

Abstract
Renal interstitial fibrosis is a common pathological feature of chronic kidney disease that may involve changes of metabolism in kidney cells. In the present study, we first showed that blockade of glycolysis with either dichloroacetate (DCA) or shikonin to target different glycolytic enzymes reduced renal fibrosis in a mouse model of unilateral ureteral obstruction (UUO). Both inhibitors evidently suppressed the induction of fibronectin and collagen type I in obstructed kidneys, with DCA also showing inhibitory effects on collagen type IV and α-smooth muscle actin (α-SMA). Histological examination also confirmed less collagen deposition in DCA-treated kidneys. Both DCA and shikonin significantly inhibited renal tubular apoptosis but not interstitial apoptosis in UUO. Macrophage infiltration after UUO injury was also suppressed. Shikonin, but not DCA, caused obvious animal weight loss during UUO. To determine whether shikonin and DCA worked on tubular cells and/or fibroblasts, we tested their effects on cultured renal proximal tubular BUMPT cells and renal NRK-49F fibroblasts during hypoxia or transforming growth factor-β1 treatment. Although both inhibitors reduced fibronectin and α-SMA production in NRK-49F cells during hypoxia or transforming growth factor-β1 treatment, they did not suppress fibronectin and α-SMA expression in BUMPT cells. Altogether, these results demonstrate the inhibitory effect of glycolysis inhibitors on renal interstitial fibrosis. In this regard, DCA is more potent for fibrosis inhibition and less toxic to animals than shikonin.
Keywords: kidney fibrosis, glycolysis, hypoxia, metabolism, transforming growth factor-β
INTRODUCTION
Chronic kidney disease (CKD) is one of the most common kidney diseases and affects more than 10% of the population in the United States. CKD may progress into end-stage kidney disease that requires dialysis or kidney transplantation for patient survival. The heavy social burden for CKD treatment makes it a pressing need for new therapeutics to prevent or delay CKD progression. CKD is characterized by glomerular and tubulointerstitial pathologies, including tubulointerstitial fibrosis. Previous studies have indicated the importance of both tubular epithelial cells and renal fibroblasts in interstitial fibrosis (11, 13, 20, 28, 30, 35). Tubular epithelial injury may initiate and promote renal fibrosis by secreting fibrogenic factors and stimulating chronic inflammation. Meanwhile, the activation and proliferation of myofibroblasts further drive the progression of fibrosis. Recently, emerging evidence from both patients with CKD as well as animal models indicates that there is a shift of metabolism from mitochondrial oxidative phosphorylation to aerobic glycolysis (Warburg effect) in kidneys (7, 16, 21, 37). The Warburg effect has been shown to promote fibrogenesis in several other organs, such as the liver and heart (2, 12, 15, 17, 19, 36). In a very recent study, Ding et al. (7) suggested that glycolysis may promote renal fibrosis by stimulating fibroblast activation and proliferation. However, whether glycolysis affects only fibroblasts or both renal tubular epithelial cells and fibroblasts in tubulointerstitial renal fibrosis is not clear.
Several pharmacological inhibitors of glycolysis have been tested in kidney disease models. They target different steps of glycolysis and may have different effects on the disease pathogenesis. Ding et al. (7) administered shikonin and 2-deoxyglucose to inhibit glycolysis to show inhibitory effects on renal fibrosis in obstructed kidneys. 2-Deoxyglucose was also reported to reduce the progression of polycystic kidney disease, although dichloroacetate (DCA; another glycolysis inhibitor) enhanced the progression of polycystic kidney disease (6, 10). Different from other glycolysis inhibitors, DCA inhibits glycolysis by switching the energy production pathway to oxidative phosphorylation and, as a result, may improve the overall energy production in cells. DCA was tested in the model of cisplatin nephrotoxicity and showed a renoprotective effect (9, 26). It is unknown whether DCA has a similar inhibitory effect on renal fibrosis as shikonin does.
To examine the function of glycolysis in renal fibrosis and compare the effects of different glycolysis inhibitors, we tested shikonin and DCA in renal fibrosis models both in vivo and in vitro. In the present study, we demonstrated significant inhibitory effects of both shikonin and DCA on renal fibrosis, tubular apoptosis, and inflammation in ureter- obstructed mouse kidneys. In this regard, DCA appears to have better efficacy and lower toxicity. We further showed that these glycolysis inhibitors have divergent effects on renal fibroblasts and renal tubular cells.
MATERIALS AND METHODS
Animals and unilateral ureteral obstruction.
C57BL/6J mice were originally purchased from Jackson Laboratory (Bar Harbor, ME) and maintained in the animal facility of Charlie Norwood Veterans Affairs (VA) Medical Center. Male mice aged 2–3 mo were used for unilateral ureteral obstruction (UUO) surgery. Briefly, mice were anesthetized with 60 mg/kg pentobarbital sodium and kept on a heating pad to maintain body temperature at 36.5°C. The left ureter was exposed by flank incision, and ligations with 5-0 silk suture were made at two points of the ureter. The ureter was then cut between the ligation points to ensure the blockade. All animal experiments were conducted according to the protocol approved by the Institutional Animal Care and Use Committee of Charlie Norwood VA Medical Center.
Cellular fibrosis models.
The mouse proximal tubular cell line (BUMPT) was originally obtained from Dr. Lieberthal and Dr. Schwartz (Boston University) (33). The rat kidney fibroblast cell line (NRK-49F) was purchased from the American Type Culture Collection (Manassas, VA). Cells were cultured in DMEM with 10% FBS. For the transforming growth factor (TGF)-β1-induced fibrosis model, cells were incubated in serum-free culture medium with or without 10 ng/ml recombinant human TGF-β1 (Millipore, Burlington, MA) for 24 h. For the hypoxia-induced fibrosis model, cells were incubated under 1% O2 or normal O2 conditions for 24 h in full cell culture medium.
Glycolysis inhibitor treatment.
Shikonin (Sigma-Aldrich, St. Louis, MO) was delivered to mice by intraperitoneal injection at a dosage of 5 mg·kg−1·day−1 every other day to compare with vehicle (5% DMSO in PBS)-treated mice. For cell treatment, 0.25 μM shikonin was used to inhibit glycolysis in the TGF-β1-induced fibrosis model and 0.5 μM shikonin was used in the hypoxia-induced fibrosis model. Sodium DCA (Sigma-Aldrich) was delivered to mice by intraperitoneal injection at a dosage of 250 mg·kg−1·day−1 daily to compare with saline-treated mice. For cell treatment, 20 mM DCA was used to inhibit glycolysis in the TGF-β1-induced fibrosis model, and 40 mM DCA was used in the hypoxia-induced fibrosis model.
Immunoblot analysis.
Mouse kidneys were collected at the end of experiments, frozen in liquid N2, and stored at −80°C. The frozen kidney tissues were blended and lysed in SDS lysis buffer [62.5 mM Tris·HCl (pH 6.8) 2% SDS, and 10% glycerol] with a Bullet Blender Storm homogenizer (Next Advance, Troy, NY). Cells were directly lysed with SDS lysis buffer in culture dishes. The protein concentration of the lysates was measured using a Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). Samples of equal protein amount were loaded for SDS-PAGE and transferred to a PVDF membrane. After being blocked in 5% milk, the membrane was incubated with primary antibody at 4°C overnight. After the membrane had been washed and incubated in horseradish peroxidase-labeled secondary antibody, the signal was developed using Clarity Western ECL Substrate (Bio-Rad, Hercules, CA) and read by MyECL imager (Thermo Scientific).
Masson’s trichrome staining.
Mouse kidneys were collected at the end of experiments and fixed in 4% paraformaldehyde overnight. Fixed kidney samples were processed for paraffin embedding by the histology facility of Augusta University (Augusta, GA). Paraffin-embedded mouse kidney sections were rehydrated and stained for collagen fibers with a Trichrome Stain (Masson) Kit (Sigma-Aldrich) according to the manufacturer’s instructions.
TUNEL assay.
Paraffin-embedded mouse kidney sections were rehydrated and stained for apoptotic cells with the In Situ Cell Death Detection Kit, TMR red (Sigma-Aldrich) according to the manufacturer’s instructions.
Immunohistochemical staining.
Paraffin-embedded mouse kidney sections were rehydrated and steamed in 10 mM sodium citrate (pH 6.0) with 0.05% Tween 20 for antigen retrieval. After being blocked, sections were incubated with primary antibody at 4°C overnight followed by horseradish peroxidase polymer-labeled anti-rabbit secondary antibody incubation (DAKO Envision+ system, Agilent Technologies, Santa Clara, CA). For immunohistochemical staining of macrophages, sections were incubated with biotin-labeled secondary antibody, and the signal was amplified with the TSA Biotin System (Perkin-Elmer, Akron, OH) and VECTASTAIN Elite ABC HRP Kit (Vector Laboratories, Burlingame, CA). Finally, the signal was developed by the DAB Peroxidase (HRP) Substrate Kit from Vector Laboratories.
Statistics.
All data are expressed as means ± SD. Adobe Photoshop CS6 was used for the densitometry analysis of immunoblots. Student’s t-test was used for comparison between two groups of data. Microsoft Excel 2017 was used for data analysis.
RESULTS
DCA and shikonin reduce renal fibrosis in unilateral ureteral obstruction.
Two separate glycolysis inhibitors, DCA and shikonin, were administered to mice subjected to UUO. At 7 days of UUO, significant renal fibrosis was induced in obstructed kidneys, as shown by the immunoblots of fibrotic factors including collagen type I, collagen type IV, α-smooth muscle actin (α-SMA), fibronectin, and vimentin (Figs. 1A and 2A). DCA (Fig. 1A) or shikonin (Fig. 2A) treatment ameliorated the induction of multiple fibrotic factors. As indicated by the immunoblot and densitometry analyses of whole kidney lysates, 250 mg·kg−1·day−1 DCA treatment significantly inhibited the increase of collagen type I, collagen type IV, fibronectin, and α-SMA in obstructed kidneys (Fig. 1, B–E). However, the inhibition of vimentin by DCA was not obvious (Fig. 1F). With shikonin treatment, the immunoblot and densitometry analyses of whole kidney lysates showed significant suppression of collagen type I and fibronectin expression (Fig. 2, B and C), although no obvious inhibition of collagen type IV, α-SMA, and vimentin was detected (Fig. 2, D–F). The suppression of interstitial collagen type I and fibronectin by DCA and shikonin in obstructed kidneys was also confirmed by immunohistochemical staining (Supplemental Figs. S1 and S2, available online at https://doi.org/10.6084/m9.figshare.7868636.v1). Meanwhile, only DCA showed an inhibitory effect on collagen type IV deposition (Supplemental Fig. S3, available online at https://doi.org/10.6084/m9.figshare.7868636.v1).
Fig. 1.

Dichloroacetate (DCA) ameliorates the induction of multiple fibrotic markers in unilateral ureteral obstruction (UUO)-injured kidneys. C57BL/6 male mice were treated with PBS or DCA and subjected to UUO for 7 days. Kidneys were collected to extract whole kidney lysate and examined by immunoblot analysis. protein expression levels from immunoblots were quantified by densitometry analysis and normalized with cyclophilin B expression. Data are expressed as means ± SD; n is the repetition number of parallel experiments. A: representative immunoblot images showing protein level changes of collagen type IV, fibronectin, collagen type I, vimentin, and α-smooth muscle actin (α-SMA). The matched cyclophilin B immunoblots indicate the protein loading. con, Contralateral kidneys; UUO7D, kidneys with 7-day UUO injury. B: densitometry analysis of the collagen type IV level (n = 5, *P = 0.016). C: densitometry analysis of the fibronectin level (n = 4, *P = 0.048). D: densitometry analysis of the collagen type I level (n = 5, *P = 0.041). E: densitometry analysis of the α-SMA level (n = 5, *P = 0.049). F: densitometry analysis of the vimentin level (n = 4).
Fig. 2.

Shikonin ameliorates the induction of multiple fibrotic markers in unilateral ureteral obstruction (UUO)-injured kidney. C57BL/6 male mice were treated with vehicle solution (VEH) or shikonin (SK) and subjected to UUO for 7 days. Kidneys were collected to extract whole kidney lysate and examined by immunoblot analysis. Protein expression levels from immunoblots were quantified by densitometry analysis and normalized with cyclophilin B expression. Data are expressed as means ± SD; n is the repetition number of parallel experiments. A: representative immunoblot images showing protein level changes of collagen type I, fibronectin, α-smooth muscle actin (α-SMA), vimentin, and collagen type IV. The matched cyclophilin B immunoblots indicate protein loading. con, Contralateral kidneys; UUO7D, kidneys with 7-day UUO injury. B: densitometry analysis of the collagen type I level (n = 5, *P = 0.005). C: densitometry analysis of the fibronectin level (n = 5, *P = 0.040). D: densitometry analysis of the collagen type IV level (n = 5). E: densitometry analysis of the vimentin level (n = 5). F: densitometry analysis of the α-SMA level (n = 5).
Furthermore, we examined the overall collagen deposition in kidneys by Masson’s trichrome staining (Fig. 3). In contralateral control kidneys, the collagen signal (blue color) localized mainly in glomeruli or around blood vessels with minimum interstitial staining. In obstructed kidneys, collagen deposition significantly increased, as shown by extensive collagen deposition in the renal interstitium (Fig. 3, A and C). With DCA treatment, the collagen deposition was significantly suppressed, as indicated by the semiquantitative analysis of the collagen deposition area (Fig. 3B). It was consistent with the inhibition of total collagen type I and IV shown by immunoblot analysis (Fig. 1, B and D). However, no obvious difference of total collagen deposition was detected after shikonin treatment by quantification (Fig. 3D). This was probably because shikonin only inhibited collagen type I, not collagen type IV, accumulation (Fig. 3, B and D, and Supplemental Figs. S1 and S3). The results indicate that both DCA and shikonin can ameliorate UUO-induced renal fibrosis; however, DCA was more efficacious than shikonin at the dosages used.
Fig. 3.

Dichloroacetate (DCA) reduces interstitial collagen deposition in unilateral ureteral obstruction (UUO)-injured kidneys. C57BL/6 male mice were treated with PBS versus DCA or vehicle solution versus shikonin and subjected to UUO for 7 days. Kidneys were fixed for paraffin-embedded sections. Kidneys were stained by Masson’s trichrome staining to detect collagen deposition. The percent area with collagen deposition in the whole kidney was analyzed by ImageJ. Data are expressed as means ± SD; n is the repetition number of parallel experiments. A: representative images of PBS- or DCA-treated kidneys by Masson’s trichrome staining. CON, contralateral kidneys; UUO7D, kidneys with 7-day UUO injury. Scale bar = 0.2 mm. B: collagen deposition area percentage in PBS- or DCA-treated kidneys after 7-day UUO injury (n = 4, *P = 0.026). C: representative images of vehicle solution- or shikonin-treated kidneys by Masson’s trichrome staining. Scale bar = 0.2 mm. D: collagen deposition area percentage in vehicle solution- or shikonin-treated kidneys after 7-day UUO injury (n = 4).
DCA and shikonin suppress renal tubular apoptosis in UUO.
In UUO, the induction of renal cell apoptosis is one of the major pathological injuries to initiate fibrosis development (3, 5). Suppression of renal apoptosis may also ameliorate fibrosis in UUO. Thus, we examined apoptosis in kidneys by TUNEL assay in mice. In contralateral kidneys, few renal apoptotic cells were detected and DCA did not induce obvious apoptosis (Fig. 4A). With shikonin treatment, there was slightly more (but statistically insignificant) interstitial apoptosis (Fig. 4C). Ureter obstruction significantly induced renal apoptosis (Fig. 4, A and C). By examination of the overlay images with red fluorescence signal (apoptosis) and green fluorescence signal (renal structure autofluorescence), we quantified the changes of both renal tubular apoptosis and interstitial apoptosis after UUO (Supplemental Fig. S4, available online at https://doi.org/10.6084/m9.figshare.7868636.v1). The results indicate that DCA or shikonin treatment significantly repressed the renal tubular apoptosis, whereas interstitial apoptosis was unaffected (Fig. 4, B and D).
Fig. 4.

Both dichloroacetate (DCA) and shikonin reduce renal cell apoptosis in unilateral ureteral obstruction (UUO)-injured kidneys. C57BL/6 male mice were treated with PBS versus DCA or vehicle solution versus shikonin and subjected to UUO for 7 days. Kidneys were fixed for paraffin-embedded sections. Kidneys were stained by a TUNEL assay to detect apoptosis. Data are expressed as means ± SD; n is the repetition number of parallel experiments. A: representative images of PBS- or DCA-treated kidneys by TUNEL staining. CON, contralateral kidneys; UUO7D, kidneys with 7-day UUO injury. Scale bar = 0.2 mm. B: TUNEL-positive cells counted in PBS- or DCA-treated kidneys after 7-day UUO injury (n = 5, *P = 0.027 and ΔP = 0.043). C: representative images of vehicle solution- or shikonin-treated kidneys by TUNEL staining. Scale bar = 0.2 mm. D: TUNEL-positive cells counted in vehicle solution- or shikonin-treated kidneys after 7-day UUO injury (n = 5, *P = 0.040 and ΔP = 0.036).
DCA and shikonin inhibit macrophage infiltration in UUO.
After tubular injury in UUO, macrophages are activated and infiltrate into the renal interstitium, promoting cytokine secretion and renal fibrosis development (5). In contralateral kidneys, little macrophage infiltration was detected, but extensive macrophage infiltration was detected at 7 days after UUO (Fig. 5). DCA treatment substantially suppressed macrophage infiltration into the kidney (Fig. 5A). Shikonin treatment also reduced renal macrophage infiltration but with a relatively milder effect (Fig. 5B).
Fig. 5.

Dichloroacetate (DCA) and shikonin reduce interstitial macrophage infiltration in unilateral ureteral obstruction (UUO)-injured kidneys. C57BL/6 male mice were treated with PBS versus DCA or vehicle solution versus shikonin and subjected to UUO for 7 days. Kidneys were fixed for paraffin-embedded sections and stained for macrophages by immunohistochemistry. A: representative fibronectin images of PBS- or DCA-treated kidneys. CON, contralateral kidneys; UUO7D, kidneys with 7-day UUO injury. Scale bar = 0.2 mm. B: representative fibronectin images of vehicle solution- or shikonin-treated kidneys. Scale bar = 0.2 mm.
Shikonin, but not DCA, induces obvious weight loss.
In UUO mice, we also noticed that sustained shikonin treatment led to an obvious loss of body weight (Fig. 6). UUO usually caused a mild animal weight loss of 5–6% at the first 3 days postsurgery, but the weight loss was fully recovered at 5–7 days postsurgery (Fig. 6 and data not shown). DCA administration did not obviously affect the animal weight change pattern from the surgery (Fig. 6A). However, with shikonin treatment, significantly more animal weight loss was detected compared with vehicle-treated groups starting at 3 days after UUO (Fig. 6B), and this weight loss did not recover even at 7 days after UUO (data not shown).
Fig. 6.
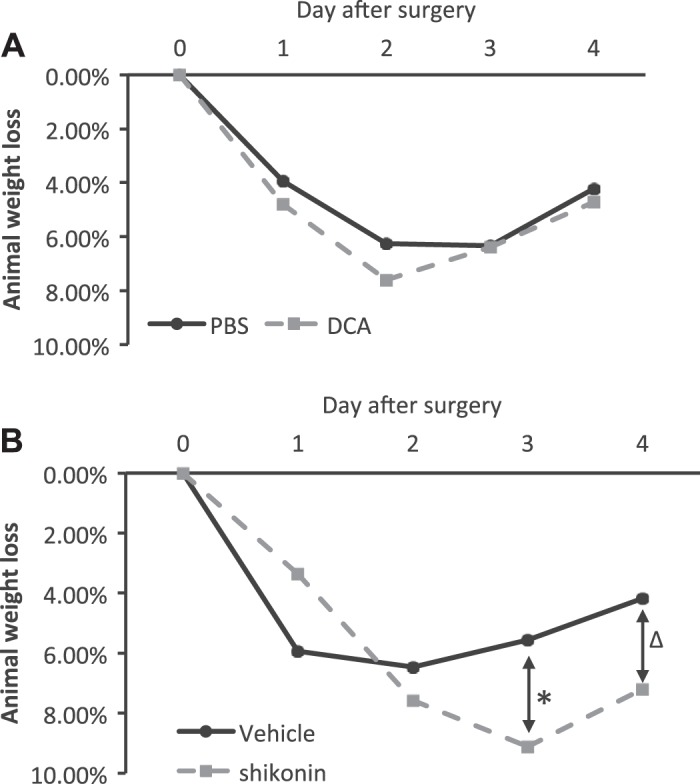
Sustained shikonin treatment, but not dichloroacetate (DCA) treatment, results in severe animal weight loss. C57BL/6 mice were treated with PBS versus DCA or vehicle solution versus shikonin and subjected to unilateral ureteral obstruction surgery. Animal weights were recorded daily and compared with their body weights before surgery to calculate weight loss percentages. Data are expressed as means; n is the number of mice. A: animal weight loss percentage for PBS- and DCA-treated mice. B: animal weight loss percentage for vehicle- and shikonin-treated mice. n = 7 for the vehicle-treated group and n = 8 for the shikonin-treated group, *P = 0.003; n = 4 for the vehicle-treated group and n = 5 for the shikonin-treated group, ΔP = 0.039.
DCA and shikonin show divergent effects on fibrotic changes of proximal tubular cells and renal fibroblasts.
Fibrotic changes of both renal fibroblasts and tubular cells contribute to renal fibrogenesis. To examine whether glycolysis inhibitors affect renal fibroblasts and/or tubular cells, we determined the effects of shikonin and DCA on fibrotic changes in cultured renal proximal tubular cells and renal fibroblast cells. To induce fibrotic changes, we treated renal proximal tubular BUMPT cells and renal fibroblast NRK-49F cells with hypoxia (1% O2) or 10 ng/ml TGF-β1. In NRK-49F cells, hypoxia or TGF-β1 treatment obviously induced fibronectin and α-SMA expression (Figs. 7A and 8A). Interestingly, the induction of fibronectin and α-SMA was significantly suppressed by shikonin, which is consistent with the in vivo inhibitory effect of shikonin on renal fibrosis (Fig. 7, A, C, and E, and Fig. 8, A, C, and E). In BUMPT cells, both hypoxia and TGF-β1 significantly induced fibronectin expression (Figs. 7A and 8A). Although BUMPT cells also expressed low levels of α-SMA, we did not detect any obvious change during hypoxia or TGF-β1 treatment (Figs. 7D and 8D). Notably, in BUMPT cells, shikonin did not inhibit fibronectin or α-SMA expression during hypoxia or TGF-β1 treatment (Fig. 7, B and D, and Fig. 8, B and D); instead, shikonin induced obvious increases of α-SMA during TGF-β1 treatment and fibronectin during hypoxia treatment in BUMPT cells (Figs. 7B and 8D).
Fig. 7.

Divergent effects of shikonin (SK) on hypoxia-induced fibrotic changes in renal epithelial cells and fibroblasts. Renal proximal tubular cells (BUMPT) or fibroblasts (NRK-49F) were treated with or without 0.5 μM SK. Cells were subjected to 1% O2 treatment for 24 h to induce fibrosis. Whole cell lysates were collected and examined by immunoblot analysis. Protein expression levels from immunoblots were quantified by densitometry analysis and normalized with cyclophilin B expression. Data are expressed as means ± SD; n is the repetition number of parallel experiments. A: representative immunoblot images showing protein level changes of fibronectin and α-smooth muscle actin (α-SMA) with matched cyclophilin B blots as an internal marker for protein loading. H, hypoxia (24-h treatment); N, normoxia (24-h treatment). B: densitometry analysis of the fibronectin level in BUMPT cells. n = 4, *P = 0.019. C: densitometry analysis of the fibronectin level in NRK-49F cells. n = 4, *P = 0.013. D: densitometry analysis of the α-SMA level in BUMPT cells. E: densitometry analysis of the α-SMA level in NRK-49F cells. n = 3, *P = 0.010.
Fig. 8.

Divergent effects of shikonin (SK) on transforming growth factor (TGF)-β1-induced fibrotic changes in renal fibroblasts (NRK-49F) and proximal tubular cells (BUMPT) cells. BUMPT cells or NRK-49F cells were treated with or without 0.25 μM SK. Cells were exposed to 10 ng/ml TGF-β1 for 24 h to induce fibrosis. Whole cell lysates were collected and examined by immunoblot analysis. Protein expression levels from immunoblots were quantified by densitometry analysis and normalized with cyclophilin B expression. Data are expressed as means ± SD; n is the repetition number of parallel experiments. A: representative immunoblot images showing protein level changes of fibronectin and α-smooth muscle actin (α-SMA) with matched cyclophilin B as an internal marker for protein loading. CON, control. B: densitometry analysis of the fibronectin level in BUMPT cells. C: densitometry analysis of the fibronectin level in NRK-49F cells. n = 3, *P = 0.026. D: densitometry analysis of the α-SMA level in BUMPT cells. n = 3, *P = 0.029. E: densitometry analysis of the α-SMA level in NRK-49F cells. n = 3, *P = 0.028.
To confirm whether the divergent effects of shikonin on BUMPT and NRK-49F cells were due to glycolysis inhibition, we further tested the effects of DCA (Figs. 9 and 10). Similarly, in NRK-49F cells, DCA significantly suppressed fibronectin and α-SMA expression during hypoxia (Fig. 9, C and E) and TGF-β1 treatments (Fig. 10, C and E). DCA also marginally induced fibronectin in BUMPT cells (Figs. 9B and 10B). Additionally, although the overall α-SMA level was not high in BUMPT cells, it was significantly induced by DCA (Figs. 9D and 10D).
Fig. 9.

Divergent effects of dichloroacetate (DCA) on hypoxia-induced fibrotic changes in renal epithelial cells and fibroblasts. Renal proximal tubular cells (BUMPT) or fibroblasts (NRK-49F) were treated with or without 40 mM DCA. Cells were subjected to 1% O2 treatment for 24 h to induce fibrosis. Whole cell lysates were collected and examined by immunoblot analysis. Protein expression levels from immunoblots were quantified by densitometry analysis and normalized with cyclophilin B expression. Data are expressed as means ± SD; n is the repetition number of parallel experiments. A: representative immunoblot images showing protein level changes of fibronectin and α-smooth muscle actin (α-SMA) with matched cyclophilin B as an internal marker for loading control. H, hypoxia (24-h treatment); N, normoxia (24-h treatment). B: densitometry analysis of the fibronectin level in BUMPT cells. C: densitometry analysis of the fibronectin level in NRK-49F cells. n = 4, *P = 0.019. D: densitometry analysis of the α-SMA level in BUMPT cells. n = 4, *P = 0.016. E: densitometry analysis of the α-SMA level in NRK-49F cells. n = 4, *P = 0.037.
Fig. 10.

Divergent effects of dichloroacetate (DCA) on transforming growth factor (TGF)-β1-induced fibrotic changes in renal fibroblasts and renal proximal tubular cells (BUMPT) cells. BUMPT cells or renal fibroblasts (NRK-49F) cells were treated with or without 20 mM DCA. Cells were exposed to 10 ng/ml TGF-β1 for 24 h to induce fibrosis. Whole cell lysates were collected and examined by immunoblot analysis. Protein expression levels from immunoblots were quantified by densitometry analysis and normalized with cyclophilin B expression. Data are expressed as means ± SD; n is the repetition number of parallel experiments. A: representative immunoblot images showing protein level changes of fibronectin and α-smooth muscle actin (α-SMA) with matched cyclophilin B as an internal marker for loading control. CON, control. B: densitometry analysis of the fibronectin level in BUMPT cells. C: densitometry analysis of the fibronectin level in NRK-49F cells. n = 4, **P = 0.019. D: densitometry analysis of the α-SMA level in BUMPT cells. n = 4, **P = 0.016. E: densitometry analysis of the α-SMA level in NRK-49F cells. n = 4, *P = 0.037.
DISCUSSION
In the present study, we showed that both shikonin and DCA significantly suppressed renal interstitial fibrosis, tubular apoptosis, and inflammation in the mouse model of UUO, suggesting a pathogenic role of glycolysis. In this regard, DCA showed better efficacy and less toxicity as indicated by more obvious inhibition of collagen deposition, less macrophage infilatration, and less animal weight loss. Interestingly, while both inhibitors reduced fibronectin and α-SMA production during hypoxia or TGF-β1 treatment of NRK-49F cells, they did not suppress fibronectin and α-SMA expression in BUMPT cells and inhibited renal tubular apoptosis instead of interstitial apoptosis. This suggests divergent effects of glycolysis on renal tubular cells and fibroblasts in renal injury and interstitial fibrosis.
By examination of the effect of glycolysis inhibitors (shikonin and DCA) in UUO, we confirmed the profibrotic role of glycolysis. This conclusion is consistent with a recent report by Ding et al. (7) supporting the therapeutic use of glycolysis inhibitors in related kidney diseases. Currently, the Warburg effect is intensively studied in cancer research, and glycolysis inhibition has been found effective for cancer treatment (18, 31). Multiple glycolysis inhibitors have been tested clinically and preclinically for cancer therapy (8, 14, 29, 32). The test of glycolysis inhibitors in kidney disease models may expand their potential medical application. Of note, both the study of Ding et al. (7) and our study used UUO as the in vivo model. Further work is warranted to investigate the role of glycolysis in renal fibrosis in other relevant kidney disease conditions (1). Additionally, both studies tested pharmacological inhibitors of glycolysis, and it is important to determine the role of glycolysis and pinpoint the cell type-specific regulation by using gene knockout models.
Although both Ding et al. (7) and our cohort demonstrated the inhibitory effects of glycolysis inhibitors on renal fibrosis, there are slight differences between the results from these two studies. For example, we detected only the inhibition of collagen type I and fibronectin by shikonin treatment but no obvious suppression of collagen type IV, α-SMA, vimentin, and total collagen staining (Figs. 2 and 3B), whereas Ding et al. (7) showed inhibitory effects on α-SMA and total collagen suppression. This discrepancy was likely caused by the dosage difference of shikonin tested. We used a lower dosage because shikonin induced obvious animal weight loss at higher doses (Fig. 6B). The reason for the toxicity caused by shikonin is unclear. We did not notice any obvious increase of inflammation because macrophage infiltration in contralateral kidneys was not changed by shikonin treatment (Fig. 5B). It is also unlikely that liver function was impaired because shikonin treatment has been reported not to significantly affect liver function at a higher dosage (22). In addition to shikonin, DCA inhibited the expression of most of the fibrotic markers except vimentin (Figs. 1 and 3A, and Supplemental Figs. S1–S3) without inducing obvious weight loss in mice (Fig. 6A). Thus, we examined the effects of shikonin and DCA and Ding et al. (7) examined shikonin and 2-deoxyglucose, but the results of these two studies are generally consistent in supporting a role of glycolysis in renal fibrosis.
We also noticed some differences between shikonin and DCA in their inhibitory effects on renal fibrosis in UUO. DCA treatment significantly inhibited collagen type I, collagen type IV, fibronectin, and α-SMA expression in obstructed kidneys (Fig. 1, B–E, and Supplemental Figs. S1–S3) and led to obvious suppression of overall collagen deposition in kidney (Fig. 3, A and B), whereas shikonin only showed obvious effects on collagen type I and fibronectin suppression (Fig. 2, B and C, and Supplemental Figs. S1 and S2). One possible reason is that at the dosage we used shikonin may not suppress glycolysis as effectively as DCA treatment; however, we cannot increase shikonin dosage because of the toxicity noticed. Also, mechanistically, shikonin inhibits pyruvate kinase-M2 to block glycolysis, which may lead to a decrease in cellular ATP production (23). In contrast, DCA inhibits pyruvate dehydrogenase kinase and may shift glycolysis to oxidative phosphorylation, which may actually enhance cellular ATP production via mitochondria (34). Although this can be a valid cause of the difference between the observed effects of shikonin and DCA in our study, we also examined a few other potential mechanisms related to renal fibrosis development. Tubular dedifferentiation and TGF-β pathway activation were not significantly changed by either DCA or shikonin treatment, as evidenced by E-cadherin decrease (Supplemental Fig. S5, available online at https://doi.org/10.6084/m9.figshare.7868636.v1) and Smad2/3 induction and phosphorylation (Supplemental Fig. S6, available online at https://doi.org/10.6084/m9.figshare.7868636.v1), respectively. However, we did notice better efficacy of DCA to suppress renal inflammation shown by macrophage infiltration compared with shikonin (Fig. 5).
In addition to fibroblasts, it is known that renal tubular cells, especially those experiencing injury and stress, play a critical role in renal fibrogenesis (4, 11, 13, 24, 25, 35, 38). Injury of proximal tubules is sufficient to initiate renal fibrosis (13). Our previous work also indicates that suppression of renal tubular apoptosis leads to the suppression of renal fibrosis in UUO (24). Tubules may also cross-talk with fibroblasts through the regulation of multiple signaling pathways such as WNT/β-catenin pathway (25, 38). Moreover, tubular injury may promote inflammation and macrophage infiltration, which will further enhance renal fibrosis (5, 27). In the present study, we examined obstructed kidneys by TUNEL and found that the inhibition of renal tubular apoptosis by shikonin or DCA was associated with less fibrosis (Fig. 4). Furthermore, both DCA and shikonin suppressed kidney macrophage infiltration after UUO injury (Fig. 5). Therefore, in addition to the suppression of fibroblast activation and proliferation (7), glycolysis inhibition may reduce fibrosis by reducing tubular injury and renal inflammation.
Our results from cell cultures further revealed the divergent effects of glycolysis inhibitors in fibroblasts and renal tubular cells. First, shikonin and DCA treatment mainly reduced renal tubular apoptosis without an obvious effect on renal interstitial apoptosis (Fig. 4, B and D), indicating the pro-cell death role of glycolysis in tubular cells but not in fibroblasts. Second, glycolysis inhibition exerted different profibrotic effects on fibroblasts and tubular cells. In renal fibroblasts, both shikonin and DCA suppressed the profibrotic changes during hypoxia or TGF-β1 treatment (Figs. 7–10), which is consistent with the results of Ding et al. (7). Importantly, we further showed that proximal tubular cells reacted quite differently to glycolysis inhibition by showing slight increases in the expression of fibrotic genes (Figs. 7–10). These results suggest that glycolysis is essential to fibroblasts in renal fibrosis, but it may reduce fibrotic changes in renal tubular cells. These observations do not distract the important role of renal tubules in fibrogenesis in kidney diseases; rather, they support the complexity of tubular regulation in renal fibrosis. The importance of proximal tubular cells in fibrosis was shown by their initial stimulation of renal fibrosis (11, 13). It is unclear to what extent proximal tubules contribute to extracellular matrix accumulation. However, it is known that proximal tubular cells may regulate fibroblasts through paracrine effects (35). Because of the divergent effects of glycolysis on tubular cells and fibroblasts, it is necessary for further study to distinguish the role of glycolysis in kidney injury and to reveal the underlying mechanisms with cell type-specific transgenic models.
GRANTS
This work was supported partly by the Augusta University Extramural Success Award, National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-058831 and DK-087843, and Department of Veterans Affairs Grant BX000319. Z. Dong is a recipient of the Senior Research Career Scientist award from the Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Q.W. and Z.D. conceived and designed research; Q.W., J.S., and G.D. performed experiments; Q.W., M.Z., Y.H., and Z.D. analyzed data; Q.W. and Z.D. interpreted results of experiments; Q.W. prepared figures; Q.W. drafted manuscript; Q.W., J.S., M.Z., Y.H., and Z.D. edited and revised manuscript; Q.W., J.S., G.D., M.Z., Y.H., and Z.D. approved final version of manuscript.
REFERENCES
- 1.Black LM, Lever JM, Traylor AM, Chen B, Yang Z, Esman SK, Jiang Y, Cutter GR, Boddu R, George JF, Agarwal A. Divergent effects of AKI to CKD models on inflammation and fibrosis. Am J Physiol Renal Physiol 315: F1107–F1118, 2018. doi: 10.1152/ajprenal.00179.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Z, Liu M, Li L, Chen L. Involvement of the Warburg effect in non-tumor diseases processes. J Cell Physiol 233: 2839–2849, 2018. doi: 10.1002/jcp.25998. [DOI] [PubMed] [Google Scholar]
- 3.Chevalier RL. Molecular and cellular pathophysiology of obstructive nephropathy. Pediatr Nephrol 13: 612–619, 1999. doi: 10.1007/s004670050756. [DOI] [PubMed] [Google Scholar]
- 4.Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol 311: F145–F161, 2016. doi: 10.1152/ajprenal.00164.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75: 1145–1152, 2009. doi: 10.1038/ki.2009.86. [DOI] [PubMed] [Google Scholar]
- 6.Chiaravalli M, Rowe I, Mannella V, Quilici G, Canu T, Bianchi V, Gurgone A, Antunes S, D’Adamo P, Esposito A, Musco G, Boletta A. 2-Deoxy-d-glucose ameliorates PKD progression. J Am Soc Nephrol 27: 1958–1969, 2016. doi: 10.1681/ASN.2015030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, Luo J, Zen K, Yang J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol 313: F561–F575, 2017. doi: 10.1152/ajprenal.00036.2017. [DOI] [PubMed] [Google Scholar]
- 8.Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs 32: 452–464, 2014. doi: 10.1007/s10637-013-0047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galgamuwa R, Hardy K, Dahlstrom JE, Blackburn AC, Wium E, Rooke M, Cappello JY, Tummala P, Patel HR, Chuah A, Tian L, McMorrow L, Board PG, Theodoratos A. Dichloroacetate prevents cisplatin-induced nephrotoxicity without compromising cisplatin anticancer properties. J Am Soc Nephrol 27: 3331–3344, 2016. doi: 10.1681/ASN.2015070827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gattone VH II, Bacallao RL. Dichloroacetate treatment accelerates the development of pathology in rodent autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol 307: F1144–F1148, 2014. doi: 10.1152/ajprenal.00009.2014. [DOI] [PubMed] [Google Scholar]
- 11.Gewin LS. Renal fibrosis: primacy of the proximal tubule. Matrix Biol 68–69: 248–262, 2018. doi: 10.1016/j.matbio.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodwin J, Choi H, Hsieh MH, Neugent ML, Ahn JM, Hayenga HN, Singh PK, Shackelford DB, Lee IK, Shulaev V, Dhar S, Takeda N, Kim JW. Targeting hypoxia-inducible factor-1α/pyruvate dehydrogenase kinase 1 axis by dichloroacetate suppresses bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 58: 216–231, 2018. doi: 10.1165/rcmb.2016-0186OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82: 172–183, 2012. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo XP, Zhang XY, Zhang SD. Clinical trial on the effects of shikonin mixture on later stage lung cancer [in Chinese]. Zhong Xi Yi Jie He Za Zhi 11: 598–599, 1991. [PubMed] [Google Scholar]
- 15.Karthikeyan S, Potter JJ, Geschwind JF, Sur S, Hamilton JP, Vogelstein B, Kinzler KW, Mezey E, Ganapathy-Kanniappan S. Deregulation of energy metabolism promotes antifibrotic effects in human hepatic stellate cells and prevents liver fibrosis in a mouse model. Biochem Biophys Res Commun 469: 463–469, 2016. doi: 10.1016/j.bbrc.2015.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, Venkatachalam MA. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 27: 3356–3367, 2016. doi: 10.1681/ASN.2015020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lian N, Jin H, Zhang F, Wu L, Shao J, Lu Y, Zheng S. Curcumin inhibits aerobic glycolysis in hepatic stellate cells associated with activation of adenosine monophosphate-activated protein kinase. IUBMB Life 68: 589–596, 2016. doi: 10.1002/iub.1518. [DOI] [PubMed] [Google Scholar]
- 18.Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41: 211–218, 2016. [Erratum in Trends Biochem Sci 41: 211, 2016.] doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin H, Angeli M, Chung KJ, Ejimadu C, Rosa AR, Lee T. sFRP2 activates Wnt/β-catenin signaling in cardiac fibroblasts: differential roles in cell growth, energy metabolism, and extracellular matrix remodeling. Am J Physiol Cell Physiol 311: C710–C719, 2016. doi: 10.1152/ajpcell.00137.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int 93: 568–579, 2018. doi: 10.1016/j.kint.2017.09.033. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Li W, He Q, Xue J, Wang J, Xiong C, Pu X, Nie Z. Mass spectrometry imaging of kidney tissue sections of rat subjected to unilateral ureteral obstruction. Sci Rep 7: 41954, 2017. doi: 10.1038/srep41954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T, Zhang Q, Mo W, Yu Q, Xu S, Li J, Li S, Feng J, Wu L, Lu X, Zhang R, Li L, Cheng K, Zhou Y, Zhou S, Kong R, Wang F, Dai W, Chen K, Xia Y, Lu J, Zhou Y, Zhao Y, Guo C. The protective effects of shikonin on hepatic ischemia/reperfusion injury are mediated by the activation of the PI3K/Akt pathway. Sci Rep 7: 44785, 2017. doi: 10.1038/srep44785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Sun G. Shikonin enhances Adriamycin antitumor effects by inhibiting efflux pumps in A549 cells. Oncol Lett 14: 4270–4276, 2017. doi: 10.3892/ol.2017.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mei S, Li L, Wei Q, Hao J, Su Y, Mei C, Dong Z. Double knockout of Bax and Bak from kidney proximal tubules reduces unilateral urethral obstruction associated apoptosis and renal interstitial fibrosis. Sci Rep 7: 44892, 2017. doi: 10.1038/srep44892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nlandu-Khodo S, Neelisetty S, Phillips M, Manolopoulou M, Bhave G, May L, Clark PE, Yang H, Fogo AB, Harris RC, Taketo MM, Lee E, Gewin LS. Blocking TGF-β and β-catenin epithelial crosstalk exacerbates CKD. J Am Soc Nephrol 28: 3490–3503, 2017. doi: 10.1681/ASN.2016121351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh CJ, Ha CM, Choi YK, Park S, Choe MS, Jeoung NH, Huh YH, Kim HJ, Kweon HS, Lee JM, Lee SJ, Jeon JH, Harris RA, Park KG, Lee IK. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int 91: 880–895, 2017. doi: 10.1016/j.kint.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 27.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest 118: 3522–3530, 2008. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato Y, Yanagita M. Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflamm Regen 37: 17, 2017. doi: 10.1186/s41232-017-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin Investig Drugs 17: 1533–1545, 2008. doi: 10.1517/13543784.17.10.1533. [DOI] [PubMed] [Google Scholar]
- 30.Shaw I, Rider S, Mullins J, Hughes J, Péault B. Pericytes in the renal vasculature: roles in health and disease. Nat Rev Nephrol 14: 521–534, 2018. doi: 10.1038/s41581-018-0032-4. [DOI] [PubMed] [Google Scholar]
- 31.Sheng H, Tang W. Glycolysis inhibitors for anticancer therapy: a review of recent patents. Recent Patents Anticancer Drug Discov 11: 297–308, 2016. doi: 10.2174/1574892811666160415160104. [DOI] [PubMed] [Google Scholar]
- 32.Shi L, Pan H, Liu Z, Xie J, Han W. Roles of PFKFB3 in cancer. Signal Transduct Target Ther 2: 17044, 2017. doi: 10.1038/sigtrans.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinha D, Wang Z, Price VR, Schwartz JH, Lieberthal W. Chemical anoxia of tubular cells induces activation of c-Src and its translocation to the zonula adherens. Am J Physiol Renal Physiol 284: F488–F497, 2003. doi: 10.1152/ajprenal.00172.2002. [DOI] [PubMed] [Google Scholar]
- 34.Subramani K, Lu S, Warren M, Chu X, Toque HA, Caldwell RW, Diamond MP, Raju R. Mitochondrial targeting by dichloroacetate improves outcome following hemorrhagic shock. Sci Rep 7: 2671, 2017. doi: 10.1038/s41598-017-02495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan RJ, Zhou D, Liu Y. Signaling crosstalk between tubular epithelial cells and interstitial fibroblasts after kidney injury. Kidney Dis (Basel) 2: 136–144, 2016. doi: 10.1159/000446336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM, Bernard K, Thannickal VJ, Liu G. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med 192: 1462–1474, 2015. doi: 10.1164/rccm.201504-0780OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang G, Darshi M, Sharma K. The Warburg effect in diabetic kidney disease. Semin Nephrol 38: 111–120, 2018. doi: 10.1016/j.semnephrol.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou D, Fu H, Zhang L, Zhang K, Min Y, Xiao L, Lin L, Bastacky SI, Liu Y. Tubule-derived Wnts are required for fibroblast activation and kidney fibrosis. J Am Soc Nephrol 28: 2322–2336, 2017. doi: 10.1681/ASN.2016080902. [DOI] [PMC free article] [PubMed] [Google Scholar]