 2A). By using a β-galactosidase reporter assay, we further determined that at both 24 and 48 h, there was a significant decrease in the expression of the biofilm matrix gene tapA in the ΔsdaAA mutant relative to the wild type (Fig. 2B). On the other hand, overexpression of serA only mildly impacted biofilm formation with a modest delay at 48 h (Fig. 2A), and changes in tapA expression were also insignificant compared with the wild type (Fig. 2B). It is possible that the increase in serine levels by serA overexpression is still below a threshold to significantly alter the timing of biofilm induction. Consistent with that idea, in the previous assay (Fig. 1B) we showed that the stationary-phase supernatant from a serA overexpression strain supported much more serine autotroph growth than the wild type (WT), but to a lesser degree than the ΔsdaAA strain (Fig. 2B). We did not test how ΔserA could impact biofilm formation simply because the growth of the auxotroph strain in MSgg required supplementation of large amounts of exogenous serine. Overall, our results supported the idea that alteration of serine levels by genetic manipulation impacted biofilm formation. However, we would not rule out other possibilities. In the case of ΔsdaAA, it is also possible that increased levels of pyruvate instead of decreasing serine levels contribute to the biofilm phenotype.
FIG 2.

Inability to convert serine into pyruvate delays biofilm formation. (A) The ΔsdaAA strain (JG08) shows a weaker biofilm robustness compared to wild type (3610) at 48 hours but not at 72 hours in MSgg, indicating a delay in biofilm formation. Scale bar, 5 mm. (B) PtapA-lacZ expression from pellicles grown in MSgg is significantly lower in the ΔsdaAA strain (JG101) at 24 and 48 hours than that in the wild type (YC755) (***, P < 0.001). Dots represent individual data points. Error bars represent standard deviations.
serA expression decreases upon entry into stationary phase.
Decreasing serine levels upon cells entering stationary phase could be due to the regulation of serine homeostasis. Amino acid biosynthesis genes are normally upregulated upon entry into stationary phase, as that is when cells resume de novo amino acid biosynthesis (37). This is the case for serA in E. coli, which is under catabolite control (21). To investigate if the regulation of serA is the same in B. subtilis as in E. coli, we constructed a PserA-lacZ reporter fusion and tested PserA expression over time. As shown in Fig. 3A and 4A, in wild-type cells, serA transcription initially increased until peaking at around 3 to 4 hours after inoculation, depending on the initial inoculum. Following this increase, serA transcription began to decrease. The peak of serA expression corresponded with the timing when cells began to transition from exponential phase to early stationary phase (OD600, ≈1.0) (Fig. 4A). The timing also corresponded to when the induction of biofilm genes occurred, based on previous studies (38, 39). A decrease in serA expression could contribute to the decrease in serine levels. More importantly, it indicates that in B. subtilis, serine biosynthesis is regulated in an unusual manner relative to biosynthesis of other amino acids. A decrease in serA expression upon entry into stationary phase in B. subtilis was also shown by transcription profiling in a published study (40).
FIG 3.

Expression of serine pathway in relation to biofilm gene expression. (A) The deletion of sinR allows cells to constitutively turn on biofilm genes (43), and to prevent clumping, epsH, a transferase gene involved in building the biofilm matrix, was also deleted (44). Beta-galactosidase assay quantification of PserA expression in wild-type cells (JG106) compared with the ΔsinR ΔepsH double mutant (ABR167) under shaking conditions in MSgg. The wild type had peak expression at 4 h, while the mutant peaked at 5 h, indicating a slight delay in expression decline. Error bars represent standard deviations. There is no significant difference in expression except at 5 and 7 h after inoculation (**, P < 0.01; ***, P < 0.001). The lack of significant difference (P > 0.05) in other time points further suggests that expression of serA is regulated independent of biofilm formation. (B) Dual reporter assay of PserA-GFP (green) and PtapA-mkate2 (76) in wild-type cells (JG157) during growth in MSgg. PserA expression was constitutively on at 48 hours but decreased significantly when cells entered mature biofilm stage (96 hours). The decrease was accompanied by the continued turn on of matrix gene expression (PtapA-mkate2). Scale bar, 10 μm. (C) Beta-galactosidase assay quantification of PsdaA expression in wild-type cells (ABR176) under shaking conditions in MSgg. Low overall expression and decreasing expression as entering biofilm stage. Error bars represent standard deviations.
FIG 4.

PserA transcription is regulated by CcpA but not intracellular serine levels. (A) Expression of serA in wild-type cells initially increased during exponential phase and then began to decrease at approximately OD600 of 1.0; the arrow indicates the time point of OD600 around 1.0. Deletion of ccpA led to constitutively low expression of serA. Error bars represent standard deviations. (B) Confirmation of the β-galactosidase assay with qRT-PCR. At all time points tested, serA mRNA levels were significantly lower than in the ΔccpA mutant (**, P ≤ 0.01; ***, P ≤ 0.001). Dots represent individual data points. Error bars represent standard deviations.
Because the timing of decrease in serA expression also coincided with the timing when induction of biofilm genes occurred, based on previous studies (38, 39), we wanted to further investigate a possible correlation between decreasing serA expression and biofilm gene induction. To do this, we constructed a dual fluorescent reporter strain. We fused the regulatory region of serA to the gfp gene and the regulatory region of the biofilm gene tapA to mKate2, which encodes a red fluorescent protein. We then tracked the expression of the two reporters in the engineered cell population over time during biofilm development. As shown in Fig. 3B, most cells were expressing serA at 48 hours (cells false-colored in purple), while only a small proportion of cells were expressing tapA (cells false-colored in cyan). The bimodal pattern of tapA expression was reported previously by several published studies (38, 41, 42). After 96 hours, cells, many of which were in the form of bundles and clumps due to matrix production, were still strongly expressing tapA, while serA expression was largely diminishing (Fig. 3B). Our results seemed to support the correlation between a decrease in serA expression and induction of biofilm genes, although it was not yet clear if decreasing serA expression proceeded the biofilm gene induction.
On the other hand, it was also possible that the biofilm pathway impacting serine homeostasis and decreasing serA expression were a result of biofilm activation. To test this possibility, we “locked” the cells into the “biofilm on” state by deleting the sinR gene for the biofilm master repressor (43). This allowed matrix genes to be constitutively expressed for biofilm formation and resulted in hyperrobust biofilms (43). The epsH gene, which encodes a sugar transferase for exopolysaccharide (EPS) biosynthesis, was also deleted to prevent cell clumping due to excess matrix production and facilitate sample collection during bioassays (44). As shown in Fig. 3A, there was no difference in serA expression up until the transition point (hour 4). serA expression was then decreased in the wild-type cells while the “biofilm on” cells (ΔepsH ΔsinR) did not show decrease in serA expression until an hour later (Fig. 3A). This result suggests that the biofilm pathway does not impact the expression of the serine biosynthetic gene serA, and it is less likely that decreasing serine levels is due to activation of the biofilm pathway. Lastly, we wanted to examine the expression of the serine deaminase genes (sdaAB-sdaAA). If serine levels are being depleted, it may be as well because the serine deaminase is activated to convert existing serine back to pyruvate for metabolism. We tested this by constructing a lacZ fusion to the promoter of the sdaA operon (sdaAB-sdaAA) PsdaAB-lacZ. Cells were grown in MSgg, and expression of the reporter was measured over time. We found that overall sdaA expression was very low and declining over time (Fig. 3C). This result does not seem to support the note of elevated serine deaminase activity through increased gene expression.
CcpA activates serA expression.
Following the observation of decreased serA expression upon entry into stationary phase, we decided to characterize potential regulators of serA. The regulation of serA is well-studied in E. coli, where serine biosynthesis is under the regulation of catabolite control. serA is activated by both CRP and Lrp (45), indicating that its production increases when cells run out of preferred carbon sources and begin to rely on secondary carbon sources. In B. subtilis, CcpA, the master regulator of catabolite control, is a functional counterpart of CRP in E. coli. While CcpA is commonly thought of as a transcriptional repressor (46), there is evidence of CcpA also serving as an activator of genes in the pathways involved in carbon utilization, such as the acetate metabolism (47) or branched-chain amino acid biosynthesis (leucine, isoleucine, and valine) (37, 48). We examined serA expression in a B. subtilis strain deleted for ccpA and found that deletion of ccpA led to lowered levels of serA expression compared with the wild-type strain (Fig. 4A). Lowered expression of serA in the ccpA mutant was also confirmed using reverse transcription-quantitative PCR (qRT-PCR) (Fig. 4B). These results suggest that CcpA positively regulates serA expression.
We could not identify a direct binding site of CcpA in the serA promoter. This, we speculated that CcpA regulates serA likely through repression of a repressor. In an effort to further elucidate the mechanism of serA regulation by CcpA, we examined two genes in the characterized CcpA regulon both encoding regulatory proteins of unknown function (49). Importantly, these two genes were shown to be upregulated under biofilm conditions (49–51). The first candidate gene ykuL encodes a putative CBS-domain containing protein. ykuL is both repressed by CcpA and activated by Spo0A (51, 52). The second candidate yvnA encodes a transcriptional repressor. yvnA is repressed by both CcpA and AbrB (49, 50). However, as shown in Fig. S1A in the supplemental material, deletion of either gene had no impact on serA expression. Thus, how CcpA activates serA is still unclear. Lastly, deletion of sdaAA also did not significantly affect expression of serA (Fig. S1B), indicating that serine levels did not affect serA transcription via any putative feedback mechanism.
tRNASer isoacceptors are expressed at lower levels in stationary phase than at exponential phase.
In our previous study, we showed that serine starvation led to ribosomes preferentially pausing on the four UCN (N being U, A, C, G) serine codons but not the AGC or AGU serine codon (16). The logical connection between intracellular serine levels and ribosome translation efficiency is the serine tRNAs that add the amino acid to the growing polypeptide chain within the ribosome. B. subtilis encodes a single seryl-tRNA synthetase to carry out the charging of all five serine tRNA isoacceptors (33). The five isoacceptors are clustered into two subgroups, with one group (trnB-ser2 and trnS-ser) recognizing AGC and AGU codons and the other (trnB-ser1, trnD-ser, and trnSL-ser1) recognizing the four UCN codons (Fig. 5). Given that there is only one synthetase for serine tRNA charging, we reasoned that differential activity/expression of tRNA isoacceptors could play a role in mediating the codon-specific ribosome pausing upon serine starvation previously observed (16). By using qRT-PCR, we examined levels of the five tRNASer isoacceptors in both exponential-phase and stationary-phase cells. We observed that the levels of all five isoacceptors were decreased in stationary phase compared with those in exponential phase (decrease ranged from ∼37% to ∼3%, stationary versus exponential), relative to the internal control of 16S rRNA (Fig. 5). More importantly, our results also showed that isoacceptors recognizing the UCN serine codons (trnB-ser1, trnD-ser, and trnSL-ser1) were expressed at far lower levels in stationary phase than those recognizing AGY codons (trnB-ser2 and trnS-ser) (Fig. 5). This significantly differentiated decrease in the levels of the tRNASer isoacceptors recognizing the four UCU serine codons and of those recognizing the AGC and AGU serine codons could contribute to the preferential pausing of ribosomes on the UCN serine codons under serine starvation that we observed previously (16).
FIG 5.
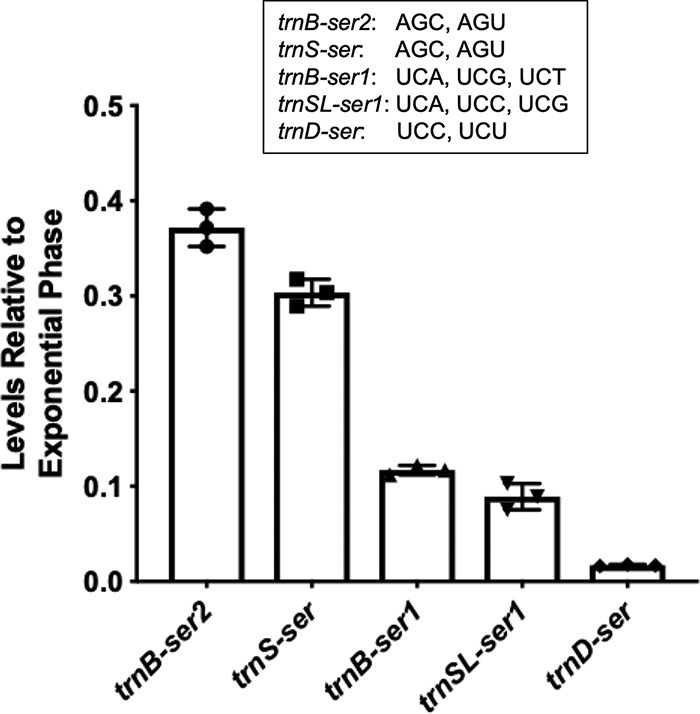
tRNASer isoacceptor transcription levels are decreased in stationary phase relative to exponential phase. tRNASer isoaccepter gene expression was quantified by qRT-PCR on cells grown to exponential and stationary phase in MSgg medium. There was a significant reduction in expression levels all five tRNASer isoacceptor genes in stationary phase relative to exponential phase. The three that recognize UCN codons (trnB-ser1, trnSL-ser1, and trnD-ser) were found to have expression levels lower than those that recognize AGY codons. All serine codons recognized by each of the five tRNASer isoacceptors are listed. Each individual symbol represents an individual data point. Error bars represent the 95% confidence interval.
Overexpression of individual tRNASer genes does not impact biofilm formation.
Following the above reasoning, we further argued that if ribosome pausing on selective serine codons was due to differentially lower production of individual tRNASer isoacceptors upon serine starvation, it was then possible to be overcome by overexpressing corresponding tRNASer isoacceptors. As a prior example, it was shown that in E. coli, amino acid starvation led to differential charging of leucine tRNA isoacceptors, which was overcome by overexpressing the leucine tRNAs that were charged at lower levels during starvation (53). To test if our argument was valid, we used a genetic approach and overexpressed each of the five serine tRNA genes from an alternative chromosomal locus in the wild-type strain. We then tested its effect on biofilm matrix gene expression. However, our results showed that overexpression of the individual serine tRNAs did not impact matrix gene expression (see Fig. S2A in the supplemental material). However, it is possible that the induction is not strong enough to be physiologically relevant, as tRNAs in general are expressed at high levels and quite stable in the cells (54, 55). Note that in the E. coli study, leucine tRNA genes were overexpressed using a high copy number plasmid system (56).
We previously showed that synonymously mutating the AGC and AGT serine codons in the sinR gene to any of the four TCN codons dramatically increased the impact of serine starvation on sinR translation and increased expression of the matrix operon (16). We speculated that strains with those synonymous substitutions in the sinR gene might become more sensitive than the wild-type strain to the alteration of specific serine tRNA levels. We repeated serine tRNA gene overexpression in selected synonymous mutants. However, overexpression of individual tRNASer genes in the two engineered strains each carrying a different type of synonymous mutations in sinR (AGY to TCC in Fig. S2B and AGY to TCT in Fig. S2C) did not significantly affect matrix gene expression.
DISCUSSION
We previously showed that serine depletion triggered biofilm induction in B. subtilis due to global ribosome pausing on selective serine codons during translation and specifically a decrease in translation of the master biofilm repressor gene sinR (16). What remained unaddressed was the regulation driving the decrease in intracellular serine levels at the entry of stationary phase and the mechanism of serine starvation-triggered preferential ribosome pausing on the UCN serine codons. In this study, we investigated both how serine depletion occurs in early stationary phase (biofilm entry) and how changes in serine tRNA levels may be playing a role in causing ribosomes to stall selectively on UCN serine codons. By using a novel microbiological assay, we showed that levels of serine decreased when cells enter stationary phase. Our results suggest that serine levels in the cell act as a signal for biofilm formation. Our result also aligns with previous evidence that serine is one of the first amino acids to be exhausted in E. coli (34–36). While using supernatant is an indirect measurement of intracellular levels, the finding that higher levels of auxotroph growth were observed when grown in the supernatant from cells overexpressing serA or lacking sdaAA supports such a positive correlation between intracellular and extracellular serine levels (Fig. 1B).
Serine depletion upon entry into stationary phase is likely due to a decrease in serine biosynthesis and the shunt pathway that converts serine to pyruvate. We showed that the key serine biosynthesis gene serA was regulated by the catabolite repressor CcpA, linking the regulation of serine homeostasis to cell global metabolic status. A previous study presented evidence suggesting that carbon metabolism and serA expression were linked, as levels of serA transcription decreased 22-fold following a minute of glucose starvation in M9 medium (57), but the mechanism was unclear. Here, we also showed that serA was subject to catabolite regulation, being activated by CcpA. This regulation is most likely indirect, due to the lack of a direct binding site of CcpA in the promoter region of serA. While most amino acid biosynthesis genes are upregulated under nutrient starvation, our data indicate that serA actually begins to decrease in expression upon entry into stationary phase. The peak expression of serA happens at approximately the same time as B. subtilis is beginning to run out of its preferred carbon sources and it switches to secondary carbon sources, known as the diauxic shift (58).
A link between glucose metabolism and biofilm formation has been previously identified in B. subtilis (27). Glucose represses biofilm formation in a CcpA-dependent manner (27). Our work fits into this model, adding a connection between a decrease in CcpA activity and an increase in biofilm formation. A lack of glucose would decrease CcpA activity, leading to a decrease in serA expression, which, in turn, decreases serine levels. This drop in serine causes a decrease in SinR protein levels due to ribosomes pausing on the enriched TCN codons in sinR (16). Decreases in free SinR derepresses matrix gene expression, leading to biofilm formation. Regulating biofilm formation in this manner allows B. subtilis to monitor its environment as well as its intracellular metabolic state.
Finally, we showed that all five tRNASer isoacceptors were accumulated at lower levels in stationary phase than in exponential phase. More interestingly, the decrease differed significantly for different isoacceptors in that the three isoacceptors recognizing the UCN serine codons decreased more substantially than the two isoacceptors recognizing the AGC and AGU serine codons. In this study, we primarily focused on the differential expression by using qRT-PCR. One can argue that shutting off transcription of tRNA genes would not be fast enough to reduce tRNA levels due to the stability of the tRNA molecules in the cells (55). One mechanism that may regulate this stability is the various modifications of tRNAs (reviewed in reference 59). Modifications of tRNAs have been well studied in both eukaryotic cells and model bacteria, such as E. coli, but less is known in B. subtilis (reviewed in references 59–61). Only three tRNA modification enzymes have been studied, namely, MiaB, YmcB, and YqeV, which includes two homologs of E. coli (62). In E. coli, MiaB is involved in methylthiolation of selected tRNAs (63). In B. subtilis, the gene that encodes YmcB is in an operon with ymcA, a regulatory gene known to be essential for biofilm formation (43, 64, 65). In contrast to ymcA, the ymcB gene is not important for biofilm formation (Y. Chai, unpublished data). YmcB is a methylthiotransferase (MTT), which modifies an already present tRNA modification (62). It is possible that the primary modification is critical for regulating the degradation of tRNAs upon nutrient starvation.
In addition to gene expression, the decrease in serine tRNA levels may also be due to a lack of charging. Recent work in E. coli suggests that tRNAs become unstable during amino acid starvation (66). Using strains auxotrophic for specific amino acids, it was shown that upon induction of starvation, levels of tRNAs decreased. This was noted for the specific amino acid cells that were starved for as well as other amino acids. It was speculated that a reduction in charging (due to amino acid starvation) leads to decreased translation, which, in turn, triggers a degradation of excess tRNA, regardless of its acylation state (66). Our data fit into this hypothesis, as the decrease in serine tRNA expression levels coincides with a decrease in serine levels.
MATERIALS AND METHODS
Strains, media, and growth conditions.
For general purposes, Bacillus subtilis strains PY79 (67) and NCBI3610 (hereafter 3610) (5) and their derivatives were grown at 37°C in lysogeny broth (LB) (10 g tryptone, 5 g yeast extract, and 10 g NaCl per liter broth) or on solid LB medium supplemented with 1.5% (wt/vol) agar. For assays of biofilm formation, LBGM and MSgg media were used. LBGM is composed of LB with supplementation of 1% (vol/vol) glycerol and 100 μM MnSO4 (68). The MSgg medium is as previously described (5). Escherichia coli DH5α was used as a host for molecular cloning and grown at 37°C in LB medium. When required, antibiotics were added at the following concentrations for growth of B. subtilis: 1 μg/ml of erythromycin, 5 μg/ml of tetracycline, 5 μg/ml of chloramphenicol, 100 μg/ml of spectinomycin, and 10 μg/ml of kanamycin. For growth of E. coli, 100 μg/ml of ampicillin was used. A list of strains used in this study is summarized in Table 1.
TABLE 1.
Strains used in this study
| Strain | Detailsa | Reference or source |
|---|---|---|
| DH5α | E. coli strain for molecular cloning | Invitrogen |
| PY79 | Laboratory strain of B. subtilis used for genetic manipulation | 67 |
| 168 | Domesticated strain of B. subtilis | 77 |
| 3610 | Undomesticated strain B. subtilis capable of robust biofilm formation | 5 |
| YC755 | Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in 3610 | 16 |
| YC913 | ΔserA::Tetr in 3610 | 16 |
| YC1173 | sinRΩKanr at native locus, Ser32 AGC>TCA, Ser76 AGT>TCA in sinR in 3610 | 16 |
| YC1174 | sinRΩKanr at native locus, Ser32 AGC>TCC, Ser76 AGT>TCC in sinR in 3610 | 16 |
| YC1175 | sinRΩKanr at native locus, Ser32 AGC>TCG, Ser76 AGT>TCG in sinR in 3610 | 16 |
| YC1176 | sinRΩKanr at native locus, Ser32 AGC>TCT, Ser76 AGT>TCT in sinR in 3610 | 16 |
| YCN095 | sacA::PtapA-mkate2 Kanr in 3610 | 74 |
| JG8 | ΔsdaAA::Mlsr in 3610 | This study |
| JG59 | amyE::Physpank-serA Specr in 3610 | This study |
| JG87 | Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG88 | Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG89 | Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG90 | Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG91 | amyE::Physpank-trnD-ser Specr in 3610 | This study |
| JG92 | amyE::Physpank-trnS-ser Specr in 3610 | This study |
| JG93 | amyE::Physpank-B-ser2 Specr in 3610 | This study |
| JG96 | amyE::Physpank-SL-ser1 Specr in 3610 | This study |
| JG97 | amyE::Physpank-B-ser1 Specr in 3610 | This study |
| JG100 | Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in JG59 | This study |
| JG101 | ΔsdaAA::Mlsr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in 3610 | This study |
| JG105 | amyE::PserA-lacZ Cmr in PY79 | This study |
| JG106 | amyE::PserA-lacZ Cmr in 3610 | This study |
| JG111 | amyE::Physpank-trnD-ser Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG112 | amyE::Physpank-trnD-ser Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1174 | This study |
| JG113 | amyE::Physpank-trnD-ser Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1175 | This study |
| JG114 | amyE::Physpank-trnD-ser Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1176 | This study |
| JG115 | amyE::Physpank-trnB-ser1 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1176 | This study |
| JG116 | amyE::Physpank-trnB-ser2 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1176 | This study |
| JG117 | amyE::Physpank-trnS-ser Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1176 | This study |
| JG118 | amyE::Physpank-trnSL-ser1 Specr Tn917::140°(ylnF/yloA)::PtapA-lacZ Cmr in YC1176 | This study |
| JG119 | amyE::Physpank-trnB-ser1 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG120 | amyE::Physpank-trnB-ser2 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG121 | amyE::Physpank-trnS-ser Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG122 | amyE::Physpank-trnSL-ser1 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1173 | This study |
| JG123 | amyE::Physpank-trnB-ser1 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1174 | This study |
| JG125 | amyE::Physpank-trnS-ser Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1174 | This study |
| JG126 | amyE::Physpank-trnSL-ser1 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1174 | This study |
| JG127 | amyE::Physpank-trnB-ser2 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1175 | This study |
| JG128 | amyE::Physpank-trnS-ser Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1175 | This study |
| JG129 | amyE::Physpank-trnSL-ser1 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1175 | This study |
| JG130 | amyE::Physpank-trnB-ser1 Specr Tn917::140°(ylnF/yloA)ΩamyE::PtapA-lacZ Cmr in YC1175 | This study |
| JG138 | ΔccpA::Mlsr in JG106 | This study |
| JG147 | ΔykuLA::Mlsr in JG106 | This study |
| JG148 | ΔyvnA::Mlsr in JG106 | This study |
| JG156 | amyE::PserA-gfp Cmr in 3610 | This study |
| JG157 | amyE::PserA-gfp Cmr sacA::PtapA-mKate2 Kanr in 3610 | This study |
| ABR167 | ΔepsH::Tetr ΔsinR::kan amyE::PserA-lacZ Cmr in 3610 | This study |
| ABR176 | amyE::PsdaAB-lacZ Specr in 3610 | This study |
140°(ylnF/yloA)ΩamyE::PtapA-lacZ, the PtapA-lacZ fusion was introduced into a locus between the ylnF and yloA genes at 140° on the chromosome; Cmr, chloramphenicol resistance; Kanr, kanamycin resistance; Mlsr, erythromycin resistance; Specr, spectinomycin resistance; Tetr, tetracycline resistance.
DNA manipulation.
General methods for molecular cloning followed the published protocols (69). Restriction enzymes (New England BioLabs) were used according to the manufacturer’s instructions. Transformation of plasmid DNA into B. subtilis strains was performed as described previously (70). SPP1 phage-mediated general transduction was also used to transfer antibiotic-marked DNA fragments among different strains (71). Plasmids used in this study are listed in Table 2 and oligonucleotides (purchased from Eurofins) are listed in Table S1 in the supplemental material.
TABLE 2.
Plasmids used in this study
| Plasmid | Detailsa | Reference or source |
|---|---|---|
| pDG268 | An amyE integration vector with a promoterless lacZ, Ampr Cmr | BGSC |
| pDG1728 | An amyE integration vector with a promoterless lacZ, Ampr Specr | BGSC |
| pDR111 | An amyE integration vector that contains Physpank, Ampr Specr | BGSC |
| pYC121 | An amyE integration vector with a promoterless gfp-mut2, Ampr Specr | 38 |
| pJG01 | amyE::Physpank-serA in pDR111, Ampr Specr | This study |
| pJG02 | amyE::PserA-lacZ in pDG268, Ampr Cmr | This study |
| pJG03 | amyE::Physpank-trnB-ser1 in pDR111, Ampr Specr | This study |
| pJG04 | amyE::Physpank-trnB-ser2 in pDR111, Ampr Specr | This study |
| pJG05 | amyE::Physpank-trnS-ser in pDR111, Ampr Specr | This study |
| pJG06 | amyE::Physpank-trnSL-ser1 in pDR111, Ampr Specr | This study |
| pJG07 | amyE::Physpank-trnD-ser in pDR111, Ampr Specr | This study |
| pJG08 | amyE::PserA-gfp Ampr Cmr | This study |
| pABR174 | amyE::PsdaAB-lacZ in pDG1728, Ampr Specr | This study |
Ampr, ampicillin resistance; Cmr, chloramphenicol resistance; Specr, spectinomycin resistance.
Strain construction.
For construction of the insertional deletion mutations of ccpA and sdaAA in 3610, the corresponding mutants in the B. subtilis strain 168 were obtained from the Bacillus Genetic Stock Center (BGSC) at Ohio State University. Antibiotic cassette-marked deletions were introduced into 3610 using SPP1 phage-mediated transduction to generate strains JG138 and JG8 (71).
To overexpress serA in B. subtilis, the serA coding sequence was amplified by PCR using 3610 genomic DNA as the template and primers serA-F and serA-R. The PCR product was then cloned into the HindIII and BamHI sites of pDR111, which contains an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible hyperspank promoter flanked by the amyE gene, generating the recombinant plasmid pJG01 (72). The recombinant plasmid was then introduced into the B. subtilis laboratory strain PY79 by genetic transformation for a double-crossover recombination of the DNA sequences at the amyE locus. The construct was then introduced into 3610 by transformation to generate strain JG59 (71). Overexpression of each of the five tRNASer isoacceptor genes was done in a similar manner, using primers listed in Table S1 to generate strains JG91 through JG97.
To compare the expression of the serA gene in the wild-type and various deletion strains, the promoter sequence of serA was amplified by PCR using primers PserA-F and PserA-R and genomic DNA of 3610 as the template. The PCR products were digested with EcoRI and HindIII and cloned into the plasmid pDG268 (73), which carries a chloramphenicol resistance marker and a polylinker upstream of the lacZ gene between two arms of the amyE gene. The resulting recombinant plasmid pJG02 contains a PserA-lacZ transcriptional fusion. For comparison of tapA expression in wild-type and various engineered strains for serine metabolism, lysate from a strain containing a PtapA-lacZ fusion (YC755) was introduced into strains JG08 and JG59 by SPP1 phage-mediated transduction. The PtapA-lacZ fusion was introduced into a locus between the ylnF and yloA genes at 140° on the chromosome, as previously described (13). This insertion keeps the amyE gene intact, which allows for additional insertions, such as the Physpank-serA or tRNA overexpression constructs, and for measurement of lacZ transcription as well as IPTG induction.
To examine single-cell fluorescence and track both serA expression and biofilm matrix gene expression, a dual reporter strain was constructed. The regulatory sequence of serA was amplified by PCR using primers PserA-F and PserA-R and genomic DNA of B. subtilis strain 3610 as the template. The PCR product was digested with EcoRI and HindIII and cloned into the plasmid pYC121, which has a promoterless gfp gene (gfp-mut2) flanked by the amyE sequence (38). This resulting plasmid pJG08 was then introduced into B. subtilis PY79 by genetic transformation for a double-crossover recombination at the amyE locus. The reporter fusion was then moved into 3610 by transduction (71), generating strain JG156. To construct the dual-reporter strain, the PserA-gfp reporter was introduced to YCN095, which contains a PtapA-mKate2 reporter fusion integrated at sacA (74), generating strain JG157.
To measure sdaAA expression, a 150-bp promoter region (PsdaAB) was amplified using primers PsdaAB-F and PsdaAB-R and 3610 genomic DNA as the template. The PCR product was digested with BamHI and HindIII restriction enzymes and inserted onto the pDG1728 vector backbone to generate plasmid pABR174. pABR174 was then transformed into B. subtilis PY79 and further into 3610 to generate strain ABR176 (71).
Serine auxotroph assay.
To harvest supernatant, wild-type 3610 cells or its derivatives were grown overnight on an LB plate at 37°C. Single colonies were then grown overnight in 3 ml of LB medium under shaking conditions at 37°C. Cells were subcultured at 1:100 into MSgg medium. Cultures were collected at OD600 of 0.6 (exponential phase) and OD600 of 1.4 (stationary phase), and cells were removed by centrifugation at 15,000 × g for 10 min. Supernatants were filter sterilized (0.2 μm) to further remove the cells, passed through a mini-sizing column (molecular weight [MW] cutoff, 5,000 Da; Qiagen) to remove large molecular weight macromolecules, such as proteins, and then diluted in MSgg. The final diluted supernatant was equivalent to the spent culture medium of 108 cell per ml. To test auxotroph growth, the ΔserA strain (YC913) was grown overnight on an LB plate at 37°C. Single colonies were grown overnight in 3 ml of LB medium under shaking conditions at 37°C. Cells were subcultured at 1:100 into MSgg medium for 6 hours. Cells were washed in fresh MSgg and then diluted to a concentration of approximately 100 CFU/ml in fresh MSgg. One milliliter of cells was added to 1 ml of supernatant. Cells were grown for 24 hours under shaking conditions at 37°C. After 24 hours of incubation, cells were plated and CFU were counted the next day. Resulting CFU were divided by starting CFU to assess relative growth ability. Experiments were performed in triplicate and repeated at least three times for each strain.
For generation of the standard curve, YC913 was grown as described above. However, after 6 h of growth, the culture was diluted to 104 cells per ml. This dilution was inoculated 1:100 into 2-ml culture tubes containing MSgg with various concentrations of serine (Sigma). An initial inoculum was plated for CFU onto LB plates. The remaining cultures grew at 37°C under shaking conditions for 24 h and were then plated for CFU. The calculation of the curves was standardized to the initial inoculum. The standard curve was carried out in triplicate for four independent experiments.
β-Galactosidase activity assay.
For assays of β-galactosidase activity, an established protocol was used (75). Briefly, cells were incubated in a flask of MSgg or LBGM medium at 37°C with shaking. One milliliter of culture was collected at each indicated time point after inoculation. Cells were spun down, and pellets were resuspended in 1 ml Z buffer (40 mM NaH2PO4, 60 mM Na2HPO4, 1 mM MgSO4, 10 mM KCl, and 38 mM β-mercaptoethanol) supplemented with 200 μg/ml freshly made lysozyme. Resuspensions were incubated at 30°C for 15 min. Reactions were started by adding 200 μl of 4 mg/ml o-nitrophenyl-β-d-galactopyranoside (ONPG) and stopped by adding 500 μl of 1 M Na2CO3. Samples were spun down to remove any cell debris. OD420 and OD550 values of the samples were recorded using a Bio-Rad SmartSpec 3000 instrument. The β-galactosidase-specific activity was calculated according to the following equation: activity (Miller units) = [OD420 − 1.75(OD550)/(time × volume × OD600)] × 1,000. Experiments were performed in triplicate and repeated at least three times. Statistical analyses were performed by using Student’s t test.
Bioassays on pellicle biofilms.
B. subtilis cells were grown in LB broth at 37°C to mid-log phase. For pellicle formation, 4 μl of the cells was mixed with 4 ml of liquid MSgg medium in 12-well plates (Corning). Plates were incubated at 30°C for up to 72 hours. Images were taken using a Leica MZ10F dissecting scope. For β-galactosidase activity assays on pellicle biofilm samples, pellicle biofilms were set up as described above and collected at 24, 48, or 72 hours. Pellicles were resuspended using the medium in the well and gently sonicated to disrupt cell chains and bundles but not individual cells. One milliliter of the pellicle was spun down and resuspended in 1 ml Z buffer. A total of 100 μl of sample was then diluted into 900 μl Z buffer due to high cell density in the pellicle. The β-galactosidase activity was assayed as described above. The activity was calculated similarly as described above. Experiments were performed in triplicate and repeated at least three times.
Quantitative real-time PCR.
Five hundred microliters of B. subtilis cells was harvested during exponential phase (OD600, ∼0.6) and stationary phase (OD600, ∼1.4) and added to 1 μl of RNAProtect bacteria reagent (Qiagen). After incubation at room temperature for 60 minutes, samples were spun down (15,000 × g for 5 min), and the cell pellet was resuspended in 1 ml of TRIzol reagent (Thermo) and incubated for 5 minutes at room temperature. Total RNA was extracted using the Zymo Direct-zol RNA kit (Zymo) following the manufacturer’s protocol. RNA concentration and purity were measured using a NanoDrop One instrument (Thermo). RNA was converted to cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosciences). Quantitative PCR (qPCR) was performed using the primers listed in Table S3 in the supplemental material using the protocol provided by the manufacturer for the Fast SYBR green master mix (Applied Biosystems) using a StepOnePlus real-time PCR machine (Applied Biosystems). Relative expression was calculated using the comparative threshold cycle (ΔΔCT) for each sample. Technical triplicates were run for each sample to calculate the ΔΔCT and biological triplicates were run for each strain, and the experiment was repeated three times, for a total of nine biological replicates. The relative expression was standardized using the endogenous 16S control with the exponential-phase samples used as the reference sample. A one-way analysis of variance (ANOVA) was used to assess statistical significance (P < 0.05).
Dual reporter assay.
For the dual reporter assay, the strain JG157 containing the PserA-gfp and PtapA-mKate2 fluorescent reporters was first grown in LB broth overnight. On the next day, pellicle formation was performed by mixing 4 μl of the overnight cells with 4 ml of liquid MSgg medium in 12-well plates (Corning). Plates were incubated at 30°C for up to 96 hours. Cells were collected at different time points for microscopic observation. To visualize cells under the microscope, 1 ml of cells was harvested, washed with 1 ml of phosphate-buffered saline (PBS) buffer to remove residual MSgg medium, and then concentrated in 100 μl of PBS buffer. A volume of 2 μl of cells was added to a 1% agarose pad (wt/vol) and covered with a coverslip. Cells were imaged using a Leica DFC3000 G camera on a Leica AF6000 microscope. Images of samples collected from different time points were taken using the same exposure settings. For GFP observation, the setting of the excitation wavelength was 450 to 490 nm, while the setting of the emission wavelength was 500 to 550 nm. For mKate2 observation, the excitation wavelength setting was at 540 to 580 nm and the emission wavelength setting was at 610 to 680 nm. The experiment was conducted in triplicate, and images taken were representatives of the three replicates.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Chai and Godoy labs for helpful discussions. We thank Leticia Lima Angelini, Christina Potter, Rachel Son, and Philip Wasson for technical assistance.
Funding for this work was provided by Northeastern University and a grant from the National Science Foundation to Y. Chai (MCB1651732).
J.G., A.R., K.G., V.G.-C., and Y.C. designed the experiments. J.G., A.R., and G.D.C. performed the experiments. T.T. provided substantial technical help. J.G., A.R., and Y.C. wrote the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00155-19.
REFERENCES
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.O'Toole G, Kaplan HB, Kolter R. 2000. Biofilm formation as microbial development. Annu Rev Microbiol 54:49–79. doi: 10.1146/annurev.micro.54.1.49. [DOI] [PubMed] [Google Scholar]
- 3.Stoodley P, Sauer K, Davies DG, Costerton JW. 2002. Biofilms as complex differentiated communities. Annu Rev Microbiol 56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- 4.Hall-Stoodley L, Stoodley P. 2009. Evolving concepts in biofilm infections. Cell Microbiol 11:1034–1043. doi: 10.1111/j.1462-5822.2009.01323.x. [DOI] [PubMed] [Google Scholar]
- 5.Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. 2001. Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci U S A 98:11621–11626. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vlamakis H, Chai Y, Beauregard P, Losick R, Kolter R. 2013. Sticking together: building a biofilm the Bacillus subtilis way. Nat Rev Microbiol 11:157–168. doi: 10.1038/nrmicro2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Cao S, Chai Y, Clardy J, Kolter R, Guo JH, Losick R. 2012. A Bacillus subtilis sensor kinase involved in triggering biofilm formation on the roots of tomato plants. Mol Microbiol 85:418–430. doi: 10.1111/j.1365-2958.2012.08109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Yan F, Chai Y, Liu H, Kolter R, Losick R, Guo JH. 2013. Biocontrol of tomato wilt disease by Bacillus subtilis isolates from natural environments depends on conserved genes mediating biofilm formation. Environ Microbiol 15:848–864. doi: 10.1111/j.1462-2920.2012.02860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beauregard PB, Chai Y, Vlamakis H, Losick R, Kolter R. 2013. Bacillus subtilis biofilm induction by plant polysaccharides. Proc Natl Acad Sci U S A 110:E1621–E1630. doi: 10.1073/pnas.1218984110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLoon AL, Kolodkin-Gal I, Rubinstein SM, Kolter R, Losick R. 2011. Spatial regulation of histidine kinases governing biofilm formation in Bacillus subtilis. J Bacteriol 193:679–685. doi: 10.1128/JB.01186-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamon MA, Lazazzera BA. 2001. The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Mol Microbiol 42:1199–1209. [DOI] [PubMed] [Google Scholar]
- 12.Burbulys D, Trach KA, Hoch JA. 1991. Initiation of sporulation in Bacillus subtilis is controlled by a multicomponent phosphorelay. Cell 64:545–552. doi: 10.1016/0092-8674(91)90238-T. [DOI] [PubMed] [Google Scholar]
- 13.Chai Y, Norman T, Kolter R, Losick R. 2011. Evidence that metabolism and chromosome copy number control mutually exclusive cell fates in Bacillus subtilis. EMBO J 30:1402–1413. doi: 10.1038/emboj.2011.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bai U, Mandic-Mulec I, Smith I. 1993. SinI modulates the activity of SinR, a developmental switch protein of Bacillus subtilis, by protein-protein interaction. Genes Dev 7:139–148. doi: 10.1101/gad.7.1.139. [DOI] [PubMed] [Google Scholar]
- 15.Newman JA, Rodrigues C, Lewis RJ. 2013. Molecular basis of the activity of SinR protein, the master regulator of biofilm formation in Bacillus subtilis. J Biol Chem 288:10766–10778. doi: 10.1074/jbc.M113.455592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramaniam AR, Deloughery A, Bradshaw N, Chen Y, O'Shea E, Losick R, Chai Y. 2013. A serine sensor for multicellularity in a bacterium. Elife 2:e01501. doi: 10.7554/eLife.01501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Townsley L, Yannarell SM, Huynh TN, Woodward JJ, Shank EA. 2018. Cyclic di-AMP acts as an extracellular signal that impacts Bacillus subtilis biofilm formation and plant attachment. mBio 9:e00341-18. doi: 10.1128/mBio.00341-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Chai Y, Guo JH, Losick R. 2012. Evidence for cyclic Di-GMP-mediated signaling in Bacillus subtilis. J Bacteriol 194:5080–5090. doi: 10.1128/JB.01092-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stauffer G. 2004. Regulation of serine, glycine, and one-carbon biosynthesis. EcoSal Plus doi: 10.1128/ecosalplus.3.6.1.2. [DOI] [PubMed] [Google Scholar]
- 20.Saski R, Pizer LI. 1975. Regulatory properties of purified 3-phosphoglycerate dehydrogenase from Bacillus subtilis. Eur J Biochem 51:415–427. doi: 10.1111/j.1432-1033.1975.tb03941.x. [DOI] [PubMed] [Google Scholar]
- 21.Rex JH, Aronson BD, Somerville RL. 1991. The tdh and serA operons of Escherichia coli: mutational analysis of the regulatory elements of leucine-responsive genes. J Bacteriol 173:5944–5953. doi: 10.1128/jb.173.19.5944-5953.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Crombrugghe B, Busby S, Buc H. 1984. Cyclic AMP receptor protein: role in transcription activation. Science 224:831–838. doi: 10.1126/science.6372090. [DOI] [PubMed] [Google Scholar]
- 23.Taniguchi T, O'Neill M, de Crombrugghe B. 1979. Interaction site of Escherichia coli cyclic AMP receptor protein on DNA of galactose operon promoters. Proc Natl Acad Sci U S A 76:5090–5094. doi: 10.1073/pnas.76.10.5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman EB, D'Ari R, Lin RT. 1992. The leucine-Lrp regulon in E. coli: a global response in search of a raison d'etre. Cell 68:617–619. doi: 10.1016/0092-8674(92)90135-Y. [DOI] [PubMed] [Google Scholar]
- 25.Henkin TM, Grundy FJ, Nicholson WL, Chambliss GH. 1991. Catabolite repression of alpha-amylase gene expression in Bacillus subtilis involves a trans-acting gene product homologous to the Escherichia coli lacl and galR repressors. Mol Microbiol 5:575–584. doi: 10.1111/j.1365-2958.1991.tb00728.x. [DOI] [PubMed] [Google Scholar]
- 26.Gorke B, Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 27.Stanley NR, Britton RA, Grossman AD, Lazazzera BA. 2003. Identification of catabolite repression as a physiological regulator of biofilm formation by Bacillus subtilis by use of DNA microarrays. J Bacteriol 185:1951–1957. doi: 10.1128/jb.185.6.1951-1957.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Gozzi K, Yan F, Chai Y. 2015. Acetic acid acts as a volatile signal to stimulate bacterial biofilm formation. mBio 6:e00392. doi: 10.1128/mBio.00392-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brinsmade SR, Alexander EL, Livny J, Stettner AI, Segrè D, Rhee KY, Sonenshein AL. 2014. Hierarchical expression of genes controlled by the Bacillus subtilis global regulatory protein CodY. Proc Natl Acad Sci U S A 111:8227–8232. doi: 10.1073/pnas.1321308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su HS, Lang BF, Newman EB. 1989. l-serine degradation in Escherichia coli K-12: cloning and sequencing of the sdaA gene. J Bacteriol 171:5095–5102. doi: 10.1128/jb.171.9.5095-5102.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tobey KL, Grant GA. 1986. The nucleotide sequence of the serA gene of Escherichia coli and the amino acid sequence of the encoded protein, D-3-phosphoglycerate dehydrogenase. J Biol Chem 261:12179–12183. [PubMed] [Google Scholar]
- 32.Zhang X, Newman E. 2008. Deficiency in l-serine deaminase results in abnormal growth and cell division of Escherichia coli K-12. Mol Microbiol 69:870–881. doi: 10.1111/j.1365-2958.2008.06315.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhu B, Stulke J. 2018. SubtiWiki in 2018: from genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res 46:D743–D748. doi: 10.1093/nar/gkx908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liebs P, Riedel K, Graba JP, Schrapel D, Tischler U. 1988. Formation of some extracellular enzymes during the exponential growth of Bacillus subtilis. Folia Microbiol (Praha) 33:88–95. doi: 10.1007/BF02928073. [DOI] [PubMed] [Google Scholar]
- 35.Sezonov G, Joseleau-Petit D, D'Ari R. 2007. Escherichia coli physiology in Luria-Bertani broth. J Bacteriol 189:8746–8749. doi: 10.1128/JB.01368-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pruss BM, Nelms JM, Park C, Wolfe AJ. 1994. Mutations in NADH:ubiquinone oxidoreductase of Escherichia coli affect growth on mixed amino acids. J Bacteriol 176:2143–2150. doi: 10.1128/jb.176.8.2143-2150.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molle V, Nakaura Y, Shivers RP, Yamaguchi H, Losick R, Fujita Y, Sonenshein AL. 2003. Additional targets of the Bacillus subtilis global regulator CodY identified by chromatin immunoprecipitation and genome-wide transcript analysis. J Bacteriol 185:1911–1922. doi: 10.1128/jb.185.6.1911-1922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chai Y, Chu F, Kolter R, Losick R. 2008. Bistability and biofilm formation in Bacillus subtilis. Mol Microbiol 67:254–263. doi: 10.1111/j.1365-2958.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu F, Kearns DB, McLoon A, Chai Y, Kolter R, Losick R. 2008. A novel regulatory protein governing biofilm formation in Bacillus subtilis. Mol Microbiol 68:1117–1127. doi: 10.1111/j.1365-2958.2008.06201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLoughery A, Lalanne J-B, Losick R, Li G-W. 2018. Maturation of polycistronic mRNAs by the endoribonuclease RNase Y and its associated Y-complex in Bacillus subtilis. Proc Natl Acad Sci U S A 115:E5585–E5594. doi: 10.1073/pnas.1803283115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vlamakis H, Aguilar C, Losick R, Kolter R. 2008. Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev 22:945–953. doi: 10.1101/gad.1645008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lopez D, Fischbach MA, Chu F, Losick R, Kolter R. 2009. Structurally diverse natural products that cause potassium leakage trigger multicellularity in Bacillus subtilis. Proc Natl Acad Sci U S A 106:280–285. doi: 10.1073/pnas.0810940106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kearns DB, Chu F, Branda SS, Kolter R, Losick R. 2005. A master regulator for biofilm formation by Bacillus subtilis. Mol Microbiol 55:739–749. doi: 10.1111/j.1365-2958.2004.04440.x. [DOI] [PubMed] [Google Scholar]
- 44.Marvasi M, Visscher PT, Casillas Martinez L. 2010. Exopolymeric substances (EPS) from Bacillus subtilis: polymers and genes encoding their synthesis. FEMS Microbiol Lett 313:1–9. doi: 10.1111/j.1574-6968.2010.02085.x. [DOI] [PubMed] [Google Scholar]
- 45.Yang L, Lin RT, Newman EB. 2002. Structure of the Lrp-regulated serA promoter of Escherichia coli K-12. Mol Microbiol 43:323–333. doi: 10.1046/j.1365-2958.2002.02744.x. [DOI] [PubMed] [Google Scholar]
- 46.Tuan LR, D'Ari R, Newman EB. 1990. The leucine regulon of Escherichia coli K-12: a mutation in rblA alters expression of L-leucine-dependent metabolic operons. J Bacteriol 172:4529–4535. doi: 10.1128/jb.172.8.4529-4535.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grundy FJ, Waters DA, Allen SH, Henkin TM. 1993. Regulation of the Bacillus subtilis acetate kinase gene by CcpA. J Bacteriol 175:7348–7355. doi: 10.1128/jb.175.22.7348-7355.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wunsche A, Hammer E, Bartholomae M, Volker U, Burkovski A, Seidel G, Hillen W. 2012. CcpA forms complexes with CodY and RpoA in Bacillus subtilis. FEBS J 279:2201–2214. doi: 10.1111/j.1742-4658.2012.08604.x. [DOI] [PubMed] [Google Scholar]
- 49.Blencke HM, Homuth G, Ludwig H, Mader U, Hecker M, Stulke J. 2003. Transcriptional profiling of gene expression in response to glucose in Bacillus subtilis: regulation of the central metabolic pathways. Metab Eng 5:133–149. doi: 10.1016/S1096-7176(03)00009-0. [DOI] [PubMed] [Google Scholar]
- 50.Chumsakul O, Takahashi H, Oshima T, Hishimoto T, Kanaya S, Ogasawara N, Ishikawa S. 2011. Genome-wide binding profiles of the Bacillus subtilis transition state regulator AbrB and its homolog Abh reveals their interactive role in transcriptional regulation. Nucleic Acids Res 39:414–428. doi: 10.1093/nar/gkq780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molle V, Fujita M, Jensen ST, Eichenberger P, Gonzalez-Pastor JE, Liu JS, Losick R. 2003. The Spo0A regulon of Bacillus subtilis. Mol Microbiol 50:1683–1701. doi: 10.1046/j.1365-2958.2003.03818.x. [DOI] [PubMed] [Google Scholar]
- 52.Blencke HM, Reif I, Commichau FM, Detsch C, Wacker I, Ludwig H, Stulke J. 2006. Regulation of citB expression in Bacillus subtilis: integration of multiple metabolic signals in the citrate pool and by the general nitrogen regulatory system. Arch Microbiol 185:136–146. doi: 10.1007/s00203-005-0078-0. [DOI] [PubMed] [Google Scholar]
- 53.Sorensen MA, Elf J, Bouakaz E, Tenson T, Sanyal S, Bjork GR, Ehrenberg M. 2005. Over expression of a tRNA(Leu) isoacceptor changes charging pattern of leucine tRNAs and reveals new codon reading. J Mol Biol 354:16–24. doi: 10.1016/j.jmb.2005.08.076. [DOI] [PubMed] [Google Scholar]
- 54.Waldron C, Lacroute F. 1975. Effect of growth rate on the amounts of ribosomal and transfer ribonucleic acids in yeast. J Bacteriology 122:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis BD, Luger SM, Tai PC. 1986. Role of ribosome degradation in the death of starved Escherichia coli cells. J Bacteriol 166:439–445. doi: 10.1128/jb.166.2.439-445.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramaniam AR, Pan T, Cluzel P. 2013. Environmental perturbations lift the degeneracy of the genetic code to regulate protein levels in bacteria. Proc Natl Acad Sci U S A 110:2419–2424. doi: 10.1073/pnas.1211077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Jong IG, Veening JW, Kuipers OP. 2012. Single cell analysis of gene expression patterns during carbon starvation in Bacillus subtilis reveals large phenotypic variation. Environ Microbiol 14:3110–3121. doi: 10.1111/j.1462-2920.2012.02892.x. [DOI] [PubMed] [Google Scholar]
- 58.Jacob F, Monod J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol 3:318–356. doi: 10.1016/S0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 59.Shepherd J, Ibba M. 2015. Bacterial transfer RNAs. FEMS Microbiol Rev 39:280–300. doi: 10.1093/femsre/fuv004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan T. 2018. Modifications and functional genomics of human transfer RNA. Cell Res 28:395–404. doi: 10.1038/s41422-018-0013-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maraia RJ, Arimbasseri AG. 2017. Factors that shape eukaryotic tRNAomes: processing, modification and anticodon-codon use. Biomolecules 7:26. doi: 10.3390/biom7010026. [DOI] [Google Scholar]
- 62.Anton BP, Russell SP, Vertrees J, Kasif S, Raleigh EA, Limbach PA, Roberts RJ. 2010. Functional characterization of the YmcB and YqeV tRNA methylthiotransferases of Bacillus subtilis. Nucleic Acids Res 38:6195–6205. doi: 10.1093/nar/gkq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Esberg B, Leung H-C, Tsui H-C, Björk GR, Winkler ME. 1999. Identification of the miaB gene, involved in methylthiolation of isopentenylated A37 derivatives in the tRNA of Salmonella Typhimurium and Escherichia coli. J Bacteriol 181:7256–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Branda SS, González-Pastor JE, Dervyn E, Ehrlich SD, Losick R, Kolter R. 2004. Genes involved in formation of structured multicellular communities by Bacillus subtilis. J Bacteriol 186:3970–3979. doi: 10.1128/JB.186.12.3970-3979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DeLoughery A, Dengler V, Chai Y, Losick R. 2016. Biofilm formation by Bacillus subtilis requires an endoribonuclease-containing multisubunit complex that controls mRNA levels for the matrix gene repressor SinR. Mol Microbiol 99:425–437. doi: 10.1111/mmi.13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Svenningsen SL, Kongstad M, Stenum TS, Muñoz-Gómez AJ, Sørensen MA. 2017. Transfer RNA is highly unstable during early amino acid starvation in Escherichia coli. Nucleic Acids Res 45:793–804. doi: 10.1093/nar/gkw1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schroeder JW, Simmons LA. 2013. Complete genome sequence of Bacillus subtilis strain PY79. Genome Announc 1:e01085-13. doi: 10.1128/genomeA.01085-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shemesh M, Chai Y. 2013. A combination of glycerol and manganese promotes biofilm formation in Bacillus subtilis via histidine kinase KinD signaling. J Bacteriol 195:2747–2754. doi: 10.1128/JB.00028-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sambrook J. 2001. Molecular cloning. a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA. [Google Scholar]
- 70.Gryczan TJ, Contente S, Dubnau D. 1978. Characterization of Staphylococcus aureus plasmids introduced by transformation into Bacillus subtilis. J Bacteriol 134:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yasbin RE, Young FE. 1974. Transduction in Bacillus subtilis by bacteriophage SPP1. J Virol 14:1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chai Y, Kolter R, Losick R. 2009. A widely conserved gene cluster required for lactate utilization in Bacillus subtilis and its involvement in biofilm formation. J Bacteriol 191:2423–2430. doi: 10.1128/JB.01464-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Antoniewski C, Savelli B, Stragier P. 1990. The spoIIJ gene, which regulates early developmental steps in Bacillus subtilis, belongs to a class of environmentally responsive genes. J Bacteriol 172:86–93. doi: 10.1128/jb.172.1.86-93.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gozzi K, Ching C, Paruthiyil S, Zhao Y, Godoy-Carter V, Chai Y. 2017. Bacillus subtilis utilizes the DNA damage response to manage multicellular development. NPJ Biofilms Microbiomes 3:8. doi: 10.1038/s41522-017-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu Y, Yan F, He Y, Qin Y, Chen Y, Chai Y, Guo J-H. 2018. The ClpY-ClpQ protease regulates multicellular development in Bacillus subtilis. Microbiology 164:848–862. doi: 10.1099/mic.0.000658. [DOI] [PubMed] [Google Scholar]
- 76.Huang X, Fredrick KL, Helmann JD. 1998. Promoter recognition by Bacillus subtilis sigmaW: autoregulation and partial overlap with the sigmaX regulon. J Bacteriol 180:3765–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessières P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Cummings NJ, Daniel RA, Denizot F, Devine KM, Düsterhöft A, Ehrlich SD, Emmerson PT, Entian KD, Errington J, Fabret C, Ferrari E, Foulger D, Fritz C, Fujita M, Fujita Y, Fuma S, Galizzi A, Galleron N, Ghim SY, Glaser P, Goffeau A, Golightly EJ, Grandi G, Guiseppi G, Guy BJ, Haga K, et al. 1997. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.